

Abstract
Sequencing of cell-free DNA (cfDNA) in cancer patients’ plasma offers a minimally-invasive solution to detect tumor cell genomic alterations to aid real-time clinical decision-making. The reliability of copy number detection decreases at lower cfDNA tumor fractions, limiting utility at earlier stages of the disease. To test a novel strategy for detection of allelic imbalance, we developed a prostate cancer bespoke assay, PCF_SELECT, that includes an innovative sequencing panel covering ∼25 000 high minor allele frequency SNPs and tailored analytical solutions to enable allele-informed evaluation. First, we assessed it on plasma samples from 50 advanced prostate cancer patients. We then confirmed improved detection of genomic alterations in samples with <10% tumor fractions when compared against an independent assay. Finally, we applied PCF_SELECT to serial plasma samples intensively collected from three patients previously characterized as harboring alterations involving DNA repair genes and consequently offered PARP inhibition. We identified more extensive pan-genome allelic imbalance than previously recognized in prostate cancer. We confirmed high sensitivity detection of BRCA2 allelic imbalance with decreasing tumor fractions resultant from treatment and identified complex ATM genomic states that may be incongruent with protein losses. Overall, we present a framework for sensitive detection of allele-specific copy number changes in cfDNA.
INTRODUCTION
Prostate cancer is a leading cause of cancer death among men and in the past few years studies investigating the genomic landscape of metastatic prostate cancer have led to the identification of targetable molecular alterations, emerging resistance mechanisms, and new therapeutic options. Following the approval of poly (ADP-ribose) polymerase inhibitors (PARPi) as a therapeutic option in metastatic castration resistant prostate cancer (mCRPC) patients with an alteration in BRCA and, in some situations, other DNA repair genes, genomic biomarker-testing is now routinely performed (1). As fresh biopsies are often impractical or unsafe to obtain, assessment is usually determined using archival tissue specimens. However, the sample taken at diagnosis and therefore prior to treatment may no longer represent the current state of advanced disease due to the acquisition of new genomic alterations and/or the clonal expansion of previously undetectable alterations (2).
Recently, analysis of plasma for circulating tumor DNA (ctDNA), popularly referred to as liquid biopsy, has proven to be a valid alternative to using a tissue biopsy for molecular subtypes characterization (3) with the added advantage of enabling serial testing during the course of therapy, through non-invasive blood draws. Liquid biopsies may also help identify possible resistance mechanisms and detect minimal residual disease (4,5). Furthermore, liquid biopsies may provide a more comprehensive characterization of the patient's tumor that is neither temporally nor spatially restricted as in the case of tissue biopsies with the added advantage of capturing tumor heterogeneity (6).
However, biological and technical issues can influence the ability of ctDNA assays to accurately stratify patients, especially at low ctDNA fractions, especially relevant for tumor types such as mCRPC with a high genomic tumor burden in terms of copy number changes and/or aneuploidy. For instance, low ctDNA fractions limit the accurate detection of copy number changes, making it challenging to discriminate between mono- and biallelic gene loss (7). Heterozygous SNPs loci could be exploited to assess a differential representation of the two alleles in a tumor cell, a feature referred to as allelic imbalance, and can therefore play a key role to distinguish mono- and biallelic gene losses (8–10). Selecting for high minor allele frequency (MAF, i.e. the frequency at which the second most common allele occurs in a population) SNPs increases the probability that a SNP will be heterozygous across diverse individuals. To this end, we posited that a dedicated platform combining a custom targeted sequencing panel and tailored computational approaches exploiting features of allelic imbalance that are patient and gene-specific would increase the quality of signal from ctDNA and ultimately improve the implementation of liquid biopsies in clinical practice for patients with mCRPC.
MATERIALS AND METHODS
Selection of target and control genes for inclusion in the panel
A consortium was supported by The Prostate Cancer Foundation (PCF) to develop a prostate cancer specific plasma assay, PCF_SELECT (Specific Evaluation in Liquid biopsies of Established prostate Cancer Targets). Target genes were selected for inclusion in the panel based on at least one of the following criteria: (i) recurrent copy number changes or point mutation in localized and/or advanced prostate cancer (PCa) based on N = 278 tumors (11–13), (ii) potential therapeutic relevance; overall leading to the selection of 70 autosomal genes and four gene on chromosome X. Further, the genomic region on 21q between TMPRSS2 and ERG that undergoes interstitial deletion as mechanism of gene rearrangement was also selected (Supplementary Table S1). To optimize data quality, processing and downstream analysis, 39 control genes were also selected across all chromosomes and having minimal aberration frequency across the 278 PCa reference dataset. Specifically, for each of the 39 chromosomal arms with available data, upon exclusion of cytobands adjacent to telomeres or centromeres, the top three genes ranked for minimal aberration frequency (abs(log2(T/N)) < 0.5, not altered in >95% of tumors) were considered and finally genes were selected based on maximization of high MAF SNPs availability (see below). In addition, UGT2B17 and ZBTB9 genes, located in complex genomic regions and encompassed by high frequency germline copy number losses were included as internal control genes (14). Throughout the manuscript we will refer to gene-regions rather than to genes, due to the inclusion of flanking regions on a gene basis for enrichment of informative SNPs.
Selection of informative SNPs per gene-region
To allow for tumor purity and ploidy estimations and to improve somatic copy number computation (5,9,15), high MAF SNPs for each target and control gene-region were included in the panel. Intronic and intergenic SNPs from dbSNP v144 with single reference/alternative bases and MAF ≥20% were considered. For each gene, an iterative selection strategy was implemented. Specifically, starting from gene coordinates, the number of high MAF SNPs in the selected genomic area is counted and, if lower than a threshold N, the genomic area is iteratively extended by 10Kbp at both ends to either converge at inclusion of N SNPs or to a maximum extension of 200 kb per side. The value of N was set to 400 for 64 target genes on autosomal chromosomes, to 1000 for target genes of special interest BRCA2, ATM, RB1, NKX3-1, TP53 and PTEN and for the 21q area; to 300 for control/other genes. To optimize the selection of SNPs presenting high MAF across different ethnic groups, genotype data of ∼2,000 samples from the 1,000 Genome Project data were considered. For each SNP, the fraction of individuals with heterozygous genotype status (HetFrac) in all 1,000 Genome project dataset and across four different major ethnic groups (Africans, Europeans, South Asian and East Asian) was computed. For each gene, a subset of M<N SNPs was selected; K<M SNPs were selected among the ones with the highest HetFrac across the different ethnic groups, while M-K SNPs were selected among the ones with the highest HetFrac in the overall dataset. M and K were respectively equal to 200 and 100 for 64 target genes, to 500 and 200 for BRCA2, ATM, RB1, NKX3-1, TP53 and PTEN and for the 21q area, and to 150 and 100 for control/other genes. The numbers of SNPs included by design per gene-region are reported in Supplementary Table S1. SNPs compatible with the selection criteria were not available for the IDH1 gene-region that was included in the panel for SNV detection only. Overall, a total of 18 723 SNPs for target genes and of 8392 SNPs for control genes were selected. Note that the number of SNPs included in the panel also allows for ethnicity inference and annotation (16) and for sample identity check (17). The final panel covers a total of 116 gene-regions and spans a total of 3.49 Mb (1.13 Mb on exonic/intronic regions of targeted genes and 2.36 Mb on flanking regions). All coordinates in Supplementary Tables refer to the reference genome hg19.
Clinical cohorts
Patients in the study were enrolled at 3 institutions on IRB-approved protocols. Patients were eligible for this study if they had confirmed prostate adenocarcinoma receiving treatment with androgen deprivation therapy and provided informed consent to sample collection. Additional criteria were required for specific experiments as detailed in the manuscript text. Overall, the current study included 66 cfDNA samples from 44 patients at Weill Cornell Medical College (WCM, IRB: 1305013903), 16 cfDNA samples from 7 patients at Vancouver Prostate Center (VPC, IRB: H14-00738 and H18-00944), 45 cfDNA samples from 17 patients at University College of London (UCL; REC approval: 20/YH/088), including one patient from whom six tumor samples were harvested post-humously in the Cancer Research UK PEACE study (Posthumous Evaluation of Advanced Cancer Environment; NCT03004755). Plasma from 4 healthy volunteers, with consent for genomic analysis, was sourced from Cambridge Bioscience. In total, 15 samples from four healthy volunteers were sequenced at WCM, UCL, VPC and from three healthy volunteers at Trento (Supplementary Table S2).
Blood collection, plasma separation and whole blood cells isolation
Blood collection was performed with EDTA tubes, kept at 4°C and processed within 2 h from collection. Plasma separation was performed with a double spin protocol—first, the whole blood was centrifuged at 1600 rcf for 15 min at 4°C, then the separated plasma was collected and centrifuged again at 3000 rcf for 10 min at 4°C. The plasma was then divided into 1.8 ml aliquots and stored at −80°C. The buffy coat was collected after the first centrifuge and divided into 250 μl aliquots and stored at −80°C.
cfDNA and gDNA extraction
cfDNA was extracted starting from 1.8 ml plasma with QIAGEN QIAamp Circulating Nucleic Acid Kit according to the manufacturer's protocol and eluted in 30 μl Tris−HCl 10 mM pH 8. The obtained cfDNA was then quantified using Qubit dsDNA High Sensitivity Assay and the quality was assessed with Agilent Bioanalyzer High Sensitivity DNA Kit. gDNA was extracted from 200 μl buffy coat with QIAGEN QIAamp DNA Mini Blood Kit and eluted in 200 μl Tris−HCl 10 mM pH 8. The extracted gDNA was quantified using NanoDrop.
Libraries preparation
gDNA for library preparation was fragmented with Covaris M220. Libraries for target sequencing were prepared starting from 25 and 100 ng cfDNA and gDNA respectively with KAPA HyperPrep Kit (Roche) following the SeqCap EZ HyperCap v2.3 protocol with a few modifications. For probes hybridization, up to 8 cfDNA/gDNA samples were pooled together to obtain a combined mass of 2 μg and incubated for capture at 47°C for 72 h. The captured DNA was then amplified for 13 cycles. Pre- and post-captured libraries were quantified using Qubit dsDNA High Sensitivity Assay and the quality was assessed with Agilent Bioanalyzer High Sensitivity DNA Kit.
ATM mutation detection and copy number estimation using Droplet Digital PCR
Droplet Digital PCR was performed on a QX200 AutoDG droplet digital PCR system (Bio-Rad). Mutation detection was performed for the ATM mutation c748t (p.R250*) using a custom-made SNP genotyping assay (Assay ID ANAAPPE, FAM/VIC, Life Technologies). For copy number estimation the reference genes NSUN3 (dHsaCP2506682, HEX, Bio-Rad) and AP3B1 (dHsaCP2500348, HEX, Bio-Rad) were used. Multiplex assays were set up using the mutation detection assay together with either of the reference genes. PCR reactions were prepared with 4 ng DNA in a total volume of 22 μl and partitioned into ∼20 000 droplets per sample using the Automated Droplet generator (Bio-Rad). The PCR reaction was performed on a Mastercycler Nexus GSX1 (Eppendorf) and read on a Bio-Rad QX100 droplet reader using FAM/HEX settings and analysed with QuantaSoft v1.3.2.0 software (Bio-Rad).
Data generation and pre-processing
Study sample sequencing was performed at institutions facilities at WCM, VPC, UCL and University of Trento with the following platforms Illumina HiSeq for WCM, VPC and, Trento and NovaSeq for UCL, respectively. Detailed information and sequencing statistics are reported in Supplementary Table S2. Paired-end reads were trimmed to remove adapters using trimmomatic (18) (version 0.32). Alignment to the humanG1Kv37 reference genome was performed using BWA-MEM (19) (version 0.7.17-r1188). Duplicate reads were marked and removed with Picard MarkDuplicates (20) (version 1.92). Realignment and recalibration were performed using GATK (20) (version 3.8.0). MD tags were calculated using samtools calmd (21) (version 1.10) and overlapping read pairs clipped using bamUtil (version 1.0.14). PaCBAM (version 1.5.0) was used to generate pileup files (22).
Reference Mapping Bias correction
To correct for the Reference Mapping Bias (23) (RMB) and to ensure proper downstream analysis of allelic fraction (AF) data of informative SNPs (iSNPs), a peak correction was applied separately to control and cfDNA samples. Specifically, a Kernel Density Estimation (KDE, performed on R using the function ‘density’ from ‘stats’ package with bw = ‘SJ’) was applied on the informative SNPs AF distribution and peaks extracted by computing the local maxima of the smoothed distribution; the closest peak to RMB (by default 0.47) is extracted and data centered to the 0.5 theoretical value. RMB correction is applied both for the generation of reference model and the computation of allelic imbalance (Supplementary Figure S1). This correction ensures a proper comparison of AF distributions from independent samples.
Read-depth estimation for wide and focal copy number aberrations
Gene-region based read-depth estimation for the detection of copy number aberrations (CNA)
Gene-region based read-depth estimation is performed similarly to as previously described (5). Briefly, mean amplicon depths of coverage are normalized for both GC content (Supplementary Figure S2) and sample mean coverage. Then, let  and
and 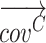 the vectors of normalized amplicon depth of coverage spanning a gene-region respectively in the tumor and matched control sample, the copy number (CN) state (in log2 scale) of the gene region is computed as:
the vectors of normalized amplicon depth of coverage spanning a gene-region respectively in the tumor and matched control sample, the copy number (CN) state (in log2 scale) of the gene region is computed as:
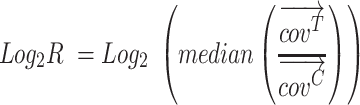 |
To improve confidence in the assessment of copy-number (CN) states, we adapted a previously developed procedure used to assess AR CN state (15). In a nutshell, the procedure measures the probability that an observed CN is compatible with the presence of aberrations accounting for stochastic noise in CN estimations. By computing control vs. control segmentation on WCM cohort, we observed that gene-region are associated with specific noise (Supplementary Figure S3A, B). Hence, we exploited the procedure to define gene-region specific cutoffs (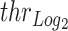 ) by integrating such information. For each gene-region, we defined
) by integrating such information. For each gene-region, we defined  as the value such that:
as the value such that:
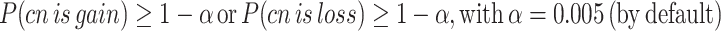 |
The complete list of estimated 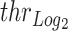 for each gene-region obtained at different levels of
for each gene-region obtained at different levels of 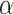 (0.01, 0.05, 0.005, 0.0001 and 0.0005) is reported in Supplementary Table S3.
(0.01, 0.05, 0.005, 0.0001 and 0.0005) is reported in Supplementary Table S3.
Detection of focal CNA
In order to detect CNAs that span areas smaller than entire gene-regions, a simple iterative process was implemented. Given the set of gene-regions  within the study panel (including control and target gene-regions,
within the study panel (including control and target gene-regions,  and
and 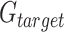 respectively), the set of amplicons within each gene-region (
respectively), the set of amplicons within each gene-region ( ) is subdivided into those spanning exonic/intronic regions (
) is subdivided into those spanning exonic/intronic regions ( ) and into those within the flanking regions (
) and into those within the flanking regions (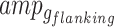 ). For each gene-region
). For each gene-region  in
in 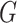 , an iterative procedure is applied to compute values of Log2Ri (Supplementary Figure S4). First, Log2R1 is computed calculating the median of all
, an iterative procedure is applied to compute values of Log2Ri (Supplementary Figure S4). First, Log2R1 is computed calculating the median of all 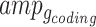 ; next, for each iteration i (for i = 1:(n − 1)), the Log2R1+i is computed by including right and left side windows of size
; next, for each iteration i (for i = 1:(n − 1)), the Log2R1+i is computed by including right and left side windows of size 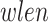 (10 kb, by default). The last iteration (n) corresponds to the inclusion of the entire gene-region.
(10 kb, by default). The last iteration (n) corresponds to the inclusion of the entire gene-region.
Gene-regions in 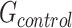 are then used to compute per-gene-regions differences between the Log2Rn and the respective Log2R1 (gene only). The parameters of the reference normal distribution
are then used to compute per-gene-regions differences between the Log2Rn and the respective Log2R1 (gene only). The parameters of the reference normal distribution 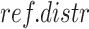 with mean equal to
with mean equal to 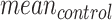 and standard deviation (s.d.) equal to
and standard deviation (s.d.) equal to 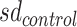 are defined by taking the mean and the s.d. of the Log2R differences, respectively. Last, for each target gene-region in
are defined by taking the mean and the s.d. of the Log2R differences, respectively. Last, for each target gene-region in 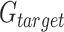 , the
, the 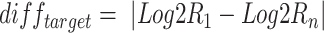 is compared
is compared  times (by default 10 000) against simulated distributions (
times (by default 10 000) against simulated distributions ( , with
, with  ), each built by sampling 10 000 times from
), each built by sampling 10 000 times from 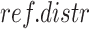 . The probability of focal lesion for each target gene is computed as:
. The probability of focal lesion for each target gene is computed as:
 |
With 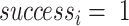 if
if 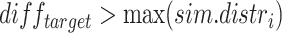 or
or 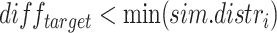 , 0 otherwise. To minimize noise-induced false positives, we only consider a lesion to be focal if
, 0 otherwise. To minimize noise-induced false positives, we only consider a lesion to be focal if 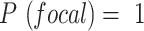 .
.
For each detected focal event, in order to define the exact boundaries, we then proceed as follows: (i) identify the lowest (for losses) and (ii) highest (for gains) Log2Ri within the corresponding gene-region and expand boundaries if two adjacent amplicons present monotonic values on both sides.
Reference model generation for allelic imbalance
For a set of N control samples (white blood cells, genomic DNA from healthy cells), SNPs that are informative (i.e. heterozygous call; 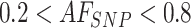 ) in at least two of the N samples are selected (Supplementary Figure S5A). Summary statistics for each informative SNP across control samples are computed. Namely, for the AF distribution, mean, coefficient of variation, and proportion of samples out of N harboring the heterozygous genotype (
) in at least two of the N samples are selected (Supplementary Figure S5A). Summary statistics for each informative SNP across control samples are computed. Namely, for the AF distribution, mean, coefficient of variation, and proportion of samples out of N harboring the heterozygous genotype ( ,
, 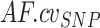 and
and 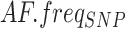 , respectively); for the local coverage distribution, mean (
, respectively); for the local coverage distribution, mean ( ). Further, AF standard deviations stratified by local coverage quantiles Q (min 0%, max 100%, step 10%, quantile interval q) are also computed (
). Further, AF standard deviations stratified by local coverage quantiles Q (min 0%, max 100%, step 10%, quantile interval q) are also computed (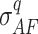 ). To exclude noisy/low quality SNPs, the following filters are applied:
). To exclude noisy/low quality SNPs, the following filters are applied:
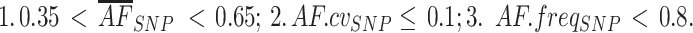 |
The collection of the summary statistics for each SNP is referred to as reference model (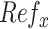 ; where
; where  is one of the statistics). Of note, the reference model needs only to be computed once and can be applied cross-platform. The use of control samples with intended sequencing coverage compared to that of plasma samples is suggested.
is one of the statistics). Of note, the reference model needs only to be computed once and can be applied cross-platform. The use of control samples with intended sequencing coverage compared to that of plasma samples is suggested.
Computation of Allelic Imbalance per gene-region
Allelic imbalance is computed independently for each gene-region and cfDNA sample (Supplementary Figure S5B). First, the set of informative SNPs spanning a gene-region (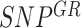 ) is defined retaining only positions with
) is defined retaining only positions with 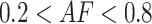 in the matched control and present in the reference model. For each SNP
in the matched control and present in the reference model. For each SNP 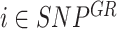 , observed local coverages, corresponding local coverage quantile and mirrored allele fractions in the cfDNA sample are defined as
, observed local coverages, corresponding local coverage quantile and mirrored allele fractions in the cfDNA sample are defined as 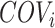 ,
, 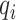 and
and  , respectively.
, respectively.
The evidence of allelic imbalance for the gene-region is computed as:
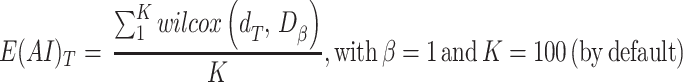 |
where 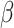 is the proportion of neutral reads (24),
is the proportion of neutral reads (24), 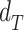 is the observed mirrored AF distribution in cfDNA sample,
is the observed mirrored AF distribution in cfDNA sample,  is a simulated AF distribution generated sampling one time for each
is a simulated AF distribution generated sampling one time for each 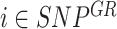 from a Normal distribution with mean
from a Normal distribution with mean  and standard deviation
and standard deviation 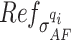 and
and 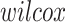 is a function returning 1 if the difference between
is a function returning 1 if the difference between 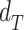 and
and 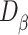 applying a Wilcoxon signed-rank test with significance cutoff of 1% is statistically significant, 0 otherwise.
applying a Wilcoxon signed-rank test with significance cutoff of 1% is statistically significant, 0 otherwise.
Finally, the beta estimate for the gene-region in cfDNA sample (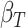 ) is computed by comparing
) is computed by comparing 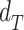 with simulated distributions mimicking levels of local admixture searching for the most similar one. Formally:
with simulated distributions mimicking levels of local admixture searching for the most similar one. Formally:
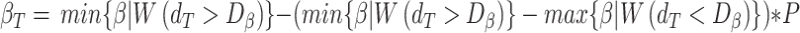 |
with
 |
and where 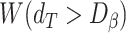 is the Wilcoxon signed-rank statistic (significance cutoff of 1%) comparing
is the Wilcoxon signed-rank statistic (significance cutoff of 1%) comparing 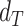 and
and  . Similarly, evidence of allelic imbalance
. Similarly, evidence of allelic imbalance  and beta (
and beta (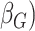 for each gene-region in the matched control sample are computed by substituting
for each gene-region in the matched control sample are computed by substituting 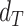 with
with 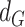 in the equations above with
in the equations above with 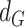 defined as the observed mirrored AF distribution in matched control sample.
defined as the observed mirrored AF distribution in matched control sample.
The allelic imbalance estimated is then coupled with the read-depth estimation to ultimately build the Log2R-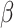 space (Supplementary Figure S5C) of a cfDNA sample.
space (Supplementary Figure S5C) of a cfDNA sample.
Tumor content and ploidy estimation
Tumor content (TC, also referred as ctDNA level) and ploidy estimations for each sample are performed integrating the outputs of the ad hoc procedures presented above within the CLONETv2 framework (24). In case of missing estimation by CLONETv2, the following procedure is applied for the estimation of TC:
Log2R of each target gene-region is corrected for ploidy;
Target gene-regions with
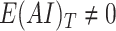 and ploidy-corrected Log2R < −0.05 are retained (i.e. target genes harboring putative hemizygous losses);
and ploidy-corrected Log2R < −0.05 are retained (i.e. target genes harboring putative hemizygous losses);Tumor content is computed as:
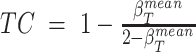 , with
, with 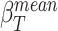 equal to the mean beta value of the selected target gene-regions.
equal to the mean beta value of the selected target gene-regions.
All CLONETv2 ploidy estimates are verified through visual inspection using the Log2R-beta space (see Figure 1D).
Figure 1.

The PCF_SELECT assay. (A) Schematic for selecting gene regions for inclusion in panel. (B) Summary of panel statistics. Genomic sizes and number of high MAF SNPs are reported for target and control gene-regions. (C) Schematics summarizing the main components of the pipeline. Copy-number assessment and gene-region allelic imbalance detection are independently performed; results are integrated and corrected by tumor ploidy and tumor content to generate allele-specific copy number status for each gene-region. (D) Integration of read-depth estimation and allelic imbalance. Top: distribution of Log2R colored by allele-specific copy-number. Bottom: Log2R-beta space obtained by integration of read-depth estimations and allelic imbalance. Each point represents a gene-region. Clusters of points are annotated with their expected allele-specific copy numbers.
Definition of gene-regions allele-specific copy numbers
In order to define the allele-specific CN status of each gene-region, we apply the decision tree depicted in Supplementary Figure S6. Briefly, allele-specific CN is defined by integrating read-depth estimations and allelic imbalance calls. First, a check for the quality of the control samples is performed. Then, the presence of allelic imbalance is assessed and Log2R corrected for ploidy and TC/ctDNA level of the sample (24). Note that ploidy/TC correction is only applied if uncorrected signal supports the presence of aberration (i.e.  ). Moreover, to be conservative if estimated
). Moreover, to be conservative if estimated 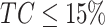 and
and  (i.e. no allelic imbalance detected), the method reports only the likely presence of aberration in a gene-region.
(i.e. no allelic imbalance detected), the method reports only the likely presence of aberration in a gene-region.
To obtain the copy number values of the two alleles, cnA and cnB (by design cnA ≥ cnB) for each gene-region, the following original equations are applied (24):
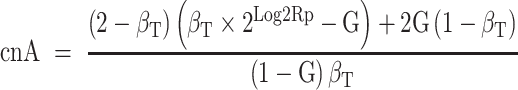 |
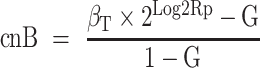 |
where 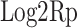 is the ploidy-corrected Log2R of the gene-region and G is the admixture of the sample (i.e. 1 − TC).
is the ploidy-corrected Log2R of the gene-region and G is the admixture of the sample (i.e. 1 − TC).
Computation of allele-specific ploidy (asP)
In low TC samples, CLONETv2 ploidy estimate does not recapitulate the actual amount of DNA per cell and, actual polyploid samples may be classified as diploid. Therefore, we adapted from a previous work an allele-specific informed ploidy (asP) measure based on the allele-specific CN profile of each sample (25), computed as the weighted mean of the allele-specific CN of each gene-region  in the panel, that is:
in the panel, that is:
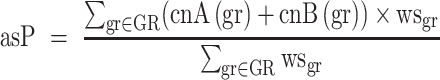 |
where 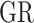 is the set of gene-regions covered by the panel and
is the set of gene-regions covered by the panel and 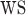 is the genomic size of the gene-region.
is the genomic size of the gene-region.
Detection of somatic and germline mutations
To detect somatic single nucleotide variants (SNVs) we applied ABEMUS (26), a recently developed method specifically designed for SNVs detection in plasma samples. We ran ABEMUS with parameters reported in Supplementary Table S4. To decrease the impact of false positives, we also applied the following filters: (i) local coverage in cfDNA sample >50; (ii) AF in control sample ≤0.01; (iii) exclude positions annotated as SNPs and with MAF >0.01 in dbSNP v144. SNVs were further annotated with Oncotator (27) (version 1.9.6.1) and only non-synonymous SNVs were retained. Germline variants were identified in control samples by looking for positions with AF ≥0.15. Only positions annotated as ‘pathogenic’ in ClinVar were retained (28).
Allele-specific informed copy number calls without matched control sample
PCF_SELECT can also be engaged to detect allele-specific CNAs in the absence of a patient's matched control sample. Specifically, the following components are needed: a panel of normal (PON) to be used as read-depth estimation control, a pre-computed reference model for allelic imbalance computation and a procedure to infer individual's informative SNPs directly from the cfDNA sample. The PON can be easily computed by pooling together a set of non-tumor samples with comparable coverages and by computing the mean coverage of each amplicon across the selected samples. iSNPs can be inferred directly from the cfDNA. In case of high ctDNA levels or specifically in gene-regions harboring loss of heterozygosity (LOH) events, loser thresholds for iSNPs selection (i.e. 0.05 < AF < 0.95) can be applied, while preserving a good proportion of iSNPs calls (Supplementary Figure S7A, B).
RESULTS
High density polymorphisms for sensitive detection of copy number alterations
To enhance the quantification of tumor signal and to enable accurate estimation of copy number changes of prostate cancer relevant genes, we designed a custom targeted sequencing panel named PCF_SELECT that aimed to leverage the patient's genetic background across gene-regions of interest for allelic imbalance estimations. We first used information from large scale prostate cancer genomic studies (11–13) to identify a set of target gene-regions that included genes recurrently (defined as present in at least 5% of tumors) aberrant in localized and/or advanced prostate cancer whole-exome or whole-genome sequencing studies and/or involved in frequently altered or targetable pathways. From the same data sets, we then identified a set of control genes observed as minimally aberrant in prostate cancer that would provide the backbone structure (e.g. wild-type status) for data analysis, including for tumor ploidy and purity estimations (24) (Figure 1A, see Methods).
Next, as allelic imbalance can only be measured through inherited heterozygous loci (here referred to as informative SNPs (iSNPs)) (10,24,29), we used the 1,000 Genome Project data. Specifically, to enrich the design for high minor allele frequency (MAF) SNPs that we hypothesized would maximize the detection sensitivity, we aimed to achieve a minimum of 100 iSNPs per gene-region of interest (including for example TP53, BRCA2, ATM, PTEN) for each patient. For each gene-region, we therefore populated the assay with high MAF SNPs across different ethnic groups in both exonic and intronic areas and increased the number by iteratively adding upstream and downstream regions, aiming for a minimum of 400 high MAF SNPs (see Materials and Methods). We then performed quality checks on individual probes and excluded ones that were predicted to have poor or non-specific binding. At completion, the panel included a total of 116 gene-regions spanning 3.49 Mb (1.13 Mb on exonic/intronic regions and 2.36 Mb on flanking regions) and covering 27 115 high MAF SNPs (Figure 1B), providing a range of iSNPs per each gene-region and individual (Supplementary Figure S8 and S9).
Finally, to accurately assess copy number states of target genes and to increase the sensitivity in detecting imbalances also in low and moderate ctDNA level samples (<15%), we tailored an ad hoc method for allele-specific copy number (asCN) assessment taking full advantage of the panel design. The method integrates (i) a read-depth estimation approach modeling gene-specific sequencing coverage noise and (ii) a gene-region based allelic imbalance detection method leveraging iSNPs upon stringent quality filters, followed by their integration and data correction for tumor ploidy and tumor content (i.e. ctDNA level) (Figure 1C). Briefly, the allelic imbalance detection step embeds the use of a pre-computed reference model that provides local (mean AF per informative SNPs) and global (variation on iSNPs AF by coverage quantiles) statistics of iSNPs and is generated as a one-time procedure using a set of pooled high coverage control samples (Supplementary Figure S5A, see Materials and Methods); for each patient and per each gene-region, the observed iSNPs AF distribution is compared against simulated AF distributions to quantify the allelic imbalance (Supplementary Figure S5C). Last, read depth and allelic imbalance results are passed into a framework for ploidy and purity estimation and adjusted values are used to estimate asCNs as described previously (24,25) Figure 1D, see Materials and Method).
Performance of customized prostate cancer assay
False positive rate (FPR)
To assess the PCF_SELECT assay performance, we sequenced plasma samples (N = 66, median coverage 560×) and white blood cells (WBCs, median coverage 779×) from 44 individuals with mCRPC (‘WCM dataset’, including serial samples) as well as plasma samples from three healthy male age-matched volunteers (median coverage 650×). Intra-patient genomic heterogeneity and tumor evolution directly affect the signal observed in circulation hindering tissue biopsies from being the optimal ground truth for performance assessment. We therefore opted for the use of WBC signal as gold standard proxy for the assessment of FPR, as no copy number changes or allelic imbalance events are expected (with minimal exceptions due to inherited structural changes); we defined each allelic imbalance call in control samples as a false positive to compute the assay FPR. When evaluated in the set of 44 individuals, the performance of the assay significantly differs compared to a coverage-based only approach (McNemar's test, P < 2.2e−16) with FPR of 0.12% and 8.5%, respectively. We next queried the impact of the reference model cardinality (i.e. number of control samples used for reference model generation) on the FPR: we iteratively built a total of 80 reference models by randomly selecting WBC samples from 10, 20, 30 and 40 unique individuals in the WCM dataset at each iteration (20 reference models per each cardinality). We observed an overall low number of false positives (FPR < 0.3%) independently of the reference model cardinality, supporting the benefit of the gene-region based approach and better performances at increasing cardinality with no substantial changes when comparing models built using 20, 30 or 40 control samples (Supplementary Figure S10A). Last, we used healthy volunteer plasma data to assess the feasibility of a cross-platform reference model (i.e. combination of sequencing platform and sequencing site). We applied reference models built with control samples sequenced at three institutions (‘WCM’, ‘UCL’ and ‘VPC’) on 3 healthy volunteer samples sequenced independently at four institutions (‘WCM’, ‘UCL’, ‘VPC’, Trento) and we checked for consistency in gene-region specific information. Inconsistencies were observed only for gene-regions with fewer than 30 iSNPs available (Supplementary Figure S10B).
Increasing number of iSNPs is associated with improved detection of allelic imbalance
To measure the impact of the high MAF SNPs-enriched design on allelic imbalance detection, we applied the approach on the WCM dataset by using for each target gene-region all the available iSNPs per individual and by considering randomly selected subsets (i.e. 20, 40, 60 and 80%). Results confirmed that a higher number of iSNPs was associated with enhanced detection of imbalance events in tumor samples (Figure 2A). Of note, when we applied our strict selection criteria (see Materials and Method) on SNPs within exonic regions, only few positions (min. 0, max. 8) met the standards making it impossible to call imbalance and suggesting that this approach would not be feasible with whole-exome sequencing data (Figure 2A). When we focused on a set of representative gene-regions, including both gene-regions of special interest (selected based on their prevalence and/or potential therapeutic relevance) such as PTEN and TP53, and other genes (e.g. CDK6, CDKN1B and CUL1), this analysis corroborated the previous results highlighting the impact of the number of iSNPs on allelic imbalance detection. In particular, detection performance decreased significantly by lowering the number of SNPs (average 6% and 30% reductions with 60% and 20% of initial SNPs, Figure 2B and Supplementary Figure S11A; Wilcoxon signed-rank test, P = 3.3 × 10–5 and P = 5.4 × 10–8). Moreover, independently of the estimated proportion of tumor reads, a higher number of SNPs led to higher confidence in calling allelic imbalance (Supplementary Figure S11B).
Figure 2.

Assessment of allelic imbalance (AI) calls using high density iSNPs achieves increased accuracy and sensitivity. (A) Percentage of AI calls as a function of number of informative SNPs (iSNPs) available for that gene-region and sequential bars showing detection at decreasing percentage of iSNPs (80%, 60%, 40%, 20%) and using only iSNPs spanning exonic region simulating whole-exome sequencing (WES) data (ranges of SNPs used are reported). Values are shown for both control germline and cfDNA samples (green and red boxes at the bottom of each bar, respectively). Reported proportions are relative to the number of AI calls obtained using all the iSNPs available for each gene in the PCF_SELECT panel (i.e. 904). The number of AI calls is shown on top of each bar. AI calls are stratified for the number of iSNPs used (bar colors). (B) Evidence of allelic imbalance (E(AIT), y-axis) for representative gene-regions at varying percentages of iSNPs (WCM cohort, N = 66). Lines color shade indicates ctDNA level of the sample. Donuts show proportion of evidence of imbalance per percentage of iSNPs used. Data for all gene-regions are shown in Supplementary Figure S10A. (C) Proportion of recovered AI calls in synthetically diluted samples. Tumor fraction levels are reported on x-axis. Calls are stratified by allele-specific CN call as established in the real cfDNA samples subjected to dilution.
Allelic imbalance detection as a function of sequencing coverage
To evaluate how the sequencing coverage impacts on allelic imbalance detection, we randomly selected five tumor samples from the WCM dataset sequenced at a median coverage of at least 500x for each of the following ctDNA level intervals: (15,25], (25,50], (50,100] for a total of 15 samples. Starting from the original bam files, we generated a total of 90 bam files at the following coverages: 500, 250, 100, 50, 25, 10×. We evaluated performances in allelic imbalance detection in terms of FPR and sensitivity using the original bam files calls. We observed an increase in FPR starting at 25×, but no difference in sensitivity at decreasing sequencing read depth (Supplementary Figure S12).
Allelic imbalance detection as a function of tumor content calculated using synthetic dilutions
To evaluate the performances of the method for allelic imbalance detection in the context of varying ctDNA fractions, we synthetically admixed tumor reads from five cfDNA samples from the WCM cohort with reads from the matched control sample. We generated a total of 100 synthetically diluted samples spanning ctDNA levels from 20% to 1%. We monitored the ability of the method to consistently detect allelic imbalance at diminishing ctDNA levels. Of note, the method was able to detect a signal of imbalance at a ctDNA level as low as 5% with more than 50% of imbalance calls recovered at 15% ctDNA level (Supplementary Figure S13). Of note, all the imbalance calls were observed in gene-regions with imbalance detected also in the undiluted sample. This result was highly consistent with the low FPR observed in previous analyses and reconfirmed the high specificity of the method in detecting allelic imbalance.
We further characterized the ability of the method to detect evidence of imbalance as a function of ctDNA level by focusing on four highly recurrent aberrant gene-regions known to be early (i.e. NKX3-1 on chromosome 8p and chromosome 21q intronic space between TMPRSS2 and ERG) and late events (i.e. TP53 on chromosome 17 and RB1 on chromosome 13q) in prostate cancer progression (Supplementary Table S1) and previously detected in the circulation of mCRPC patients. The detection of imbalance varied across patients and across gene-regions (Supplementary Figure S14). This detection variability was also observed when we stratified allelic imbalance calls by allele-specific CN states of the gene-regions. For instance, we obtained a higher sensitivity for detection of copy neutral loss of heterozygosity (CNNL, i.e. copies of allele A and B equal to 2 and 0, respectively; down to 5% ctDNA level) when compared with hemizygous deletions (i.e. copies of alleles A and B equal to 1 and 0, respectively; down to 7% ctDNA level) (Figure 2C). Together, these results support the presence of a gene- and patient-specific signal affecting the ability of the method to detect imbalance. In addition, the number of available iSNPs together with lesion clonality (associated with intra-patient heterogeneity), and sequencing coverage depth are all factors that could affect the ability to detect aberrations.
Comparison of serial samples using PCF_SELECT and an independent assay
To then assess the sensitivity of PCF_SELECT in human samples with a range of ctDNA fractions, we studied serial samples (N = 9, median coverage 633×) from three patients starting first-line second-generation hormone treatment (line 1) for mCRPC in a clinical trial (NCT02125357) and selected based on high ctDNA fraction at the first timepoint, a reduction in fraction at the second timepoint due to treatment response and a third timepoint characterized by a rise following clinical progression. Analysis was first performed using Log2R calls (primarily in exonic regions) to estimate copy number change, originally reported in Annala et al. (30), and hereafter referred to as the standard assay. We first compared the copy number calls from the standard assay with the PCF_SELECT assay and observed overall concordant results (105/109 copy number aberrations, 96.33%). However, notably, we also observed some differences as a result of the PCF_SELECT panel specific design.
Patient #110
PCF_SELECT but not the standard assay detected hemizygous deletions of TP53 and CHD1 at the earliest time point (Timepoint-1), both potentially clinically relevant calls. Both calls were confirmed by both assays at Timepoint-3 when the estimated ctDNA level was higher (51% versus 34%) (Figure 3A, B). PCF_SELECT, but not the standard assay, detected copy number changes in NKX3.1, CDK12 and BRCA1 at Timepoints-1 and -3 but not at Timepoint 2 when ctDNA was not detectable. The PCF_SELECT BRCA2 call at Timepoint-2 did not find support in other timepoints.
Figure 3.

Comparative overview of somatic copy-number aberrations (SCNA) calls on three serial samples from two CRPC patients using two independent assays (A). SCNA and SNVs calls detected by the two assays (Standard Log2ratio assay from Annala et al. upper row; PCF_SELECT, lower row) are reported for serial samples with varying ctDNA levels as determined by PCF_SELECT. Gene-regions are grouped by pathways and sorted alphabetically. (B). Log2R-beta spaces for patient #110. CHD1 and TP53 are highlighted in red and blue, respectively. Bottom panels show the mirrored allelic fraction distribution of the informative SNPs (iSNPs) spanning CHD1 and TP53 for both cfDNA (red) and matched control (green) samples. (C) Log2R-beta spaces for patient #134. PTEN and RB1 are highlighted in red and blue, respectively. Bottom panels show the mirrored allelic fraction distribution of the iSNPs spanning PTEN and RB1 for both cfDNA (red) and matched control (green) samples.
Patient #134
PCF_SELECT detected copy number changes in MYC, RB1, and PTEN at all three timepoints (estimated ctDNA fractions: 28%, 7% and 17% for the three respective time-points) whilst the standard assay detected gain of MYC and loss of RB1 only at Timepoint-1 and loss of PTEN only at Timepoint-1 and -3 (Figure 3A). PCF_SELECT also detected deletion and a non-synonymous SNV (p.M237I, VAF = 0.13) at Timepoint-1 and -3 of TP53, suggesting biallelic loss and consequent complete loss of protein, whilst the standard assay detected the SNV but did not detect the copy number loss (Supplementary Figure S15). PCF_SELECT also consistently detected a PTEN homozygous deletion at all time points whilst the standard assay detected a hemizygous deletion at Timepoint-1 and -3 but not Timepoint-2. Close inspection of the Log2R-beta space and of the distribution of iSNPs allelic fractions supports deletion of both alleles: no significant shift was observed in the distribution across neither of the 3 timepoints when comparing cfDNA samples against the matched control samples (Figure 3C).
Patient #55
Using PCF_SELECT we detected a polyploidy signal in each of the three timepoints (ploidy of 2.78, 2.19 and 2.51 for Timepoints-1, -2 and -3, respectively) (Supplementary Figure S16A–C). Different attributions of ploidy resulted in discordance of multiple calls with the standard assay, including hemizygous deletion in MSH2, MSH6 and FOXP1 in the standard assay that were not compatible with the PCF_SELECT ploidy assumptions. Detailed visual analysis of the three regions supported the PCF_SELECT copy number neutral call (Supplementary Figure S16D).
Detection in plasma DNA of allelic imbalance at mutated DNA repair genes in serial samples with decreasing tumor fraction
We then applied PCF_SELECT to serially-collected samples (median coverage 960×) from three patients treated with a PARPi (niraparib 300mg once daily) based on prior detection of a qualifying alteration in BRCA2 (one patient) or ATM (2 patients) in a previously reported Phase II clinical trial (31). From the BRCA2-aberrant patient (TR029), we profiled 8 samples collected over 250 days from prior to treatment initiation to clinical progression (Figure 4A). In 4/8 (50%) sequenced samples with assessable ctDNA (levels ranging from 7 to 56%), PCF_SELECT identified the presence of allelic imbalance in the BRCA2 gene-region (Figure 4C). From these, in 3/4 samples with sufficient ctDNA for allele-specific analysis PCF_SELECT consistently identified a hemizygous deletion (cnA = 1, cnB = 0) of the BRCA2 gene-region. The deletion was accompanied by a pathogenic missense germline mutation (p.E2663V, VAF = 0.47 in the control sample; Figure 4B-D). Through CN-based correction of VAF, we determined deletion of the BRCA2 non-mutated allele in the tumor (CN-corrected VAF = 1; Figure 4D) with predicted loss of function. Clinically, the patient experienced a PSA decline of 20% along with ctDNA. Treatment was interrupted after 90 days due to toxicity and the next plasma sample collected after 14 days showed a rebound in ctDNA levels from undetectable to 20% and a rise in serum PSA. ctDNA levels decreased to undetectable after treatment re-initiation. The patient progressed clinically at a central nervous system metastasis after 250 days treatment whilst remaining stable on whole-body CT and bone scans and ctDNA remained undetectable. Our in vivo serial dilution following response to niraparib confirmed detection of allelic imbalance involving BRCA2 at tumor fractions as low as 7%. As ctDNA decreased further, we were unable to consistently confirm allelic imbalance at this locus.
Figure 4.

In vivo serial sampling in a BRCA2-mutant CRPC patient (TR029) treated with niraparib confirms enhanced sensitivity of detection of BRCA2 allelic imbalance using PCF_SELECT. (A) Changes in serum prostate specific antigen (PSA) and ctDNA levels over 250-day period of niraparib treatment for mCRPC. – dotted line refers to treatment interruption for toxicity. (B) BRCA2 copy number (CN) status. The allele-specific CN space of plasma samples with estimated ctDNA level > 15% is shown; BRCA2 gene-region corresponding status is highlighted. (C) Mirrored allelic fractions (AF) distribution of informative SNPs (iSNPs) within the BRCA2 gene-region. Dashed lines represent the mirrored AF median. Left panel corresponds to germline signal derived from peripheral blood mononuclear cells (green); subsequent panels show signal in sequential plasma samples and are ordered by time from first to last. (D) Variant allele frequency (VAF) of BRCA2 p.E2663V germline mutation from plasma samples. Dotted green line represents the VAF observed in the matched control sample. *Indicates manually imputed CN-corrected VAF.
From the first ATM-aberrant patient (TR067), we profiled 9 plasma samples collected prior to start of and after progression with docetaxel chemotherapy (six 3-weekly cycles), repeatedly whilst receiving his next line of treatment with niraparib and then before and after his next (and last) line of treatment, with dexamethasone (0.5 mg daily). At death, fresh frozen tissue samples from the prostate and metastatic sites were harvested in the PEACE posthumous study. We subjected nine plasma and six tissue samples to high-coverage targeted capture using the PCF_SELECT assay (Figure 5A). Allele-specific analysis revealed the presence of three ATM copies (two copies of one allele and one of the other) within aneuploid genomes (min. asPloidy = 3.17, max. asPloidy = 4.09) accompanied by a non-sense mutation harbored in the non-gained allele (CN-corrected VAF ≈ 33%) (Figure 5B–D). These results were consistently confirmed across all assessable plasma samples (ctDNA level ≥ 15%) and metastatic tissue samples. Of note, the complex ATM CN status was not confirmed in the prostate sample, that showed wild-type, non-mutant ATM without allelic imbalance and a diploid genome (asP = 2.2, Figure 5B–D). In contrast, we observed the same allelic imbalance breakpoints between ERG and TMPRSS2, known to be an early event in prostate cancer (32,33), in both the prostate and metastases. These data suggest that the clone dominant in metastases at death, characterized by a tetraploid genome and an ATM mutation on the non-gained allele, evolved from the prostate tumor biopsied posthumously that remained in situ but with no evidence of distant spread to plasma or sampled metastases (Supplementary Figure S17).
Figure 5.

PCF_SELECT design identifies allelic imbalance secondary to gain of ATM wild-type allele in ATM-mutant CRPC patients. (A, E) Changes in serum prostate specific antigen (PSA) and ctDNA levels over a 550-day period of treatment with docetaxel followed by niraparib (TR067 and TR081) and radium223 (TR081); black diamond indicates time of death. The inset in A shows the summary of the tissue samples collected after death and profiled with PCF_SELECT from TR067 who participated in the PEACE trial. (B) ATM copy number (CN) status in patient TR067. The allele-specific CN space of plasma and tissue samples is shown; ATM gene-region corresponding status is highlighted; for plasma samples average CN and standard deviations are reported. (C) Mirrored allelic fractions (AF) distribution of patient's ATM gene-region informative SNPs (iSNPs). Dashed lines represent the mirrored AF median. Allele-specific ploidy and ctDNA level/TC of each sample are reported on top of the plot. TC: tumor content. (D) CN-corrected variant allele frequency (VAF) of ATM p.R2763* mutation. (F) ATM and TP53 copy number (CN) status in patient TR081. The allele-specific CN space of plasma samples is shown; ATM and TP53 gene-regions corresponding statuses are highlighted; average CN and standard deviations are reported. (G) CN-corrected variant allele frequency (VAF) of ATM p.R2763* and TP53 p.R175G mutations. (H) Observed events in TR081 plasma samples using bespoke droplet digital PCR multiplex assays with probes for ATM p.R250* mutation and wild type and two control genes (AP3B1, top panel or NSUN3, bottom panel).
From the second ATM-aberrant patient (TR081), we profiled nine samples collected before and during three sequential lines of treatments, including PARP inhibition with niraparib (Figure 5E). Allele-specific analysis on each sample similarly as for TR067 revealed complex ATM status. The gene region consistently presents three copies (two copies of one allele and one of the other), within aneuploid genomes (min. asPloidy = 2.51, max. asPloidy = 3.80; Supplementary Table S5) and a non-sense mutation (p.R250*) harbored on the non-gained allele: the CN-corrected VAF was consistently ≈ 33% and the mutant allele frequency tracked changes in ctDNA level (Figure 5F-G). Despite no decrease in serum PSA, we observed a transient decrease in ctDNA fraction and ATM mutant VAF after starting niraparib. We developed bespoke multiplex droplet digital PCR assays and confirmed in TR081 plasma samples the decrease in ATM mutant VAF following treatment initiation (Figure 5H, Supplementary Figure S18A–D). We also confirmed that at high ctDNA fractions, the sum of ATM wild-type and mutant copies was equivalent to reference gene copies, supporting the prediction of ATM copy number made by the PCF_SELECT assay. We also consistently identified CNNL (2 copies of one allele and 0 of the other) spanning TP53 and accompanied by a clonal missense mutation on both allele copies (CN-corrected VAF = 100%), suggesting full-impairment of TP53 (Figure 5F–G). Clinically, neither ATM-altered patient showed evidence of benefit from PARPi, with no decline in PSA and the absence of a radiological response. Although the complex ATM copy number state does not necessarily explain the absence of tumor response, it is likely to be incongruent with loss of ATM protein.
DISCUSSION
By accounting for estimated tumor content and ploidy, well-established and intertwined confounding factors for the accurate detection of the CN state of a gene (8,9,24), and further integrating CN with deleterious SNVs detected, the combination of the PCF_SELECT sequencing panel and computational method showed enhanced ability to detect allelic imbalance events, a crucial step to enable accurate estimation of asCNAs. This provides a framework to discriminate between complex asCNAs in samples with low ctDNA level, for example homozygous deletions (no imbalance) and hemizygous deletion (in which imbalance is present). Compared to other methods designed to infer CNs, such as PureCN (34) and CNVkit (35), our approach combines a specifically designed targeted assay to exploit the high number of SNPs and tailored computations that exploit the informative SNPs. Using synthetic simulations, serial samples from responding patients (in vivo dilutions) and comparisons with previously reported assays (30), we verified an increased ability to detect lesions at low ctDNA level and with complex copy number states. We observed more extensive pan-genome allelic imbalance than previously recognized in prostate cancer, in keeping with a recent pan-cancer study of >4000 tissue samples (25). Previous work suggests that LOH involving certain tumor suppressor genes is more likely to result in reduced mRNA expression (25). The functional impact of LOH observed at specific genes in ctDNA requires more investigation but could prove relevant for improved patient selection for targeted treatments.
The strategy was designed to include multiple genes of relevance to precision medicine for prostate cancer. By focusing only on prostate cancer relevant target genes and with the aim to enrich the assay for high MAF SNPs, we did not include telomeric regions, albeit they could be of interest in prostate cancer (36). Thus, our assay is not suited for the investigation of telomeres length. Of note, low complexity regions were excluded at the time of design by the manufacturers’ (Roche) stringency filters. Also noteworthy, we accounted for racial disparities by optimizing the SNPs selection for the four major ethnicities. By testing serial samples from a BRCA2 mutant mCRPC patient, we performed an in vivo serial dilution experiment and established lower thresholds for detection of allelic imbalance in BRCA2. The enhanced sensitivity could expand the utility and accuracy of liquid biopsy for patient selection for PARPi treatment. The efficacy of PARPi in ATM mutant CRPC is uncertain with limited responses reported (1,37). Most clinical trials in prostate cancer have selected patients based on the presence of a pathogenic mutation in ATM with or without CN change. Complex CN changes with intra-patient inter-tumor differences in ATM status, such as in the two patients presented in our study, may have led to patient accrual in the absence of gene loss. Future studies could evaluate how common this genomic feature is in mCRPC populations and its impact on ATM protein and PARPi response. Moreover, since our method relies on the theoretical framework proposed by CLONETv2 (24), our approach is suited for the detection of subclonal events that could potentially be of interest to better understand patient's response to targeted treatment such as PARPi. AKT inhibitors are being developed for mCRPC (38) and similarly our approach could be implemented for improved selection of patients harbouring genomic loss of PTEN detected in plasma. This could have several advantages over archived tissue analysis. Similarly, assessment of RB1 and TP53 CN status could be implemented for detection of aggressive variants of prostate cancer that require bespoke treatment changes (39). As allelic imbalance estimates utilize information from large numbers of iSNPs, they are not subject to bias caused by single base position changes in WBC clones (clonal haematopoiesis of indeterminate potential, CHIP).
Despite the specificity achieved in detecting genomic aberrations, insufficient ctDNA may be observed in patients responding to systemic therapy or those with low burden of disease. Given the low false positive rate observed on serial plasma samples, a tumor-informed approach could be applied to mitigate the problem by lowering the threshold on the  ; this is evident for instance for the confirmation of aberrations observed at prior time-points as in the case of BRCA2 (third sample from patient TR029). Moreover, to either reduce the running cost of patient management or to comply with guideline indications, PCF_SELECT could in principle be run without a matched control sample upon previous generation of a panel of normal to be used as control for read-depth estimation and by determining the informative SNPs directly on the cfDNA sample. However, since no matched control is available, the integration of CN and SNVs could be potentially confounded by CHIP mutations leading to misinterpretations of the genomic status of a gene. The exclusion of genomic positions known to be recurrently affected by CHIP mutations could represent a way to partially overcome this problem.
; this is evident for instance for the confirmation of aberrations observed at prior time-points as in the case of BRCA2 (third sample from patient TR029). Moreover, to either reduce the running cost of patient management or to comply with guideline indications, PCF_SELECT could in principle be run without a matched control sample upon previous generation of a panel of normal to be used as control for read-depth estimation and by determining the informative SNPs directly on the cfDNA sample. However, since no matched control is available, the integration of CN and SNVs could be potentially confounded by CHIP mutations leading to misinterpretations of the genomic status of a gene. The exclusion of genomic positions known to be recurrently affected by CHIP mutations could represent a way to partially overcome this problem.
Enhanced sensitivity with our approach for detecting allelic imbalance events is not specific to prostate cancer or males. Gene regions that are relevant to other cancer types such as genes involved in DNA repair for breast cancer could be incorporated in other cancer specific or pan-cancer panels for testing of liquid biopsies or other material such as tissue or urine. In summary, our work provides a comprehensive assay for prostate cancer patient management suitable for diverse purposes from stratification to monitoring and prognostication. Currently, our approach is specifically designed for prostate cancer, but it could be generalized to other cancer types with appropriate tailoring of target selection.
DATA AVAILABILITY
Processed data will be provided following review of request to the corresponding authors.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the valuable support of the PEACE consortium and the UCH Biobank team. We also thank our patient representative (Vernon Trafford) who gave input to the applicability of the assay, Caterina Nardella for support and co-ordination, and Yari Ciani for critical input on the assay. We also thank the patients and their families who through their gift allowed this research.
Author contributions: F.D., G.A., H.B., A.W. and M.A.R. conceived the study and obtained funding. F.D., A.R. and F.O. designed the analytical and computational solutions and performed the computational analysis with input from G.A., H.B., A.W., M.A.R., M.S. and D.W. B.T., D.W., M.S., G.M., O.Q., J.Z.X. performed NGS. D.W. performed droplet digital PCR experiments. B.T., S.T., M.L., H.B., G.A. provided patient samples. M.J-H. and C.S. are the PEACE chief investigators. F.O., B.T., G.A. and F.D. drafted the manuscript. All authors commented on and accepted the manuscript.
Contributor Information
Francesco Orlando, Department of Cellular, Computational and Integrative Biology, University of Trento, Trento, Italy.
Alessandro Romanel, Department of Cellular, Computational and Integrative Biology, University of Trento, Trento, Italy.
Blanca Trujillo, UCL Cancer Institute, University College London, London, UK; Department of Medical Oncology, University College London Hospitals, London NW1 2BU, UK.
Michael Sigouros, Englander Institute for Precision Medicine, Presbyterian Hospital, Weill Cornell Medicine, NY, USA.
Daniel Wetterskog, UCL Cancer Institute, University College London, London, UK.
Orsetta Quaini, Department of Cellular, Computational and Integrative Biology, University of Trento, Trento, Italy.
Gianmarco Leone, UCL Cancer Institute, University College London, London, UK; Department of Medical Oncology, University College London Hospitals, London NW1 2BU, UK.
Jenny Z Xiang, The Genomics Resources Core Facility, Department of Microbiology and Immunology, Weill Cornell Medicine. NY, NY, USA.
Anna Wingate, UCL Cancer Institute, University College London, London, UK.
Scott Tagawa, Department of Medicine, Division of Hematology and Medical Oncology, Weill Cornell Medicine. NY, NY, USA.
Anuradha Jayaram, UCL Cancer Institute, University College London, London, UK; Department of Medical Oncology, University College London Hospitals, London NW1 2BU, UK.
Mark Linch, UCL Cancer Institute, University College London, London, UK; Department of Medical Oncology, University College London Hospitals, London NW1 2BU, UK.
Mariam Jamal-Hanjani, Department of Medical Oncology, University College London Hospitals, London NW1 2BU, UK; Cancer Metastasis Laboratory, University College London Cancer Institute, London, UK; Cancer Research UK Lung Cancer Centre of Excellence, UCL Cancer Institute, London, UK.
Charles Swanton, UCL Cancer Institute, University College London, London, UK; Department of Medical Oncology, University College London Hospitals, London NW1 2BU, UK; The Francis Crick Institute, London NW1 1AT, UK.
Mark A Rubin, Department for BioMedical Research and Bern Center of Precision Medicine, University of Bern and Inselspital, Bern, Switzerland.
Alexander W Wyatt, Vancouver Prostate Centre, Department of Urologic Sciences, University of British Columbia, Vancouver, BC, Canada.
Himisha Beltran, Englander Institute for Precision Medicine, Presbyterian Hospital, Weill Cornell Medicine, NY, USA; Department of Medical Oncology, Dana Farber Cancer Institute, Boston, MA, USA.
Gerhardt Attard, UCL Cancer Institute, University College London, London, UK; Department of Medical Oncology, University College London Hospitals, London NW1 2BU, UK.
Francesca Demichelis, Department of Cellular, Computational and Integrative Biology, University of Trento, Trento, Italy; Englander Institute for Precision Medicine, Presbyterian Hospital, Weill Cornell Medicine, NY, USA.
PEACE Consortium:
Charles Swanton, Mariam Jamal-Hanjani, Chris Abbosh, Simone Zaccaria, Sonya Hessey, Kai-Keen Shiu, John Bridgewater, Daniel Hochhauser, Martin Forster, Siow-Ming Lee, Tanya Ahmad, Dionysis Papadatos-Pastos, Sam Janes, Peter Van Loo, Katey Enfield, Nicholas McGranahan, Ariana Huebner, Sergio Quezada, Stephan Beck, Peter Parker, Tariq Enver, Robert E Hynds, Krijn Dijkstra, David R Pearce, Mary Falzon, Ian Proctor, Ron Sinclair, Chi-wah Lok, Zoe Rhodes, David Moore, Teresa Marafioti, Miriam Mitchison, Peter Ellery, Monica Sivakumar, Mark Linch, Sebastian Brandner, Andrew Rowan, Crispin Hiley, Selvaraju Veeriah, Heather Shaw, Gert Attard, Cristina Naceur-Lombardelli, Antonia Toncheva, Paulina Prymas, Tom Watkins, Chris Bailey, Carlos Martinez Ruiz, Kevin Litchfield, Maise Al-Bakir, Nnenna Kanu, Sophie Ward, Emilia Lim, James Reading, Benny Chain, Blanca Trujillo Alba, Tom Watkins, Melek Akay, Adrienne Flanagan, Dhruva Biswas, Oriol Pich, Michelle Dietzen, Clare Puttick, Emma Colliver, Alistair Magness, Mihaela Angelova, James Black, Olivia Lucas, William Hill, Wing-Kin Liu, Alexander Frankell, Neil Magno, Foteini Athanasopoulou, Gareth Wilson, Rachel Rosenthal, Roberto Salgado, Claudia Lee, Kristiana Grigoriadis, Othman Al-Sawaf, Takahiro Karasaki, Abigail Bunkum, Imran Noorani, Sarah Benafif, Vittorio Barbe, Supreet Bola, Osvaldas Vainauskas, Anna Wingate, Daniel Wetterskog, Mahedi Hasan, Stefano Lise, GianMarco Leone, Anuradha Jayaram, Constantine Alifrangis, Ursula McGovern, Kerstin Thol, Samuel Gamble, Seng Kuong Ung, Teerapon Sahwangarrom, Claudia Peinador Marin, Sophia Wong, Piotr Pawlik, Faye Gishen, Adrian Tookman, Paddy Stone, Caroline Stirling, Samra Turajlic, James Larkin, Lisa Pickering, Andrew Furness, Kate Young, Will Drake, Kim Edmonds, Nikki Hunter, Mary Mangwende, Karla Pearce, Lauren Grostate, Lewis Au, Lavinia Spain, Scott Shepherd, Haixi Yan, Ben Shum, Zayd Tippu, Brian Hanley, Charlotte Spencer, Max Emmerich, Camille Gerard, Andreas Michael Schmitt, Lyra Del Rosario, Eleanor Carlyle, Charlotte Lewis, Lucy Holt, Analyn Lucanas, Molly O'Flaherty, Steve Hazell, Hardeep Mudhar, Christina Messiou, Arash Latifoltojar, Annika Fendler, Fiona Byrne, Husayn Pallinkonda, Irene Lobon, Alex Coulton, Anne Laure Cattin, Daqi Deng, Geoffrey Hugang Feng, Andew Rowan, Nadia Yousaf, Sanjay Popat, Olivia Curtis, Charlotte Milner-Watts, Gordon Stamp, Antonia Toncheva, Emma Nye, Aida Murra, Justine Korteweg, Denise Kelly, Lauren Terry, Jennifer Biano, Kema Peat, Kayleigh Kelly, Peter Hill, Debra Josephs, Sheeba Irshad, Ashish Chandra, James Spicer, Ula Mahadeva, Anna Green, Ruby Stewart, Lara-Rose Iredale, Tina Mackay, Ben Deakin, Debra Enting, Sarah Rudman, Sharmistha Ghosh, Lena Karapagniotou, Elias Pintus, Andrew Tutt, Sarah Howlett, Vasiliki Michalarea, James Brenton, Carlos Caldas, Rebecca Fitzgerald, Merche Jimenez-Linan, Elena Provenzano, Alison Cluroe, Anna Paterson, Sarah Aitken, Kieren Allinson, Grant Stewart, Ultan McDermott, Emma Beddowes, Tim Maughan, Olaf Ansorge, Peter Campbell, Patricia Roxburgh, Sioban Fraser, Andrew Kidd, Kevin Blyth, John Le Quesne, Matthew Krebs, Fiona Blackhall, Yvonne Summers, Pedro Oliveira, Ana Ortega-Franco, Caroline Dive, Fabio Gomes, Mat Carter, Jo Dransfield, Anne Thomas, Dean Fennell, Jacqui Shaw, Babu Naidu, Shobhit Baijal, Bruce Tanchel, Gerald Langman, Andrew Robinson, Martin Collard, Peter Cockcroft, Charlotte Ferris, Hollie Bancroft, Amy Kerr, Gary Middleton, Joanne Webb, Salma Kadiri, Dr Peter Colloby, Bernard Olisemeke, Rodelaine Wilson, Ian Tomlinson, Iain McNeish, Sanjay Jogai, Samantha Holden, Tania Fernandes, Blanche Hampton, Mairead McKenzie, Allan Hackshaw, Abby Sharp, Kitty Chan, Laura Farrelly, Hayley Bridger, and Rachel Leslie
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
Challenge Award from the Prostate Cancer Foundation (to G.A., H.B., F.D., A.W., M.A.R.); Accelerator Award from AIRC [22792 to F.D.]; CRUK [A26822 to G.A.]; the PEACE study is supported by a CRUK Accelerator Award [C416/A21999 to C.S., M.J-H.]; National Institute for Health Research (NIHR) Biomedical Research Centre at University College London Hospital (G.A., M.L., C.S., M.J-H.); G.A. was supported by a Cancer Research UK advanced clinician scientist fellowship [A22744]; M.J-H. is a Cancer Research UK Career Establishment Awardee; C.S. is Royal Society Napier Research Professor [RSRP\R\210001].
Conflict of interest statement. A.R., M.A.R., A.W., H.B., G.A. and F.D. are listed as co-inventors on a patent application pending approval related to this work (P032355GB). G.A. reports personal fees, grants and/or travel support from Janssen and Astellas; personal fees and/or travel support from Pfizer, Novartis, ESSA Pharmaceuticals, Bayer Healthcare Pharmaceuticals, Takeda, Astra Zeneca and Sanofi-Aventis; in addition, G.A.’s former employer, The Institute of Cancer Research, receives royalty income from abiraterone and G.A. receives a share of this income through the ICR’s Rewards to Discoverers Scheme. G.A. and D.W. are co-inventors on a ctDNA methylation patent (GB2020/052706) and G.A. is a co-founder of a company for ctDNA analysis (Cansor Ltd). H.B. has served as consultant/advisory board member for Janssen, Sanofi Genzyme, Astellas, Astra Zeneca, Merck, Pfizer, Foundation Medicine, Blue Earth Diagnostics, Amgen, Oncorus, Eli Lilly/Loxo and has received research funding from Janssen, AbbVie/Stemcentrx, Eli Lilly, Millennium, Bristol Myers Squibb. A.W. has received research funding from Janssen and ESSA Pharma, and served as a consultant or advisory board member for AstraZeneca, Astellas, Janssen and Merck. M.A.R. is on the scientific board of advisors for Neogenomics. M.J-H. has consulted, and is a member of the Scientific Advisory Board and Steering Committee, for Achilles Therapeutics, has received speaker honoraria from Astex Pharmaceuticals, Oslo Cancer Cluster, and holds a patent PCT/US2017/028013 relating to methods for lung cancer detection. C.S. acknowledges grant support from AstraZeneca,Boehringer-Ingelheim, Bristol Myers Squibb, Pfizer, Roche-Ventana, Invitae (previously Archer Dx Inc - collaboration in minimal residual disease sequencing technologies), and Ono Pharmaceutical. He is an AstraZeneca Advisory Board member and Chief Investigator for the AZ MeRmaiD 1 and 2 clinical trials and is also chief investigator of the NHS Galleri trial. He has consulted for Achilles Therapeutics, Amgen, AstraZeneca, Pfizer, Novartis, GlaxoSmithKline, MSD, Bristol Myers Squibb, Illumina, Genentech, Roche-Ventana, GRAIL, Medicxi, Metabomed, Bicycle Therapeutics, Roche Innovation Centre Shanghai, and the Sarah Cannon Research Institute, C.S. had stock options in Apogen Biotechnologies and GRAIL until June 2021, and currently has stock options in Epic Bioscience, Bicycle Therapeutics, and has stock options and is co-founder of Achilles Therapeutics. C.S. holds patents relating to assay technology to detect tumour recurrence (PCT/GB2017/053289); to targeting neoantigens (PCT/EP2016/059401), identifying patent response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients who respond to cancer treatment (PCT/GB2018/051912), US patent relating to detecting tumour mutations (PCT/US2017/28013), methods for lung cancer detection (US20190106751A1) and both a European and US patent related to identifying insertion/deletion mutation targets (PCT/GB2018/051892). M.L. reports grants or contracts from BMS, Shionogi, and AstraZeneca; consulting fees from BioNTech, Bicycle Therapeutics, Janssen, Merck Sorano, Pfizer, and ADC Therapeutics; honoraria from AstraZeneca and Pfizer; and support for attending meetings or travel from MSD, Janssen, and Bayer.
REFERENCES
- 1. de Bono J., Mateo J., Fizazi K., Saad F., Shore N., Sandhu S., Chi K.N., Sartor O., Agarwal N., Olmos D.et al.. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020; 382:2091–2102. [DOI] [PubMed] [Google Scholar]
- 2. Colomer R., Mondejar R., Romero-Laorden N., Alfranca A., Sanchez-Madrid F., Quintela-Fandino M.. When should we order a next generation sequencing test in a patient with cancer?. EClinicalMedicine. 2020; 25:100487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maia M.C., Salgia M., Pal S.K.. Harnessing cell-free DNA: plasma circulating tumour DNA for liquid biopsy in genitourinary cancers. Nat. Rev. Urol. 2020; 17:271–291. [DOI] [PubMed] [Google Scholar]
- 4. Abbosh C., Birkbak N.J., Wilson G.A., Jamal-Hanjani M., Constantin T., Salari R., Le Quesne J., Moore D.A., Veeriah S., Rosenthal R.et al.. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017; 545:446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carreira S., Romanel A., Goodall J., Grist E., Ferraldeschi R., Miranda S., Prandi D., Lorente D., Frenel J.S., Pezaro C.et al.. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 2014; 6:254ra125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wyatt A.W., Annala M., Aggarwal R., Beja K., Feng F., Youngren J., Foye A., Lloyd P., Nykter M., Beer T.M.et al.. Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. J. Natl. Cancer. Inst. 2017; 109:djx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Warner E., Herberts C., Fu S., Yip S., Wong A., Wang G., Ritch E., Murtha A.J., Vandekerkhove G., Fonseca N.M.et al.. BRCA2, ATM, and CDK12 defects differentially shape prostate tumor driver genomics and clinical aggression. Clin. Cancer Res. 2021; 27:1650–1662. [DOI] [PubMed] [Google Scholar]
- 8. Carter S.L., Cibulskis K., Helman E., McKenna A., Shen H., Zack T., Laird P.W., Onofrio R.C., Winckler W., Weir B.A.et al.. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012; 30:413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prandi D., Baca S.C., Romanel A., Barbieri C.E., Mosquera J.M., Fontugne J., Beltran H., Sboner A., Garraway L.A., Rubin M.A.et al.. Unraveling the clonal hierarchy of somatic genomic aberrations. Genome Biol. 2014; 15:439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Loo P., Nordgard S.H., Lingjaerde O.C., Russnes H.G., Rye I.H., Sun W., Weigman V.J., Marynen P., Zetterberg A., Naume B.et al.. Allele-Specific Copy Number Analysis of Tumors. 2010; 107:Proceedings of the National Academy of Sciences of the United States of America; 16910–16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beltran H., Prandi D., Mosquera J.M., Benelli M., Puca L., Cyrta J., Marotz C., Giannopoulou E., Chakravarthi B.V., Varambally S.et al.. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016; 22:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. The Cancer Genome Atlas Research Network The molecular taxonomy of primary prostate cancer. Cell. 2015; 163:1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robinson D., Van Allen E.M., Wu Y.M., Schultz N., Lonigro R.J., Mosquera J.M., Montgomery B., Taplin M.E., Pritchard C.C., Attard G.et al.. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Setlur S.R., Chen C.X., Hossain R.R., Ha J.S., Van Doren V.E., Stenzel B., Steiner E., Oldridge D., Kitabayashi N., Banerjee S.et al.. Genetic variation of genes involved in dihydrotestosterone metabolism and the risk of prostate cancer. Cancer Epidemiol. Biomarkers Prev. 2010; 19:229–239. [DOI] [PubMed] [Google Scholar]
- 15. Romanel A., Tandefelt D.G., Conteduca V., Jayaram A., Casiraghi N., Wetterskog D., Salvi S., Amadori D., Zafeiriou Z., Rescigno P.et al.. Plasma AR and abiraterone-resistant prostate cancer. Sci. Transl. Med. 2015; 7:312re310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Romanel A., Zhang T., Elemento O., Demichelis F.. EthSEQ: ethnicity annotation from whole exome sequencing data. Bioinformatics. 2017; 33:2402–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Demichelis F., Greulich H., Macoska J.A., Beroukhim R., Sellers W.R., Garraway L., Rubin M.A.. SNP panel identification assay (SPIA): a genetic-based assay for the identification of cell lines. Nucleic Acids Res. 2008; 36:2446–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M.et al.. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.1000 Genome Project Data Processing Subgroup . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Valentini S., Fedrizzi T., Demichelis F., Romanel A.. PaCBAM: fast and scalable processing of whole exome and targeted sequencing data. BMC Genomics. 2019; 20:1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Degner J.F., Marioni J.C., Pai A.A., Pickrell J.K., Nkadori E., Gilad Y., Pritchard J.K.. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2009; 25:3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Prandi D., Demichelis F.. Ploidy- and purity-adjusted allele-specific DNA analysis using CLONETv2. Curr. Protoc. Bioinformatics. 2019; 67:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ciani Y., Fedrizzi T., Prandi D., Lorenzin F., Locallo A., Gasperini P., Franceschini G.M., Benelli M., Elemento O., Fava L.L.et al.. Allele-specific genomic data elucidate the role of somatic gain and copy-number neutral loss of heterozygosity in cancer. Cell Syst. 2021; 13:183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Casiraghi N., Orlando F., Ciani Y., Xiang J., Sboner A., Elemento O., Attard G., Beltran H., Demichelis F., Romanel A.. ABEMUS: platform-specific and data-informed detection of somatic SNVs in cfDNA. Bioinformatics. 2020; 36:2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ramos A.H., Lichtenstein L., Gupta M., Lawrence M.S., Pugh T.J., Saksena G., Meyerson M., Getz G.. Oncotator: cancer variant annotation tool. Hum. Mutat. 2015; 36:E2423–E2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R.. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014; 42:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shen R., Seshan V.E.. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016; 44:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Annala M., Vandekerkhove G., Khalaf D., Taavitsainen S., Beja K., Warner E.W., Sunderland K., Kollmannsberger C., Eigl B.J., Finch D.et al.. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 2018; 8:444–457. [DOI] [PubMed] [Google Scholar]
- 31. Smith M.R., Scher H.I., Sandhu S., Efstathiou E., Lara P.N., Yu E.Y., George D.J., Chi K.N., Saad F., Stahl Oet al.. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2022; 23:362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Demichelis F., Rubin M.A.. TMPRSS2-ETS fusion prostate cancer: biological and clinical implications. J. Clin. Pathol. 2007; 60:1185–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perner S., Mosquera J.M., Demichelis F., Hofer M.D., Paris P.L., Simko J., Collins C., Bismar T.A., Chinnaiyan A.M., De Marzo A.M.et al.. TMPRSS2-ERG fusion prostate cancer: an early molecular event associated with invasion. Am. J. Surg. Pathol. 2007; 31:882–888. [DOI] [PubMed] [Google Scholar]
- 34. Riester M., Singh A.P., Brannon A.R., Yu K., Campbell C.D., Chiang D.Y., Morrissey M.P.. PureCN: copy number calling and SNV classification using targeted short read sequencing. Source Code Biol. Med. 2016; 11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Talevich E., Shain A.H., Botton T., Bastian B.C.. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016; 12:e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Livingstone J., Shiah Y.J., Yamaguchi T.N., Heisler L.E., Huang V., Lesurf R., Gebo T., Carlin B., Eng S., Drysdale E.et al.. The telomere length landscape of prostate cancer. Nat. Commun. 2021; 12:6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abida W., Campbell D., Patnaik A., Shapiro J.D., Sautois B., Vogelzang N.J., Voog E.G., Bryce A.H., McDermott R., Ricci F.et al.. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the Phase II TRITON2 study. Clin. Cancer Res. 2020; 26:2487–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sweeney C., Bracarda S., Sternberg C.N., Chi K.N., Olmos D., Sandhu S., Massard C., Matsubara N., Alekseev B., Parnis F.et al.. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet. 2021; 398:131–142. [DOI] [PubMed] [Google Scholar]
- 39. Corn P.G., Heath E.I., Zurita A., Ramesh N., Xiao L., Sei E., Li-Ning-Tapia E., Tu S.M., Subudhi S.K., Wang J.et al.. Cabazitaxel plus carboplatin for the treatment of men with metastatic castration-resistant prostate cancers: a randomised, open-label, phase 1-2 trial. Lancet Oncol. 2019; 20:1432–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed data will be provided following review of request to the corresponding authors.