

Abstract
Motivation
The chemical modification of ribonucleotides regulates the structure, stability and interactions of RNAs. Profiling of these modifications using short-read (Illumina) sequencing techniques provides high sensitivity but low-to-medium resolution i.e. modifications cannot be assigned to specific transcript isoforms in regions of sequence overlap. An alternative strategy uses current fluctuations in nanopore-based long read direct RNA sequencing (DRS) to infer the location and identity of nucleotides that differ between two experimental conditions. While highly sensitive, these signal-level analyses require high-quality transcriptome annotations and thus are best suited to the study of model organisms. By contrast, the detection of RNA modifications in microbial organisms which typically have no or low-quality annotations requires an alternative strategy. Here, we demonstrate that signal fluctuations directly influence error rates during base-calling and thus provides an alternative approach for identifying modified nucleotides.
Results
DRUMMER (Detection of Ribonucleic acid Modifications Manifested in Error Rates) (i) utilizes a range of statistical tests and background noise correction to identify modified nucleotides with high confidence, (ii) operates with similar sensitivity to signal-level analysis approaches and (iii) correlates very well with orthogonal approaches. Using well-characterized DRS datasets supported by independent meRIP-Seq and miCLIP-Seq datasets we demonstrate that DRUMMER operates with high sensitivity and specificity.
Availability and implementation
DRUMMER is written in Python 3 and is available as open source in the GitHub repository: https://github.com/DepledgeLab/DRUMMER.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The selective chemical modification of an RNA transcript impacts its splicing, stability, structure, translation and turnover (Barbieri and Kouzarides, 2020; He and He, 2021; Shi et al., 2020). Over 170 distinct chemical modifications of RNA have been reported; however, only a minority are well characterized (Boccaletto et al., 2018). Key challenges in RNA modification studies include the precise mapping of modified bases at nucleotide resolution and on the level of individual RNAs. These challenges are exacerbated where transcriptome annotation quality is low and/or the defined genome contains multitudes of overlapping transcription units (i.e. viruses) (Depledge et al., 2019b). While antibody-based (e.g. meRIP-Seq, miCLIP-Seq) and antibody-independent (e.g. DART-Seq, MAZTER-Seq) have significantly advanced our understanding of RNA modifications such as the methylation of adenosine at the N6 position (m6A) (Dominissini et al., 2012; Garcia-Campos et al., 2019; Linder et al., 2015; Meyer, 2019; Zhang et al., 2019), they remain constrained by the biases of short-read sequencing approaches which include technical and biological variation leading to noise and affecting reproducibility (Kukurba and Montgomery, 2015).
Alternative approaches to RNA modification detection are centered on the analysis of datasets generated using Oxford Nanopore Technologies (ONT) direct RNA sequencing (DRS) methodology (Depledge et al., 2019a; Workman et al., 2019). Here, sequencing of polyadenylated RNAs in their native state using a molecular motor that ratchets the RNA through a membrane-embedded protein pore, disrupting the flow of ions through the nanopore. These changes in current are subsequently interpreted using neural networks to predict the sequence of nucleotides within the RNA. The reader-head, which records these signals, is positioned within a region of the nanopore in which an average of five nucleotides of the RNA is present. Thus, the signal change is a reflection of this 5-mer. The structural changes to a ribonucleotide resulting from modification will thus alter the current measurements during its entire time in the reader-head region (Garalde et al., 2018). During subsequent base-calling, the neural networks, trained on unmodified nucleotides, are prone to misinterpreting the signal and calling an incorrect base.
Multiple tools applying discrete approaches have been developed to exploit DRS data ranging from classifiers (Liu et al., 2019; Lorenz et al., 2020), comparative signal-level analysis tools (Leger et al., 2019; Pratanwanich et al., 2020), comparative error-rate analysis tools (Jenjaroenpun et al., 2021; Parker et al., 2020; Price et al., 2020), and most recently, direct signal-level analysis (Begik et al., 2021; Hendra et al., 2021). The utility of each approach varies according to the experimental questions and datasets in question. For instance, signal-level analyses predominantly operate at the transcriptome level and their success is thus dependent on having high-quality transcriptome annotations. While powerful, their utility is often limited to well-characterized datasets and requires a higher level of computational expertise. By contrast, organisms for which genome but not high-quality transcriptome annotation are available (e.g. microbes) require alternative strategies for analysis. Here, the use of comparative error-rate analysis tools provides a simpler alternative to screen for RNA modifications.
Here, we introduce DRUMMER (Detection of Ribonucleic acid Modifications Manifested in Error Rates—https://github.com/DepledgeLab/DRUMMER); an RNA modification detection package that predicts modified nucleotides via a comparative assessment of base-call error rates in two or more datasets. While designed primarily for the detection and analysis of RNA modifications in diverse viruses, we use well-characterized DRS datasets, supported by meRIP and miCLIP, to demonstrate DRUMMER’s ability to map m6A modifications at high resolution across both mammalian and viral transcriptomes.
2 Materials and methods
DRUMMER is implemented in Python 3 and allows for up to three biological replicates per condition to be processed using a round-robin approach (where each control sample is compared against each of the treatment samples). One condition is represented by datasets from a control sample in which the RNA modification(s) of interest is present, while the second condition is represented by datasets from a treatment sample in which one or more RNA modifications have been ablated using inhibitors, gene knockdown, gene knockout or in vitro transcription strategies (Fig. 1). The choice of approach dictates the specificity of the resulting analysis i.e. a broad analysis revealing the location of all modified ribonucleotides or a narrow analysis showing the location of a specific modification, such as m6A.
Fig. 1.
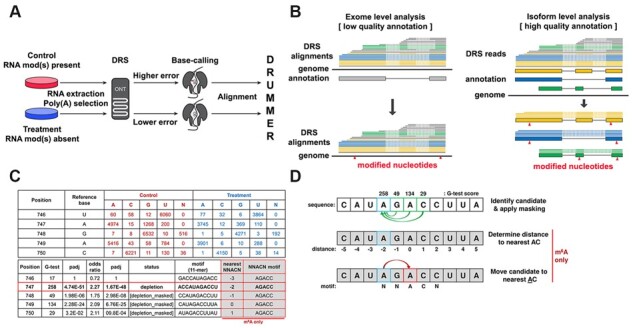
Schematic overview of DRUMMER. (A) DRUMMER identifies putative RNA modifications through comparative analysis of nanopore DRS datasets. The presence or increased abundance of a modified ribonucleotide is more likely to result in an incorrect nucleotide being reported during base-calling (i.e. a higher error rate). (B) DRUMMER can process both genome-level and transcriptome-level alignments. In ‘exome’ mode DRUMMER uses sequence read alignments against a genome to predict the location of putative RNA modifications (triangles) in a genomic context. In ‘isoform’ mode, DRUMMER relies on sequence read alignments (blue, yellow, green lines) against a (high-quality) transcriptome and predicts the location of putative RNA modifications (red triangles) at the level of individual transcript isoforms (large blue, yellow, green boxes) and in a genomic context. Note that low-quality sequence read alignments (grey lines) should be filtered prior to analysis. (C and D) DRUMMER parses BAM files using bamreadcount to generate per nucleotide counts of A, C, G, U and N (indels) base-calls in both treatment and control datasets. A G-test (2 × 5 contingency table) is used to determine whether a significant difference in erroneous base-calls is observed between the two datasets at a given position, supported by an odds ratio test to determine whether an increased error rate is observed in the control (depletion of RNA modification abundance in treatment relative to control) or treatment (accumulation of RNA modification abundance in treatment relative to control) dataset. A given site is reported (by default) as a depletion/accumulation candidate if G-test padj < 0.05 and O/R > 1.5. Where multiple sites within a five-nucleotide window are classed as candidates, only the site with the largest G-test score is retained with all others reported as [masked]. Additional reporting shows 11-nt sequence windows centered on the candidate site that can be used for sequence motif/context discovery. When specifically run in m6A detection mode (−m6A), DRUMMER also reports the distance (nt) between a given candidate site and the nearest AC dinucleotide along with the 5-nt sequence motif centered on that nearest AC dinucleotide. Data shown in C and D are derived from isoform-level analysis of the Adenovirus L2-Penton transcript
To prepare data for DRUMMER analysis after nanopore sequencing, all input datasets should be base-called using the same version of Guppy (or equivalent), and aligned against either a representative genome (exome mode) or transcriptome (isoform mode) to produce sorted BAM files (Fig. 1). The specific choice of exome versus isoform mode is guided by the available data for the transcriptome of interest. Exome mode is primarily suited for smaller genomes with low-quality transcriptome annotations. Here, it is more prudent to align reads directly to the genome prior to identifying putative modified ribonucleotides using DRUMMER. While this approach reduces sensitivity compared to isoform-level alignments (see Supplementary Materials) and may be more affected by misalignments around splice junctions, it broadens the accessibility of RNA modification detection such that analyses can be performed in the absence of high-quality transcriptome annotations. By contrast, where high-quality reference transcriptomes are available, alignments of nanopore reads against a transcriptome in fasta format and comprising all documented transcript isoforms can be further parsed to remove noise from 5ʹ truncated and/or multi-mapping alignments. This filtering notably increases the sensitivity compared to exome level analyses (see Supplementary Materials).
DRUMMER processes each genome/transcript isoform individually, parsing alignments from the input BAM files to generate base-call distributions (i.e. the number of A, C, G, U and indels) for each position along the genome/transcript. Each position is then subject to a 2 × 5 G-test and an Odds Ratio (O/R) test with resultant P-values undergoing multiple testing Bonferroni correction. Putative RNA modification positions are labeled as candidate sites if both G-test and O/R adjusted P-values are less than the user-specified input (default < 0.05) and the O/R test result exceeds a user-specified input (default > 1.5) (Fig. 1C). Candidate sites within 5 nt of each other are masked, leaving only a single candidate possessing the highest G-test score. This increases the specificity of downstream analyses and prevents the inclusion of false positives that may occur due to influence of modified nucleotides on neighboring unmodified nucleotides. Note, however, that this function can be disabled within DRUMMER if neighboring modifications are expected.
Additional information collected on a per site basis includes an 11-base sequence motif centered on the position of interest, and a determination of whether a homopolymer (≥3 nt) is present in the 11-base motif. When run specifically in m6A detection mode (−m6A), DRUMMER also determines the distance to the nearest AC dinucleotide and the 5-base sequence centered upon that motif (i.e. NNACN) (Fig. 1D).
Finally, DRUMMER classifies candidate sites according to the direction of the odds ratio result. Accumulation sites are defined as having a higher error rate in the treatment versus control sample where depletion sites have a higher error rate in the control versus treatment. This specificity allows users to identify RNA modifications that either increase or decrease in frequency according to the experimental design (e.g. depletion of a methyltransferase or depletion of a demethylase). Importantly, the presence of accumulation sites when a specific modification is depleted allows DRUMMER to establish a baseline for false-positive detection that leads to increasingly stringent filtering and higher specificity for true-positives (see Supplementary Materials).
Upon completion, DRUMMER outputs a report table containing detailed lists of all putative modified nucleotides that can be filtered and visualized using bundled scripts (see Supplementary Materials). Two case studies demonstrating the ability of DRUMMER to detect m6A modifications in viral and murine datasets are showcased in the Supplementary Materials with further examples found in recent publications on adenovirus, SARS-CoV-2 and herpes simplex virus type 1 (Burgess et al., 2021; Price et al., 2020; Srinivas et al., 2021).
3 Conclusions
The sensitive detection and precise mapping of RNA modifications to the transcriptomes of model and non-model organisms remains challenging in most scenarios. Where read depth is sufficiently high, DRUMMER operates with high specificity and sensitivity, allowing the identification of modified ribonucleotides on individual transcript isoforms. While primarily designed for the analysis of microbial transcriptomes (and in particular, viruses), DRUMMER is also capable of providing rapid analyses of larger eukaryotic transcriptomes.
Supplementary Material
Acknowledgements
We thank members of the Depledge, Weitzman and Mohr/Wilson Labs for insightful discussions and input.
Data availability
The data underlying this article are available from the European Nucleotide Archive (ENA), Sequence Read Archive (SRA), and Gene Expression Omnibus (GEO) and can be accessed with the following study accessions: PRJEB35652 (ENA), SRP166020 (SRA), and GSE52681 (GEO).
Funding
This work was supported in part through National Institute of Health [R01-AI073898 and R01-GM056927] (I.M., D.P.D.), [AI130618 and AI147163] (A.C.W.) and [AI145266, AI121321 and AI118891] (M.D.W.). Additional support came from the National Cancer Institute T32 Training Grant in Tumor Virology [CA115299] (A.M.P.) and Individual National Research Service Award AI138432 (A.M.P.).
Conflict of Interest: none declared.
Contributor Information
Jonathan S Abebe, Department of Microbiology, New York University School of Medicine, New York, NY 10016, USA.
Alexander M Price, Division of Protective Immunity, Department of Pathology and Laboratory Medicine, The Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA.
Katharina E Hayer, Department of Biomedical and Health Informatics, The Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA.
Ian Mohr, Department of Microbiology, New York University School of Medicine, New York, NY 10016, USA.
Matthew D Weitzman, Division of Protective Immunity, Department of Pathology and Laboratory Medicine, The Children's Hospital of Philadelphia, Philadelphia, PA 19104, USA; Department of Pathology and Laboratory Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104, USA.
Angus C Wilson, Department of Microbiology, New York University School of Medicine, New York, NY 10016, USA.
Daniel P Depledge, Department of Microbiology, New York University School of Medicine, New York, NY 10016, USA; Institute of Virology, Hannover Medical School, Hannover 30625, Germany; German Center for Infection Research (DZIF), Partner Site Hannover-Braunschweig, Hannover, Germany.
References
- Barbieri I., Kouzarides T. (2020) Role of RNA modifications in cancer. Nat. Rev. Cancer, 20, 303–322. [DOI] [PubMed] [Google Scholar]
- Begik O. et al. (2021) Quantitative profiling of pseudouridylation dynamics in native RNAs with nanopore sequencing. Nat. Biotechnol., 39, 1278–1291. [DOI] [PubMed] [Google Scholar]
- Boccaletto P. et al. (2018) MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res., 46, D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess H.M. et al. (2021) Targeting the m6A RNA modification pathway blocks SARS-CoV-2 and HCoV-OC43 replication. Genes Dev., 35, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depledge D.P. et al. (2019a) Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat. Commun., 10, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depledge D.P. et al. (2019b) Going the distance: optimizing RNA-Seq strategies for transcriptomic analysis of complex viral genomes. J. Virol., 93, e01342–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D. et al. (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature, 485, 201–206. [DOI] [PubMed] [Google Scholar]
- Garalde D.R. et al. (2018) Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods, 15, 201–206. [DOI] [PubMed] [Google Scholar]
- Garcia-Campos M.A. et al. (2019) Deciphering the ‘m6A code’ via antibody-independent quantitative profiling. Cell, 178, 731–747.e16. [DOI] [PubMed] [Google Scholar]
- He P.C., He C. (2021) m6A RNA methylation: from mechanisms to therapeutic potential. EMBO J., 40, e105977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendra C. et al. (2021) Detection of m6A from direct RNA sequencing using a multiple instance learning framework. bioRxiv, 2021.09.20.461055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenjaroenpun P. et al. (2021) Decoding the epitranscriptional landscape from native RNA sequences. Nucleic Acids Res., 49, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukurba K.R., Montgomery S.B. (2015) RNA sequencing and analysis. Cold Spring Harb. Protoc., 2015, 951–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger A. et al. (2021) RNA modifications detection by comparative nanopore direct RNA sequencing . Nat. Commun., 12, 7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B. et al. (2015) Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods, 12, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. et al. (2019) Accurate detection of m6A RNA modifications in native RNA sequences. Nat. Commun., 10, 4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz D.A. et al. (2020) Direct RNA sequencing enables m6A detection in endogenous transcript isoforms at base-specific resolution. RNA, 26, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K.D. (2019) DART-seq: an antibody-free method for global m6A detection. Nat. Methods, 16, 1275–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker M.T. et al. (2020) Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. eLife, 9, e49658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratanwanich,P.N. et al. (2021) Identification of differential RNA modifications from nanopore direct RNA sequencing with xpore. Nat. Biotechnol., 39, 1394–1402. 10.1038/s41587-021-00949-w. [DOI] [PubMed] [Google Scholar]
- Price A.M. et al. (2020) Direct RNA sequencing reveals m6A modifications on adenovirus RNA are necessary for efficient splicing. Nat. Commun., 11, 6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H. et al. (2020) Novel insight into the regulatory roles of diverse RNA modifications: re-defining the bridge between transcription and translation. Mol. Cancer, 19, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas K.P. et al. (2021) Widespread remodeling of the m6A RNA-modification landscape by a viral regulator of RNA processing and export. Proc. Natl. Acad. Sci. USA, 118, e2104805118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman R.E. et al. (2019) Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods, 16, 1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. et al. (2019) Single-base mapping of m6A by an antibody-independent method. Sci. Adv., 5, eaax0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available from the European Nucleotide Archive (ENA), Sequence Read Archive (SRA), and Gene Expression Omnibus (GEO) and can be accessed with the following study accessions: PRJEB35652 (ENA), SRP166020 (SRA), and GSE52681 (GEO).