

Abstract
Cancer cells are challenged by a myriad of microenvironmental stresses but their ability to efficiently adapt to the constantly changing nutrient, energy, oxidative and/or immune landscape allows them to survive and proliferate. Such adaptations, however, result in distinct vulnerabilities that are attractive therapeutic targets. PI5P4Ks, a family of phosphoinositide kinases, are druggable stress-regulated kinases that become conditionally essential as a metabolic adaptation, paving the way to targeting cancer cell dependencies. Further, PI5P4Ks have a synthetic lethal interaction with the tumor suppressor p53, the loss of which is one of the most prevalent genetic drivers of malignant transformation in many cancers. PI5P4K’s emergence as a crucial axis in the expanding landscape of phosphoinositide signaling in cancer has already stimulated the development of inhibitors targeting these enzymes. Thus, future investigations to fully understand the functional biology of the PI5P4Ks will allow for targeted and effective therapeutic interventions. Here we will attempt to summarize the mounting roles of the PI5P4Ks in cancer, including evidence that targeting them is a therapeutic vulnerability and promising next in line treatment for multiple cancer subtypes.
Introduction
Phosphoinositides comprise only a small group of phospholipids however are fundamental signaling molecules that are involved in numerous essential cellular processes such as membrane trafficking, cytoskeletal organization, cell survival and proliferation [1]. Understandably, dysregulation of phosphoinositide metabolism is associated with prevalent conditions, such as obesity, type 2 diabetes, neurodegenerative disorders, and cancer [2–4]. Recently, they have also drawn increased attention due to the ability of infectious agents to utilize phosphoinositides for their trafficking, replication, and assembly [5]. Phosphoinositides are derived by the phosphorylation of the third, fourth and fifth positions of the inositol headgroup of phosphatidylinositol (PI), resulting in the generation of seven distinct phosphoinositide derivatives [4]. In eukaryotic cells, these seven phosphoinositide species are interconverted into each other through the precise and spatially regulated activity of phosphoinositide kinases and phosphatases [4]. In fact, the importance of maintaining a tight balance between the activities of the various phosphoinositide-metabolizing enzymes, to control the cellular phosphoinositide pools, is underscored by the diseases, including cancers, that arise from dysregulation. One of the most established phosphoinositide kinases linked to cancer is phosphoinositide 3-kinase (PI3K). PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PI-4,5-P2) to generate phosphatidylinositol-3,4,5-triphosphate (PI-3,4,5-P3) leading to the subsequent activation of Akt and its downstream signaling cascades, which has set the stage for phosphoinositide signaling in cancer research [6]. On the other hand, the tumor suppressor protein, phosphatase and tensin homolog (PTEN) acts as a major inhibitory regulator of the PI3K-Akt oncogenic pathway, by dephosphorylating PI-3,4,5-P3 to PI-4,5-P2, and is thus a common target of inactivation in cancer [7]. Notably PI3K is also one of the most frequently mutated genes in cancer which has led to the emergence of inhibitors targeting PI3K entering the clinic for cancer therapy. Interestingly, the PI3K product PI-3,4,5-P3 is practically nonexistent from quiescent cells but is rapidly stimulated by extracellular factors, whereas PI-4,5-P2 is abundant in cells [8]. PI-4,5-P2 is an established regulator of multiple cellular processes, such as being a precursor to generate the second messengers diacylglycerol and inositol-1,4,5-trisphosphate, signaling, vesicular trafficking, membrane dynamics, gene expression, and importantly there are increasing reports that now connect PI-4,5-P2 signaling to diseases, such as cancer, including leukemia, melanoma, and breast cancer [4, 9]. Yet, there is still a great deal to learn about how PI-4,5-P2 is regulated and in particularly about the enzymes that produce PI-4,5-P2. Besides being generated by the dephosphorylation of PI-3,4,5-P3, there are other pathways for PI-4,5-P2 generation, involving the activity of two divergent families of lipid kinases, which generate the lipid through phosphorylation of monophosphoinositides.
These phosphoinositide kinases that generate PI-4,5-P2 were first purified from erythrocytes in the late 1980s and were separated into two groups, Type I and Type II kinases, based on their biochemical and immunogenic phenotypes [10, 11]. However, in 1997 the Cantley lab made the unexpected finding that the Type II kinases actually made PI-4,5-P2 by phosphorylating the 4-position of phosphatidylinositol 5-phosphate (PI-5-P), a contaminate which was present in the commercial phosphatidylinositol 4-phosphate (PI-4-P) [12]. Not only did this discovery show for the first time that PI-4,5-P2 could be generated from two independent pathways, but most importantly it revealed PI-5-P, which was not known to exist. Further, these groundbreaking observations defined the two pathways that generate PI-4,5-P2 in mammalian cells, as the Type I canonical pathway in which the phosphatidylinositol 4-phosphate 5-kinases (PI4P5Ks) phosphorylate the 5-position of PI-4-P to generate PI-4,5-P2, and the non-canonical pathway where the phosphatidylinositol 5-phosphate 4-kinases (PI5P4Ks) phosphorylate the 4-position of PI-5-P to make PI-4,5-P2. Despite the fact Type I and Type II kinases generate the same lipid product; over the years it is becoming increasingly more apparent that these distinct families have diverse biological and metabolic functions. Further, the cellular location of where these enzyme families produce PI-4,5-P2 is crucial. Whereas the Type I canonical pathway generates PI-4,5-P2 predominantly at the plasma membrane, the Type II non-canonical produced PI-4,5-P2 occurs largely on intracellular membranes, such as lysosomes [13] and peroxisomes [14]. Here in this review, we will focus on the non-canonical PI5P4Ks which are emerging into the limelight as cancer targets especially for cancers with metabolic liabilities.
PI5P4K family
In mammals the PI5P4K family consists of three isoforms (α, β, and γ), and the genes encoding the PI5P4Ks are called PIP4K2A, PIP4K2B, and PIP4K2C [15–18]. The importance of this alternative pathway to generate PI-4,5-P2 is emphasized by the fact that the PI5P4K family is conserved back to Drosophila and C. elegans, each having a single catalytically active PI5P4K that is essential for survival during metabolic stress [13, 18–20].
At the protein level, the PI5P4Kα and β isoforms are 83% identical and PI5P4Kγ is approximately 60% identical to PI5P4Kα and β [21]. The distinct features of their structural organization are a dimerization domain and the kinase domain which includes the catalytic activity residues, substrate binding site and a specificity loop that together determine the preference for PI-5-P binding (Figure 1) [22, 23]. Of the three isoforms, PI5P4Kα is the most catalytically active, displaying significantly more activity compared to PI5P4Kβ, whereas PI5P4Kγ has very little inherent activity [18, 24]. Noteworthy is that these are dimeric enzymes, thus are capable of forming heterodimers with each other which has been speculated to be fundamental for their functional relevance at specific cellular locations, particularly for the less active isoforms [25, 26].
Figure 1. Domain organization of PI5P4Ks.
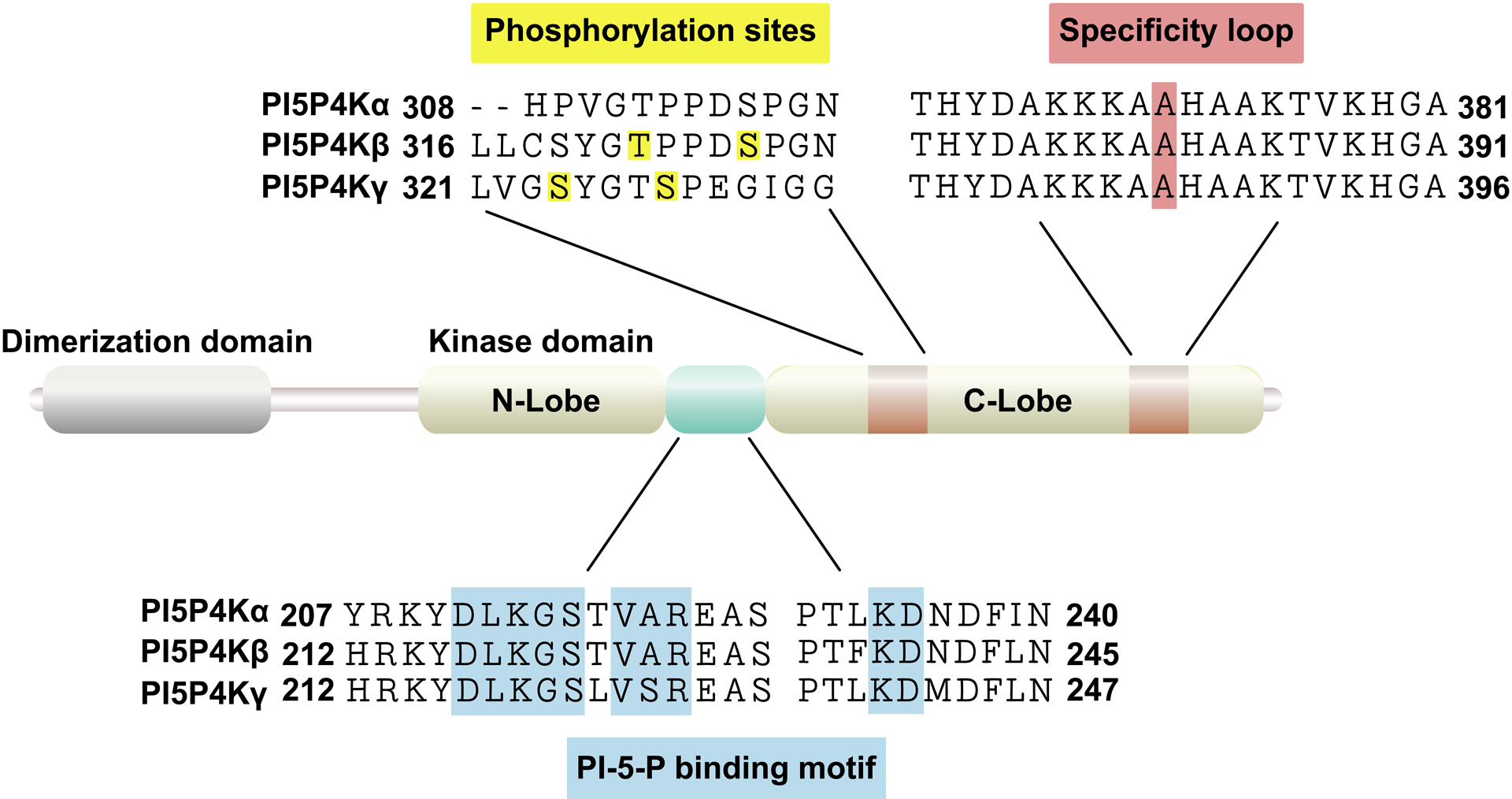
The schematic representation illustrates the similarity between the three isoforms and depicts the dimerization and kinase domain. It also includes the phosphorylation sites on the isoforms for p38 MAP kinase and mTORC1.
All isoforms are ubiquitously expressed. As for the particular subcellular locations and functions of the PI5P4K isoforms, there still remains gaps in our knowledge. However, to date what definitely can be said is that the individual isoforms are indeed present at distinct cellular locations, thus providing a rationalization of why higher organisms evolved to have three isoforms [27]. Endogenous PI5P4Kα has been shown to be in the cytosol and recent overexpression and genetic studies have demonstrated that this isoform can localize to autophagosomes, lysosomes, and peroxisomes [13, 14, 28]. PI5P4Kβ predominantly localizes to the nucleus, yet overexpression experiments have indicated that both the PI5P4Kβ and PI5P4Kγ isoforms are present at autophagosomes [26, 28]. Further, PI5P4Kγ has been shown to be reside within the endomembrane compartment and partially localize with the Golgi [29].
Upstream regulation of the lipid kinases has remained unexplored. The regulation is most likely driven by cellular requirements for the substrate, PI-5-P, or for the product, PI-4,5-P2. Like PI-3,4,5-P3, PI-5-P, the last phosphoinositide to be discovered, is minimal in abundance, however levels are increased upon cellular stress [30–32]. There is some evidence that the increased levels of PI-5-P, under stress, are achieved by inhibitory phosphorylation of PI5P4Kβ by MAP kinase p38 at Ser326 [30] (Figure 1). Further, phosphorylation of the kinases at Ser326 by p38 or at Thr322 by an unidentified kinase (Figure 1) was found to be responsible for the interaction of both PI5P4Kα and PI5P4Kβ with Pin1, allowing for inhibitory regulation of the lipid kinase activity [33] to regulate PI-5-P pools. Pin 1, or peptidyl-prolyl cis/trans isomerase (PPIase), is known to regulate the function of multiple proteins due to its ability to isomerize the Serine/Threonine-Proline motif [34]. To expand on phosphorylation-dependent regulation of PI5P4Ks, there has also been evidence for mTORC1 dependent phosphorylation of PI5P4Kγ at Ser324 and Ser328 [35] (Figure 1) for maintaining optimal activation of mTORC1 through a feedback loop. The amount of PI-4,5-P2 synthesized from PI-5-P is minor compared to the amount generated by the canonical PI4P5Ks, as their lipid substrate PI-4-P is one of the most abundant phosphoinositide. Due to these discrepancies, it was assumed that the PI5P4Ks evolved to solely control PI-5-P levels under stress conditions instead of producing PI-4,5-P2 [30, 33, 36]. However, recent studies have suggested that the specialized PI-4,5-P2 pools generated by the PI5P4Ks by phosphorylating PI-5-P on intracellular membranes in fact are critical for processes, such as peroxisome function, cholesterol trafficking, and tumorigenesis [13, 14, 37]. Furthermore, these findings highlight the importance of PI5P4K activity and the increasing relevance for these enzymes in a variety of pathological processes, including cancer.
PI5P4Ks in cancer
In this section we will focus on recent advances made in elucidating the roles of all the PI5P4K isoforms in the pathogenesis of multiple cancer types (Table 1). PI5P4Ks are not usually mutated in cancers but have been shown to have altered expression in most cancers. Different cancers are characterized with dissimilar cellular and microenvironmental requirements. In this context, changes in expression of PI5P4Ks would be consistent with their role in predominantly acting as an interface between the various intracellular organelles and their environment, making them sensitive to alterations in the demands of a tumor. As an initial glimpse into the expression of PIP4K2A, PIP4K2B and PIP4K2C in different cancers, Figure 2 illustrates comparative expression data of normal versus tumor samples from tools that utilize publicly available datasets in the GTEx portal and The Cancer Genome Atlas (TCGA) [38]. The three isoforms are upregulated in some cancers and downregulated in others. This can be potentially explained by the fact that different cancers are characterized by unique molecular, genetic, metabolic, and immune landscape, which results in personalized requirements and thus in distinct rewiring of gene expression profiles. It is interesting to note that the α and β isoforms have a similar expression profile in multiple cancer types and γ has its own distinct profile, suggesting that they might exhibit different functions in cancers. Given the expression analysis tool does not carefully take into consideration the complexity and heterogeneity of tumors, this information should be considered as an initial guiding platform. In the future, not only are additional datasets required but there is a need for them to be segregated and profiled more appropriately so that valuable information can be obtained through such comparative studies. For PI5P4Ks, in fact, there is evidence suggesting they act both as oncogenes as well as tumor suppressors, and these studies are mostly consistent with the expression profile obtained through the public datasets (Figure 2). Nonetheless, most studies favor oncogenic roles for the PI5P4Ks, and we will discuss some of these studies in details below.
Table 1. PI5P4Ks in cancer.
Summary of the multiple cancer types in which PI5P4Ks have been implicated. Alongside with each cancer type is the information about the associated isoform and a brief description of the evidence, function, and putative mechanism in cancer.
| Cancer Type | Implicated Isoform | Evidence / Potential Mechanism |
|---|---|---|
| Breast | PI5P4Kα, PI5P4Kβ | |
| AML | PI5P4Kα, PI5P4Kγ | |
| ALL | PI5P4Kα | |
| Sarcoma | PI5P4Kα, PI5P4Kβ | |
| Glioblastoma | PI5P4Kα |
Figure 2. Comparative overview of PI5P4K isoform expression in multiple cancers.
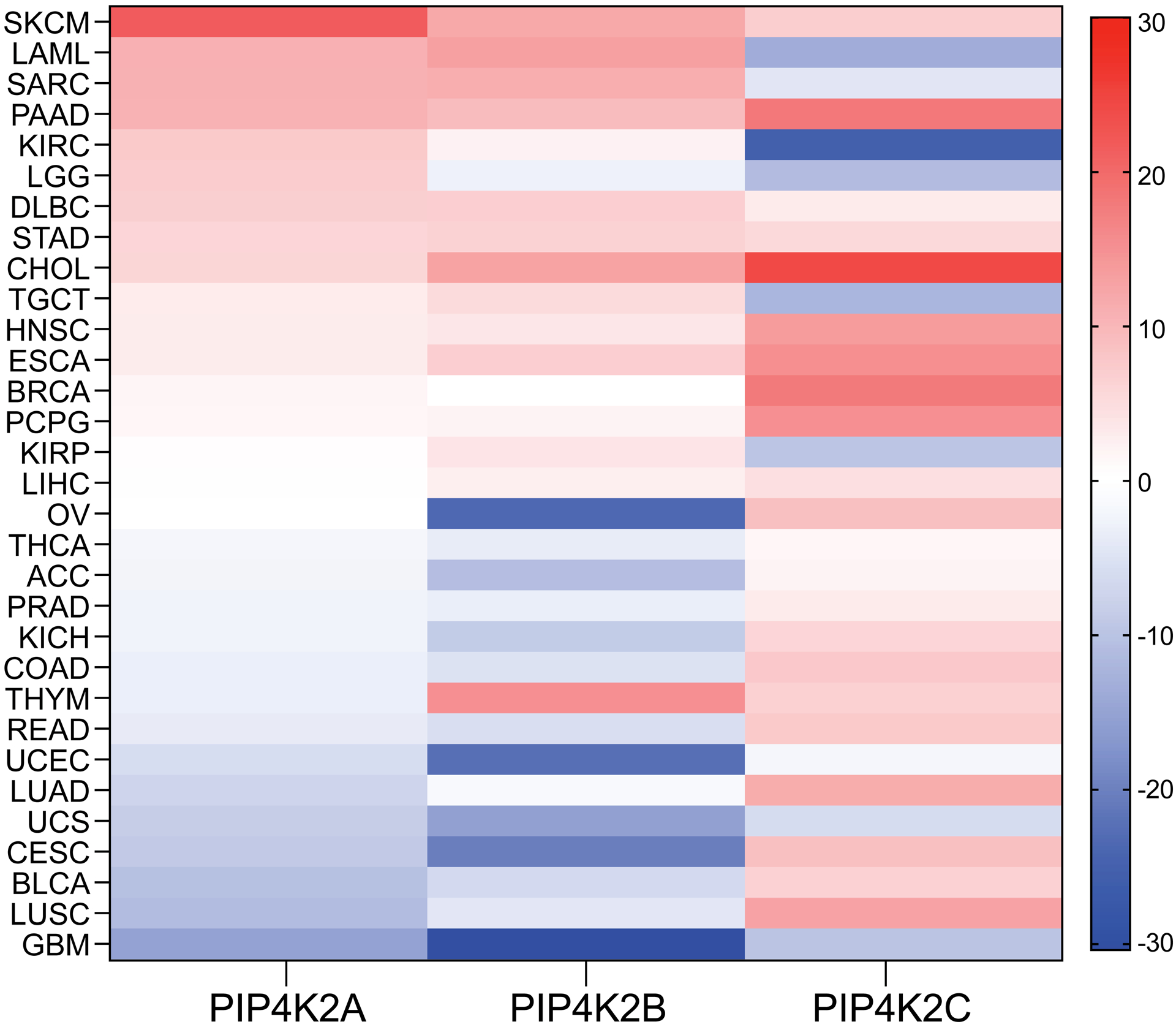
The heat map is organized based on the difference in median expression between the tumor samples (TCGA datasets) and the corresponding normal tissue samples (from GTEx and TCGA datasets[38]) and the cancer types are ranked based on PIP4K2A expression, going from cancers in which it is most upregulated to the ones it is most downregulated. Cancers highlighted in this review are LAML (Acute Myeloid Leukemia), SARC (Sarcoma), BRCA (Breast invasive carcinoma) and GBM (Glioblastoma multiforme).
Breast cancer
Breast cancer is a predominant cause of cancer-related mortality among women worldwide. It is a heterogeneous disease that has been classified into immunohistochemistry-based subtypes based on the expression of estrogen or progesterone receptors and human epidermal growth factor 2 (ERBB2; formerly HER2) amplification. The first study to implicate the PI5P4Ks in cancer and for setting the stage for oncological research was in breast cancer [15]. Elevated expression of both PI5P4Kα and PI5P4Kβ was observed in breast tumors compared with normal breast tissue (Table 1) [15]. The gene encoding PIP4K2B, which is located at 17q21.2, lies in close proximity to the proto-oncogene ERBB2 gene, thus often resulting in co-amplification [15, 39, 40]. However, high expression of PIP4K2B was also observed to be independent of ERRB2 expression levels, raising the possibility of potential benefit of PI5P4Kβ inhibitors to patients that display resistance to HER2 targeted therapy [15].
Remarkably, the same study elucidated a striking co-occurrence between high PIP4K2A expression as well as PIP4K2B amplification with TP53 mutation/deletion. This led to the identification of a synthetic lethal interaction between PI5P4Ks and TP53, an important breakthrough in the search for vulnerabilities to target cancers with p53 loss/mutation. p53 is a tumor suppressor and thus its inactivation is a central genetic driver of malignancies, rendering it difficult to target with drugs. Emerling et al further identified that the unique PI5P4Ks create a synthetic lethality when deleted or inhibited in ERBB2 amplified TP53 mutant breast cancer cells but not in breast cancer cells or normal mammary epithelial cells that express wild-type TP53. This was attributed to increased oxidative stress and suppressed glucose metabolism. Furthermore, the kinase depleted TP53 mutant breast cancer cell line failed to from xenograft tumors in nude mice [15] (Table 2). Importantly, the study went on to utilize the Trp53−/− genetically engineered model (GEM), which forms spontaneous tumors and has a survival span of about 4–6 months. Loss of PI5P4Ks (Pip4k2a−/−, Pip4k2b+/−, Trp53−/−) in this well-established mouse tumor model resulted in a significant increase in tumor-free survival (Table 2) and in embryonic lethality upon loss of Trp53 and Pipk2b [15] (Table 2). Not much progress has been made in the translation of synthetic lethal therapies to the clinic yet, despite the extensive research efforts in this promising nature of cancer treatment. PARP inhibitors, which target the inhibition of DNA repair pathways, have been the only FDA approved drug based on the synthetic lethal approach to target BRCA1/2-mutated tumors [41]. Given that tumors are mostly fueled by multiple mutations, identification of synthetic lethal partners for different drivers can be utilized to develop powerful combination therapies to concurrently target multiple adaptations that are generated by these genomic alterations. This could also help to overcome the commonly faced issues with drug resistance in clinical practice. Why there is a synthetic lethal interaction between p53 and PI5P4Ks is briefly discussed in the next section but is definitely a question that warrants future investigation.
Table 2. In vivo targeting of PI5P4Ks.
Summary of the in vivo evidence of targeting PI5P4Ks in mouse models. The top half of the table comprises the phenotypes of the genetic models of kinase deletion, including germline deletions and the GEM model of sarcoma. The bottom half includes the outcomes of the multiple tumor implantation models in which PI5P4Ks were manipulated.
| Genetic Mouse Models | Implicated Isoform | Phenotype |
|---|---|---|
| Pip4k2a −/− | PI5P4Kα | Normal health and life span [15] |
| Pip4k2b −/− | PI5P4Kβ | Normal life span; retarded growth; Insulin sensitivity [16] |
| Pip4k2c −/− | PI5P4Kγ | Hyperinflammation; increased helper T cells and reduced regulatory T cells [17] |
| Pip4k2a−/− Pip4k2b−/− | PI5P4Kα, PI5P4Kβ | Perinatal lethal [15] |
| Pip4k2b−/− Trp53−/− | PI5P4Kβ | Embryonic lethal [15] |
| Pip4k2a−/− Pip4k2b+/− Trp53−/− | PI5P4Kα, PI5P4Kβ | Increased tumor free survival compared to Trp53−/− [15] |
| KrasLSL-G12D/+ Trp53flx/flx Pip4k2aflx/flx Pip4k2b−/− | PI5P4Kα, PI5P4Kβ | Restricted tumor formation and increased survival compared to KrasLSL-Gi2D/+ Trp53flx/flx [37] |
| Tumor Implantation Models | Implicated Isoform | Phenotype |
| Xenograft | ||
| BT474 Breast cancer p53 mutant cell line | PI5P4Kα, PI5P4Kβ | Impaired tumor growth with kinase knockdown [15] |
| THP1 AML cell line | PI5P4Kα | No evidence of leukemic engraftment with kinase knockdown [47] |
| SV40 transformed MEF | PI5P4Kβ | Decreased tumor growth with GTP insensitive Pip4k2b [61] |
| LN428 GBM cell line | PI5P4Kα | Increased tumor growth with kinase knockdown [53] |
| Orthotopic | ||
| GBM cell line | PI5P4Kα | Decreased tumor growth with kinase overexpression [53] |
| KPS-142 Sarcoma cell line | PI5P4Kα, PI5P4Kβ | Decreased tumor growth with kinase knockdown [37] |
In another recent study by the Emerling lab, the role of PI5P4Ks in breast cancer was expanded to the triple negative breast cancer subtype (TNBC), which uncovered a role of PI5P4Ks in cellular energetics. Downregulation of both the α and β isoforms of PI5P4Ks suppressed the proliferative capacity of multiple TNBC cell lines and exposure to metabolic stress dramatically diminished the cellular viability [37], expanding the critical role of PI5P4Ks in multiple breast cancer subtypes. It will be imperative to investigate a larger panel of TNBC models, given the poor prognosis and limited therapeutic success for this subtype. Also, current studies suggest implication of both the α and β isoforms, making it an important consideration for drug development. Further, in a recent comprehensive analysis of the secretome of breast cancer, the tumor interstitial fluid (TIF) proteome was characterized and identified PI5P4Kβ as one of the 10 proteins that can be used as a reliable biomarker to stratify the breast cancer tumor-subtypes, all of which were also analyzed by immunohistochemistry (IHC) on matched tumor tissue samples [42]. In summary, all of the above data points to clinical relevance of PI5P4Ks in breast cancer and to a promising outcome with the targeting of PI5P4Ks in a broad population of breast cancer patients, particularly including those that harbor altered TP53 status.
Acute leukemias
Acute leukemias are common malignant disorders worldwide and are classified into myeloid or lymphoid according to the predominant lineage of the hematopoietic cell involved. AML (Acute Myeloid Leukemia) is prevalent mostly in adults whereas ALL (Acute Lymphocytic Leukemia), is one of the most common pediatric cancers. PI5P4Kα has been implicated in both of the acute leukemias in multiple studies [43–45] (Table 1). Phosphoinositide signaling plays an important role in hematological malignancies [46] and this motivated a study in which a knockdown based screening of phosphoinositide signaling regulators was performed in AML cells and interestingly PI5P4Kα was found to be required for survival and proliferation of the AML cells [47]. They further demonstrated that PI5P4Kα was important for the proliferation potential of both human as well as mouse AML cells but not for normal hematopoietic stem and progenitor cells [47], indicating that the alpha isoform is essential under rewired signaling of cancer cells. Reassuringly, alpha isoform depleted AML cells did not result in leukemia in the tumor implantation study [47] (Table 2). In vitro, the phenotype was attributed to a mTOR dependent increase in CDKN1A accumulation. High expression of not only PI5P4Kα but also of PI5P4Kγ was found to be associated with unfavorable clinical outcomes for AML patients [43]. Genome-wide association studies have also identified several single nucleotide polymorphisms (SNPs) in the PIP4K2A locus that have been associated with a genetic predisposition in ALL [48–50] however the contribution of the SNPs to PIP4K2A expression or the functional involvement of PI5P4Kα to the leukemogenesis of ALL is still unexplored. Nevertheless, targeting PI5P4Ks in acute leukemias could be a promising treatment either by itself or as a combination therapy [45].
Glioblastoma
Glioblastoma (GBM) is the most prevalent and lethal malignant brain tumor with extremely low survival rate in the range of few months to less than three years [51]. Loss of PTEN is observed in approximately 60% of GBM patients and is associated with poor prognosis and drug resistance. Given the antagonizing role of PTEN to the oncogenic PI3K – Akt signaling, loss of PTEN is associated with increased Akt activation in GBM. However, pharmacological inhibition of PI3K inhibitors have not been very successful in improving the outcome in GBM [52]. In a recent study in GBM, Shin et al performed an in vivo RNAi screen to identify putative tumor suppressors and established PI5P4Kα as a robust candidate [53]. Interestingly, both PIP4K2A and PTEN are located on chromosome 10, and by analyzing the TCGA dataset for GBM, they found that a large majority of the patients exhibited genomic deletion of both, and this associated with poor survival outcomes. However, patients with high PIP4K2A expression demonstrated a significant survival advantage when compared with patients lacking PIP4K2A in PTEN-deleted GBM patients, highlighting the clinical relevance of PI5P4Kα in GBM. Using both in vitro and in vivo approaches (Tables 1 and 2) they demonstrate that PI5P4Kα attenuates cell proliferation and tumorigenesis in PTEN-deficient GBMs [53]. PI5P4K’s role as a tumor suppressor in GBM is attributed to its ability to negatively regulate PI3K-Akt signaling by targeting the p85 adaptor subunit of class IA PI3Ks for proteasome-mediated degradation [53]. The group further elucidated that PI5P4Kα regulates the stability of the PI3K complex independent of its kinase activity, broadening the scope of these lipid enzymes in cancer beyond the modulation of phosphoinositide cellular pools.
Soft tissue sarcomas
Soft-tissue sarcomas are a very heterogenous group of malignancies and encompass more than 80 different subtypes. Current standard of care is limited to surgical resection and radio-chemotherapy for localized cases and metastatic disease is usually treated with cytotoxic chemotherapy [54]. Sarcomas have been underexplored and thus also lack safe and effective therapeutic interventions. Intriguingly, lymphomas as well as soft tissue sarcomas are the most frequently occurring tumor types in the Trp53−/− mouse cancer model, raising the possibility of PI5P4Ks involvement in sarcomas. Indeed, inhibition of both PI5P4Kα and PI5P4Kβ in a panel of sarcoma cell lines resulted in a proliferation and survival disadvantage, similar to the TNBC panel of cells described above [37]. Using a tissue microarray representing 50 different soft tissue entities, spanning from benign tumors to highly aggressive, treatment-naïve sarcomas, we showed strong correlation of high PI5P4Kα expression with high-grade sarcomas with no to low expression in benign/low grade sarcomas, indicating that high expression of PI5P4Ks might be associated with poor prognosis[37]. Relevance of PI5P4Ks in sarcoma pathogenesis was further established using the preclinical KrasLSL-G12D/+Trp53flx/flx (KP) model [55] (Table 2), which successfully recapitulates the human disease. Using this model, it was demonstrated for the first time that PI5P4Ks control tumor formation since genetic deletion of the kinases resulted in diminished tumor formation and increased survival [37]. Furthermore, the kinases were silenced in a cell line that was established from a KP tumor. Tumor implantation studies using these cells resulted in drastically diminished tumor growth supporting the essentiality of PI5P4Ks for tumor maintenance [37] (Table 1 and 2). This comprehensive study offers a glimpse of hope for difficult to treat sarcoma subtypes and has expanded the cancer relevance of the PI5P4Ks. The functional requirement of PI5P4Ks in sarcomas are discussed in further detail in the next section.
What are the functional roles of the PI5P4Ks in cancer?
As from the above cancer studies, there is indeed growing evidence for functional requirement of PI5P4Ks in controlling tumorigenesis. In this section we describe recent studies in various in vitro and in vivo models that can enable us to infer potential functional roles of PI5P4Ks that make them vital in cancer.
Genetic Models
Here we will first discuss some of the genetic models of targeting the different isoforms of PI5P4Ks and their phenotypes (Table 2). Mice lacking the most catalytically active isoform, Pip4k2a−/−, have a normal lifespan and did not manifest any striking metabolic or developmental phenotypes, however it will be interesting to see if challenged with metabolic or nutrient stresses how these mice would cope with such adversities. Mice with homozygous germline deletion of Pip4k2b, Pip4k2b−/−, were found to demonstrate increased insulin sensitivity, were growth retarded and protected from obesity when subjected to a high fat diet [16]. Similarly, inactivation of the only PI5P4K isoform in Drosophila, dPIP4K29 , also resulted in developmentally delayed animals possibly due to decreased mTOR signaling [20], and in modulation of insulin receptor signaling along with partial protection from the effects of exposure to high sugar diet [56]. Germline deletion of the least catalytically active isoform, Pip4k2c (Pip4k2c−/−), resulted in viable mice but they exhibited a mTOR-dependent increase in inflammation with an increase in proinflammatory cytokines and T helper cells, as well as a decrease in regulatory T cells [17]. Mice lacking both of the most catalytically active isoforms, Pip4k2α−/−Pip4k2b−/−, develop into normal embryos, but die within ten-twelve hours after birth resulting in perinatal lethality [15]. This is phenotypically similar to observations in mice with impaired metabolic and nutrient signaling, including mTOR signaling, insulin/IGF signaling, and autophagy [13]. In vivo targeting of PI5P4Ks in the Trp53−/− and KP sarcoma models have revealed the importance of the kinases in both tumor initiation and maintenance [15, 37]. It is not evident as to what functions of PI5P4Ks make them crucial for tumorigenesis in these in vivo models yet the in vitro results from the same studies point towards crucial influence on cellular energetics. Performing metabolomic profiling in tumor models might provide the necessary insights into the pathways through which the lipid kinases provide an advantage to tumors. Moreover, it will be exciting to evaluate the consequences of targeting PI5P4Ks in more sophisticated preclinical mouse models, including breast, ovarian and pancreatic cancer, all of which have high mutational burden of TP53. Investigation of interactions with other genetic drivers of malignancy will be another exciting prospect that might allow teasing out the metabolic adaptations under which targeting the PI5P4Ks would have the most meaningful affect in tumor control. In summary, all of the in vivo mouse models to date point broadly towards functional engagement of PI5P4Ks in the regulation of cellular energetics as well as in immune modulation, both of which are crucial hallmarks of cancer (Figure 3).
Figure 3. Multipronged effect of pharmacological targeting of PI5P4Ks in the fight against cancer.
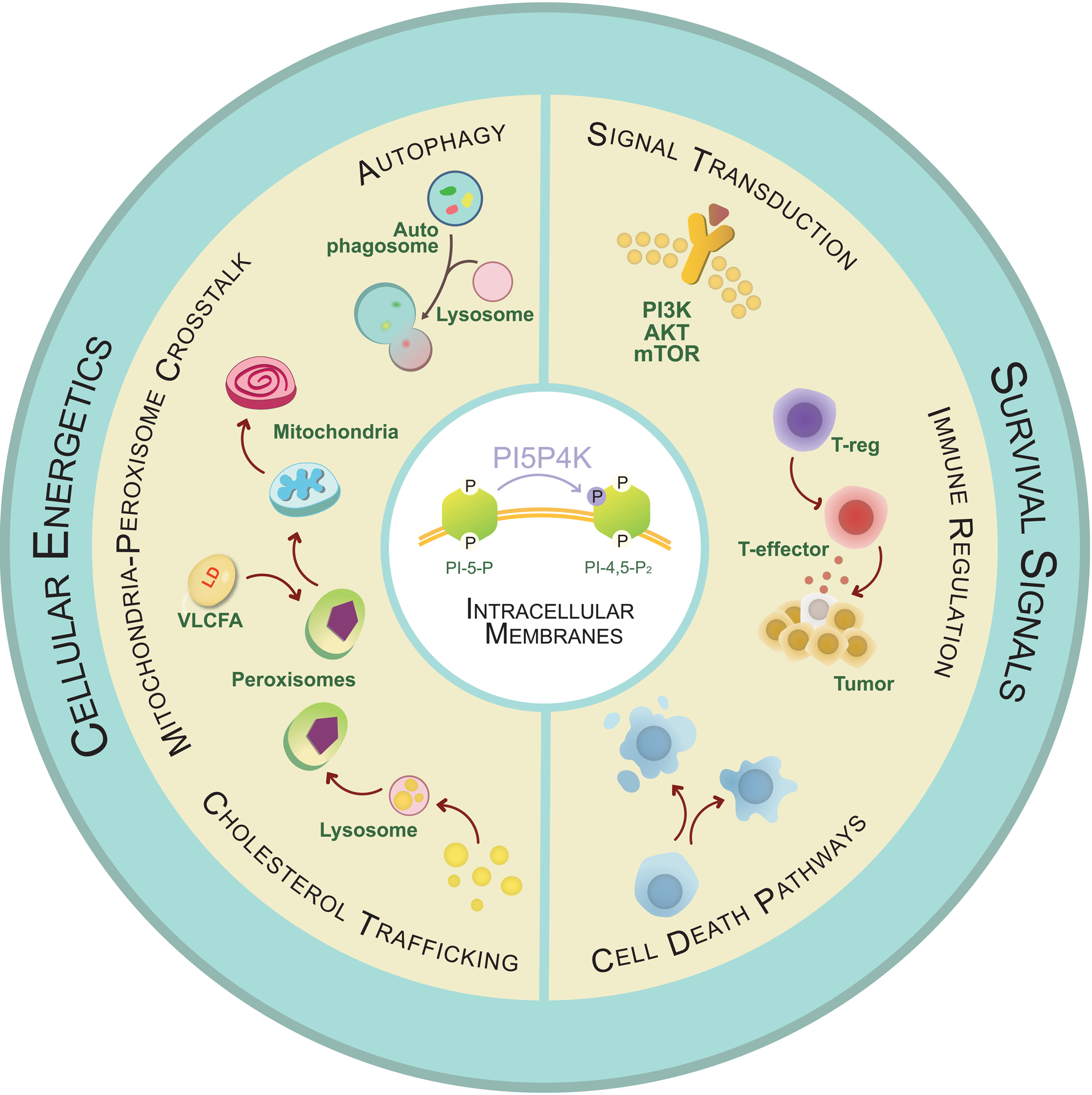
PI5P4Ks perform a central role in influencing multiple cancer hallmarks due to its dual role in cell intrinsic pathways as well as in modulation of the immune environment. PI5P4Ks contribute to the classic hallmark of evading cell-death pathways since targeting them selectively decreases survival of cancer cells. PI5P4Ks potentially regulates specialized phosphoinositide pools in subcellular organelles and modulates the dynamic contacts between organelles, exerting a far-reaching impact on cellular energetics as demonstrated by disruption of autophagy, cholesterol trafficking, ROS homeostasis and efficient peroxisomal-mitochondrial cross talk with the manipulation of the kinases. Several of the above-described functional implications arise as a result of alteration in signaling of mTOR, PI3K-Akt and AMPK networks, all of which have been crucial clinical targets for cancer treatment modalities. PI5P4Ks extend their relevance in tumor biology beyond the boundaries of the cells and into the microenvironment by selectively affecting the activity of the Tregs, allowing for the PI5P4K inhibitors to act as a combination of chemo- and immunotherapy.
Recent insights
Alteration in the activities of PI5P4Ks will vary the cellular pool of phosphoinositides and such changes can govern the proliferative and invasive capacity of cancer cells [57, 58]. Mostly, due to the changes in these pools, PI5P4Ks have been demonstrated to play important roles in growth control [20, 59], cellular energetics and metabolism [13, 37, 60], oxidative stress regulation [31, 32], cholesterol trafficking [14], GTP sensing [61] and immune function [17, 62]. Here we will highlight some of the recent advancements in our understanding of the cellular functions of PI5P4Ks which would be relevant in the context of tumorigenesis (Figure 3). We will not be discussing some of the studies that have been described in previous reviews [22, 27, 63, 64].
Metabolic dependencies
Cancer cells generate context dependent metabolic patterns to meet their excessive demands for membrane components and nutrients. Autophagy serves as one of the adaptive metabolic responses and provides access to metabolites to meet the demands of the highly proliferative cancer cells [65]. PI-5-P was revealed to be a regulator of autophagosome biogenesis and changes in its pool by silencing of the PI5P4Ks resulted in increased number of autophagosomes [28]. PI5P4Ks’s role in autophagy, under metabolic stress, has been further elucidated in the study by Lundquist and colleagues which took advantage of multiple genetic models from worms to mice [13]. Inhibition of the α and β isoforms of PI5P4K, results in a striking accumulation of autophagic vesicles due to a defect in autophagosome-lysosome fusion (Figure 3), leading to metabolic stress which in turn suppressed mTORC1 signaling and upregulated the transcription factor EB (TFEB) regulated transcriptional program [13] Notably, the autophagy defect in mouse embryonic fibroblasts (MEFs) was observed only when p53 was inhibited but not in MEFs with functional p53. p53 signaling has been implicated to be a prominent mediator of the autophagy pathway in tumors and this study provides a functional interaction between p53 and PI5P4K. Whether this ability of PI5P4Ks to mediate autophagy applies to cancer cells, and whether dysregulation of autophagy can explain the synthetic lethal phenotype remains to be determined. Multiple pharmacological approaches are targeted towards disruption of autophagy either alone or in combination with other therapies but have not been so successful clinically, largely due to metabolic differences in cancers [66]. Therefore, targeting PI5P4Ks might potentially have better efficacy, at least in the p53 disrupted patient population.
Cholesterol is a crucial membrane component and is also important for generating metabolites that play an integral role in supporting cancer progression. Reprogrammed cholesterol metabolism is driving the need to develop therapeutic targets to alter different aspects of cholesterol metabolism including synthesis, uptake, efflux and trafficking [67]. PI5P4Kα was shown to be important for intracellular cholesterol transport since its depletion resulted in accumulation of cholesterol in lysosomes, a phenotype associated with lysosomal storage disorders [14]. The trafficking defect was a consequence of decreased membrane contacts between the lysosomes and peroxisomes due to altered PI-4,5-P2 homeostasis on peroxisome membranes [68] (Figure 3). In this study, cholesterol trafficking did not seem to depend on the beta or the gamma isoforms. Evidence in support of targeting cholesterol trafficking for inhibition of tumor progression has been shown by a study with an approved drug, Cepharanthine, which inhibited endolysosomal trafficking of free-cholesterol and suppressed tumor growth [69], raising the possibility of a similar outcome with PI5P4Kα inhibitors.
PI5P4Ks role in inter organelle communication has been expanded to include peroxisomes and mitochondria, both of which are very intimately connected. Dysregulated organelle communication has pathological implications, including cancer. Mitochondria is the most metabolically active organelle and a critical hub for cellular energy requirements and is therefore vital in sustaining the growth and survival of cancer cells [70]. Therefore, targeting mitochondrial metabolism is an attractive concept to suppress cancers that have been characterized as having profound metabolic alterations and are with limited treatment options, such as TNBC [71] and soft tissue sarcomas [72]. Interestingly, alterations in peroxisomal PI-4,5-P2 due to loss of both α and β isoforms impairs trafficking of very long chain fatty acids (VLCFAs) to peroxisomes leading to mitochondrial defects that are manifested as decreased mitochondrial membrane potential and disrupted structural integrity [37] (Figure 3). These PI5P4Ks depleted cells are able to maintain viability under normal conditions but collapse when placed under nutrient stress, a situation commonly faced by cancer cells, indicating that the kinases play an integral role in metabolic addictions of cancer cells, presenting them as a promising candidate for therapeutic targeting in TNBC and sarcomas.
Besides being stress sensors, PI5P4Ks, specifically the β isoform was also shown to be a sensor of cellular GTP due to its ability to preferentially utilize GTP over ATP to generate PI-4,5-P2 from PI-5-P. This GTP sensing ability was further elucidated to be important for tumorigenesis since expression of GTP-insensitive mutant Pip4k2b in SV40 T-antigen immortalized MEFs resulted in the absence of tumor growth in nude mice [61]. This suggested that changes in GTP concentration can be sensed by PI5P4Kβ and are transduced to PI-5-P signaling, allowing for metabolic adaptations and cancer growth, making this feature an important consideration when developing inhibitors of the β isoform.
Lastly, a minimally explored dimension of PI5P4Ks function is whether its catalytic activity is the sole executor of its in vivo roles. PI5P4Kα-mediated degradation of the PI3K complex subunits in the GBM model is independent of the kinase activity [53]. Another couple of recent studies also suggested a non-catalytic function in the regulation of insulin signaling and PI-3,4,5-P3 turnover [56, 73]. These findings certainly warrant further investigation as many kinases are found to have such moonlighting functions.
Summarizing the metabolic roles of PI5P4Ks it can be concluded that these enzymes become conditionally essential during energy stress and can regulate various nodes of metabolism. The mechanisms by which PI5P4Ks play a role in these dependencies are just beginning to be understood and a deeper knowledge of how PI5P4Ks govern the metabolic networks, will allow stratification of patients into metabolic profiles that would benefit the most from drugs targeting PI5P4Ks.
Immune modulation
Tumor microenvironment modulation involves evasive maneuvers by cancer cells to avoid assault from the host immune response. Given the exploding research into the immune microenvironment in cancer, it is exciting to see evidence for PI5P4Ks in immune modulation. As mentioned above, mice lacking PI5P4Kγ have increased inflammation characterized by decreased regulatory effectors cells [17]. Highlighting PI5P4Ks role in T cell signaling, a recent study demonstrated that PI5P4Ks are expressed in both T effector as well as in T regulatory cells (Tregs), but their inhibition restricted the functions of only the Tregs but not the T effector cells which target the cancer cells [62] (Figure 3). This study implicated the β and γ isoforms in the transcriptional reprogramming of the Tregs through alteration in PI3K signaling and consequent changes in the expression of FOXP3, which is the Tregs master regulator. Infiltration of tumors with the Tregs is often associated with poor prognosis [74] and thus, targeting PI5P4Ks could allow for improved immunotherapy treatment through preferential inhibition of the immunosuppressive Treg cell population.
Targeting PI5P4Ks with small molecule inhibitors
With the expanding implications of the PI5P4Ks in multiple cancers and a better understanding of the functional biology of this family of lipid kinases, it is not surprising that there is an increasing focus towards therapeutic exploitation of these enzymes. Given the current roles of the PI5P4Ks in cancer cell-intrinsic metabolism as well as in immune response, it is motivating to surmise that PI5P4K inhibitors will exercise a multipronged impact on tumor control, allowing for better handling of pharmacological challenges arising due to tumor heterogeneity (Figure 3).
In fact, efforts have been made to identify and characterize pan inhibitors [33, 34] as well as isoform specific inhibitors of PI5P4Ks [75, 76] and over the years there have been reports of more potent and selective small molecule inhibitors [60, 77–79], strengthening the evidence for efficient druggability of the PI5P4Ks. Recently in an attempt to identify drugs that are selectively lethal to cancer cells but not to normal cells, a novel compound, a131, was identified and interestingly, the group discovered that the target of the a131 compound is indeed the PI5P4Ks and it selectively regulated the cell cycle entry of Ras-activated cancer cells resulting in their death through mitotic catastrophe, while causing reversible growth arrest in normal cells [77]. This study provides further support for the critical role of PI5P4Ks in cancer cell dependencies. Later, PI5P4Kα was also identified in a genome wide RNAi screen using a colorectal cancer cell line to identify synthetic lethal interactions with KRAS, further supporting its interaction with activated Ras signaling [80]. Another recently identified inhibitor is a pan-PI5P4K inhibitor, THZ-P1–2, which covalently modifies the cysteines in a loop close to the ATP-binding kinase domain of all the three isoforms. Treatment with THZ-P1–2 successfully phenocopied the impairment of autophagy flux as seen with genetic ablation of the kinases and also resulted in increased survival sensitivity in six AML/ALL cell lines. With regards to off target effects, the inhibitor exerted a potent effect on PIKfyve, and also on BRK and ABL1, underscoring the need for further improvement [79]. In a follow up study by the same group, they sought to find reversible inhibitors of PI5P4Ks so that issues associated with acquired resistance of tumor cells could be avoided. Among a library of approximately 6,000 kinase inhibitors, they identified a potent inhibitor and further developed a pan-PI5P4K inhibitor with better selectivity [78]. Most recently a noncovalent dual PI5P4Kα/β inhibitor, CC260, was identified using a high throughput screen among 5,759 small molecules. CC260 treatment was shown to affect AKT phosphorylation, impair cellular energetics as demonstrated by changes in AMPK activation, mTOR inhibition and decreased mitochondrial ATP production [60]. CC260 treatment increased the sensitivity of cancer cells to nutrient stress and was also able to selectively kill p53 mutant breast cancer cells [60], confirming several published data from our laboratory and others. Even though the C260 compound appears to engage with the intended target, it does have significant off-target activity against PI3K-δ, indicating that there is still enough scope to improve the specificity and overall pharmacokinetics of the inhibitor. Nonetheless, this study strengthens the role of PI5P4Ks in cellular energetics (Figure 3) and given the role of p53 in cellular energy metabolism, it is most likely that dual loss of the kinase and p53 function creates excessive energy stress specially for the metabolically constrained tumor cells. With further improvements in specificity of the inhibitors, the next steps will be to test their safety, efficacy, and dosage requirements in preclinical mouse models, including the Trp53−/− GEM model, following which it would be more apparent if these inhibitors could be beneficial on their own or would require a combinatorial approach. Further, it remains to be tested if PI5P4Kα/β inhibitors could also be used to exploit the vulnerabilities created by the frequently occurring p53 missense mutations in human cancers. It is promising though that the inhibitor candidates thus far, especially the dual CC260 PI5P4Kα/β inhibitor, can phenocopy the some of the effects seen with targeting PI5P4Ks using genetic tools and cancer models. Additionally, it is also important to consider the fact that the kinases can exert non catalytic activity and if such a role is crucial for tumorigenesis, then a PROTAC (Proteolysis targeting chimeras)-based strategy [81] to degrade the entire protein could be effective and thus should be taken into consideration for developing targeting approaches for PI5P4Ks.
In the end, to date most of the cellular and in vivo evidence points towards oncogenic roles of PI5P4Ks and thus fuels the need for inhibitors. It is possible that in the near future there might be more indications for a tumor suppressor role of PI5P4Ks as seen in the GBM study and that would indicate that these enzymes are regulated differently depending on metabolic or tumor microenvironment cues with the ultimate objective to benefit tumor cells.
Conclusion and perspective
From a therapeutic perspective it is evident that pharmacological manipulation of PI5P4Ks will have beneficial effects for cancer patients but clearly there is a requirement for experimental studies on different cancer subtypes to identify the genetic and metabolic contexts under which will be most vulnerable to PI5P4K inhibitors. Further in this context, there is a need for more high-throughput analysis of patient data in multiple cancers to get an overview of association between distribution and expression of the PI5P4K isoforms with stages and subtypes to successfully foresee the patient pool who would be most responsive to PI5P4K targeted therapy. Also, importantly it will be crucial to identify the specific isoforms that cancer subtypes are dependent on for tumor growth. Currently, what we can infer from published work is that p53 deficient cancers, especially breast tumors, and high-grade soft tissue sarcomas will require dual inhibitors that target both PI5P4K isoforms, α and β, whereas AML seems to require inhibition of only the α isoform.
Further functional understanding of the PI5P4Ks in oncogenic dependencies requires additional investigation into the regulation of their cellular distribution and kinase activity, identification of downstream effectors and interaction partners and finally the function(s) of their substrate PI-5-P and product PI-4,5-P2 in the pathogenesis of cancer. It will be crucial to assess whether it is PI5P4K’s role in suppressing PI-5-P, or its ability to generate specialized local pools of PI-4,5-P2 at subcellular compartments or both, that is required in the context of tumorigenesis. With advancements in lipidomics and improved imaging techniques and abilities to visualize the phosphoinositide pools in vivo as well as the distribution of endogenous kinases we might have better insights into how cells respond to variations in these pools [82]. Nonetheless, with the emerging evidence of PI5P4Ks in cancer, it is important to leverage our current knowledge to already assess PI5P4K inhibitors in cancer models to allow for quicker bench to clinical transition.
References
- 1.Hammond GRV and Burke JE, Novel roles of phosphoinositides in signaling, lipid transport, and disease. Curr Opin Cell Biol, 2020. 63: p. 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thapa N, et al. , The Hidden Conundrum of Phosphoinositide Signaling in Cancer. Trends Cancer, 2016. 2(7): p. 378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pendaries C, et al. , Phosphoinositide signaling disorders in human diseases. FEBS Lett, 2003. 546(1): p. 25–31. [DOI] [PubMed] [Google Scholar]
- 4.Balla T, Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev, 2013. 93(3): p. 1019–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beziau A, Brand D, and Piver E, The Role of Phosphatidylinositol Phosphate Kinases during Viral Infection. Viruses, 2020. 12(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanhaesebroeck B, et al. , PI3K inhibitors are finally coming of age. Nat Rev Drug Discov, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maehama T and Dixon JE, The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem, 1998. 273(22): p. 13375–8. [DOI] [PubMed] [Google Scholar]
- 8.Czech MP, PIP2 and PIP3: complex roles at the cell surface. Cell, 2000. 100(6): p. 603–6. [DOI] [PubMed] [Google Scholar]
- 9.Sun Y, et al. , Phosphatidylinositol 4,5-bisphosphate: targeted production and signaling. Bioessays, 2013. 35(6): p. 513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bazenet CE, et al. , The human erythrocyte contains two forms of phosphatidylinositol-4-phosphate 5-kinase which are differentially active toward membranes. J Biol Chem, 1990. 265(29): p. 18012–22. [PubMed] [Google Scholar]
- 11.Ling LE, Schulz JT, and Cantley LC, Characterization and purification of membrane-associated phosphatidylinositol-4-phosphate kinase from human red blood cells. J Biol Chem, 1989. 264(9): p. 5080–8. [PubMed] [Google Scholar]
- 12.Rameh LE, et al. , A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature, 1997. 390(6656): p. 192–6. [DOI] [PubMed] [Google Scholar]
- 13.Lundquist MR, et al. , Phosphatidylinositol-5-Phosphate 4-Kinases Regulate Cellular Lipid Metabolism By Facilitating Autophagy. Mol Cell, 2018. 70(3): p. 531–544 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu A, et al. , PIP4K2A regulates intracellular cholesterol transport through modulating PI(4,5)P2 homeostasis. J Lipid Res, 2018. 59(3): p. 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emerling BM, et al. , Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors. Cell, 2013. 155(4): p. 844–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamia KA, et al. , Increased insulin sensitivity and reduced adiposity in phosphatidylinositol 5-phosphate 4-kinase beta−/− mice. Mol Cell Biol, 2004. 24(11): p. 5080–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shim H, et al. , Deletion of the gene Pip4k2c, a novel phosphatidylinositol kinase, results in hyperactivation of the immune system. Proc Natl Acad Sci U S A, 2016. 113(27): p. 7596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clarke JH and Irvine RF, Evolutionarily conserved structural changes in phosphatidylinositol 5-phosphate 4-kinase (PI5P4K) isoforms are responsible for differences in enzyme activity and localization. Biochem J, 2013. 454(1): p. 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lecompte O, Poch O, and Laporte J, PtdIns5P regulation through evolution: roles in membrane trafficking? Trends Biochem Sci, 2008. 33(10): p. 453–60. [DOI] [PubMed] [Google Scholar]
- 20.Gupta A, et al. , Phosphatidylinositol 5-phosphate 4-kinase (PIP4K) regulates TOR signaling and cell growth during Drosophila development. Proc Natl Acad Sci U S A, 2013. 110(15): p. 5963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clarke JH and Irvine RF, The activity, evolution and association of phosphatidylinositol 5-phosphate 4-kinases. Adv Biol Regul, 2012. 52(1): p. 40–5. [DOI] [PubMed] [Google Scholar]
- 22.Burke JE, Structural Basis for Regulation of Phosphoinositide Kinases and Their Involvement in Human Disease. Mol Cell, 2018. 71(5): p. 653–673. [DOI] [PubMed] [Google Scholar]
- 23.Muftuoglu Y, et al. , Mechanism of substrate specificity of phosphatidylinositol phosphate kinases. Proc Natl Acad Sci U S A, 2016. 113(31): p. 8711–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clarke JH, Wang M, and Irvine RF, Localization, regulation and function of type II phosphatidylinositol 5-phosphate 4-kinases. Adv Enzyme Regul, 2010. 50(1): p. 12–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang M, et al. , Genomic tagging reveals a random association of endogenous PtdIns5P 4-kinases IIalpha and IIbeta and a partial nuclear localization of the IIalpha isoform. Biochem J, 2010. 430(2): p. 215–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bultsma Y, Keune WJ, and Divecha N, PIP4Kbeta interacts with and modulates nuclear localization of the high-activity PtdIns5P-4-kinase isoform PIP4Kalpha. Biochem J, 2010. 430(2): p. 223–35. [DOI] [PubMed] [Google Scholar]
- 27.Fiume R, et al. , PIP4K and the role of nuclear phosphoinositides in tumour suppression. Biochim Biophys Acta, 2015. 1851(6): p. 898–910. [DOI] [PubMed] [Google Scholar]
- 28.Vicinanza M, et al. , PI(5)P regulates autophagosome biogenesis. Mol Cell, 2015. 57(2): p. 219–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clarke JH, Emson PC, and Irvine RF, Localization of phosphatidylinositol phosphate kinase IIgamma in kidney to a membrane trafficking compartment within specialized cells of the nephron. Am J Physiol Renal Physiol, 2008. 295(5): p. F1422–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones DR, et al. , Nuclear PtdIns5P as a transducer of stress signaling: an in vivo role for PIP4Kbeta. Mol Cell, 2006. 23(5): p. 685–95. [DOI] [PubMed] [Google Scholar]
- 31.Jones DR, et al. , PtdIns5P is an oxidative stress-induced second messenger that regulates PKB activation. FASEB J, 2013. 27(4): p. 1644–56. [DOI] [PubMed] [Google Scholar]
- 32.Keune WJ, Jones DR, and Divecha N, PtdIns5P and Pin1 in oxidative stress signaling. Adv Biol Regul, 2013. 53(2): p. 179–89. [DOI] [PubMed] [Google Scholar]
- 33.Keune WJ, et al. , Regulation of phosphatidylinositol-5-phosphate signaling by Pin1 determines sensitivity to oxidative stress. Sci Signal, 2012. 5(252): p. ra86. [DOI] [PubMed] [Google Scholar]
- 34.Chen Y, et al. , Prolyl isomerase Pin1: a promoter of cancer and a target for therapy. Cell Death Dis, 2018. 9(9): p. 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mackey AM, et al. , PIP4kgamma is a substrate for mTORC1 that maintains basal mTORC1 signaling during starvation. Sci Signal, 2014. 7(350): p. ra104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilcox A and Hinchliffe KA, Regulation of extranuclear PtdIns5P production by phosphatidylinositol phosphate 4-kinase 2alpha. FEBS Lett, 2008. 582(9): p. 1391–4. [DOI] [PubMed] [Google Scholar]
- 37.Ravi A, et al. , PI5P4Ks drive metabolic homeostasis through peroxisome-mitochondria interplay. Dev Cell, 2021. 56(11): p. 1661–1676 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang Z, et al. , GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res, 2017. 45(W1): p. W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luoh SW, Venkatesan N, and Tripathi R, Overexpression of the amplified Pip4k2beta gene from 17q11–12 in breast cancer cells confers proliferation advantage. Oncogene, 2004. 23(7): p. 1354–63. [DOI] [PubMed] [Google Scholar]
- 40.Keune WJ, et al. , Low PIP4K2B expression in human breast tumors correlates with reduced patient survival: A role for PIP4K2B in the regulation of E-cadherin expression. Cancer Res, 2013. 73(23): p. 6913–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lord CJ and Ashworth A, PARP inhibitors: Synthetic lethality in the clinic. Science, 2017. 355(6330): p. 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terkelsen T, et al. , High-throughput proteomics of breast cancer interstitial fluid: identification of tumor subtype-specific serologically relevant biomarkers. Mol Oncol, 2021. 15(2): p. 429–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lima K, et al. , PIP4K2A and PIP4K2C transcript levels are associated with cytogenetic risk and survival outcomes in acute myeloid leukemia. Cancer Genet, 2019. 233-234: p. 56–66. [DOI] [PubMed] [Google Scholar]
- 44.Lima K, et al. , Differential profile of PIP4K2A expression in hematological malignancies. Blood Cells Mol Dis, 2015. 55(3): p. 228–35. [DOI] [PubMed] [Google Scholar]
- 45.Ratti S, et al. , “Modulating Phosphoinositide Profiles as a Roadmap for Treatment in Acute Myeloid Leukemia”. Front Oncol, 2021. 11: p. 678824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jabbour E, et al. , Targeting the phosphoinositide 3-kinase pathway in hematologic malignancies. Haematologica, 2014. 99(1): p. 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jude JG, et al. , A targeted knockdown screen of genes coding for phosphoinositide modulators identifies PIP4K2A as required for acute myeloid leukemia cell proliferation and survival. Oncogene, 2015. 34(10): p. 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu H, et al. , Novel susceptibility variants at 10p12.31–12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst, 2013. 105(10): p. 733–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang S, et al. , Regulatory Network and Prognostic Effect Investigation of PIP4K2 A in Leukemia and Solid Cancers. Front Genet, 2018. 9: p. 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Migliorini G, et al. , Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood, 2013. 122(19): p. 3298–307. [DOI] [PubMed] [Google Scholar]
- 51.Ostrom QT, et al. , CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol, 2017. 19(suppl_5): p. v1–v88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eckerdt FD, et al. , Combined PI3Kalpha-mTOR Targeting of Glioma Stem Cells. Sci Rep, 2020. 10(1): p. 21873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shin YJ, et al. , PIP4K2A as a negative regulator of PI3K in PTEN-deficient glioblastoma. J Exp Med, 2019. 216(5): p. 1120–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gamboa AC, Gronchi A, and Cardona K, Soft-tissue sarcoma in adults: An update on the current state of histiotype-specific management in an era of personalized medicine. CA Cancer J Clin, 2020. 70(3): p. 200–229. [DOI] [PubMed] [Google Scholar]
- 55.Kirsch DG, et al. , A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat Med, 2007. 13(8): p. 992–7. [DOI] [PubMed] [Google Scholar]
- 56.Sharma S, et al. , Phosphatidylinositol 5 Phosphate 4-Kinase Regulates Plasma-Membrane PIP3 Turnover and Insulin Signaling. Cell Rep, 2019. 27(7): p. 1979–1990 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Viaud J, et al. , Phosphatidylinositol 5-phosphate regulates invasion through binding and activation of Tiam1. Nat Commun, 2014. 5: p. 4080. [DOI] [PubMed] [Google Scholar]
- 58.Oppelt A, et al. , Production of phosphatidylinositol 5-phosphate via PIKfyve and MTMR3 regulates cell migration. EMBO Rep, 2013. 14(1): p. 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Elouarrat D, et al. , Role of phosphatidylinositol 5-phosphate 4-kinase alpha in zebrafish development. Int J Biochem Cell Biol, 2013. 45(7): p. 1293–301. [DOI] [PubMed] [Google Scholar]
- 60.Chen S, et al. , Pharmacological inhibition of PI5P4Kalpha/beta disrupts cell energy metabolism and selectively kills p53-null tumor cells. Proc Natl Acad Sci U S A, 2021. 118(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sumita K, et al. , The Lipid Kinase PI5P4Kbeta Is an Intracellular GTP Sensor for Metabolism and Tumorigenesis. Mol Cell, 2016. 61(2): p. 187–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poli A, et al. , PIP4Ks impact on PI3K, FOXP3, and UHRF1 signaling and modulate human regulatory T cell proliferation and immunosuppressive activity. Proc Natl Acad Sci U S A, 2021. 118(31). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bulley SJ, et al. , Exploring phosphatidylinositol 5-phosphate 4-kinase function. Adv Biol Regul, 2015. 57: p. 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sasaki T, et al. , Mammalian phosphoinositide kinases and phosphatases. Prog Lipid Res, 2009. 48(6): p. 307–43. [DOI] [PubMed] [Google Scholar]
- 65.Mulcahy Levy JM and Thorburn A, Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ, 2020. 27(3): p. 843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Levy JMM, Towers CG, and Thorburn A, Targeting autophagy in cancer. Nat Rev Cancer, 2017. 17(9): p. 528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang B, Song BL, and Xu C, Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab, 2020. 2(2): p. 132–141. [DOI] [PubMed] [Google Scholar]
- 68.Chu BB, et al. , Cholesterol transport through lysosome-peroxisome membrane contacts. Cell, 2015. 161(2): p. 291–306. [DOI] [PubMed] [Google Scholar]
- 69.Lyu J, et al. , Pharmacological blockade of cholesterol trafficking by cepharanthine in endothelial cells suppresses angiogenesis and tumor growth. Cancer Lett, 2017. 409: p. 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wallace DC, Mitochondria and cancer. Nat Rev Cancer, 2012. 12(10): p. 685–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mehanna J, et al. , Triple-negative breast cancer: current perspective on the evolving therapeutic landscape. Int J Womens Health, 2019. 11: p. 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huangyang P, et al. , Fructose-1,6-Bisphosphatase 2 Inhibits Sarcoma Progression by Restraining Mitochondrial Biogenesis. Cell Metab, 2020. 31(5): p. 1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang DG, et al. , PIP4Ks Suppress Insulin Signaling through a Catalytic-Independent Mechanism. Cell Rep, 2019. 27(7): p. 1991–2001 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tanaka A and Sakaguchi S, Regulatory T cells in cancer immunotherapy. Cell Res, 2017. 27(1): p. 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Voss MD, et al. , Discovery and pharmacological characterization of a novel small molecule inhibitor of phosphatidylinositol-5-phosphate 4-kinase, type II, beta. Biochem Biophys Res Commun, 2014. 449(3): p. 327–31. [DOI] [PubMed] [Google Scholar]
- 76.Davis MI, et al. , A homogeneous, high-throughput assay for phosphatidylinositol 5-phosphate 4-kinase with a novel, rapid substrate preparation. PLoS One, 2013. 8(1): p. e54127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kitagawa M, et al. , Dual blockade of the lipid kinase PIP4Ks and mitotic pathways leads to cancer-selective lethality. Nat Commun, 2017. 8(1): p. 2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Manz TD, et al. , Discovery and Structure-Activity Relationship Study of (Z)-5-Methylenethiazolidin-4-one Derivatives as Potent and Selective Pan-phosphatidylinositol 5-Phosphate 4-Kinase Inhibitors. J Med Chem, 2020. 63(9): p. 4880–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sivakumaren SC, et al. , Targeting the PI5P4K Lipid Kinase Family in Cancer Using Covalent Inhibitors. Cell Chem Biol, 2020. 27(5): p. 525–537 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luo J, et al. , A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell, 2009. 137(5): p. 835–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sun X, et al. , PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther, 2019. 4: p. 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Montano-Rendon F, Grinstein S, and Walpole GFW, Monitoring Phosphoinositide Fluxes and Effectors During Leukocyte Chemotaxis and Phagocytosis. Front CellDev Biol, 2021. 9: p. 626136. [DOI] [PMC free article] [PubMed] [Google Scholar]