

ABSTRACT
Background
Polycystic kidney diseases (PKD) are an important cause of chronic kidney disease (CKD). Autosomal dominant polycystic kidney disease (ADPKD) due to PKD1 or PKD2 mutations is the most common form, but other genes can be responsible for ADPKD and its phenocopies. Among them, a form of atypical ADPKD caused by DNAJB11 mutations (DNAJB11-PKD) has been recently described.
Methods
We retrospectively recruited a cohort of 27 patients from six different families sharing common ancestries and harboring the same DNAJB11 mutation (c.100C>T, p.Arg34*) and we compared it with a cohort of 42 typical ADPKD patients.
Results
DNAJB11-PKD patients show small/normal-sized kidneys, with significantly smaller cysts and a slower progression to end-stage kidney disease (ESKD) than ADPKD patients. In the DNAJB11-PKD cohort, the cystic phenotype could not be detected by ultrasound in about half of the patients, but all cases with available computed tomography/magnetic resonance scans displayed cysts. Clinically, DNAJB11-PKD patients displayed proteinuria (mostly albuminuria). Compared with ADPKD, DNAJB11-PKD patients were older and had a higher prevalence of type 2 diabetes mellitus (19% versus 0%; P = 0.007) and nephrolithiasis (62% versus 29%; P = 0.01), whereas the prevalence of cardiac valvular defects was lower (4% versus 51%; P < 0.001).
Conclusions
Overall, clinical features of DNAJB11-PKD were more subtle compared with those of ADPKD. DNAJB11-PKD shows a unique renal and extrarenal phenotype, clinical presentation and natural history. Therefore our data support that this genetic disease is classified separately from ADPKD.
Keywords: ADPKD, chronic kidney disease, cystic kidney disease, DNAJB11, genetics
Graphical Abstract
Graphical Abstract.

INTRODUCTION
Monogenic inherited diseases are underestimated but important causes of chronic kidney disease (CKD) and despite the rapid increase in knowledge, they are identified in <10% of CKD patients [1, 2]. Among monogenic causes of CKD, polycystic kidney disease (PKD) represents a group of disorders with a clinical and genetic heterogeneity and a variable phenotype, from early manifestations during pregnancy or childhood to oligo-symptomatic cases until adulthood [1, 3–8].
Among different PKDs, autosomal dominant polycystic kidney disease (ADPKD) is recognized as the most common inherited kidney disease, with an estimated prevalence of 1:2500 [6]. ADPKD is a genetically heterogeneous disease, with two main causative genes, PKD1 (chromosome 16.p13.3) and PKD2 (chromosome 4p21) that account for the 72–78% and 15% of affected patients [3–7, 9, 10], respectively. There are some other genes that are rarely described in ADPKD, particularly GANAB (chromosome 11q12.3) and the causative genes of autosomal dominant polycystic liver disease (ADPLD) (such as PRKCSH, SEC63, LRP5, PMM2, ALG8, ALG9 and SEC61B) that can mildly affect the kidneys [4, 10]. PKD phenocopies due to genetic variants in numerous other genes (such as HNF1β, UMOD, PKHD1, COL4A1 or TSC1/2) have been reported as well [11, 12]. Nevertheless, ∼7–10% of all ADPKD patients remain genetically unresolved despite a clinical and radiological diagnosis [13]. Recently a large cohort of ADPKD patients negative for all known genes was examined [13] and a new gene (DNAJB11 in chromosome region 3q27) was described as being responsible of an atypical dominant form of ADPKD (DNAJB11-PKD).
DNAJB11 encodes a co-chaperone of the endoplasmic reticulum (ER), also called ERdj3. It is a part of the HSP40 protein family and plays a central role in both intracellular and extracellular proteomic homeostasis (proteostasis) [14–18]. It also acts in the pathway of the unfolded protein response (UPR), binding misfolded proteins and activating BiP, an HSP70 of the ER whose function is to correct the misfolding. DNAJB11 malfunction disrupts its interaction with BiP and impairs both proteostasis and upregulation of the UPR in response to stress [14–18].
Herein we report the largest retrospective single-center cohort of DNAJB11-PKD patients carrying a single point nonsense DNAJB11 mutation and belonging to a small village with less than 2000 inhabitants perched in the Apennine Mountains near Parma, Italy. This allowed us to precisely define the natural history of the DNAJB11-PKD as well as the renal and extra-renal phenotypes. Additionally, we compared DNAJB11-PKD patients with a cohort of ADPKD patients (carrying either PKD1 or PKD2 mutations) to describe the differences between the two diseases.
MATERIALS AND METHODS
In April 2018 we identified two sisters with end-stage kidney disease (ESKD) on chronic hemodialysis carrying the same single point nonsense pathogenic variant of DNAJB11 gene (c.100C>T, pArg34*). This variant leads to a stop codon, resulting in the production of a shortened, and likely nonfunctional, protein. Subsequently, an in-depth analysis of the family history revealed other patients followed for CKD/ESKD at our outpatient clinic and dialysis service who shared common ancestors with the index family. These patients underwent genetic analysis in the suspicion of DNAJB11-PKD.
From April 2020, we conducted a retrospective analysis of patients affected by DNAJB11-PKD and ADPKD (PKD1 and PKD2 genes) followed at the Nephrology Unit of the Parma University Hospital from 2000 to today. We reviewed both the outpatient clinic data and the dialysis registry.
We included all the patients whose diagnoses were established in a proband with age-specific renal imaging criteria and an affected first-degree relative with genetically proven ADPKD/DNAJB11-PKD or with the identification of a heterozygous pathogenic variant of DNAJB11 or other ADPKD genes in the proband [19]. No other mutations in genes known to be involved in PKD have been detected through the whole exome sequencing (WES) used in the original genetic analysis. A panel of 109 genes created from the database Genomics England PanelApp (https://panelapp.genomicsengland.co.uk/) was used (Supplementary data, Table S1) with an average coverage at 20X from 94 to 100; the gene LRP5 was added to the panel, because it is reported in association with ADPKD, whereas the gene MUC1, even if included in the panel could not be considered because the variable number tandem repeat domain usually harboring mutations could not be correctly analyzed with WES.
Data analyzed for both cohorts (ADPKD cohort and DNAJ11-PKD one) were as follows: clinical history, imaging findings [renal size, defined as pole-to-pole diameter on renal ultrasound or as pole-to-pole diameter on the sagittal plane on computed tomography (CT) or magnetic resonance (MR) imaging when the kidneys’ diameter exceeded the scanning area on ultrasound, as in some ADPKD patients, renal cyst diameter, cyst localization in other organs such as liver and pancreas (detected with ultrasound, CT and/or MRI), presence of valvular defects on echocardiography (mitral valve stenosis and prolapse or mitral valve insufficiency, aortic valve insufficiency or stenosis, tricuspid valve insufficiency or stenosis and pulmonary valve insufficiency or stenosis]. Each defect was considered independently of its grade––mild, moderate or severe—but valve calcification was considered only in association with one of the above defects, presence of vascular aneurisms or arteries dissection and laboratory data (serum creatinine, creatinine clearance, blood glucose level, hemoglobin A1c, serum and urinary electrolytes, serum uric acid levels and urinary protein levels). Urinary electrolytes, particularly sodium, potassium, calcium, phosphate, citrate and oxalate, were analyzed as factors predisposing to lithiasis.
Estimated glomerular filtration rate (eGFR) was calculated using the Chronic Kidney Disease Epidemiology Collaboration equation (CKD-EPI creatinine formula) [20]. Proteinuria was defined as the presence of >0.5 g of protein in a 24-h urine collection or the presence of a urinary protein: creatinine ratio >0.5 g/g. Comorbidities were recorded at the last follow-up visit or at the beginning of dialysis in order to reduce confounding factors linked to dialysis or to renal transplantation.
The study protocol conformed to the Declaration of Helsinki and was approved by a local research ethics committee (protocol number 14721/2020). Informed consent was obtained from the patients.
Statistical analysis
We used Stata version 17 (StataCorp, College Station, TX, USA) for all the analyses. A two-sided P-value <0.05 was regarded as statistically significant. We compared two-sample differences in continuous variables using the Mann–Whitney U test and those in categorical variables using Fisher's exact test.
We used the least absolute shrinkage and selection operator (LASSO) [21] to identify which clinical characteristics helped in discriminating between ADPKD and DNAJB11-PKD as the dependent dichotomous variable (1 if DNAJB11-PKD and 0 if ADPKD). LASSO is meant for high-dimensional models having too many potential covariates (possibly highly correlated) for the sample size. It is meant to avoid overfitting the dataset by preventing the inclusion of spurious predictors that would likely not be confirmed upon validation in an external dataset. LASSO is a procedure for selecting covariates: it does not provide P-values. LASSO estimates coefficients of the regression model as a function of a tuning parameter that ‘shrinks’ the coefficient toward zero as its value gets larger. In other words, the value of the tuning parameter acts as a volume knob: large values of the parameter penalize model complexity (i.e. a large number of predictors), whereas smaller values weakly penalize model complexity. By setting some coefficient to zero, the tuning parameter determines which variables the LASSO will eventually exclude. The potential values of the tuning parameter cannot be estimated by the data, therefore their candidate values need to be calculated by cross-validation. We chose the final optimal value of the tuning parameter based on the Bayesian information criterion. We fitted LASSO via logistic regression on the 61 patients with all clinical variates available.
We reported how the rates of ESKD and death vary with the patient's age using cumulative failure rate (hazard) functions. The advantage of using cumulative hazard rates over Kaplan–Meier estimates was 2-fold: first, unlike Kaplan–Meier estimates, the cumulative hazard function does not rely on the assumption of ESKD and death being independent from each other; second, the cumulative hazard estimates cannot immediately be interpreted as a conditional probability of failure from birth, which would be misleading since we could not retrospectively select and follow-up all affected patients since time of birth. The interpretation of the cumulative hazard rate function is as follows: if the hazard (of ESKD or death) is constant over age bands, then the cumulative hazard will increase linearly with age; if the hazard increases with age, the cumulative hazard will increase nonlinearly, showing an increase in slope with increasing age; if the hazard decreases with age, the cumulative hazard will still increase, but now with a decrease in the slope [22]. We tested the differences in hazard functions between ADPKD and DNAJB11-PKD using Mantel–Cox method.
RESULTS
We identified 42 patients deriving from 23 families with genetically proven PKD1-PKD2 ADPKD diagnosis (35 harboring a PKD1 mutation and 7 a PKD2 mutation; stratified data are shown in Supplementary Data, Table S2) and 27 patients with DNAJB11-PKD from six related families. No other mutations in genes known to be involved in PKD have been detected through the WES used in the original genetic analysis.
Patients’ characteristics for both cohorts are reported in Table 1.
Table 1.
Patients’ clinical data
| Cystic disease | |||||||
|---|---|---|---|---|---|---|---|
| ADPKD | DNAJB11-PKD | ||||||
| Characteristics | n | Mean ± SD | % | n | Mean ± SD | % | P-value |
| Patients | 42 | 27 | |||||
| Age at last follow-up (years) | 42 | 55.0 ± 16.8 | 27 | 74.8 ± 12.3 | 0.000 | ||
| Males/females | 26/16 | 38.1 | 12/15 | 55.6 | 0.22 | ||
| ESKD (y/n) | 16/26 | 16/26 | 38.1 | 12/15 | 44.4 | 0.62 | |
| Age at ESKD (years) | 15 | 59.9 ± 11.4 | 12 | 71.1 ± 4.5 | 0.006 | ||
| Kidney diameter (cm)a | 33 | 16.4 ± 4.6 | 22 | 10.3 ± 1.6 | <0.001 | ||
| Larger kidney cyst (cm) | 29 | 5.7 ± 2.9 | 22 | 3.0 ± 2.5 | <0.001 | ||
| Pancreatic cysts (y/n) | 2/38 | 5.0 | 1/22 | 4.3 | 1.00 | ||
| Liver cysts (y/n) | 30/10 | 75.0 | 11/12 | 47.8 | 0.06 | ||
| Kidney stones (y/n) | 12/29 | 29.3 | 16/10 | 61.5 | 0.01 | ||
| Hb (g/dL)b | 29 | 12.9 ± 2.3 | 23 | 11.0 ± 2.6 | 0.006 | ||
| PTH (pg/mL)c | 19 | 188.5 ± 196.5 | 14 | 305.8 ± 276.8 | 0.09 | ||
| 24-h proteinuria >00.5 g (y/n) | 6/28 | 17.6 | 12/10 | 54.5 | 0.007 | ||
| ESA use | 5/28 | 15.2 | 9/16 | 36 | 0.12 | ||
| Diabetes type 2 (y/n) | 0/41 | 0.0 | 5/21 | 19.2 | 0.007 | ||
| Cancer (y/n) | 4/37 | 9.8 | 8/16 | 33.3 | 0.02 | ||
| Valvular defectsd (y/n) | 21/20 | 51.2 | 1/22 | 4.3 | <0.001 | ||
| Cerebrovascular disease (y/n) | 0/41 | 0 | 2/21 | 8.7 | 0.12 | ||
| Cardiovascular disease (y/n) | 1/39 | 2.5 | 5/18 | 21.7 | 0.02 | ||
| Hypertension | 25/15 | 62.5 | 16/7 | 69.6 | 0.78 | ||
aKidney size is assessed using pole-to-pole diameter on ultrasound or pole-to-pole diameter on a sagittal scan on CT or MRI.
bFor 10 ADPKD patients, data were collected just before the beginning of dialysis; the other 19 patients had a mean eGFR of 80 mL/min/1.73 m2 using the CKD-EPI equation. Among 5 DNAJB11-PKD patients, data were collected just before dialysis; the other 10 had a mean eGFR of 50 mL/min/1.73 m2.
cSeven ADPKD patients were starting dialysis; among others, the mean eGFR was 71 mL/min/1.73 m2. Five DNAJB11-PKD patients were starting dialysis and the other eight had a mean eGFR of 51 mL/min/1.73 m2.
dThe valvular defects considered were mitral valve insufficiency, mitral valve stenosis, aortic valve insufficiency, aortic valve stenosis and tricuspid valve insufficiency. Each of these defects could be mild, moderate or severe in different patients. Tricuspid valve stenosis and pulmonary valve defects were not reported among patients.
Hb, hemoglobin; PTH, parathormone; ESA, erythropoietin-stimulating agent.
Values in bold are statistically significant.
DNAJB11-PKD patients
The six different DNAJB11-PKD families acknowledge parentage with a great and enlarged family in the Parma Appennines [23] (Figure 1). Among them, 14 patients carried the reported mutation of the DNAJB11 gene. The other 13 patients were identified retrospectively as DNAJB11-PKD first-degree relatives from highly suggestive imaging. In 11 patients (41%), the peculiar radiological renal appearance was described by CT or MRI, while ultrasound was not conclusive for diagnosis. Among them, three patients displayed ESKD. Overall, 12 of 27 (44.4%) patients with DNAJB11-PKD reached ESKD at a mean age of 71.1 ± 4.5 years. Only one patient had a pancreatic cyst, while liver cysts were documented in 47.8% of the cohort. Imaging findings or a suggestive clinical history of nephrolithiasis was identified in 16 patients (61.5%), but urinary electrolyte analyses were available only for a minority of patients and a specific excretory profile could not be defined. The mean renal diameter was 10.3 ± 1.6 cm, while the mean diameter of the bigger cyst measured 3 ± 2.5 cm. Among comorbidities, 5/26 (19.2%) were affected by type 2 diabetes mellitus (T2DM) and 8 (33.3%) patients by malignant neoplasms. Specifically, malignancy diagnoses were two breast cancers, two prostate cancers, one melanoma, one hepatocellular carcinoma, one bladder cancer and one acute myeloid leukemia. About 70% of the patients were hypertensive; two patients had a stroke and five a myocardial infarction, whereas only one patient displayed a cardiac valvular defect. Moreover, >50% of patients with proteinuria available showed 24-h proteinuria >1 g/day, mainly represented by albuminuria. Of them, four patients had diabetes while two patients without T2DM presented with nephrotic-range proteinuria (renal biopsy was not performed for the presence of renal cysts) (Figure 2). A total of 36% of cases had a combination of lithiasis/proteinuria. Finally, we analyzed how patients were initially diagnosed before genetic analysis. Seven of 27 (26%) patients were diagnosed as having ADPKD; 6 patients (22%) had a previous diagnosis of medullary sponge kidney disease (MSKD) and 10 patients (41%) did not have a definite diagnosis. Curiously, four patients (15%) were reported as suspected diabetic nephropathy. Five of six patients diagnosed as having MSKD belonged to the same family; among other family members, one was recognized as having MSKD. The diagnosis in affected patients was based on ultrasonography without the use of intravenous urography.
FIGURE 1:

Family trees. (A–F) Family trees of the six families carrying the same DNAJB11 gene mutation from a restricted area in the Parma Appennines are reported. All the families recognize parentage with a local great and enlarged family. Black squares and circles are genetically confirmed patients or first-degree relatives with identical clinical/radiological presentations, while grey squares and gray circles are obliged carriers.
FIGURE 2:
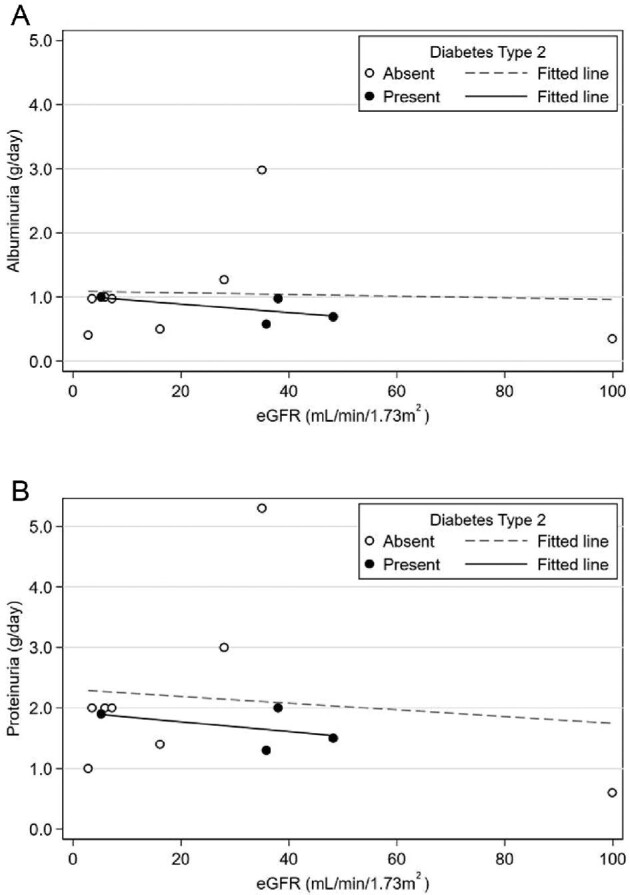
(A) Plot of albuminuria or (B) proteinuria against eGFR at the time of urine collection. 24-h proteinuria/albuminuria appearance was unrelated with eGFR or the presence of diabetes.
ADPKD patients
In contrast, in the ADPKD cohort the clinical diagnosis was achieved before genetics in the whole cohort. A total of 39 patients carried a mutation of PKD1 and 3 of PKD2. Overall, 16 of 42 patients (38.1%) reached ESKD at a mean age of 59.9 ± 11.4 years. Only 2 patients displayed pancreatic cysts, while liver cysts were detected in 29 (72.5%). Nephrolithiasis was identified in 29.3% of cases. The mean renal size was 16.4 ± 4.6 cm, with a median diameter of the bigger cyst of 5.5 cm (range 2–13.5). No patient had T2DM and four (9.8%) had a neoplasia (an ovarian cancer, a breast cancer and two cases of skin cancer). Hypertension was present in 62.5% of the patients, while cerebrovascular disease was not diagnosed in the whole cohort, and only one patient reported a myocardial infarction. Cardiac valvular defects were reported in 21 of 41 (51.8%) patients. No intracranial aneurysm was detected; however, only 25.5% of patients underwent magnetic resonance imaging (MRI) angiography of cerebral vessels as a screening exam.
Comparison between DNAJB11-PKD versus ADPKD patients
An ADPKD diagnosis was suspected before genetic testing in a quarter of DNAJB11-PKD patients (26% versus 100% of the ADPKD cohort; P < 0.001). The DNAJB11-PKD cohort presented a significantly smaller renal size (mean 10.3 versus 16.4 cm; P < 0.001) and smaller renal cysts (mean 3 versus 5.7 cm; P < 0.001). Therefore, whereas ultrasound could detect the presence of cysts in all ADPKD patients, it frequently missed the presence of cysts in patients with DNAJB11-PKD, and almost half of them eventually required MR/CT studies (supplementary data, Figure S1).
We used the LASSO statistical approach [21] to select among the 61 patients with all clinical variates available (reported in Table 1) the clinical characteristics that independently predicted DNAJB11-PKD (Figure 3). Compared with ADPKD, DNAJB11-PKD patients had a higher prevalence of T2DM (19% versus 0%) and kidney stones (62% versus 29%), whereas the prevalence of cardiac valvular defects was lower (4% versus 51%).
FIGURE 3:

Variables characterizing DNAJB11-PKD patients. LASSO for variable selection via logistic regression for discriminating between ADPKD and DNAJB11-PKD. The plot shows standardized coefficient estimates (y-axis) as a function of the ‘L1-norm’ of the standardized coefficients (i.e. the maximum allowed sum of the absolute values of the coefficients) (x-axis). The tuning parameter (not shown in the plot) ‘shrinks’ the coefficient toward zero as its value gets larger. By setting some coefficient to zero, the tuning parameter determines which variables the LASSO will eventually exclude. The final value of the L1-norm (vertical dash line) resulted from the selection among the potential candidates of the tuning parameters that were estimated by cross-validation (see text) of the one value that minimized the Bayesian information criterion. Age at last follow-up, valvular defects (negative association with DNAJB11-PKD), kidney stones and type 2 diabetes were the variables with the largest standardized coefficients and that were eventually selected by LASSO.
The risk of ESKD manifested after the age of 40 years in ADPKD patients and after 60 years of age in DNAJB11-PKD patients, being the median time to dialysis start (respectively, 64.4 and 76.5 years; P = 0.0006, by Mantel–Cox test). In contrast, the hazard of death was not significantly different between the groups (Figure 4).
FIGURE 4:
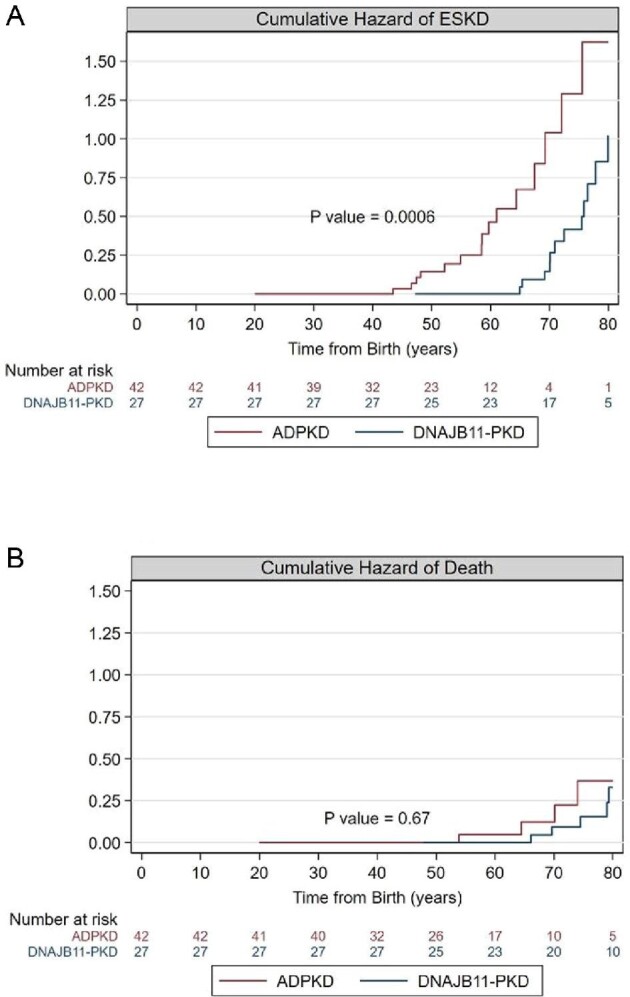
Differences in renal outcome and death between ADPKD and DNAB11-PKD patients. (A) Cumulative failure rate (hazard) functions of ESKD in ADPKD (red line) and in DNAJB11-PKD (blue line). The interpretation of the cumulative hazard rate function is as follows: if the hazard of ESKD is constant over age bands, then the cumulative hazard will increase linearly with age; if the hazard increases with age, the cumulative hazard will increase nonlinearly, showing an increase in slope with increasing age; if the hazard decreases with age, the cumulative hazard will still increase, but now with a decrease in the slope. P-value refers to the Mantel–Cox method. (B) Cumulative failure rate (hazard) functions of death in ADPKD (red line) and in DNAJB11-PKD (blue line). P-value refers to the Mantel–Cox method.
DISCUSSION
The presence of a DNAJB11 mutation as a cause of PKD was first described in May 2018 by Cornec-Le-Gall et al. [13], who were analyzing patients with genetically unresolved ADPKD. Even if this first cohort was recruited among families with an established diagnosis of ADPKD or ADPLD, researchers described a quite different disease, characterized by the development of multiple small renal cysts, nonenlarged kidneys, the presence of chronic interstitial fibrosis and development of ESKD in the sixth decade of life [13]. They recognized this new entity as an atypical form of ADPKD or a disease that combines features of ADPKD and ADTKD (autosomal dominant tubulointerstitial kidney disease) [24–26]. The subsequent work by Huynh et al. [27] better defined the clinical spectrum of DNAJB11 kidney disease. It confirmed the presence of nonenlarged kidneys with small cysts and the development of ESKD between the fifth and eighth decade of life and underlined the presence of liver cysts in ∼49% of patients [13, 27].
Our data confirm prior findings in a well-characterized cohort. First, we showed that DNAJB11-PKD is characterized by significantly low-normal-/normal-size kidneys with cysts smaller than those of ADPKD counterparts. Another relevant issue is that ultrasound for individuation of cystic disease in our DNAJB11 cohort seems to be inadequate, whereas MRI allows an early identification of patients, showing smaller cysts (Figure 5).
FIGURE 5:

Differences in kidney imaging in DNAJB11 and ADPKD. (A) Ultrasound shows in DNAJB11-PKD the presence of multiple small cysts distributed both in the cortex and in the medulla with a normal-sized kidney. Hyperechoic spots (microcalcifications or small kidney stones) could also be detected. In ADPKD, ultrasound shows enlarged kidneys with bigger cysts and the corticomedullary differentiation is not recognizable because of extensive parenchymal substitution with cysts. (B) Contrast-enhanced abdomen CT does not add useful information in the diagnostic process of ADPKD. It is often crucial in DNAJB11-PKD diagnosis because it improves the visualization of small cysts, whereas in ADPKD, cyst size is usually big enough to be detected without contrast in the context of enlarged kidneys. (C) The goal of MRI for DNAJB11-PKD is detection of simple cysts with T2-weighted images and complicated hematic debris-filled cysts with T1-weighted fat saturation.
Interestingly, only a quarter of our DNAJB11-PKD cases had a clinical diagnosis of ADPKD before genetic testing. Indeed, our DNAJB11-PKD cohort belongs to a peculiar geographical area and not, as in previous studies, to a quite undeniable ADPKD phenotype; therefore, we captured more DNAJB11-PKD cases in which the cystic phenotype does not seem to be so suggestive.
The second most represented diagnosis in our cohort was MSKD. Patients were diagnosed using ultrasonography, so even if we have peculiar characteristics that addressed this diagnosis on imaging, we lacked what is recognized as the gold standard in the literature, intravenous urography. On the one hand, the lack of the gold standard imaging technique could have led to an incorrect diagnosis; on the other hand, considering that an MSKD diagnosis mainly relies on an imaging study and that only a small percentage of cases are genetically resolved, it could be that different phenocopies of MSKD exist with different gene mutations supporting a similar expression.
The hazard of ESKD in the DNAJB11-PKD cohort starts ∼15 years later than in the ADPKD cohort. The normal-sized kidneys, smaller cysts, difficult diagnosis with ultrasound and later onset of ESKD justify the frequent lack of a definite diagnosis, even in the face of a suggestive family history of nephropathy.
From a clinical point of view, we documented that ∼20% of DNAJB11 patients have T2DM, while our typical ADPKD cohort has no cases. A unique characteristic highlighted by our DNAJB11-PKD cohort is the presence of proteinuria ranging from mild levels to the nephrotic range; this finding is totally unexpected in a patient with ADPKD. Interestingly, albumin contributed the vast majority of urinary proteins, suggesting a glomerular origin rather than a tubular one, despite the tubular dysfunction that is supposed to be associated with DNAJB11-PKD [13, 24–26]. Moreover, albuminuria was usually documented at the time of diagnosis and in one PKD-DNAJB11 patient was reported despite normal renal function. Proteinuria in DNAJB11-PKD patients was not reported in previous studies; however, it is unclear whether the data were available in the whole group [13]. Our data set the basis for further studies aimed at understanding the pathophysiology of proteinuria/albuminuria in DNAJB11-PKD patients.
The impaired proteostasis of the ER secondary to loss of function of DNAJB11 co-chaperone would explain the various clinical expressions of DNAJB11-PKD. First, cystogenesis seems to be caused by the disruption of proteostasis due to the defect in the appropriate localization or secretion of different proteins involved in cyst development such as polycystin 1, uromodulin and MUC1 [13, 27]. Second, since UPR is involved in different metabolic diseases and, specifically, regulates insulin resistance, this pathologic variation would determine an augmented insulin synthesis from β-cells leading to β-cell apoptosis and T2DM, as the UPR is impaired [15, 28, 29]. Finally, ER stress and UPR activation would both explain the appearance of relevant proteinuria.
Kidney stones seem to be a characterizing feature of DNAJB11-PKD. This could be related to the more relevant involvement of the distal nephron rather than the proximal one in DNAJB11-PKD compared with ADPKD. In fact, DNAJB11 is predominantly expressed in the thick ascending limb of Henle's loop, in the distal tubule and in the collecting duct. However, considering the small number of patients and that the analysis of urine for factors predisposing to lithiasis was available in only a few patients in our retrospective study, this observation should be tested in a larger cohort. The LASSO analysis did not detect a significant difference between cohorts in the risk of malignancies, as the higher number of events in the DNAJB11-PKD patients is probably due to the older age in this cohort.
Previous studies on DNAJB11-PKD gave a general description of clinical and imaging features, but they are mainly based on clinical databases of unsolved genetic cases of ADPKD, lacking a direct comparison with a typical ADPKD cohort. A major strength of our study is that it includes the largest single mutation cohort of DNAJB11-PKD, which allowed us to perform a comparison with an ADPKD cohort to identify affinities and divergences between the two diseases.
However, there are some caveats that should be considered in data interpretation. First, the two cohorts are relatively small and some established comorbidities, such as intracranial aneurysms, were not represented in the ADPKD cohort. Moreover, the DNAJB11-PKD cohort was older than the ADPKD cohort. Nevertheless, the reported clinical findings such as T2DM, kidney stones and proteinuria/albuminuria seem to be peculiar to DNAJB11-PKD versus ADPKD. Importantly, both DNAJB11-PKD and ADPKD patients were followed at the same institutions for years and came from the same area of origin, limiting potential confounders.
The depicted DNAJB11-PKD disease is quite different from ADPKD and has a slower and apparently benign course favoring its local spread, particularly in the context of geographic isolation, like in our cohort; indeed, biallelic mutations give rise to severe neonatal cases with poor outcomes [30, 31]. Consequently, its recognition should prompt family screening even if clinical manifestations arise in adulthood.
In conclusion, DNAJB11-PKD is characterized by normal-/low-normal-sized kidneys, small renal cysts, significant proteinuria and slowly developing CKD, with ∼30% of patients reaching ESKD. It seems to be associated with important comorbidities, such as T2DM and kidney stones.
Increased awareness among clinicians should be advised to avoid misdiagnosis and improve patient management. Moreover, our work could provide a strong rationale for future studies evaluating the pathogenic role of malfunctioning ER co-chaperone DNAJB11 in the different manifestations of the disease.
Supplementary Material
Contributor Information
Isabella Pisani, Unità Operativa Nefrologia, Azienda-Ospedaliero Universitaria di Parma & Dipartimento di Medicina e Chirurgia, Università di Parma, Parma, Italy.
Marco Allinovi, Nephrology, Dialysis and Transplantation Unit, Careggi University Hospital, Florence, Italy.
Viviana Palazzo, Medical Genetics Unit, Meyer Children's University Hospital, Florence, Italy.
Paola Zanelli, Unità di Immunogenetica dei Trapianti, Azienda-Ospedaliero Universitaria di Parma, Parma, Italy.
Micaela Gentile, Unità Operativa Nefrologia, Azienda-Ospedaliero Universitaria di Parma & Dipartimento di Medicina e Chirurgia, Università di Parma, Parma, Italy.
Maria Teresa Farina, Unità Operativa Nefrologia, Azienda-Ospedaliero Universitaria di Parma & Dipartimento di Medicina e Chirurgia, Università di Parma, Parma, Italy.
Sara Giuliotti, Unità Operativa Radiologia, Azienda Ospedaliero-Universitaria di Parma, Parma, Italy.
Paolo Cravedi, Department of Medicine, Renal Division, Icahn School of Medicine at Mount Sinai, NY, USA.
Marco Delsante, Unità Operativa Nefrologia, Azienda-Ospedaliero Universitaria di Parma & Dipartimento di Medicina e Chirurgia, Università di Parma, Parma, Italy.
Umberto Maggiore, Unità Operativa Nefrologia, Azienda-Ospedaliero Universitaria di Parma & Dipartimento di Medicina e Chirurgia, Università di Parma, Parma, Italy.
Enrico Fiaccadori, Unità Operativa Nefrologia, Azienda-Ospedaliero Universitaria di Parma & Dipartimento di Medicina e Chirurgia, Università di Parma, Parma, Italy.
Lucio Manenti, Unità Operativa Nefrologia, Azienda-Ospedaliero Universitaria di Parma & Dipartimento di Medicina e Chirurgia, Università di Parma, Parma, Italy.
CONFLICT OF INTEREST STATEMENT
The authors have declared no conflicts of interest. The results presented in this article have not been published previously in whole or part, except in abstract format.
REFERENCES
- 1. Armstrong ME, Thomas CP. Diagnosis of monogenic chronic kidney diseases. Curr Opin Nephrol Hypertens 2019; 28: 183–194 [DOI] [PubMed] [Google Scholar]
- 2. Cornec-Le Gall E, Harris PC. The underestimated burden of monogenic diseases in adult-onset ESRD. J Am Soc Nephrol 2018; 29: 1583–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet 2019; 393: 919–935 [DOI] [PubMed] [Google Scholar]
- 4. Cornec-Le Gall E, Torres VE, Harris PC. Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol 2018; 29: 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bergmann C. Recent advances in the molecular diagnosis of polycystic kidney disease. Expert Rev Mol Diagn 2017; 17: 1037–1054 [DOI] [PubMed] [Google Scholar]
- 6. Lanktree MB, Iliuta IA, Haghighi Aet al. . Evolving role of genetic testing for the clinical management of autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2019; 34: 1453–1460 [DOI] [PubMed] [Google Scholar]
- 7. Lanktree MB, Chapman AB. New treatment paradigms for ADPKD: moving towards precision medicine. Nat Rev Nephrol 2017; 13: 750–768 [DOI] [PubMed] [Google Scholar]
- 8. Mallawaarachchi AC, Lundie B, Hort Yet al. . Genomic diagnostics in polycystic kidney disease: an assessment of real-world use of whole-genome sequencing. Eur J Hum Genet 2021; 29: 760–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lanktree MB, Chapman AB. Autosomal dominant polycystic kidney disease. CMAJ 2017; 189: E1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lanktree MB, Haghighi A, Guiard Eet al. . Prevalence estimates of polycystic kidney and liver disease by population sequencing. J Am Soc Nephrol 2018; 29: 2593–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Izzi C, Dordoni C, Econimo Let al. . Variable expressivity of HNF1B nephropathy, from renal cysts and diabetes to medullary sponge kidney through tubulo-interstitial kidney disease. Kidney Int Rep 2020; 5: 2341–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schönauer R, Baatz S, Nemitz-Kliemchen Met al. . Matching clinical and genetic diagnoses in autosomal dominant polycystic kidney disease reveals novel phenocopies and potential candidate genes. Genet Med 2020; 22: 1374–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cornec-Le Gall E, Olson RJ, Besse Wet al. . Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet 2018; 102: 832–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park SJ, Kim Y, Chen YM. Endoplasmic reticulum stress and monogenic kidney diseases in precision nephrology. Pediatr Nephrol 2019; 34: 1493–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Buck TM, Brodsky JL. Escaping the endoplasmic reticulum: why does a molecular chaperone leave home for greener pastures? EMBO J 2015; 34: 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shen Y, Hendershot LM. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP's interactions with unfolded substrates. Mol Biol Cell 2005; 16: 40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Genereux JC, Qu S, Zhou Met al. . Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis. EMBO J 2015; 34: 4–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Genereux JC, Wiseman RL. Regulating extracellular proteostasis capacity through the unfolded protein response. Prion 2015; 9: 10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adam MP, Ardinger HH, Pagon RAet al. . GeneReviews [Internet]. Seattle, WA: University of Washington, 1993 [Google Scholar]
- 20. Levey AS, Stevens LA, Schmid CHet al. . A new equation to estimate glomerular filtration rate. Ann Intern Med 2009; 150: 604–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. James G, Witten D, Hastie Tet al. . An Introduction to Statistical Learning with Applications in R. New York: Springer, 2017 [Google Scholar]
- 22. Clayton D, Hills M. Statistical Models in Epidemiology. New York: Oxford University Press, 1993 [Google Scholar]
- 23. Ablondi W, Venturini E, Stefanini Ablondi V. Una grande e complessa famiglia in alta Val Baganza. In: Baganza CSV (ed.), Per La Val Baganza. Riccò: Studio Guidotta SNC, 2018 [Google Scholar]
- 24. Eckardt KU, Alper SL, Antignac Cet al. . Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management—a KDIGO consensus report. Kidney Int 2015; 88: 676–683 [DOI] [PubMed] [Google Scholar]
- 25. Bleyer AJ, Kidd K, Živná Met al. . Autosomal dominant tubulointerstitial kidney disease. Adv Chronic Kidney Dis 2017; 24: 86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ayasreh Fierro N, Miquel Rodríguez R, Matamala Gastón Aet al. . A review on autosomal dominant tubulointerstitial kidney disease. Nefrologia 2017; 37: 235–243 [DOI] [PubMed] [Google Scholar]
- 27. Huynh VT, Audrézet MP, Sayer JAet al. . Clinical spectrum, prognosis and estimated prevalence of DNAJB11-kidney disease. Kidney Int 2020; 98: 476–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016; 529: 326–335 [DOI] [PubMed] [Google Scholar]
- 29. Gallazzini M, Pallet N. Endoplasmic reticulum stress and kidney dysfunction. Biol Cell 2018; 110: 205–216 [DOI] [PubMed] [Google Scholar]
- 30. Jordan P, Arrondel C, Bessières Bet al. . Bi-allelic pathogenic variations in DNAJB11 cause Ivemark II syndrome, a renal-hepatic-pancreatic dysplasia. Kidney Int 2021; 99: 405–409 [DOI] [PubMed] [Google Scholar]
- 31. Ateş EA, Turkyilmaz A, Delil Ket al. . Biallelic mutations in DNAJB11 are associated with prenatal polycystic kidney disease in a Turkish family. Mol Syndromol 2021; 12: 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.