

ABSTRACT
Antibody-drug conjugates (ADCs) are increasingly powerful medicines for targeted cancer therapy. Inspired by the trend to further improve their therapeutic index by generation of homogenous ADCs, we report here how the clinical-stage GlycoConnect™ technology uses the globally conserved N-glycosylation site to generate stable and site-specific ADCs based on enzymatic remodeling and metal-free click chemistry. We demonstrate how an engineered endoglycosidase and a native glycosyl transferase enable highly efficient, one-pot glycan remodeling, incorporating a novel sugar substrate 6-azidoGalNAc. Metal-free click attachment of an array of cytotoxic payloads was highly optimized, in particular by inclusion of anionic surfactants. The therapeutic potential of GlycoConnect™, in combination with HydraSpace™ polar spacer technology, was compared to that of Kadcyla® (ado-trastuzumab emtansine), showing significantly improved efficacy and tolerability.
KEYWORDS: Antibody-drug conjugates (ADCs), chemoenzymatic, metal-free click chemistry, non-genetic, therapeutic index, glycan remodeling
Graphical Abstract
Introduction
Antibody-drug conjugates (ADCs) have firmly established themselves as a valuable class of chemotherapeutics for targeted cancer therapy, with 11 market approvals and more than 100 in various stages of clinical development.1 Empowered by cytotoxic payloads spanning multiple mode-of-actions,2 including tubulin inhibitors, DNA damaging agents and topoisomerase 1 inhibitors, ADCs have been approved for numerous types of solid tumors and hematological cancers. Structurally, an ADC consists of a monoclonal antibody (mAb) covalently attached to a highly potent toxin. Whereas most ADCs are produced without strict control of linker-drug attachment, heterogenous mixtures of ADCs can display suboptimal performance. As a consequence, an important trend in the ADC field is to generate the drug substance as a single species by application of a site-specific conjugation technology.3 One approach involves re-engineering of the antibody prior to conjugation, to introduce at a defined site either: 1) an additional cysteine (THIOMAB™ technology),4 2) an amino acid sequence for enzymatic modification,5 or 3) a non-natural amino acid.6 Other site-specific approaches have been developed, but these are less frequently used.3
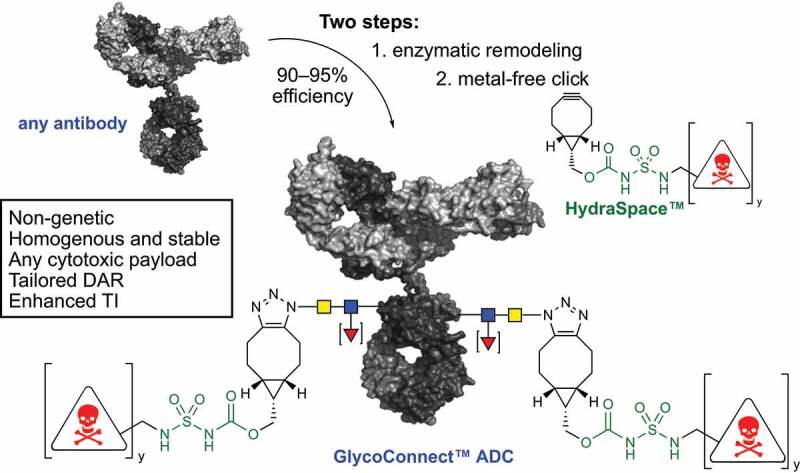
We earlier demonstrated that the native antibody glycan at asparagine-297 in the CH2 domain can be used as a natural anchor point for payload attachment (Figure 1).7 Building from the glycan, we developed a technology called GlycoConnect™, based on enzymatic remodeling of the glycan for introduction of azidosugar (a⟶b), followed by linker-drug conjugation with metal-free click chemistry (b⟶c). The resulting ADCs are homogenous and stable, but more importantly show a significantly enhanced therapeutic window versus conventional conjugation technologies.7 Another advantage of ADCs generated with GlycoConnect™ is the concomitant annihilation of binding to CD16/CD32 (Fc-γ receptor III and II) and significant reduction (<30% remaining) of binding to CD64 (Fc-γ receptor I), which is generally undesirable, given the potential Fc-γ receptor-mediated uptake in healthy tissue. We later demonstrated8 how GlycoConnect™ can be empowered by introduction of a highly polar spacer unit (HydraSpace™) based on a carbamoyl sulfamide group (Figure 1). Three GlycoConnect™ ADCs are currently in Phase 1 clinical trials (ADCT-601, XMT-1592, and MRG004a), with more than a dozen additional ADCs in various stages of preclinical development,9 thereby rendering the GlycoConnect™ approach the most prevalent (chemo)enzymatic antibody modification technology in the clinic.10
Figure 1.
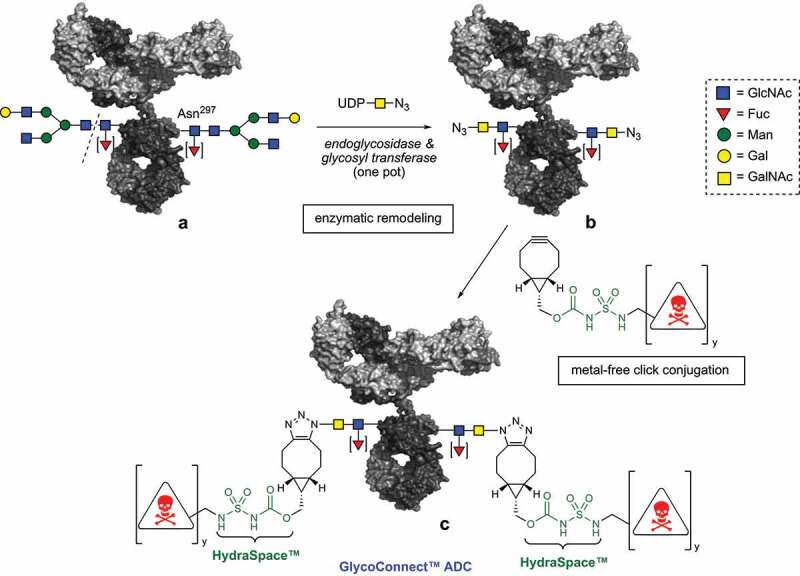
General scheme for enzymatic remodeling of antibody glycan (a⟶b) followed by metal-free click chemistry conjugation of payload (b⟶c). The drug-to-antibody ratio (DAR) can be tailored (DAR2 or DAR4) by using a linear of branched BCN-linker-drug construct (y = 1 or 2).
GlycoConnect™ technology encompasses a two-step process to convert a monoclonal antibody into an antibody-drug conjugate, abbreviated as ADC. In the first step two enzymes work together to trim the antibody glycan down to the core GlcNAc, followed by attachment of a monosaccharide functionalized with an azido group. In the second step a cyclooctyne-linker-drug is attached by means of metal-free click chemistry of the cyclooctyne – in this case BCN – with the azide. The linker-drug also features a highly polar HydraSpace™ moiety for solubility.
Although GlycoConnect™ ADCs were readily prepared at a laboratory scale, it became clear to us that significant improvement of several of the components (enzymes, azidosugar, remodeling and conjugation conditions) was mandatory to enable clinical manufacturing and potentially further elevate the therapeutic index. Here, we report on essential advancements on our previously reported technology achieved by: 1) reducing the number of process steps from antibody to ADC; 2) yield optimization of isolated ADCs; 3) employing de novo generated enzymes (endoglycosidase and glycosyl transferase) and an improved azidosugar substrate; and 4) significant reduction of linker-drug stoichiometry during final conjugation step. Furthermore, the resulting ADCs exhibited excellent efficacy and tolerability, as demonstrated by direct comparison with the marketed drug Kadcyla® (ado-trastuzumab emtansine).
Results
Our first focus was on a more efficient cleavage step of the heterogeneous mixture of glycoforms (Figure 2), present on an antibody obtained by recombinant expression in a mammalian expression system (e.g., Chinese hamster ovary (CHO)).11 Clearly, enzymatic trimming of all different glycoforms (amongst others complex, hybrid and high-mannose) to the core GlcNAc typically requires multiple endoglycosidases.12 For example, endo H is known to cleave N-linked high-mannose and hybrid glycoforms, but not complex type glycans. In contrast, endo S, an IgG- selective endoglycosidase from Streptococcus pyogenes,13 hydrolyzes complex, but not high-mannose glycans. We were intrigued to investigate if a pan-selective trimming enzyme could potentially be generated by head-to-head fusion of two complementary endoglycosidases. To this end, we designed a range of different endoglycosidase fusions, including endo S–endo H, endo F2–endo H, endo F2–endo F1 and endo F3–endo 18A (see Supplementary Tables S1 and S2). After expression and isolation from E. coli, each fusion enzyme was evaluated for hydrolysis of the full spectrum of antibody glycans to the core-GlcNAc (see Supplementary Table S3 and Figures S1–S3). Indeed, we found that while all fusions effectively led to trimmed antibody, the endo S–endo H fusion enzyme was particularly suitable: 1% (w/w) of fusion protein achieved full glycan hydrolysis in various buffers (e.g., Tris, TRIS-buffered saline, histidine and citrate), at various temperatures (room temperature–37°C) and different pH values (pH 5–8). It became apparent during our investigations that endo S2, another endoglycosidase from S. pyrogenes, also hydrolyzes all antibody N-linked glycoforms,14 which prompted us to evaluate how endo S2 would compare to endo S-endo H fusion enzyme. Interestingly, we determined that at pH 7.5 our endo S–endo H fusion (endo SH) showed enhanced hydrolytic activity to complex/hybrid glycoforms15 in comparison to endo S2 (see Supplementary Figure S4). In addition, it was intriguing to find that the endo H part of the fusion construct showed enhanced activity toward high-mannose glycans compared to endo H as separate enzyme (see Supplementary Figure S5), potentially due to enhanced stability caused by the fusion element. Given its favorable activity profile, a stable clone (master cell bank) of endo SH was generated in E. coli, and repeat large-scale bioreactor runs (≥15 L, titers >1 g/L) have now been performed to obtain sufficient endo SH for trimming of multiple kilograms of mAb.
Figure 2.
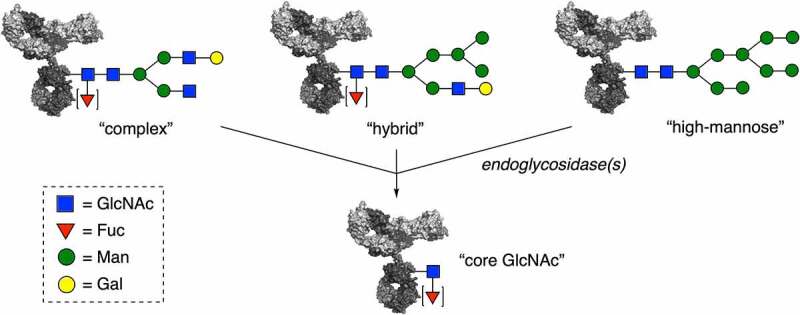
Endoglycosidase trimming of various antibody glycoforms to core GlcNAc attached to Asn297. Potential substitution of the core GlcNAc with a (1⟶6)-α-fucosyl moiety [▼] does not affect the endoglycosidase efficiency.
Monoclonal antibodies are glycosylated in the CH2 domain with a glycan that can take different forms, such a complex, hybrid or high-mannose. Each glycosylation profile can be trimmed down to the core GlcNAc with a specific endoglycosidase.
Having generated an efficient and pan-selective endoglycosidase, attention was focused on the glycosyltransferase enzyme. We7 and others16,17 have previously reported that mutant galactosyltransferase GalT(Y289L)18 efficiently transfers GalNAz (1, Figure 3) to terminal GlcNAc residues. However, recombinant expression of mutant GalT(Y289L) in E. coli exclusively results in inclusion bodies, thus requiring cumbersome refolding.11 In order to obtain mutant GalT(Y289L) in large quantities, we explored multiple strategies, including the expression of mutants (e.g., Y289A,C342T and Y289G,C342T), various GalT(Y289L) fusions (MBP or SUMO) and periplasmic expression, but to no avail.
Figure 3.
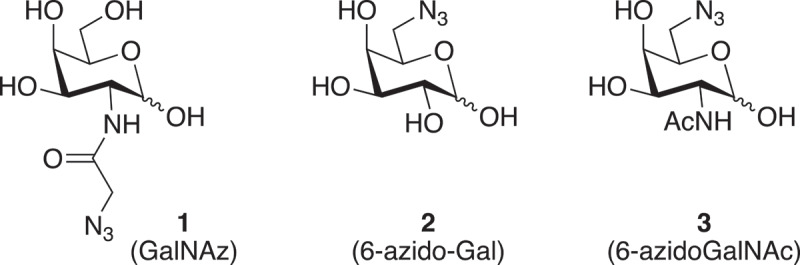
Various azidosugars 1–3 for glycosyl transfer to core GlcNAc.
Various monosaccharides with a azido group. Structure 1 is N-azidoacetyl galactosamine, structure 2 is 6-azido-galactose, structure 3 is 6-azido-GalNAc.
As a potential alternative, we turned our attention to the expression of native N-acetylgalactosaminyltransferases (GalNAc-transferases). Although β-(1,4)-GalNAc-transferases are considered highly substrate-specific,19 O-linked glycans on mucins in C. elegans have been metabolically labeled with GalNAz (1) under the action of a polypeptide-O-GalNAc-transferase.20 Similarly, a β-(1,4)-GalNAc transferase was likely responsible for the metabolic labeling N-glycoproteins with GalNAz.21 However, to the best of our knowledge, β-(1,4)-GalNAc-transferases have hitherto not been used for in vitro antibody glycoengineering. To this end, we focused our attention on wild-type GalNAc-transferases from Caenorhabditis elegans,21 Drosophila melanogaster,22 Ascaris suum and Trichoplusia ni23 and the respective enzymes CeGalNAc-T, DmGalNAc-T, AsGalNAc-T and TnGalNAc-T (see Supplementary Table S4). Because in this case recombinant expression in E. coli only provided inclusions, we turned our attention to mammalian CHO-K1. Gratifyingly, the GalNAc-transferases as well as GalT(Y289L) could be isolated in pure form after transient expression, cation exchange chromatography and size exclusion chromatography (SEC) (see Supplementary Table S5). All produced enzymes were found to be active based on a standard glycosyltransferase assay using UDP-GalNAc as donor-substrate (see Supplementary Figure S6), thereby setting the stage for azidosugar remodeling of antibodies. We first focused our attention on the incorporation of GalNAz (1), a well-known azidosugar derivative of GalNAc applied earlier in our first generation GlycoConnect™ technology. Indeed, we found that, similar to GalT(Y289L), all of the GalNAc-transferases effectively incorporated GalNAz (1) onto trimmed trastuzumab. We decided to also include in our screening other azidosugar substrates (2 and 3), given the reported ability of native β-(1,4)-galactosyltransferase (GalT) to transfer 6-biotinylated galactose24 or 6-azidogalactose (2)25 to an acceptor GlcNAc substrate. First of all, we found that reacting UDP 6-azido-galactose (UDP-2) in the presence of β-(1,4)-galactosyltransferase with trastuzumab-GlcNAc failed to lead to incorporation of 2 under all conditions (see Supplementary Figure S7), which is not surprising given the lack of the 2-NHAc functionality in 2. Gratifyingly, TnGalNAc-T, and to a minor extent AsGalNAc-T, effectively transferred the 6-azido-derivative of GalNAc (3) onto the core-GlcNAc of trastuzumab and other mAbs (see Supplementary Figure S8). Efficient transfer was observed in the presence of only 5% (w/w) of enzyme and 5 µM of UDP-3 (37.5 equiv.) at 15 mg/mL antibody concentration, leading to an incorporation efficiency of ≥90% (Table 1, entry 1).
Table 1.
Subset of conditions screened to optimize the enzymatic remodeling process. Efficiency was determined by conversion of remodeled trastuzumab-3 into ADC and determination of drug-to-antibody ratio (DAR) with RP-HPLC. In all cases buffers were set at pH 7.5 and remodeling was performed at 15 mg/mL antibody concentration (100 µM) in the presence of 1% (w/w) endo SH. For full set of conditions, including other pH values (see ESI†)
| Entry | Buffer* | UDP-3 | GalNAcT | MnCl2 | AP | efficiency |
|---|---|---|---|---|---|---|
| 1 | tris | 37.5 equiv. | 5% | 10 mM | – | ≥90% |
| 2 | tris | 20 equiv. | 0.5% | 10 mM | – | 63% |
| 3 | histidine | 75%–95% | ||||
| 4 | HEPES | 63% | ||||
| 5 | tricine | 73%–93% | ||||
| 6 | histidine | 25 equiv. | 1% | 6 mM | 79% | |
| 7 | 1.5% | 88% | ||||
| 8 | 2% | 95% | ||||
| 9 | 20 equiv. | 3% | 91% | |||
| 10 | 15 equiv. | 3% | 91% | |||
| 11 | 2% | 80% | ||||
| 12 | 10 equiv. | 3% | 89% | |||
| 13 | 15 equiv. | 2% | 0.01% | 94% | ||
| 14 | 1.5% | 93% | ||||
| 15 | 10 equiv. | 3% | 97% |
A table with screening conditions to optimize conversion of antibody into azido-remodeled antibody based on variation of buffer, quantity of UDP-3, quantity of GalNAc-transferase, quantity of alkaline phosphatase and quantity of MnCl2
Since TnGalNAcT showed highly efficient incorporation of 6-azidoGalNAc (3), we continued with the production optimization. Use of an N-terminal histidine tag allowed direct purification from CHO-K1 supernatant by immobilized metal affinity chromatography (using Ni Sepharose® Excel) from transient expressions up to 5 L scale, with isolated yields of 125–140 mg/L (see Supplementary Table S5). Supported by these results, a master cell bank was generated allowing for further scale-up of TnGalNAcT production with production runs up to 200 L (titer >1.2 g/L) producing ~120 g of isolated TnGalNAcT (>90% purity).
Having established a successful protocol for enzymatic incorporation of either azidosugar 1 or 3, we realized that the ADC properties, after subsequent linker-drug conjugation, would eventually be decisive in selection of the preferred azidosugar substrate. Thus, we remodeled two antibodies (HER2-targeting trastuzumab and CD30-targeting brentuximab) with either UDP-1 or UDP-3 to give a 2 × 2 matrix of azidosugar-remodeled antibodies. All four azido-antibodies were subsequently conjugated with either linker-drug 4 or linker-drug 5 (see Supplementary Figure S9–S11). Both linker-drugs comprised bicyclononyne (BCN)26 for metal-free click conjugation, our earlier reported8 polar spacer technology based on carbamoyl sulfamide (HydraSpace™, green), a short PEG spacer, and either maytansine or MMAE tubulin inhibitor payload (Figure 4a). Further, to maximize variability in drug-to-antibody ratio (DAR), linker-drug 4 contains a linear linker, while compound 5 contains a branched linker.
Figure 4.
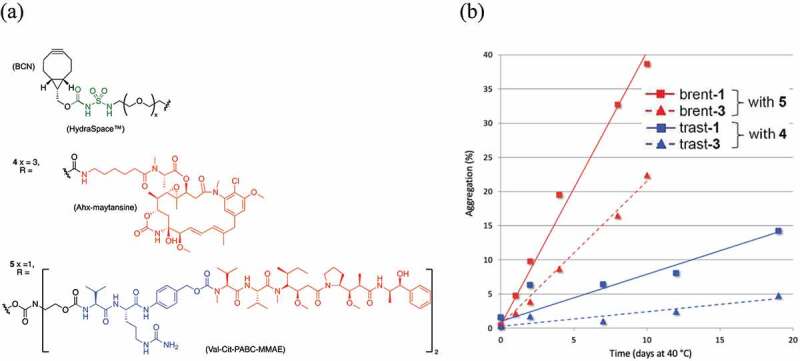
Stability of antibody-drug-conjugates based on azidosugar 1 (GalNAz) or 3 (6-azidoGalNAc). (a) Structures of BCN-HydraSpace™-linker-drugs 4 and 5. (b) Aggregation levels of ADCs derived from brentuximab (red lines) or trastuzumab (blue lines), remodeled with azidosugar 1 (solid lines) or 3 (dashed lines). Both azidosugar-remodeled derivatives of brentuximab were conjugated to linker-drug 5 (⟶DAR4 ADC), while trastuzumab azidosugar derivatives were conjugated to linker-drug 4 (⟶DAR2 ADC).
Figure 4a shows the structures of two different linker-drugs, compound 4 has a linear linker and a maytansinoid payload, to make DAR2 ADCs, while compound 5 with a branched linker and MMAE payload, to make DAR4 ADCs. Figure 4b is a plot aggregation against time of ADCs based on antibodies trastuzumab or brentuximab and with linker-drugs4 or 5, in each case showing a pronounced enhanced aggregation profile in case azidosugar GalNAz is used versus use of azidosugar 6-azidoGalNAc.
The resulting GlycoConnect™ ADCs obtained from trastuzumab-1/3 conjugated to 4 or from brentuximab-1/3 conjugated to 5 were assessed head-to-head for in vitro stability with regards to aggregation (pH 5, 40°C). The enhanced aggregation levels of DAR4 ADCs based on 5 (red lines in Figure 4b) versus the DAR2 ADCs based 4 (blue lines) were expected,27 given the two-fold higher number of lipophilic payloads. However, it was surprisingly found that under these forcing conditions both ADCs obtained by remodeling with azidosugar 6-azidoGalNAc (3) showed reduced aggregation versus analogues ADCs remodeled with GalNAz (1), irrespective of whether they were conjugated to 4 or 5. Gratifyingly, no aggregation was observed in phosphate-buffered saline (PBS) at 37°C or during the process of enzymatic remodeling and conjugation (see Supplementary Figure S12 and S14). In addition, DAR was measured over time for a model antibody conjugated to 5 or 15 in various plasmas, showing full retention of drug loading in human plasma, and only a marginal decrease in mouse and rat plasma, likely due to plasma proteases like Ces1c (see Supplementary Figure S13). Possibly the conformation and substitution attachment site of azidosugar 3 positively impacts binding of the linker-payload in the hydrophobic cavity encompassed by amino acids L233, F237, L238, F239 and Y296, as reported by Tumey et al.28 In light of the fact that trastuzumab and brentuximab are particularly stable in comparison to most other antibodies, 6-azidoGalNAc 3 was selected above 1 as the preferred azidosugar substrate for application in ADCs.
To fulfill the objective to enable the GlycoConnect™ process for production of clinical grade ADCs, an efficient, multi-gram scale manufacturing process of the requisite UDP-derivative of 6-azidoGalNAc (3) was also indispensable. To this end, we thought that the tri-O-acetyl derivative of d-galactosamine (6), accessible in near quantitative yield from GalNAc,29 would be a useful starting material for the regioselective introduction of an azide group. However, like others,30–33 we found that nucleophilic azide substitution at C-6, using different leaving groups and conditions (not shown in Scheme 1), led to low conversion, likely caused by steric hindrance of the axial 4-OH group. We reasoned that the nucleophilic approach of azide could be facilitated by locking the 6-OH in pseudo-axial orientation by means of 4,6-O-cyclic sulfate. Indeed, room temperature NaN3 treatment of compound 7, obtained from 6 by a standard protocol (SOCl2, then RuCl3/NaIO4),34 led to smooth formation of compound 8 after acidic work-up. Subsequent 4-O-acetylation (8⟶9) and anomeric deacetylation (9⟶10) proceeded seamlessly, and was followed by anomeric phosphitylation, then oxidation, to provide the desired phosphate 11 with exclusive α-anomeric selectivity. After concomitant deprotection of O-cyanoethyl and O-acetyl groups (11⟶12), the requisite UDP-α-3 was obtained upon coupling to sodium UMP-imidazolide 13 separately prepared from UMP bis-sodium salt (see Supplementary section 5), in two steps and an overall yield of 97%. Notably, only a few chromatographic purification steps were required to obtain the final UDP-α-3 in high purity (>95%) (see Supplementary section 5). The route has proven to be scalable to ≥150 gram of intermediate 7 and up to 15 gram of pure UDP-3 (further scale-up ongoing).
Having a sufficient quantity of UDP-3 at hand, a thorough and elaborate screen was performed to optimize the efficiency of enzymatic incorporation (Table 1 and Supplementary Tables S6–S7). Thereto, first a set of different buffers was evaluated (entries 2–5) under ‘suboptimal’ conditions (0.5% w/w GalNAc-T, 20 equiv. UDP-3). Clearly, histidine and tricine at pH 7.5 provided significantly higher efficiency than tris or HEPES under identical conditions. Given that histidine buffer is most common in large-scale manufacturing, the quantities of UDP-3 and GalNAc-T, as well as the quantity of MnCl2, were varied next in this buffer (entries 6–12). Gratifyingly, it was found that efficiencies ~ 90% could be attained with significantly reduced enzyme/UDP-3 stoichiometries (e.g., entries 8 and 12). The quantity of MnCl2 was also lowered (to 6 mM) to avoid enzyme inhibition and minimize potential undesired antibody oxidation. Finally, addition of alkaline phosphatase (AP) to the broth (entries 13–15), to avoid feed-back inhibition of liberated UDP, enabled further reduction of UDP-3 while overall efficiencies approached quantitative (entry 15). The latter conditions were also corroborated at higher antibody scale (300 mg of trastuzumab) and by application to various other antibodies, including brentuximab, rituximab, B12, and enfortumab (see Supplementary Figures S15–S16 and Table S8). In addition, we found that GalNAcT could be applied concomitantly with endo SH without compromising overall remodeling efficiency under mild conditions (histidine buffer, pH 7.5, 30°C), thereby avoiding one unit operation, which is clearly undesirable from a manufacturing perspective.
Scheme 1.
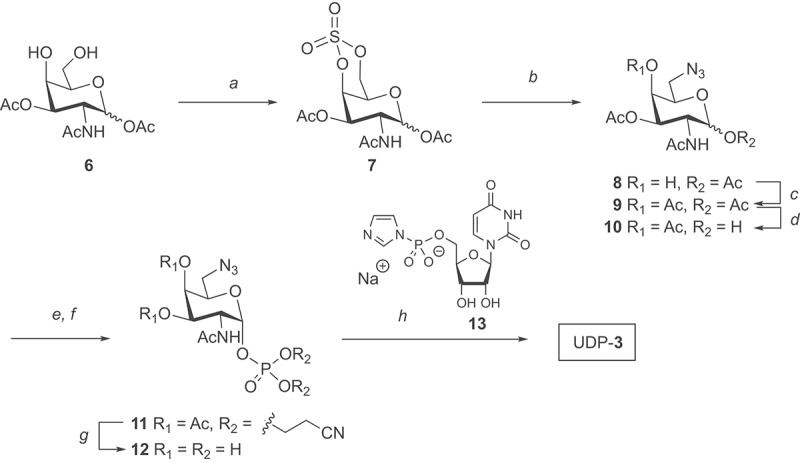
Synthetic preparation of UDP 6-azido-6-deoxy-GalNAc (UDP-3). Reagents and conditions: a) 1. SOCl2, Et3N, CH2Cl2, 0°C. 2. RuO4, NaIO4, CH2Cl2, CH3CN, water, 83% over 2 steps; b) 1. NaN3, DMF, rt, 2. H2SO4, THF, water; c) pyridine, Ac2O, 80% over two steps; d) 1-propylamine, THF; e) 5-(ethylthio)-1 H-tetrazole, bis(2-cyanoethyl)-N,N-diisopropyl phosphoramidite, CH2Cl2, MeCN; f) m-CPBA, 54% over three steps; g) Et3N, MeOH, H2O, 50°C, quantitative; h) sodium UMP-imidazolide (13), MgCl2, DMF, 52% yield. A chemical scheme showing how N-acetyl-D-galactosamine can first be converted into its 6-azido derivative and finally into UDP 6-azidoGalNAc, compound UDP-3.
With suitable protocols established for enzymatic remodeling and generation of ADCs with payloads maytansinoid (4) or MMAE (5), a range of BCN-linker-constructs was synthesized and conjugated (Table 2 and Supplementary Figures S17–S23 and S27–S32) based on other cytotoxic molecules common in the field of ADCs, including calicheamicin variants (14 and 15), a pyrrolobenzodiazepine dimer (16), PNU-159,682 (17) and duocarmycin (18). In each case, conjugations were performed on trastuzumab remodeled with 6-azidoGalNAc (trast-3) and monovalent BCN-linker-drugs (y = 1) were used to generate DAR2 ADCs (entry 1–5). Two branched BCN-linker constructs (y = 2) were also applied, one based on MMAE (5) and one based on maytansinoid payload (19, see Figure 6), each of which was successfully conjugated to trast-3 to obtain DAR4 ADCs (entries 6 and 7).
Table 2.
Conjugation of various BCN-linker-drugs to azidosugar-remodeled trastuzumab leading to DAR2 ADCs (14–18) or DAR4 ADCs (19 and 5). PG = propylene glycol. For structure of BCN-linker-payload 5, see Figure 4, for structure of 19, see Figure 6, for other structures see ESI
| Entry | Cmpd | R | x | y | Payload | Equiv. | co-solvent | Yield | DAR |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 14 | Cit | 0 | 1 | calicheamicin | 8 | 50% PG | 78% | 1.81 |
| 2 | 15 | Cit | 0 | 1 | Gly-calicheamicin | 6 | 50% PG | 66% | 1.86 |
| 3 | 16 | Ala | 0 | 1 | PBD dimer | 5 | 15% DMF | 79% | 1.79 |
| 4 | 17 | Cit | 1 | 1 | PNU-159,682 | 7 | 25% DMF | 83% | 1.81 |
| 5 | 18 | Cit | 1 | 1 | duocarmycin | 10 | 25% DMF | 79% | 1.82 |
| 6 | 19 | Cit | 0 | 2 | Ahx-maytansine | 8 | 25% DMF | 96% | 3.70 |
| 7 | 5 | Cit | 0 | 2 | MMAE | 7 | 25% DMF | 81% | 3.60 |
A table showing how an antibody can be converted into DAR2 or DAR4 ADC using GlycoConnect™ technology and conjugation of linker-drugs with payloads calicheamicin, PBD dimer, PNU-159,682, duocarmycin, Ahx-maytansine, or MMAE.
Figure 6.
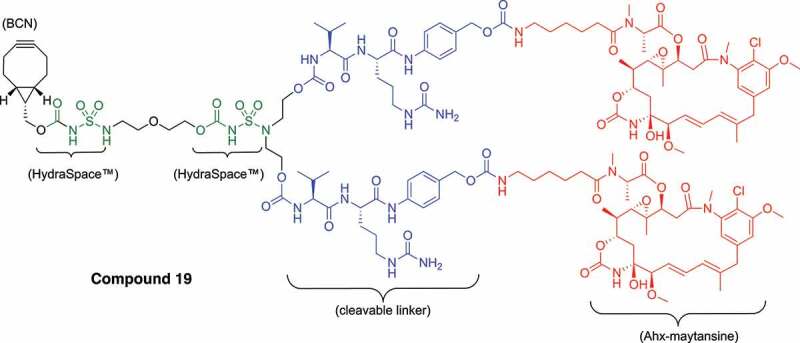
Structure of branched BCN-HydraSpace™-vc-PABC-Ahx-maytansine 19 (SYNtansine™) for the preparation of DAR4 ADC.
The figure consists of four bar plots to compare how addition of various surfactants during metal-free click conjugation of linker-drug impact the drug-to-antibody ratio, showing clearly the benefit of inclusion of sodium deoxycholate.
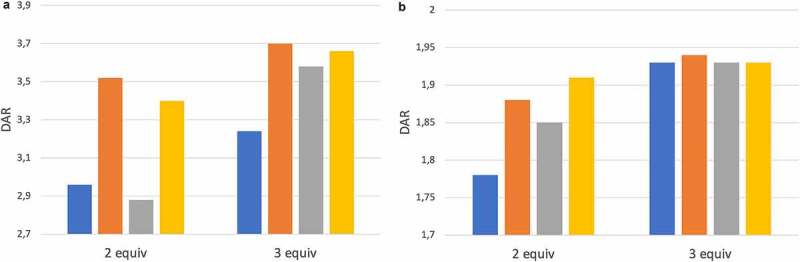
While all ADCs were obtained in high yield and with desired DAR, we have found that 25% dimethyl formamide (DMF) or 50% propylene glycol (PG) as co-solvent can induce significant in-process aggregation with some antibodies. Moreover, the rather large stoichiometry of linker-drug (5–10 equiv.) contributes to high cost-of-goods of the resulting ADCs. Therefore, we evaluated whether surfactants, reported earlier35 to facilitate the acylation of lysine side-chain in the preparation of Besponsa® (inotuzumab ozogamicin), would also impart a beneficial effect on the metal-free click conjugation. Thus, conjugations were performed in the presence of anionic surfactants (37.5 mM sodium decanoate or 11 mM sodium deoxycholate) or zwitterionic surfactant CHAPS (12 mM) with only 10% DMF co-solvent, using branched linker-drug 5 (for DAR4 ADC) at minimal stoichiometry (2–3 equiv.), and DARs were determined (Figure 5a and Supplementary Figure S24). A clear beneficial impact of anionic surfactants, but not CHAPS, was noted versus control (no additive), enhancing the DAR from <3 to close to 3.6 with only 2 equivalents of 5. Upon increase of linker-drug stoichiometry to 3 equivalents, DAR further improved to >3.6 in particular for sodium deoxycholate. The optimal conditions (10% DMF, 11 mM sodium deoxycholate) seamlessly translated to other payloads such as calicheamicin-based linker-drug 14 (Figure 5b and Supplementary Figure S25–26) to consistently provide the desired ADCs with high yield and DAR.
Figure 5.
Optimization of metal-free click conjugation in the presence of surfactants. (a) Conjugation of branched MMAE-based linker-drug 5 for generation of DAR4 ADC. (b) Conjugation of linear calicheamicin-based linker-drug 15 for generation of DAR2 ADC. Surfactant concentrations: sodium deoxycholate (11 mM), sodium decanoate (37.5 mM), CHAPS (12 mM).
A chemical structure is depicted of the branched BCN-linker-drug, with two maytansinoid payloads per cyclooctyne, used in the preparation of DAR4 ADCs for head-to-head comparison with Kadcyla®.
Finally, we were keen to evaluate the in vivo potential of GlycoConnect™/HydraSpace™ ADCs in comparison to a marketed ADC. Thereto (Figure 6), we conjugated trast-3 to branched BCN-linker-drug 19 based on Ahx-maytansinoid payload (SYNtansine™). For increased stability (aggregation) of the ADC, two occurrences of HydraSpace™ are included in the linker. Thus, trastuzumab-SYNtansine™ (trast-3 conjugated to 19) with DAR 3.70 was compared head-to-head with Kadcyla® (DAR 3.50), based on the same antibody component (trastuzumab). At the same time, some differences remain between trastuzumab-SYNtansine™ and Kadcyla®, such as the mode of attachment (glycan versus lysine conjugation), nature of the linker (cleavable versus non-cleavable) and payload (Ahx-maytansine and DM1). Therefore, to accurately assess the potential of both ADCs head-to-head in terms of therapeutic window, we prepared sufficient material (90 mg) of trastuzumab-SYNtansine™ for both efficacy and tolerability studies in rodents.
To assess the in vivo efficacy of trastuzumab-SYNtansine™ and Kadcyla®, an efficacy study was performed in the T226 mouse PDX model (Figure 7). Pronounced tumor regression was noted as early as D5 in the group treated with trastuzumab-SYNtansine™ at either dose levels, as well as for Kadcyla® at high dose (9 mg/kg). However, tumors continued to grow for mice treated with Kadcyla® at low dose (3 mg/kg). Interestingly, tumors never fully regressed with high dose Kadcyla® or low dose trastuzumab-SYNtansine™ and slow regrowth was observed after approximately 2 weeks for both groups. Gratifyingly, complete and durable tumor regression was observed in the group for trastuzumab-SYNtansine™ at the high dose level (for individual tumor volumes, see Supplementary Figure S33). Based on these data, the minimal effective dose (MED) of trastuzumab-SYNtansine™ is distinctly lower (~3-fold) than that of Kadcyla®.
Figure 7.
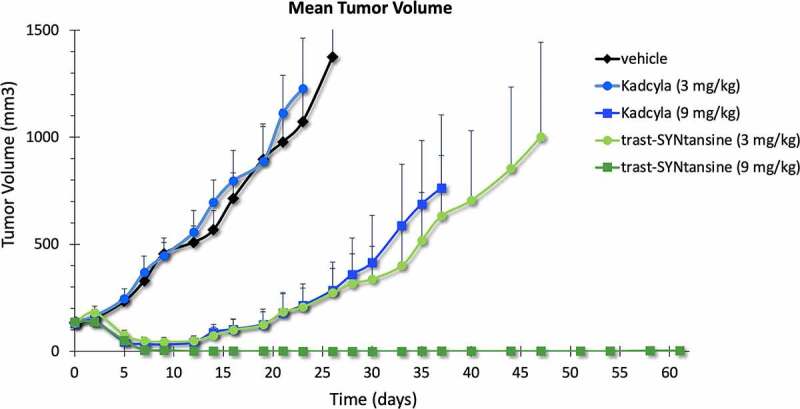
Tumor volume over time of mouse PDX T226 treated with Kadcyla® or trastuzumab-SYNtansine™ at low or high dose (3 and 9 mg/kg, respectively).
A plot shows the tumor volume over time of a mouse PDX model treated with Kadcyla® or similar DAR4 ADC based on GlycoConnect™ and HydraSpace™ technologies, showing a pronounced improvement in tumor growth inhibition for the latter.
The pronounced improvement in efficacy for trastuzumab-SYNtansine™ versus Kadcyla® is remarkable given both are based on the same antibody and same maytansinoid payload (core structure). At the same time, we were aware that the structural differences between the linkers, in the cleavage mechanism and in the released active catabolite contribute to the higher efficacy of the SYNtansine™-based ADC. Therefore, in order to fully assess the therapeutic potential of the GlycoConnect™ technology, a rodent tolerability study was also performed in rats at 20–35–50–60 mg/kg. Based on body weight over time (12 days, Figure 8), the SYNtansine™-based ADC is better tolerated than Kadcyla®, despite the cleavable linker,36 at all doses above 20 mg/kg, which becomes particularly apparent at the 35 mg/kg dose level (no weight loss for SYNtansine™ ADC, >10% for Kadcyla®) and at the highest dose of 60 mg/kg (maximum 20% weight reduction for SYNtansine™ ADC, followed by fast recovery, and up to 30% weight loss for Kadcyla®), which is in line with the reported37 maximum tolerated dose of 46 mg/kg for Kadcyla®.
Figure 8.
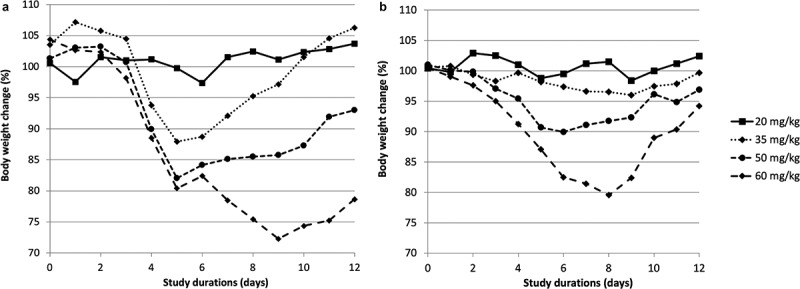
Monitoring of body weight over time of Sprague-Dawley rats treated with (a) Kadcyla® or (b) GlycoConnect™ ADC with SYNtansine™ at 20–35–50–60 mg/kg.
Two plots of time-dependent body weight of Sprague-Dawley rats clearly show that rats better tolerate DAR4 ADC based on GlycoConnect™ and HydraSpace™ with maytansinoid payload than Kadcyla®.
Discussion
We have demonstrated that GlycoConnect™ ADCs can be obtained with excellent overall efficiency (90–95%) from any antibody by enzymatic remodeling in a single step (trimming and glycosyl transfer), followed by metal-free click conjugation of cytotoxic payload. We discovered that 6-azidoGalNAc derivative 3 can be efficiently incorporated by native GalNAc-transferases, but not by the broadly applied galactosyltransferase mutant GalT(Y289L). Under optimized process conditions, full conversion into the azido-modified antibody is achieved with only 3% of GalNAc-transferase, 0.01% alkaline phosphatase and 10 equivalents of azidosugar 3. Further, we found that ADCs based on 3 were significantly less aggregation-prone than GalNAz-based ADC upon head-to-head comparison. The 3-modified antibodies reacted smoothly with an array of validated payloads, thereby demonstrating the versatility of the technology. Today, the optimized process has been applied in the manufacture of ADCs exceeding 0.5 kilogram antibody scale, demonstrating excellent scalability. Most importantly, it was found that a GlycoConnect™ ADC based on trastuzumab and a maytansinoid payload displayed a significantly enhanced therapeutic index (3–5 fold) versus the marketed ADC product Kadcyla®, with enhanced efficacy as well as tolerability. In fact, such improvement of therapeutic index has been established for a large number of scientific collaborations. This has led to the adaptation of the GlycoConnect™ technology, typically in combination with the polar spacer HydraSpace™ technology, by numerous biotechnology companies, including ADC Therapeutics, Mersana Therapeutics, Shanghai Miracogen, Innovent Biologics, Kyowa Kirin and Genmab. GlycoConnect™ is currently being clinically evaluated in three programs (ADCT-601, XMT-1592, and MRG004a) for various oncology indications and is anticipated to become one of the most prevalent technologies of new clinical ADCs. We hope to see cancer patients benefit from GlycoConnect™ technology with anticipated enhanced in-human therapeutic index in the near future.
Materials and methods
Endoglycosidases and glycosyl transferases were expressed from E. coli or CHO, respectively, and activity and stability studies were performed. Enzymatic remodeling of antibodies and metal-free click conjugation of linker-drugs was optimized by screening of various buffers and conditions. Synthesis of UDP 6-azidoGalNAc (3) and linker-payloads was performed by standard organic chemistry procedures.
Enzymatic glycan remodeling
Enzymatic glycan remodeling was performed by incubating antibody (15 mg/mL) with endo SH (0.15 mg/mL), AP (0.0015 mg/mL), UDP-6-N3-GalNAc (UDP-3, 1 mM) and TnGalNAcT (0.45 mg/mL) in 20 mM histidine pH 7.5, 150 mM NaCl and 6 mM MnCl2 for 16 hours at 30°C. Conversion into the azido-modified antibody was confirmed by mass spectral analysis. The azido-antibody was purified by protein A followed by buffer exchange to PBS pH 7.4 by dialysis or a desalting column.
Metal-free click conjugation
Conjugation conditions were optimized for each linker-payload (see Table 2 and Supplementary section 6). In general, azido-antibody (10–15 mg/mL) was incubated with linker-payload (3–10 equiv.) in the presence of co-solvent (either ≤25% DMF or ≤50% PG). Optionally, additives such as sodium deoxycholate (11 mM) were added. After overnight incubation at room temperature, the average DAR was determined by RP-HPLC analysis. ADCs were purified by SEC.
In vivo efficacy
Female SCID hairless outbred mice (6–9 weeks old) were anesthetized with ketamine/xylazine. A 20 mm3 tumor fragment of a T226 breast cancer patient-derived xenograft model was placed in the subcutaneous tissue. Next, 25 mice with T226 tumor (P12.1.4/0) between 75 and 196 mm3 were allocated, according to their tumor volume to give homogenous mean and median tumor volume in each treatment arm (5 mice/group). Treatments were initiated when the median tumor volume was 126 mm3 by intravenous injection with either vehicle (control), trastuzumab-3 coupled with 19 (3 or 9 mg/kg) or Kadcyla® (3 or 9 mg/kg). Tumor volume was measured twice weekly.
In vivo tolerability
Sprague-Dawley female rats, 6–7 weeks old and within a weight range of 150–174 grams, were placed into treatment groups of 2 female animals each. All animals were weighed and allocated to groups by computerized stratified randomization. Animals were dosed by injection into the tail vein and monitored for 12 days for mortality, morbidity, clinical signs, body weight and food consumption.
Supplementary Material
Acknowledgments
We kindly acknowledge Nazli Hilal Türkmen and our former colleagues Anna Wasiel and Ryan Heesbeen for their contributions to the work described in this manuscript. Symeres and Biosynth-Carbosynth are kindly acknowledged for their contribution in the scale-up of UDP-3 synthesis. Aravis, BioGeneration Ventures, BOM Capital, and Merck Ventures, the strategic corporate venture capital fund of Merck, are kindly acknowledged for financial support.
Funding Statement
The authors reported there is no funding associated with the work featured in this article.
Abbreviations
| ADC | Antibody-drug conjugate |
| AP | Alkaline phosphatase |
| As | Ascaris suum |
| BCN | Bicyclononyne |
| Ce | Caenorhabditis elegans |
| CHAPS | 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate |
| CHO | Chinese hamster ovary |
| DAR | Drug-to-antibody ratio |
| Dm | Drosophila melanogaster |
| DMF | N,N-dimethylformamide |
| GalNAcT | GalNAc transferase |
| GalT | Galactosyl transferase |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| IgG | Immunoglobulin |
| mAb | Monoclonal antibody |
| MBP | Maltose-binding protein |
| PBS | Phosphate-buffered saline |
| PDX | Patient-derived xenograft |
| PG | Propylene glycol |
| SEC | Size-exclusion chromatography |
| SHO | SCID hairless outbred |
| SUMO | Small ubiquitin-like modifier |
| TBS | TRIS-buffered saline |
| Tn | Trichoplusia ni |
| TRIS | Tris-(hydroxymethyl)aminomethane |
| UDP | Uridine diphosphate |
| UMP | Uridine monophosphate |
Disclosure statement
All authors are employed by Synaffix BV or were employed at the time of contribution to the scientific work described.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2022.2078466
References
- 1.Lambert JM, van Delft FL.. Introduction to antibody-drug conjugates. In: van Delft FL, Lambert JM, editors. Chemical linkers inantibody-drugconjugates, Royal Society of Chemistry; 2021, chapter 1. p. 1–10. doi: 10.1039/9781839165153. [DOI] [Google Scholar]
- 2.Thurston DE, Jackson PJM, editors. Cytotoxic payloads for antibody–drug conjugates.. Royal Society of Chemistry; 2019. doi: 10.1039/9781788012898. [DOI] [Google Scholar]
- 3.Walsh SJ, Bargh JD, Dannheim FM, Hanby AR, Seki H, Counsell AJ, Ou X, Fowler E, Ashman N, Takada Y, et al. Site-selective modification strategies in antibody-drug conjugates. Chem Soc Rev. 2021;50(2):1305–53. doi: 10.1039/d0cs00310g. [DOI] [PubMed] [Google Scholar]
- 4.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–32. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 5.van Berkel SS, van Delft FL.. Enzymatic strategies for (near) clinical development of antibody-drug conjugates. Drug Discov Today. 2018:30. doi: 10.1016/j.ddtec.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 6.Chin JW, Cropp T, Anderson JC, Mukherji M, Zhang Z, Schultz PG. An expanded eukaryotic genetic code. Science. 2003;301:964–67. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]
- 7.van Geel R, Wijdeven MA, Heesbeen R, Verkade JMM, Wasiel AA, van Berkel SS, van Delft FL. Chemoenzymatic conjugation of toxic payloads to the globally conserved N‐glycan of native mAbs provides homogeneous and highly efficacious antibody−drug conjugates. Bioconj Chem. 2015;26:2233–42. doi: 10.1021/acs.bioconjchem.5b00224. [DOI] [PubMed] [Google Scholar]
- 8.Verkade JMM, Wijdeven MA, van GR, Janssen BMG, van BSS, van DFL. A polar sulfamide spacer significantly enhances the manufacturability, stability, and therapeutic index of antibody–drug conjugates. Antibodies. 2018;7(1):12. doi: 10.3390/antib7010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.See https://synaffix.com/partnered-pipeline/, accessed Jan. 9th 2022 .
- 10.Beacon targeted therapies ADC database. [accessed2022Jan.9]. https://www.beacon-intelligence.com.
- 11.Reusch D, Tejada ML. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology. 2015;25:1325–34. doi: 10.1093/glycob/cwv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeze HH, Kranz C. Endoglycosidase and glycoamidase release of N-linked glycans. Curr Protoc Mol Biol. 2010;89:17.13A.1. doi: 10.1002/0471142735.im0815s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collin M, Olsén A. EndoS, a novel secreted protein from Streptococcus pyogenes with endoglycosidase activity on human IgG. EMBO J. 2000;20:3046–55. doi: 10.1093/emboj/20.12.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sjögren J, Struwe WB, Cosgrave EFJ, Rudd PM, Stervander M, Allhorn M, Holland SA, Nizet V, Collin M. EndoS2 is a unique and conserved enzyme of serotype M49 group A streptococcus that hydrolyses N-linked glycans on IgG and α1-acid glycoprotein. Biochem J. 2013;455:107–18. doi: 10.1042/BJ20130126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Q, Shankara S, Roy A, Qiu H, Estes S, McVie-Wylie A, Culm-Merdek K, Park A, Pan C, Edmunds T. Development of a simple and rapid method for producing non‐fucosylated oligomannose containing antibodies with increased effector function. Biotechn Bioengin. 2008;99:652–65. doi: 10.1002/bit.21598. [DOI] [PubMed] [Google Scholar]
- 16.Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Agnew BJ, Hsieh-Wilson LC. Direct in-gel fluorescence detection and cellular imaging of o-glcnac-modified proteins. J Am Chem Soc. 2008;130:11576–77. doi: 10.1021/ja8030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeglis BM, Davis CB, Aggeler R, Kang HC, Chen A, Agnew B, Lewis JS. Enzyme-mediated methodology for the site-specific radiolabeling of antibodies based on catalyst-free click chemistry. Bioconj Chem. 2013;24:1057–67. doi: 10.1021/bc400122c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramakrishnan B, Qasba PK. Structure-based design of β−1,4-galactosyltransferase I (β 4Gal-T1) with equally efficient N-Acetylgalactosaminyltransferase activity. J Biol Chem. 2002;277:20833–39. doi: 10.1074/jbc.M111183200. [DOI] [PubMed] [Google Scholar]
- 19.Kawar ZS, Van Die I, Cummings RD. Molecular cloning and enzymatic characterization of a UDP-GalNAc: glcNAcβ-R β1,4-N-acetylgalactosaminyltransferase from caenorhabditis elegans. J Biol Chem. 2002;277:34924–32. doi: 10.1074/jbc.M206112200. [DOI] [PubMed] [Google Scholar]
- 20.Laughlin ST, Bertozzi CR. In vivo imaging of Caenorhabditis elegans glycans. ACS Chem Biol. 2009;4:1068–72. doi: 10.1021/cb900254y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burnham-Marusich AR, Snodgrass CJ, Johnson AM, Kiyoshi CM, Buzby SE, Gruner MR, Berninsone PM. Metabolic labeling of Caenorhabditis elegans primary embryonic cells with azido-sugars as a tool for glycoprotein discovery. Plos One. 2012;7:e49020. doi: 10.1371/journal.pone.0049020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoskins RA, Carlson JW, Kennedy C, Acevedo D, Evans-Holm M, Frise E, Wan KH, Park S, Mendez-Lago M, Rossi F, et al. Sequence finishing and mapping of Drosophila melanogaster heterochromatin. Science. 2007;316:1625–28. doi: 10.1126/science.1139816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vadaie N, Jarvis DL. Molecular cloning and functional characterization of a Lepidopteran insect β4-N-acetylgalactosaminyltransferase with broad substrate specificity, a functional role in glycoprotein biosynthesis, and a potential functional role in glycolipid biosynthesis. J Biol Chem. 2004;279:33501–18. doi: 10.1074/jbc.M404925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bülter T, Schumacher T, Namdjou DJ, Gutiérrez Gallego R, Clausen H, Elling L. Chemoenzymatic synthesis of biotinylated nucleotide sugars as substrates for glycosyltransferases. ChemBioChem. 2001;2:884–94. doi:. [DOI] [PubMed] [Google Scholar]
- 25.Bosco M, Le Gall S, Rihouey C, S C-B, Bardor M, Lerouge P, Pannecoucke X. 6-azido-d-galactose transfer to N-acetyl-D--glucosamine derivative using commercially available β-1,4-galactosyltransferase. Tetrahedron Lett. 2008;49:2294–97. doi: 10.1016/j.tetlet.2008.02.018. [DOI] [Google Scholar]
- 26.Dommerholt J, Schmidt S, Temming R, Hendriks LJA, Rutjes FPJT, van HJCM, Lefeber DJ, Friedl P, van DFL. Readily accessible bicyclononynes for bioorthogonal labeling and three‐dimensional imaging of living cells. Angew Chem Int Ed. 2010;49:9422–25. doi: 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujii T, Reiling C, Quinn C, Kliman M, Mendelsohn BA, Matsuda Y. Physical characteristics comparison between maytansinoid-based and auristatin-based antibody-drug conjugates. Explor Target Antitumor Ther. 2021;2:576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benjamin SR, Jackson CP, Fang S, Carlson DP, Guo Z, Tumey LN. Thiolation of Q295: site-specific conjugation of hydrophobic payloads without the need for genetic engineering. Mol Pharmaceut. 2019;16:2795–807. doi: 10.1021/acs.molpharmaceut.9b00323. [DOI] [PubMed] [Google Scholar]
- 29.Nishimura SI, Hato M, Hyugaji S, Feng F, Amano M. Glycomics for drug discovery: metabolic perturbation in androgen‐independent prostate cancer cells induced by unnatural hexosamine mimics. Angew Chem Int Ed. 2012;51:3386–90. doi: 10.1002/anie.201108742. [DOI] [PubMed] [Google Scholar]
- 30.Hang HC, Yu C, Kato DL, Bertozzi CR. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc Nat Acad Sci. 2003;100:14846–51. doi: 10.1073/pnas.2335201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarabia F, Martín-Ortiz L, López-Herrera FJ. Synthetic studies towards the tunicamycins and analogues based on diazo chemistry. total synthesis of tunicaminyl uracil. Org Biomol Chem. 2003;1:3716–25. doi: 10.1039/B307674A. [DOI] [PubMed] [Google Scholar]
- 32.Cai L, Guan W, Wang W, Zhao W, Kitaoka M, Shen J, O’Neil C, Wang PG. Substrate specificity of N-acetylhexosamine kinase towards N-acetylgalactosamine derivatives. Bioorg Med Chem Lett. 2009;19:5433–35. doi: 10.1016/j.bmcl.2009.07.104. [DOI] [PubMed] [Google Scholar]
- 33.Mamidyala SK, Dutta S, Chrunyk BA, Préville C, Wang H, Withka JM, McColl A, Subashi TA, Hawrylik SJ, Griffor MC, et al. Glycomimetic ligands for the human asialoglycoprotein receptor. J Am Chem Soc. 2012;134:1978–81. doi: 10.1021/ja2104679. [DOI] [PubMed] [Google Scholar]
- 34.Megia-Fernandez A, Morales-Sanfrutos J, Hernandez-Mateo F, Santoyo-Gonzalez F. Synthetic applications of cyclic sulfites, sulfates and sulfamidates in carbohydrate chemistry. Curr Org Chem. 2011;15:401–32. doi: 10.2174/138527211794072542. [DOI] [Google Scholar]
- 35.Hu X, Lerch TF, Xu A. Efficient and selective bioconjugation using surfactants. Bioconj Chem. 2018;29:3667–76. doi: 10.1021/acs.bioconjchem.8b00594. [DOI] [PubMed] [Google Scholar]
- 36.Doronina SO, Mendelsohn BA, Bovee TD, Cerveny CG, Alley SC, Meyer DL, Oflazoglu E, Toki BE, Sanderson RJ, Zabinski RF, et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconj Chem. 2006;17:114–24. doi: 10.1021/bc0502917. [DOI] [PubMed] [Google Scholar]
- 37.Poon KA, Flagella A, Beyer J, Tibbitts J, Kaur S, Saada O, Yi JH, Girish S, Dybdal N, Reynolds T. Preclinical safety profile of trastuzumab emtansine (T-DM1): mechanism of action of its cytotoxic component retained with improved tolerability. Toxicol Appl Pharmacol. 2013;273:298–313. doi: 10.1016/j.taap.2013.09.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.