

Abstract
The aim of this study was to establish the use of the fluorescent probes carboxyfluorescein (cF) and propidium iodide (PI) for rapid assessment of viability, using Lactococcus lactis subsp. lactis ML3 exposed to different stress treatments. The cF labeling indicated the reproductive capacity of mixtures of nontreated cells and cells killed at 70°C very well. However, after treatment up to 60°C the fraction of cF-labeled cells remained high, whereas the survival decreased for cells treated at above 50°C and was completely lost for those treated at 60°C. In an extended series of experiments, cell suspensions were exposed to heating, freezing, low pH, or bile salts, after which the colony counts, acidification capacity, glycolytic activity, PI exclusion, cF labeling, and cF efflux were measured and compared. The acidification capacity corresponded with the number of CFU. The glycolytic activity, which is an indicator of vitality, was more sensitive to the stress conditions than the reproduction, acidification, and fluorescence parameters. The cF labeling depended on membrane integrity, as was confirmed by PI exclusion. The fraction of cF-labeled cells was not a general indicator of reproduction or acidification, nor was PI exclusion or cF labeling capacity (the internal cF concentration). When the cells were labeled by cF, a subsequent lactose-energized efflux assay was needed for decisive viability assessment. This novel assay proved to be a good and rapid indicator of the reproduction and acidification capacities of stressed L. lactis and has potential for physiological research and dairy applications related to lactic acid bacteria.
Lactic acid bacteria (LAB) are the most important group of bacteria encountered in the food industry. They are used as starter cultures for fermentation of milk, vegetables, and meat. In addition, LAB are used as probiotics and as silage inoculants. The reproduction of LAB and the activities of (starter) cultures containing LAB are important for the success of these fermentations. The production, storage, and use of LAB impose environmental stresses on the bacterial cells, such as freezing and drying of starter cultures, low pH during fermentations, and low temperatures and high salt concentrations during cheese ripening (38). Bacteria that are used as probiotics have to survive the low pH of the stomach and the high bile salt concentrations in the intestine to be effective in the gastrointestinal tract (22). Development of rapid and reliable methods for measuring viability is of the utmost importance for studies on bacterial physiology. Other important criteria for the use of new techniques in the dairy industry are the degree of accordance with established methods and the applicability for starter cultures subjected to different stresses.
The defined aspects of microbial viability are reproduction, vitality, and membrane integrity (27). Reproductive (living) cells are able to proliferate, whereas nonreproductive (dead) cells are not. This status is conventionally assayed by colony counting. Vital cells are metabolically active. Criteria that are used for vitality assessment include nucleic acid synthesis, rate of fermentation, heat production, ATP content, dye extrusion, dye reduction, and maintenance of membrane potential and pH gradients (21, 24). Intact cells have a cytoplasmic membrane with selective permeability. This membrane integrity can be determined by dye exclusion (14, 36). The membrane integrity, the degree of vitality, and the ability to reproduce depend on environmental conditions and the physiological status of the cell at the moment of investigation. Microorganisms can adopt different states, such as a dormant or a latent state, from which they may be resuscitated, i.e., induced to return to a physiologically active state (16).
Techniques that are used in the dairy industry to evaluate culture viability are colony counts and acidification tests (12, 13). The colony count method is the standard method for assessment of viability, but its disadvantages are the long incubation times needed to form countable colonies and underestimation of the viable cell count caused by cell clumping and chain formation. Acidification assays, for example like that described by Pearce (29), are often applied in the dairy industry in addition to plate counts. These assays are empirical ways of testing the acidification capacity of cultures and are supposed to be generally applicable, but they require long incubation times. The acidification test that is commonly used in the Dutch dairy industry takes an entire day, including 6 h of incubation.
A number of fluorescence techniques have been introduced over the past decades for assessment of viability and vitality of microorganisms (7, 21, 24, 32, 44). However, their use in applied food microbiology is still limited. Few fluorescence methods have been made applicable for practical food research and food industry situations. The described applications mainly concern detection and complete enumeration of microorganisms in food samples, such as milk, fixed cheese slides, and cryosectioned sausages (5, 6, 15, 18, 30, 39, 43, 47).
We investigated if the fluorescent probes carboxyfluorescein (cF) and propidium iodide (PI) are good indicators for viability of Lactococcus lactis subsp. lactis ML3. L. lactis is an important organism in dairy fermentations and a model organism for genetic and physiological studies (9, 17, 28, 34, 45). PI is a nucleotide-binding probe, supposed to enter only cells with damaged membranes. Therefore, it is used frequently to label dead cells (10, 11, 14, 27). Fluorescein and fluorescein derivatives have been used as viability probes for a wide range of microorganisms (1, 4, 10, 32).
To facilitate passive diffusion into the cell, nonfluorescent fluorescein (derivative) esters, such as 5(6)-cF diacetate (cFDA), are used for fluorescence labeling. The ester bonds are hydrolyzed by enzymes with esterase activity, yielding the green fluorescent dye molecules. Because enzyme activity is needed for hydrolysis, and membrane integrity is needed for the retention inside cells, it is supposed that viable cells accumulate fluorescein (derivatives) but dead cells are not able to do so (37). However, it was shown that the fluorescein derivatives cF and 2′,7′-bis(2-carboxyethyl)-5(6)-cF (BCECF) are actively extruded by L. lactis cells upon energizing with lactose (2, 17). We hypothesized that this probe efflux could be put to use as an additional measure of cell viability. cF labeling and, subsequently, the efflux could be measured to assess multiple aspects of cell viability: enzyme activity, membrane integrity, and metabolic activity upon energizing. These combined methods could give more information about the physiological condition than cF labeling alone does.
The approach of our study was to measure fluorescence-related parameters and glycolytic activity by fast assays, and also by the traditional but time-consuming plate count and acidification capacity test methods, and to compare these with each other. In dairy industry practice LAB are exposed to different types of environmental stress. Therefore, the applicability of the fluorescence-based methods was tested after exposure to a range of different stress conditions, comprising heating to 60 or 70°C, freezing at −20°C with or without glycerol, exposure to low pH, exposure to conjugated bile salts (CBS), and exposure to deconjugated bile salts (DBS). The results indicated shortcomings of cF labeling and PI exclusion for viability assessment. Combining cF labeling with subsequent cF efflux resulted in a novel assay, which proved to be a good and rapid indicator for the reproductive and acidifying capacities of L. lactis.
MATERIALS AND METHODS
Bacterial strain and culture conditions.
L. lactis subsp. lactis ML3 (NCDO 763) was grown at 30°C in M17 broth (Unipath Oxoid, Basingstoke, United Kingdom) supplemented with 0.5% (wt/vol) lactose (42). Overnight cultures were diluted 10-fold in fresh medium, incubated for approximately 2 h, and harvested in mid-exponential phase after they had reached an optical density at 620 nm (OD620) of approximately 0.7 by centrifugation at 4,000 × g for 10 min at 10°C. Cell suspensions were then centrifuged with an Eppendorf centrifuge (Biofuge Fresco; Heraeus Instruments, Ostrode, Germany) at 10,000 rpm for 2 min at 10°C. Harvested cells were washed twice with 50 mM potassium phosphate (KPi) buffer (pH 7.0) and concentrated in 50 mM KPi buffer (pH 7.0) to an OD620 of 20.
Treatments.
The concentrated cell suspensions were exposed to different types of stress: heat, freezing, low pH, and bile salts. Nontreated cell suspensions served as a positive control. All treatments were done with 400-μl portions of concentrated cell suspension (OD620 = 20). Exposure to heat was done at 70°C for 10 min or at 60°C for 90 s. Exposure to freezing was done at −20°C for 24 h with or without 30% glycerol. Exposure to low pH was done by incubation in 10 mM KPi, adjusted to pH 2.0 or 5.0 with hydrochloric acid, at 30°C for 60 min. Exposure to bile salts was done by incubation either with CBS, 0.2 or 1.0% (wt/wt) in 50 mM KPi (pH 6.0), or with DBS, 0.02, 0.06, or 1.0% (wt/wt) in 50 mM KPi (pH 7.0) at 30°C for 60 min. The CBS (Unipath Oxoid) contained mainly sodium glycocholate and sodium taurocholate. The DBS (Sigma-Aldrich, Steinheim, Germany) contained 50% sodium cholate and 50% sodium deoxycholate. For comparison, cell suspensions incubated in KPi buffer (pH 7.0) at 30°C for 60 min were used. After the treatments the cells were spun down, resuspended in KPi buffer (pH 7.0), and put on ice until use.
Plate counts.
The reproductive capacity was determined by plate counting. Cell suspensions were serially diluted with 50 mM KPi buffer (pH 7.0), and 100-μl portions of the appropriate dilution were spread out in triplicate on M17 plates containing 0.5% (wt/vol) lactose and 1.5% agar. After incubation for 3 days at 30°C the colonies from plates containing 40 to 300 colonies were counted.
Acidification capacity.
The acidification capacity was determined by the standard assay that is used in the dairy industry in The Netherlands. This assay resembles the Pearce test (29). The milk medium, a 10% (wt/wt) suspension of Nilac milk powder (NIZO, Ede, The Netherlands) in sterile water, was equilibrated at 30°C and inoculated to a final microbial protein concentration of 3.6 μg/ml. This is equal to 107 CFU/ml for a nontreated cell suspension. Then, 50 ml of the inoculated milk medium was incubated at exactly 30°C for 6 h in the dark. After incubation, the pH was measured and the acidification capacity was determined by titrating 15 ml of milk with 100 mM NaOH to pH 7.0 (determinations were made in triplicate).
Glycolytic activity.
The glycolytic activity was assessed by measuring the initial rate of acidification. The applied method was adapted from Gatto et al. (8). Cells were spun down and resuspended in 9.4 ml of 0.5 mM potassium MES (morpholineethanesulfonic acid)–50 mM KCl buffer at pH 6.5 to a final protein concentration of 0.15 mg/ml. After equilibration at 30°C, 200 μl of lactose was added to a final concentration of 6 mM. Acidification of the medium was monitored with a thin (diameter, 5 mm) Schott pH electrode and a pH meter connected to a recorder. Changes in pH values were converted into nanomoles of H+ by calibration of the cell suspension with 10-μl portions of 100 mM NaOH. The glycolytic activity was expressed in nanomoles of H+ per minute per milligram of protein.
Fluorescence labeling with cF.
Stock solutions of cF were prepared by dissolving cF (Molecular Probes, Eugene, Oreg.) in acetone (4.6 mg/ml) and immediately diluting 10- or 100-fold with 50 mM KPi buffer (pH 7.0). These solutions of 1.0 mM concentration and 100 μM concentration were divided into 0.5-ml aliquots to avoid repeated thawing and freezing and stored at −20°C in the dark. Cell suspensions (OD620 = 20) were diluted 1:1 with 100 μM cF and incubated at 30°C for exactly 10 min. Immediately after this labeling, the cells were spun down, washed once, and resuspended in 50 mM KPi (pH 7.0) to an OD620 of 4.0 for microscopic analyses or to an OD620 of 2.0 for fluorimetrical analyses.
Cell suspensions were microscopically analyzed with an Axioskop epifluorescence microscope equipped with a 50-W mercury arc lamp, a fluorescein isothiocyanate filter set (excitation wavelength, 450 to 490 nm; emission wavelength, >520 nm), an ×100 1.3 numerical-aperture Plan-Neofluar objective lens, and a camera (Carl Zeiss, Oberkochen, Germany). Photomicrographs were made with simultaneous light and epifluorescence microscopy, a low light intensity, a magnification of ×1,000, and an exposure time of 15 s, on Kodak 400 ASA color films. In these photomicrographs both the cF-labeled cells and the nonlabeled cells can be counted. In each experiment four photomicrographs were made, each depicting 100 to 400 cells.
To measure the cF labeling capacity, i.e., the average internal cF concentration (cFin), labeled cells were lysed by incubation at 70°C for 15 min and the debris was removed with a Biofuge (13,000 rpm, 2 min at 10°C) (Heraeus Instruments GmbH, Hanau, Germany). The fluorescence of the supernatant was measured fluorimetrically (excitation at 490 ± 5 nm and emission at 515 ± 5 nm), with a Perkin-Elmer LS 50B luminescence spectrometer equipped with a plate reader by using computer-controlled data acquisition. The intracellular cF concentration was calculated by using a calibration curve for cF (concentration range, 0 to 1.5 μM) in 50 mM KPi buffer (pH 7.0).
Fluorescence labeling with PI.
To test whether a treatment caused membrane damage, cells were incubated with the impermeant nucleotide binding probe PI. Stock solutions of 1.0 mg of PI (Molecular Probes) per ml were prepared in distilled water, stored in the refrigerator, and kept in the dark. PI was added to a concentration of 44 μM to a cell suspension with an OD620 of 2 and incubated at 30°C for 10 min. Photomicrographs were made with the same settings as used for the cFDA-labeled cell suspensions, and the red-labeled and nonlabeled cells were counted.
cFDA hydrolysis activity of cell extract.
Cell extracts were prepared by disrupting 600-μl portions of cell suspension (OD620 = 40) by sonication (10 times for 15 s with 45-s intervals; amplitude intensity of 15 μm). The cell debris was removed by centrifugation. The cFDA hydrolysis activity of cell extract was determined by incubation of 100 μl of 1.0 mM cFDA and 250 μl of the cell extract in 50 mM KPi buffer (pH 7.0) in a total volume of 1.0 ml at 30°C. The increase of cF concentration over time was monitored by measuring A490 every 5 min for 20 min. The measurements were corrected for the chemical hydrolysis of cFDA.
cF efflux activity.
Cells were labeled by cF as described above. The labeled cell suspensions (OD620 of 2.0) were incubated at 30°C with or without lactose (final concentration, 20 mM). Samples (200 μl) were withdrawn at specific time points and immediately centrifuged to remove the cells. From the fluorimetrically measured fluorescence of the supernatants and the total labeling capacity, the intracellular concentrations of cF at the sampling time points were calculated.
ATP concentration.
ATP concentration measurements were made under the same conditions as cF efflux measurements, and samples were withdrawn at the same time points. For measurement of total ATP concentration, a 20-μl sample was mixed with 80 μl of dimethyl sulfoxide and diluted with 5 ml of deionized water. For measurement of external ATP concentration, 20 μl of supernatant from a 80-μl, immediately centrifuged sample was used in the same way. Internal ATP concentration was calculated by subtracting the external ATP concentration from the total ATP concentration. The ATP concentrations were measured in an M 2500 biocounter (Lumac, Landgraaf, The Netherlands), with the Lumac luciferin/luciferase assay.
Estimation of cell protein content.
Protein concentrations were assayed by the method of Lowry et al. (20). Cellular volumes were calculated from the protein concentrations, assuming a ratio of 2.8 μl per mg of cell protein (31).
Experimental design and statistical analyses.
The experimental discrimination of viable and nonviable bacteria in mixtures of nontreated and heat (70°C)-treated cell suspensions by plate counts and that by cF labeling were performed with two batches of cells. The correlation between the methods was tested at a significance level of 0.05. In the series of experiments testing various physiological indicators after 13 different treatments, nearly all experiments were performed with at least three batches of cells. The effect of each stress treatment was tested for significance with the Student’s t test at the levels of 0.01 and 0.10. Furthermore, comparison of each pair of indicators was made for each treatment by calculating the probability values associated with the Student’s t test (P values). These P values, and the plot of the indicators against each other, were taken into consideration to evaluate the general correspondence between the two indicators.
RESULTS
Accumulation and retention of cF.
L. lactis can easily be labeled with cF and retains the probe well when not energized, but it extrudes cF rapidly upon lactose addition. Nearly all L. lactis cells of a nontreated cell suspension were labeled within a few minutes of incubation with cFDA (Fig. 1A). When labeled cells were stored on ice for up to 2 h, a gradual decrease of intracellular cF at the rate of 8% per hour was observed. When the cells were kept at 30°C the rate of leakage was 18% per hour. The dissipation of the proton motive force (PMF) caused by addition of the ionophores valinomycin and nigericin to nonenergized cells resulted in an additional and immediate loss of 10%, after which the rate of leakage was the same as that for the other cells kept at 30°C (Fig. 2A). These rates of cF leakage were negligible compared to the immediate and rapid extrusion upon energizing by lactose (Figs. 1B and 1C). The time needed to extrude 50% of accumulated cF (t1/2) was less than 2 min (Fig. 2B). Also, when the PMF was dissipated there was an immediate and rapid extrusion upon energizing (Fig. 2B). Since cells treated with valinomycin and nigericin also produce ATP upon addition of lactose, this indicates that the extrusion is most likely mediated by an ATP-driven transport system.
FIG. 1.

Labeling of L. lactis by cF and subsequent active extrusion. A nontreated cell suspension was labeled with cF, by incubation with 50 μM cFDA at 30°C and pH 7.0 for 10 min, and washed once (A). This labeled cell suspension was incubated with 20 mM lactose at 30°C for 2 min (B) and for 15 min (C). Cell suspensions were photographed with simultaneous light and epifluorescence microscopy (excitation wavelength, 450 to 490 nm; emission wavelength, >520 nm) to visualize both stained and nonstained cells. Bar represents 10 μm for all micrographs.
FIG. 2.

Retention of cF by L. lactis. Cells were loaded with cF and resuspended in 50 mM KPi buffer (pH 7.0). (A) Retention of cF in the absence of an energy source when cells were kept on ice (∗), at 30°C (■), and at 30°C with the addition of 2 μM valinomycin and nigericin (▴). (B) Retention of cF in cells with the addition of 20 mM lactose (■) and with the addition of 20 mM lactose after dissipation of the PMF by adding 2 μM valinomycin and nigericin (▴). The internal ATP levels in cells with the addition of lactose (□) and with the addition of lactose after dissipation of the PMF (▵) were also measured.
Correspondence between cF labeling and reproduction after temperature treatments.
Living bacteria could easily be distinguished from dead bacteria in mixtures of nontreated and heat (70°C)-treated cell suspensions by staining with cFDA and analyzing with fluorescence microscopy. This was confirmed by counterstaining the cell suspensions with the membrane-impermeant DNA stain PI. When a nontreated cell suspension was incubated with cFDA and PI, nearly all (97%) of the cells showed bright green fluorescence and very few (3%) showed red fluorescence (Fig. 3A). The small fraction that was not labeled by cF but was labeled by PI is presumably the fraction dead cells present in the cell culture at harvest. Treatment of a cell suspension at 70°C for 10 min resulted in a total loss of reproduction, as determined with standard plate counts (no colonies were detected after plating of ≥109 cells). When such a heat-treated cell suspension was incubated with cFDA and PI, not a single green fluorescent cell could be detected but all cells were brightly labeled red (Fig. 3C). This PI labeling indicated that the cells were killed because of major membrane damage. In mixtures of nontreated and heat (70°C)-treated cell suspensions, live and dead cells could be distinguished by double labeling with cF and PI (Fig. 3B). This double labeling gave the same fractions of green- and red-labeled cells as the cF labeling and PI labeling performed separately. This reflects that these probes act in a complementary manner.
FIG. 3.

Fluorescence microscopy of L. lactis cells double labeled with cF and PI. Suspensions containing 100% nontreated cells (A), 50% nontreated cells mixed with 50% heat-killed cells (B), and 100% heat-killed cells (C) were incubated with 50 μM cFDA and 44 μM PI. Cell suspensions were photographed with simultaneous light and epifluorescence microscopy (excitation wavelength, 450 to 490 nm; emission wavelength, >520 nm) to visualize both stained and nonstained cells. Bar represents 10 μm for all micrographs.
cF labeling was compared with standard plate counts in order to validate the labeling as an alternative quantitative assay for viability assessment. The number of reproductive cells determined by plate counts was linearly related to the proportion of nontreated cells (Fig. 4A). The fraction cF-labeled cells and the labeling capacity (the average intracellular cF concentration of the mixed cell suspensions) were also linearly related to the proportion of nontreated cells (Fig. 4B and C). The fluorescence microscopy method has an especially high precision as indicated by the value of R2. The coefficients of correlation between the microscopic counts and the plate counts and between the labeling capacity and the plate counts were both 0.96 (P < 0.005). Thus, cF labeling provides a valid alternative for determining the viability of these mixed cell suspensions, whether it is examined by fluorescence microscopy or by spectrofluorimetry.
FIG. 4.

Experimental discrimination of viable and nonviable bacteria. An L. lactis cell suspension (1010 CFU/ml) was divided into two portions. One was not treated and the other was exposed for 10 min to 70°C. The nontreated and the heat-treated portions were mixed in various proportions and plated on M17 agar supplemented with 0.5% (wt/vol) lactose or labeled for 10 min with 50 μM cFDA, washed, and analyzed by fluorescence microscopy and spectrofluorimetry. The experiment was performed with two batches of cells. Plate counts (A), fraction cF-labeled cells (B), and average intracellular cF concentration (C) are all plotted against the known fraction of nontreated cells.
However, after the suspensions were exposed to temperatures of 50 to 65°C the cF labeling results were discordant with the plate count results (Fig. 5). No colonies were detected after 90-s exposure to 65°C or higher, less than 0.1% of the cells survived heating to 60°C, 46% survived heating to 55°C, and 69% survived heating to 50°C. Nevertheless, after exposure to 60°C or lower, more than 95% of the cells were labeled by cF. The cF labeling capacity (cFin) decreased with increasing temperature, but the results for this parameter as well were not in accord with those for reproduction. However, when cell suspensions exposed to 60°C were tested for cF efflux, no active transport was detected (Fig. 6). Thus, in this case, the lack of efflux is a good indicator for the lack of reproduction while cF labeling is not. This supports our hypothesis of the additional value of cF efflux for assessment of viability.
FIG. 5.

Effects of temperature treatment on L. lactis survival and cF labeling. L. lactis cell suspensions (1010 CFU/ml) were subjected to elevated temperatures for 90 s. After the treatment the suspensions were plated on M17 agar supplemented with 0.5% (wt/vol) lactose and tested for cF labeling. Both the fraction cF-labeled cells and the cF labeling capacity were determined. Data are the means and standard deviations of three experiments and are expressed as values relative to that for nontreated cells (taken as 100). RT, room temperature.
FIG. 6.

Effect of stresses on the cF efflux by lactose-energized L. lactis. Cell suspensions were pretreated under different stress conditions, loaded with cF, and resuspended in 50 mM KPi buffer (pH 7.0). Five minutes after the start of the incubation at 30°C, lactose was added to a final concentration of 20 mM. Results of representative experiments with a non-treated-cell suspension and five of the stressed cell suspensions are given. No treatment, 100% = 364 μM (■); exposure to 60°C for 90 s, 100% = 149 μM (□); exposure to −20°C for 24 h, 100% = 36 μM (∗); exposure to pH 5.0, 100% = 137 μM (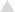 ); exposure to 1.0% CBS, 100% = 105 μM (●); and exposure to 0.02% DBS, 100% = 136 μM (◊).
); exposure to 1.0% CBS, 100% = 105 μM (●); and exposure to 0.02% DBS, 100% = 136 μM (◊).
Comparison of various physiological indicators after stress.
L. lactis cell suspensions were exposed to 13 different treatments, including heat, freezing, low pH, and addition of bile salts. Nontreated cell suspensions served as the control. The effects of the stress exposures on the reproduction (plate counts), acidification capacity, glycolytic activity, and various fluorescence-related parameters including the cF efflux are presented in Table 1.
TABLE 1.
Effects of stress on various physiological parametersa
| Treatment | Plate counts, CFU · mg of protein−1 (%) | Acidification capacity, mmol H+ · 6 h−1 · mg of protein−1 (%) | Glycolytic activity, μmol H+ · min−1 · mg of protein−1 (%) | Fraction, cF-labeled cells (%) |
|---|---|---|---|---|
| No treatment | 5.35 × 109 ± 9.71 × 108 (100) | 20.72 ± 1.55 (100) | 1.02 ± 0.08 (100) | 0.97 ± 0.02 (100) |
| 70°C, 10 min | 0.00 ± 0.00 (0) | 0.53 ± 0.46 (3)** | 0.00 ± 0.00 (0)** | 0.00 ± 0.00 (0)** |
| 60°C, 90 s | 1.24 × 106 ± 2.20 × 106 (0) | 0.61 ± 0.16 (3)** | 0.00 ± 0.00 (0)** | 0.90 ± 0.06 (92)** |
| −20°C, 24 hr, 0% glycerol | 8.11 × 108 ± 6.83 × 108 (15) | 5.40 ± 0.79 (26)** | 0.01 ± 0.01 (1)** | 0.31 ± 0.09 (32)** |
| −20°C, 24 hr, 30% glycerol | 4.19 × 109 ± 6.99 × 108 (78) | 11.79 ± 2.34 (57)** | 0.28 ± 0.17 (27)** | 0.86 ± 0.08 (89) |
| 30°C, 60 min, pH 7.0 | 4.98 × 109 ± 1.23 × 109 (93) | 18.21 ± 2.60 (88) | 0.69 ± 0.21 (67) | 0.97 ± 0.01 (100) |
| 30°C, 60 min, pH 5.0 | 4.89 × 109 ± 1.11 × 109 (91) | 16.91 ± 3.12 (82) | 0.54 ± 0.07 (53)** | 0.95 ± 0.03 (98) |
| 30°C, 60 min, pH 2.0 | 4.40 × 108 ± 7.26 × 107 (8)** | 5.24 ± 2.29 (25)** | 0.00 ± 0.00 (0)** | 0.47 ± 0.44 (48) |
| 30°C, 60 min, CBS 0.2% | 4.40 × 109 ± 1.58 × 109 (82) | 17.00 ± 3.54 (82) | 0.56 ± 0.01 (55)** | 0.95 ± 0.03 (97) |
| 30°C, 60 min, CBS 1.0% | 2.21 × 109 ± 9.97 × 108 (41)** | 16.26 ± 2.65 (78)* | 0.47 ± 0.05 (46)** | 0.86 ± 0.10 (89) |
| 30°C, 60 min, DBS 0.02% | 4.12 × 109 ± 1.12 × 109 (77) | 18.00 ± 2.62 (87) | 0.45 ± 0.08 (44)** | 0.85 ± 0.06 (88)** |
| 30°C, 60 min, DBS 0.06% | 2.61 × 109 ± 1.32 × 109 (49)* | 17.35 ± 1.99 (84)* | 0.36 ± 0.06 (35)** | 0.40 ± 0.31 (41)** |
| 30°C, 60 min, DBS 1.0% | 2.20 × 103 ± 1.93 × 103 (0)** | 0.58 ± 0.09 (3)** | 0.00 ± 0.00 (0)** | 0.00 ± 0.00 (0)** |
| Fraction, PI-excluding cells (%) | cF labeling capacity, μM (%) | cFDA hydrolysis activity, nM · min−1 · mg of protein−1 (%) | Fraction cF efflux (%) | Product of cF labeling capacity and efflux fraction (%) |
|---|---|---|---|---|
| 0.97 ± 0.02 (100) | 293 ± 48 (100) | 67.85 ± 8.82 (100) | 0.93 ± 0.05 (100) | 0.90 ± 0.05 (100) |
| 0.01 ± 0.02 (1)** | 7 ± 6 (3)** | 10.51 ± 2.57 (15)** | 0.00 (0) | |
| 0.67 ± 0.29 (69)* | 134 ± 46 (46)** | 33.76 ± 6.25 (50)** | 0.07 ± 0.03 (7)** | 0.06 ± 0.02 (6) |
| 0.27 ± 0.11 (28)** | 37 ± 15 (13)** | 55.38 ± 12.33 (82) | 0.32 ± 0.04 (34)** | 0.10 ± 0.02 (11) |
| 0.84 ± 0.10 (87) | 133 ± 28 (45)** | 47.56 ± 13.40 (70) | 0.50 ± 0.15 (54)** | 0.43 ± 0.11 (48) |
| 0.97 ± 0.01 (101) | 243 ± 51 (83)* | 49.38 ± 7.84 (73)* | 0.91 ± 0.04 (98) | 0.88 ± 0.04 (98) |
| 0.95 ± 0.01 (98) | 126 ± 39 (43)** | 48.12 ± 3.72 (71)* | 0.94 ± 0.07 (101) | 0.89 ± 0.07 (99) |
| 0.19 ± 0.20 (19)** | 32 ± 33 (11)** | 20.49 ± 10.10 (30)** | 0.30 ± 0.02 (32)** | 0.14 ± 0.11 (15) |
| 0.95 ± 0.01 (98) | 96 ± 27 (33)** | 38.37 ± 5.18 (57)* | 0.82 ± 0.07 (89) | 0.78 ± 0.07 (86) |
| 0.70 ± 0.26 (73) | 103 ± 36 (35)** | 40.83 ± 4.54 (60)* | 0.68 ± 0.20 (73) | 0.59 ± 0.18 (65) |
| 0.88 ± 0.11 (91) | 149 ± 29 (51)** | 42.39 ± 3.69 (62)* | 0.90 ± 0.06 (97) | 0.77 ± 0.07 (85) |
| 0.46 ± 0.35 (48)* | 52 ± 49 (18)** | 48.35 ± 7.85 (71)* | 0.92 (99) | 0.36 (41) |
| 0.00 ± 0.00 (0)** | 7 ± 6 (2)** | 36.71 ± 8.09 (54)* | 0.00 (0) |
Results given are the average values ± the standard deviations, and the percentages relative to that for cells no treatment given (set at 100) are shown in parentheses. In addition, the asterisks mark the stress conditions that have significant effects as determined by comparison with the value for cells given no treatment (Student’s t test; ∗∗, P < 01; ∗, P < 0.1) for all parameters except for the product of cF labeling and efflux. For this combined parameter, Student’s t test could not be performed because the fraction of cF-labeled cells and the fraction of cF efflux were measured with different cell batches. All experiments were repeated at least two times. In some efflux experiments the labeling was too low (<25 μM) to enable measurements. No efflux could be measured after exposure to 70°C or to 1% DBS, only one experiment could be performed after exposure to 0.06% DBS (therefore, no standard deviation is given), and only two experiments could be performed after exposure to pH 2.
The stress conditions were designated as severe, intermediate, or mild based on their effects on the survival (plate counts). Exposure to 70°C for 10 min resulted in a total loss of reproduction, and exposure to 60°C for 90 s or to 1.0% DBS concentration resulted in a loss of more than 99% of the population. Therefore, these three conditions were classified as severe stress conditions. Exposure to freezing with or without glycerol, to pH 2.0, to 1% CBS, or to 0.06% DBS caused loss of reproduction of part of the population, but more than 5% survived. These were classified as intermediate stress conditions. The other treatments did not decrease the plate counts significantly and were therefore classified as mild treatments.
The fraction of cF-labeled cells showed some important deviations from the (in most cases) reasonable to good correspondence (Fig. 7A). After treatment at 60°C, 90% of the cells were able to accumulate cF, but there was hardly any reproduction. Also, after exposure to 1% CBS, the labeling was significantly (P < 0.05) higher than plate counts. The cF labeling of cells exposed to low pH (pH 2) had variable results, while the plate counts were reasonably consistent. These results indicate that cF labeling is not a general indicator for reproduction. After exposure to the mild stress conditions most cells were labeled by cF, but the labeling capacity (Fig. 7B), i.e., the cFin, was lower than that of nontreated cell suspensions. The other stress conditions also decreased the labeling capacity more than they decreased the fraction cF-labeled cells. Under all conditions tested there was at least some cFDA hydrolysis activity (Fig. 7C), but it did not correspond with the cF labeling capacity. Neither the hydrolysis activity nor the cF labeling capacity corresponded with the plate counts. The fractions of PI-excluding cells corresponded with the fractions of cF-labeled cells, indicating that the cF labeling depended on the integrity of the membrane. Under most stress conditions the PI exclusion corresponded with the plate counts (Fig. 7D). However, the PI exclusion by cell suspensions treated at 60°C shows that cells can lose their reproductive capacity but still have an intact membrane.
FIG. 7.

Comparisons of fluorescence parameters, plate counts, acidification capacity, and glycolytic activity of L. lactis after exposure to different types of stress. Comparisons are shown for the fraction cF-labeled cells with plate counts (A), the cF labeling capacity with plate counts (B), the cFDA hydrolysis activity with plate counts (C), the fraction PI-excluding cells with plate counts (D), the product of cF labeling and efflux with plate counts (E), the plate counts with acidification capacity (F), the product of cF labeling and efflux with acidification capacity (G), and the glycolytic activity with acidification capacity (H). For each panel the data are extracted from Table 1. The symbols indicate no treatment (■), exposure to 70°C for 10 min ( ), exposure to 60°C for 90 s (□), exposure to −20°C for 24 h (∗), exposure to −20°C and 30% glycerol for 24 h (+), exposure to pH 7.0 (▵), exposure to pH 5.0 (), exposure to pH 2.0 (▴), exposure to 0.2% CBS (○), exposure to 1.0% CBS (●), exposure to 0.02% DBS (◊), exposure to 0.06% DBS (
), exposure to 60°C for 90 s (□), exposure to −20°C for 24 h (∗), exposure to −20°C and 30% glycerol for 24 h (+), exposure to pH 7.0 (▵), exposure to pH 5.0 (), exposure to pH 2.0 (▴), exposure to 0.2% CBS (○), exposure to 1.0% CBS (●), exposure to 0.02% DBS (◊), exposure to 0.06% DBS (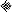 ), and exposure to 1.0% DBS (⧫). The error bars indicate the standard deviations. The bold lines indicate the optimal situation, that is, linear regression through the origin and the point for the non-treated-cell suspensions.
), and exposure to 1.0% DBS (⧫). The error bars indicate the standard deviations. The bold lines indicate the optimal situation, that is, linear regression through the origin and the point for the non-treated-cell suspensions.
The two stress conditions that caused complete loss of cF labeling (70°C and 1.0% DBS) also caused complete loss of reproductive capacity. For these conditions no further investigation was needed; absence of cF labeling indicates absence of reproduction. Cell suspensions of which (part of) the cells were labeled were investigated further by efflux assays. After exposure to pH 7.0, pH 5.0, and bile salts, cell suspensions showed efflux behavior similar to that of nontreated cell suspensions, although the cFin (the labeling capacity) was lower. After exposure to pH 2.0 and to freezing the efflux was not complete, and after exposure to 60°C there was hardly any efflux (Fig. 6). In all cases the extrusion or leakage of cF without energizing was negligible for the duration of the experiment, so no corrections were necessary. Labeling and efflux were combined into one parameter by multiplying the fraction of cells labeled after incubation with cFDA by the fraction of cells showing efflux up to 15 min after lactose addition. The consequent efflux assay improved the estimation of reproductive capacity given by cF labeling (Fig. 7A and E). In general, the results for the combined cF labeling and efflux parameter corresponded well with the plate counts.
The effects of the stresses on acidification corresponded with the effects on plate counts, and the outcome of the comparisons of fluorescence-related parameters with acidification resembled that obtained with plate counts. The acidification capacity is the viability parameter that is used in the dairy industry to estimate the success of fermentations. The acidification capacity corresponded reasonably well with the plate counts (Fig. 7F). Apparently, the results of the industrial acidification tests were largely dependent on the reproductive capacity. Comparison of the combined cF labeling and efflux parameter with the acidification assay revealed a good correspondence (Fig. 7G), whereas none of the other fluorescence-related parameters did. This combined parameter is the best fluorescence indicator for the industrially relevant characteristic of fermentation capacity.
The effects of stress on vitality were assessed by the glycolytic activity assay, which measures the physiological condition of the cell suspensions immediately after the treatments. The glycolytic activity is generally more sensitive to the stress conditions than the reproduction and acidification capacity are (Fig. 7H). Therefore, glycolytic activity is not a good indicator for acidification capacity. None of the fluorescence-related parameters corresponded with the glycolytic activity.
DISCUSSION
In this study we investigated the use of the fluorochromes cF and PI for viability assessment of L. lactis subsp. lactis ML3. The aim was to develop a rapid assay that provides a generally valid indicator for reproduction. Therefore, cell suspensions were exposed to different stress conditions. Furthermore, fluorescence-related parameters were compared not only with plate counts but also with acidification capacity and with glycolytic activity.
In mixtures of heat (70°C)-treated cells and nontreated cells, live and dead cells could be distinguished clearly by cF or PI labeling. For cF, this was reflected by the high coefficient of correlation between the number of CFU and the labeling, determined either by fluorescence microscopy or by spectrofluorimetry. Given the precision of the experimental results, fluorescence microscopy combined with photography is the preferred method. For a rapid judgment of viability, the fractions of living and dead cells can be estimated directly with fluorescence microscopy (without photography). When many samples are to be assayed, spectrofluorimetry is preferred. Because these experiments take an hour or less, they are appealing and time-saving alternatives to the classic plate count method.
However, cF labeling is not a general indicator for viability. For cells exposed to temperatures from 50 to 60°C or to high concentrations of CBS, the fractions for cF labeling were higher than the reproductive capacity. In addition, the results for PI exclusion by cells treated at 60°C were discordant with reproductive capacity. A disagreement between viability labeling with a fluorescein derivative and plate counts when the cells were incubated at low temperatures (10 and 4°C) for up to 30 days was also reported for Escherichia coli (33). This stress induced a so-called viable but nonculturable status, as shown by reduced ability to form colonies even though the cells remained intact and showed intracellular enzyme activity. PI staining is very dependent on the incubation conditions. Under suboptimal conditions staining of fresh and heat (80°C)-killed cells can already give false-positive results, especially when faintly red-labeled cells are interpreted as being dead (46). When they are exposed to milder stress conditions correct distinction of live and dead cells might be even more difficult. For mammalian cells cF fluorescence and PI exclusion were also found to be unreliable indices of viability under stress (23).
Because of the complexity of the physiological status and heterogeneity of bacterial cells in a culture, especially after stress, multiparameter analysis is preferable (3, 27). Examples indicate that after exposure to stress, cultures may contain dormant and injured subpopulations. Dormant cells may regain growth by resuscitation, while damaged cells may recover from injury and regain growth (16, 19). Study of growth, recovery, dormancy, and adaptation is important for understanding bacterial physiology. Consistent terminology and logical concepts are indeed needed to avoid confusion. Furthermore, we agree with Kell et al. (16) that the validity of a cytological assay can be confirmed only by correlation with culture assays for a specific mechanism of cell death. Therefore, it was our approach to evaluate the validity of the fluorescence assays as indicators for particular practical aspects of viability by comparing the fluorescence-related parameters with plate counts, acidification capacity, and glycolytic activity after different types of stress treatment.
The glycolytic activity assay does not appear to be a good indicator for the acidification capacity. The assay was adapted from the acidification power (AP) test, developed by Gatto et al. (8), which measures the pH decrease during 10 min of spontaneous acidification followed by 10 min of substrate-induced acidification. They suggested that there is a linear correlation between the AP test and the Pearce test (29) for Lactobacillus delbrueckii subsp. bulgaricus. The results reported by Riis et al. (35) do not indicate a linear correlation. Nevertheless, they evaluated the AP test as promising for use in the dairy industry because it detects minor differences in starter cultures, it is rapid, and it can be automated and standardized. Both the AP test and our glycolytic activity assay measure the ability to utilize exogenous carbohydrates within a short time. From the comparison with the industrial acidification test it appears that glycolytic activity is not a valid quantitative indicator for fermentation capacity. During a fermentation, such as that mimicked by the industrial acidification test, recovery and damage repair processes might result in a higher fermentation capacity than one would predict based on the vitality assessed directly after exposure to stress.
It has been suggested that the rate of FDA hydrolysis can be used to determine bacterial numbers (40) and to monitor microbial activity (41). However, our results show that the effects of the stresses on cFDA hydrolysis do not accord with the effects on plate counts, acidification, or glycolytic activity. The cFDA hydrolysis activity was decreased by all treatments, but it never became limiting for labeling; even dead cell populations still showed hydrolysis activity. Therefore, we conclude that cFDA or FDA hydrolysis is not a valid viability indicator for dairy applications.
Labeled L. lactis cells actively extrude the accumulated cF upon energizing, as they do after dissipation of the PMF by valinomycin and nigericin. Under these conditions the internal ATP levels remained high. This suggests that the cF efflux takes place via a primary transport system, which is most probably ATP dependent. The same was found for BCECF efflux (26). However, the rate of cF efflux is much higher than the rate of BCECF efflux. For cF efflux at pH 7 we found a t1/2 of less than 2 min, while for BCECF efflux t1/2 values of 6 min (pH 6) and 11 min (pH 8) were reported (25). Of the known extrusion systems only multidrug resistance transport systems have demonstrated broad substrate ranges, and since BCECF does not resemble any naturally occurring compound, it was suggested that BCECF is extruded by such an extrusion system (26). cF might be transported by the same or a similar extrusion system. The rapid efflux indicates a high affinity of the extrusion system for cF.
Our experiments showed that cells that had no cF labeling capacity also had no reproductive capacity or acidification capacity, but that the opposite was not generally true. When cF-labeled cell suspensions were subsequently tested for efflux, nonreproductive but labeled cell suspensions proved to be noneffluxing. Cell suspensions with a high labeled fraction and a high rate of efflux also had high relative reproductive capacity and acidification. The heat (60°C)-treated cell suspensions had a high fraction of labeling but hardly any efflux. They also had hardly any reproduction or acidification. The loss of the ability to fully efflux the cF might reflect the inactivation of the transport system or the loss of glycolytic energy generation when cells are killed. The labeling and the efflux ability of a culture were combined into one parameter by multiplying the fraction of cells labeled after incubation with cFDA by the fraction of cF efflux after 15 min of incubation with lactose, with cFin after labeling set at 1.0. This combined cF labeling and efflux parameter gave the best indication of reproduction and acidification capacities. It proved applicable for L. lactis after exposure to the tested stress conditions (heat, freezing, low pH, addition of CBS, and addition of DBS) and appears to be a general indicator of L. lactis reproduction and fermentation capacities.
This novel assay has potential for physiological research on LAB and for applications in the dairy industry. One application might be the assessment of the acidification capacity of cheese starters by fast fluorescence microscopic examination of cF labeling and efflux prior to the start of the fermentation. In addition, combined cF labeling and efflux assays could be used in the selection of strains of LAB to test, for example, the effect of freezing and storage on cheese starters or the effect of low pH and high bile salt concentrations on probiotics. Furthermore, combination of a cF labeling and efflux assay with strain-specific fluorescent markers could be used to study the viability and population dynamics of (stressed) mixed cultures of LAB. Finally, combining the fluorescence assays with flow cytometry may enable fast measurement of the physiological status of LAB present in cultures and of subpopulations and individual cells of LAB in dairy industry research and applications.
ACKNOWLEDGMENTS
We thank Riske Meewisse for doing preliminary experiments, Jeroen Hugenholtz from the NIZO for stimulating discussions, and Wieger Wamelink for advice on the statistical analyses and critical reading of the earlier version of the manuscript.
This research was financially supported by The Netherlands Technology Foundation (STW).
REFERENCES
- 1.Bascomb S. Enzyme tests in bacterial identifications. Methods Microbiol. 1987;19:105–160. [Google Scholar]
- 2.Breeuwer P, Drocourt J-L, Rombouts F M, Abee T. A novel method for continuous determination of the intracellular pH in bacteria with the internally conjugated fluorescent probe 5 (and 6-)-carboxyfluorescein succinimidyl ester. Appl Environ Microbiol. 1996;62:178–183. doi: 10.1128/aem.62.1.178-183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davey H M, Kell D K. Flow cytometry and cell sorting of heterogeneous microbial populations: the importance of single-cell analyses. Microbiol Rev. 1996;60:641–696. doi: 10.1128/mr.60.4.641-696.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diaper J P, Edwards C. The use of fluorogenic esters to detect viable bacteria by flow cytometry. J Appl Bacteriol. 1994;77:221–228. [Google Scholar]
- 5.Dodd C E R, Waites W M. The use of cryosectioning and histological staining to detect the sites of microbiological growth in British fresh sausages. J Appl Bacteriol. 1989;67:xvii. [Google Scholar]
- 6.Fernandes M A, Jackman P J H, Clark S A, Gunard S R. Detection and quantification of micro-organisms in a heterogenous food stuff by image analysis. CABIOS. 1988;4:291–295. doi: 10.1093/bioinformatics/4.2.291. [DOI] [PubMed] [Google Scholar]
- 7.Fouchet P, Jayat C, Héchard Y, Ratinaud M-H, Frelat G. Recent advances of flow cytometry in fundamental and applied microbiology. Biol Cell. 1993;78:95–109. doi: 10.1016/0248-4900(93)90120-4. [DOI] [PubMed] [Google Scholar]
- 8.Gatto E, Peddie F, Andrews S. Acidification power: performance evaluation of freeze-dried lactic acid bacteria. Food Aust. 1993;45:124–128. [Google Scholar]
- 9.Hartke A, Bouché S, Giard J-C, Benachour A, Boutibonnes P, Auffray Y. The lactic acid stress response of Lactococcus lactis subsp. lactis. Curr Microbiol. 1996;33:194–199. doi: 10.1007/s002849900099. [DOI] [PubMed] [Google Scholar]
- 10.Haugland R P. Handbook of fluorescent probes and research chemicals. 6th ed. Eugene, Oreg: Molecular Probes, Inc.; 1996. p. 679. [Google Scholar]
- 11.Humphreys M J, Allman R, Lloyd D. Determination of the viability of Trichomonas vaginalis using flow cytometry. Cytometry. 1994;15:343–348. doi: 10.1002/cyto.990150410. [DOI] [PubMed] [Google Scholar]
- 12.International Dairy Federation. IDF guideline: determination of acidifying activity of dairy cultures. Bull Int Dairy Fed. 1995;306:34–36. [Google Scholar]
- 13.International Dairy Federation. Dairy starter cultures of lactic acid bacteria (LAB). Standard of identity. Int Dairy Fed Standard. 1997;149A:1–8. [Google Scholar]
- 14.Kaneshiro E S, Wyder M A, Wu Y-P, Cushion M T. Reliability of calcein acetoxy methyl ester and ethidium homodimer or propidium iodide for viability assessment of microbes. J Microbiol Methods. 1993;17:1–16. [Google Scholar]
- 15.Karwoski M, Venelampi O, Linko P, Mattila-Sandholmen T. A staining procedure for viability assessment of starter culture cells. Food Microbiol. 1995;12:21–29. [Google Scholar]
- 16.Kell D B, Kaprelyants A S, Weichart D H, Harwood C R, Barer M R. Viability and activity in readily culturable bacteria: a review and discussion of the practical issues. Antonie Leeuwenhoek. 1998;73:169–187. doi: 10.1023/a:1000664013047. [DOI] [PubMed] [Google Scholar]
- 17.Konings W N, Lolkema J S, Bolhuis H, van Veen H W, Poolman B, Driessen A J M. The role of transport processes in survival of lactic acid bacteria. Antonie Leeuwenhoek. 1997;71:117–128. doi: 10.1023/a:1000143525601. [DOI] [PubMed] [Google Scholar]
- 18.Laplace-Builhe C, Hahne K, Tirilly Y, Drocourt J-L. Applications of flow cytometry to rapid microbial analysis in food and drinks industries. Biol Cell. 1993;78:123–128. doi: 10.1016/0248-4900(93)90122-u. [DOI] [PubMed] [Google Scholar]
- 19.Lloyd D, Hayes A J. Vigour, vitality and viability of microorganisms. FEMS Microbiol Lett. 1995;133:1–7. [Google Scholar]
- 20.Lowry O H, Rosebrough N J, Farr A L, Randall R J. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 21.Manafi M, Kneifel W, Bascomb S. Fluorogenic and chromogenic substrates used in bacterial diagnostics. Microbiol Rev. 1991;55:335–348. doi: 10.1128/mr.55.3.335-348.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marteau P, Minekus M, Havenaar R, Huis in’t Veld J H J. Survival of lactic acid bacteria in a dynamic model of the stomach and small intestine: validation and the effects of bile. J Dairy Sci. 1997;80:1031–1037. doi: 10.3168/jds.S0022-0302(97)76027-2. [DOI] [PubMed] [Google Scholar]
- 23.Massaro E J, Elstein K H, Zucker R M, Bair K W. Limitations of the fluorescent probe viability assay. Mol Toxicol. 1989;2:271–284. [PubMed] [Google Scholar]
- 24.McFeters G A, Yu F P, Pyle B H, Stewart P S. Physiological assessment of bacteria using fluorochromes. J Microbiol Methods. 1995;21:1–13. doi: 10.1016/0167-7012(94)00027-5. [DOI] [PubMed] [Google Scholar]
- 25.Molenaar D, Abee T, Konings W N. Continuous measurement of the cytoplasmatic pH in Lactococcus lactis with a fluorescent pH indicator. Biochim Biophys Acta. 1991;115:75–83. doi: 10.1016/0304-4165(91)90014-8. [DOI] [PubMed] [Google Scholar]
- 26.Molenaar D, Bolhuis H, Abee T, Poolman B, Konings W N. The efflux of a fluorescent probe is catalyzed by an ATP-driven extrusion system in Lactococcus lactis. J Bacteriol. 1992;174:3118–3124. doi: 10.1128/jb.174.10.3118-3124.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nebe-von Caron G, Badley R A. Viability assessment of bacteria in mixed populations using flow cytometry. J Microsc. 1995;179:55–66. [Google Scholar]
- 28.Panoff J-M, Legrand S, Thammavongs B, Boutibonnes P. The cold shock response in Lactococcus lactis subsp. lactis. Curr Microbiol. 1994;29:213–216. [Google Scholar]
- 29.Pearce L E. Activity tests for cheese starter cultures. N Z J Dairy Sci Technol. 1969;4:246–247. [Google Scholar]
- 30.Pettipher G L, Rodrigues U M. Rapid enumeration of bacteria in heat-treated milk and milk products using a membrane filtration-epifluorescent microscopy technique. J Appl Bacteriol. 1981;50:157–166. [Google Scholar]
- 31.Poolman B, Hellingwerf K J, Konings W N. Regulation of the glutamate-glutamine transport system by intracellular pH in Streptococcus lactis. J Bacteriol. 1987;169:2272–2276. doi: 10.1128/jb.169.5.2272-2276.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Porter J, Deere D, Pickup R, Edwards C. Fluorescent probes and flow cytometry: new insights into environmental bacteriology. Cytometry. 1996;23:91–96. doi: 10.1002/(SICI)1097-0320(19960201)23:2<91::AID-CYTO1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 33.Porter J, Edwards C, Pickup R W. Rapid assessment of physiological status in Escherichia coli using fluorescent probes. J Appl Bacteriol. 1995;79:399–408. doi: 10.1111/j.1365-2672.1995.tb03154.x. [DOI] [PubMed] [Google Scholar]
- 34.Rallu F, Gruss A, Maguin E. Lactococcus lactis and stress. Antonie Leeuwenhoek. 1996;70:243–251. doi: 10.1007/BF00395935. [DOI] [PubMed] [Google Scholar]
- 35.Riis S B, Pedersen H M, Sørensen N K, Jakobsen M. Flow cytometry and acidification power test as rapid techniques for determination of the activity of starter cultures of Lactobacillus delbrueckii ssp. bulgaricus. Food Microbiol. 1995;12:245–250. [Google Scholar]
- 36.Roth B L, Poot M, Yue S T, Millard P J. Bacterial viability and antibiotic susceptibility testing with SYTOX Green nucleic acid stain. Appl Environ Microbiol. 1997;63:2421–2431. doi: 10.1128/aem.63.6.2421-2431.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rotman B, Papermaster B W. Membrane properties of living mammalian cells as studied by enzymatic hydrolysis of fluorogenic esters. Proc Natl Acad Sci USA. 1966;55:134–141. doi: 10.1073/pnas.55.1.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sandine W E. Commercial production of dairy starter cultures. In: Cogan T M, Accolas J-P, editors. Dairy starter cultures. New York, N.Y: VCH Publishers, Inc.; 1996. pp. 191–206. [Google Scholar]
- 39.Sigsgaard P. Fluorescence microscopy techniques in the practice of food microbiology. In: Munck L, editor. Fluorescence analysis in foods. Singapore: Longman Singapore Publishers, Ltd.; 1989. pp. 131–141. [Google Scholar]
- 40.Snyder A P, Greenberg D B. Viable microorganism detection by induced fluorescence. Biotechnol Bioeng. 1984;26:1395–1397. doi: 10.1002/bit.260261119. [DOI] [PubMed] [Google Scholar]
- 41.Stubberfield L C F, Shaw P J A. A comparison of tetrazolium reduction and FDA hydrolysis with other measures of microbial activity. J Microbiol Methods. 1990;12:151–162. [Google Scholar]
- 42.Terzaghi B E, Sandine W E. Improved medium for lactic streptococci and their bacteriophages. J Appl Microbiol. 1975;29:807–813. doi: 10.1128/am.29.6.807-813.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ueckert J, Breeuwer P, Abee T, Stephens P, Nebe von Caron G, ter Steeg P F. Flow cytometry applications in physiological study and detection of foodborne microorganisms. Int J Food Microbiol. 1995;28:317–326. doi: 10.1016/0168-1605(95)00066-6. [DOI] [PubMed] [Google Scholar]
- 44.Waggoner A S. Fluorescent probes for cytometry. In: Melamed M R, Lindmo T, Mendelsohn M L, editors. Flow cytometry and sorting. 2nd ed. New York, N.Y: Wiley-Liss, Inc.; 1990. pp. 209–225. [Google Scholar]
- 45.Whitaker R D, Batt C A. Characterization of the heat shock response in Lactococcus lactis subsp. lactis. Appl Environ Microbiol. 1991;57:1408–1412. doi: 10.1128/aem.57.5.1408-1412.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams S C, Hong Y, Danavall D C A, Howard-Jones M H, Gibson D, Frischer M E, Verity P G. Distinguishing between living and nonliving bacteria: evaluation of the vital stain propidium iodide and its combined use with molecular probes in aquatic samples. J Microbiol Methods. 1998;32:225–236. [Google Scholar]
- 47.Yiu S H. A fluorescence microscopic study of cheese. Food Microstruct. 1985;4:99–106. [Google Scholar]