

Abstract
Ramp sequences occur when the average translational efficiency of codons near the 5′ end of highly expressed genes is significantly lower than the rest of the gene sequence, which counterintuitively increases translational efficiency by decreasing downstream ribosomal collisions. Here, we show that the relative codon adaptiveness within different tissues changes the existence of a ramp sequence without altering the underlying genetic code. We present the first comprehensive analysis of tissue and cell type-specific ramp sequences and report 3108 genes with ramp sequences that change between tissues and cell types, which corresponds with increased gene expression within those tissues and cells. The Ramp Atlas (https://ramps.byu.edu/) allows researchers to query precomputed ramp sequences in 18 388 genes across 62 tissues and 66 cell types and calculate tissue-specific ramp sequences from user-uploaded FASTA files through an intuitive web interface. We used The Ramp Atlas to identify seven SARS-CoV-2 genes and seven human SARS-CoV-2 entry factor genes with tissue-specific ramp sequences that may help explain viral proliferation within those tissues. We anticipate that The Ramp Atlas will facilitate personalized and creative tissue-specific ramp sequence analyses for both human and viral genes that will increase our ability to utilize this often-overlooked regulatory region.
INTRODUCTION
Tissue-specific regulation and resource availability directly affect gene transcription and translation, which results in widespread tissue-specific differential gene expression (1–5) that can be analyzed in the Human Protein Atlas (6), The Genotype-Tissue Expression (GTEx) Project (7) and the Functional Annotation of Mammalian Genomes (FANTOM5) (8,9) and leveraged to identify disease biomarkers (6,10–20) for a variety of diseases such as cancers (1–5), drug resistance (18), cystic fibrosis (14), cardiovascular disease (16) and Alzheimer's disease (20).
Since codon usage biases correlate with local tRNA pools and tissue-specific RNA binding proteins (21,22), codon usage biases often change between different tissues (21,23). Variations in tRNA abundances affect gene expression by altering tRNA competition and codon optimality, which changes the translational efficiency of various codons (24,25). Therefore, although the mRNA transcript remains unchanged between tissues, the efficiency at which those codons can be translated differs between tissues and cell types (22,26–30). While genome-wide codon usage biases are highly correlated with genome-wide guanine-cytosine (GC) content (31), translational selection may play an important role in maintaining gene-specific codon usage biases (32–34) because codon usage biases alter translational efficiency (32,35,36), gene expression (35,37,38), mRNA secondary structure (34,39), and protein structure and function (36). However, selection for tissue-specific codon adaptations in humans has been highly controversial: Doherty and McInerney (40) and Chamary et al. (32) argue that tRNA gene families undergo precise regulation to generate anticodon pools that directly correlate with available mRNA, while Pouyet et al. (41) concludes that global GC-content biases affect meiotic recombination and preclude tRNA optimization. Translational selection can also significantly shape codon usage biases across vertebrates, even when high GC-content biases are present (42). Regardless of selection or GC-content biases shaping the underlying tRNA pool within each tissue, gene expression can be significantly impacted by how well the mRNA codons are adapted to that available tRNA pool.
Often, tissue-specific tRNA levels correlate with the codon usage biases of highly expressed genes, suggesting that tRNA abundances play a role in tissue-specific gene and protein expression (21). The tRNA pools within tissues and cell types can differ significantly (21,43–50), and unique tRNA pools cluster into subsets of proliferating and differentiating cells when characterized by cell type (30). These cell types maintain tRNA levels that match the codon usage biases and increase the expression of genes that promote proliferation or differentiation (30). Similarly, cellular tRNA levels may dynamically change to increase the expression of certain genes in response to stress. For example, heterologous protein overexpression, diminished growth rate, and translational imbalances trigger tRNA pool readjustments to increase expression of needed genes (51–55). Additionally, cells significantly increase tRNA levels in the G2 phase of the cell cycle to temporarily increase the translational efficiency and expression of cycle-regulated genes with non-optimal codon usage bias (56).
Various diseases also trigger tRNA pool alterations. For example, the tRNA pool in breast cancer cells is altered to increase expression of genes that promote metastasis (57) and methionine misacylation increases in mammalian cells when exposed to viruses or oxidative stress, which can have a protective effect on the cells (58). The interplay between tissue and cell type-specific codon usage biases, tissue and cell type-specific tRNA abundances, and synonymous mutations can have a significant effect on various human diseases (30,43–45,57,59–64). During the later stages of infection, HIV-1 changes cellular tRNA levels to increase expression of viral genes with codon usage biases that are poorly adapted to the original host tRNA pool (65), and human protein SLFN11 counters virus-induced changes to tRNA levels by interacting with tRNA in the host cell to limit viral protein production (66). Similarly, viral genes in SARS-CoV-2 alter tRNA levels and deprive tRNA from normally highly-expressed host genes by monopolizing the tRNA supply, thereby decreasing host gene expression and inducing additional damage (67).
Viruses that infect humans often use similar codon usage biases as highly expressed genes within the tissues that they infect to facilitate co-opting available cellular machinery (67–69). For example, genes in SARS-CoV-2 utilize codon usage biases that match very highly expressed genes in lung tissue, likely contributing to increased viral gene expression, decreased host gene expression, and infection in the lungs (67,70). In SARS-CoV-2, codon usage similarities between viral genes and highly-expressed host genes likely improve viral translation and replication, which may help predict which human genes will be downregulated during an infection (71). Similarly, codon usage biases in Papillomavirus genes promote gene expression in differentiated epithelial cells to more effectively spread infection (72).
Although many tissue-specific codon usage biases have previously been reported, the tissue-specific effects of ramp sequences have yet to be characterized. Ramp sequences increase overall translational efficiency by utilizing slowly-translated codons at the beginning of genes to evenly space ribosomes and reduce downstream ribosomal collisions. Ramp sequences occur when 20–40 suboptimal codons concentrate at the beginning (5′ end) of highly-expressed gene sequences and occur in approximately 10% of genes (73). Ramp sequences generally increase gene expression by counterintuitively slowing initial translation so that ribosomes can evenly distribute (21,49,73–76) and more efficiently translate the remaining transcript without ribosomal traffic jams (77,78). Increased translational efficiency increases mRNA stability and gene and protein expression, especially in genes that have higher ribosome density, higher mRNA levels, and a strong correlation between mRNA and protein expression (77,79). The correlation between ramp sequences and increased gene expression has been shown in several species. Park and Subramaniam (79) demonstrate in Saccharomyces cerevisiae that ramp sequences increase gene expression and mRNA stability by preventing ribosome collisions at downstream stall sites (i.e. sequences within an mRNA that slow or ‘stall’ ribosomes during translation) that would otherwise halt translation, cleave the mRNA, and decrease expression. Miller et al. (73) report that ramp sequences exist in all domains of life, and highly expressed genes have a greater proportion of ramp sequences than genes with low expression in Drosophila melanogaster. They later showed that ramp sequences are conserved in all domains of life (80), but can change between different human populations (81). Goodman et al. (82) show that ramp sequences in Escherichia coli increase gene expression ∼14-fold. However, the mechanisms by which ramp sequences affect gene expression have yet to be fully characterized. Ramp sequences may increase protein expression by reducing mRNA secondary structure (82), or impact transcriptional efficiency by requiring fewer hydrogen bonds in dsDNA to be broken (83). Regardless, orthologous ramp sequences are evolutionarily conserved across all domains of life (82,84,85), which suggests that they play an important functional role in regulating gene expression.
While ramp sequences have been analyzed across several species (73,79) and populations (81) at a genomic level, they have never previously been characterized within human tissues and cell types using single cell codon efficiencies. Because tissues and cell types have distinct tRNA levels (21,43–50) and codon usage biases (21,23), we hypothesized that they would also have distinct ramp sequences, despite having no differences in the underlying genetic code. We propose that characterizing ramp sequences in tissues and cell types will more accurately describe ramp sequence role in basic biology, ramp sequence prevalence and distribution across all genes, their correlation with tissue and cell type-specific gene expression, and aid in elucidating their role in human health and disease.
Here, we present The Ramp Atlas, which is the first comprehensive analysis of tissue and cell type-specific ramp sequences and how they relate to tissue-specific gene expression and SARS-CoV-2 infection. We identified 3108 genes with ramp sequences that were alternatively present, or absent, depending on the relative translational efficiencies in tissues and tissue-stratified cell types calculated from gene expression in the Human Protein Atlas (86–88), GTEx Project (7) and FANTOM5 datasets (8,9). We allow researchers to query ramp sequences in 18 388 genes spanning 62 tissues and 66 cell types with embedded Tableau charts that allow user-specific querying to facilitate targeted ramp sequence analyses. We also present an online version of our ramp sequence identification algorithm, ExtRamp (73), to facilitate tissue-specific ramp sequence analyses online through a user-friendly interface. To explore the role of ramp sequences in disease, we used ExtRamp Online to identify ramp sequences in seven SARS-CoV-2 genes and seven human SARS-CoV-2 entry factor genes, both present in a variety of tissues. The SARS-CoV-2 analyses indicate that virus-specific ramp sequences may play an integral role in viral proliferation because ramp sequences were present significantly more often in tissues that SARS-CoV-2 is known to infect. We anticipate that The Ramp Atlas will facilitate future ramp sequence analyses on tissue or cell type-specific gene expression impacted by ramp sequences, genetic variant effects on tissue-specific gene expression, viral adaptations to specific tissues or cell types, and therapeutic developments that aim to modulate tissue-specific gene expression.
MATERIALS AND METHODS
Dataset selection
We used all available tissue and cell type expression data in the Human Protein Atlas (87–89), as well as the GTEx Project (7) and FANTOM5 (8,9) datasets, which were previously normalized to the Human Protein Atlas in a consensus dataset. We chose these specific gene expression databases because they report comprehensive gene expression profiles across various human tissues and cell types and are widely used, which allows our findings to be directly integrated and interpreted in the context of other research exploring human genetics and disease.
Identifying ramp sequences
Ramp sequences are calculated by identifying statistical outliers of codon efficiencies concentrated at the 5′ end of gene coding sequences using the software package, ExtRamp (73). Codon efficiencies are measured by the relative synonymous codon usage metric, which ranges from 0–1 and is calculated by dividing the number of occurrences for a specific codon by the number of occurrences for the most common codon that encodes the same amino acid. We calculated the relative synonymous codon usage values in April 2021 from genes with high expression in 43 tissues in the Human Protein Atlas (86–88), 34 tissues in the GTEx Project (7), 45 tissues in the FANTOM5 datasets (8,9) and 62 tissues in the consensus dataset found in the Human Protein Atlas, which combines gene expression from all three databases. We also computed the relative synonymous codon usages in 66 tissue-stratified cell types, which were also downloaded in April 2021. For each tissue or cell type in each data set, we separated genes into quartiles based on their reported expression level (Q1: ‘not detected;’ Q2: ‘low;’ Q3: ‘medium;’ Q4: ‘high’). We used default parameters in ExtRamp to identify ramp sequences in each tissue and cell type for 18 388 genes (including protein-coding open reading frames), where all annotated gene isoforms were required to have similar gene expression (i.e. gene expression was within the same quartile for each isoform). The longest isoform from the GRCh38 reference genome was considered representative for each gene sequence. Ramp sequence identification was performed for each tissue and cell type separately. Two output files were generated for each run: (i) ramp sequences identified by ExtRamp and (ii) genes that did not contain a predicted ramp sequence within the tissue or cell type. The original data files downloaded from the Human Protein Atlas were modified to include an extra column, ‘Ramp presence,’ with values of ‘Ramp’ or ‘No ramp’ for each gene in each tissue and cell type. These comma-separated values (CSV) files are available at https://ramps.byu.edu/Downloads. We also present these data in Tableau charts on the ‘Search Database’ page of The Ramp Atlas, which allows users to view tissues where ramp sequences exist for a gene and compare the presence or absence of a ramp sequence to the normalized gene expression for each tissue.
Chi-squared analysis: overall correlation between ramp sequences and gene expression
To determine if genes with ramp sequences tend to be highly expressed, we first grouped all genes across all tissues by whether or not they contained a ramp sequence (e.g. if gene A had a ramp sequence in 30 tissues and did not have a ramp sequence in 32 tissues, then it would be included 30 times in the ‘ramp’ group and 32 times in the ‘no ramp’ group with the corresponding gene expression for each tissue). We then calculated the odds of having ‘medium’ or ‘high’ expression versus ‘not detected’ or ‘low’ expression for each group. We calculated the relative odds of having increased gene expression with a ramp by performing a chi-squared analysis of the number of instances where a gene with a ramp sequence had ‘medium’ or ‘high’ expression compared to the number of instances where a gene without a ramp sequence had ‘medium’ or ‘high’ expression. That test determined the extent to which the presence or absence of a ramp sequence, in general, made a gene more likely to have ‘medium’ or ‘high’ expression compared to ‘not detected’ or ‘low’ expression.
Chi-squared analysis: tissue-specific correlation between variable ramp sequences and gene expression
We performed the following calculations in each dataset to determine the extent to which genes with a ramp sequence in some tissues and not others are differentially expressed. First, we ensured that each gene had a ramp sequence in at least 5% of the tissues and did not have a ramp sequence at least 5% of the tissues. The average gene expression (geometric mean) in the ‘ramps’ and ‘no ramps’ groups were calculated for each gene separately to limit the potential effects of sampling. The expression bins were transformed to follow a linear progression between ‘not detected’ and ‘high expression’ (i.e. ‘not detected’ was given a weight of 1×, ‘low expression’ was given a weight of 2×, ‘medium’ expression was given a weight of 3× and ‘high expression’ was given a weight of 4×). The geometric mean was then calculated across all tissues for each gene in each group (e.g. ‘ramps’ and ‘no ramps’) to determine the ‘average’ expression across all tissues for a gene when it has a ramp sequence and when it does not have a ramp sequence. The geometric mean was used to limit the potential effects of a few higher values from skewing the means while maintaining all data points in the mean calculations. Finally, we counted the number of genes that had a higher geometric mean of their gene expression when they had a ramp sequence compared to when they did not have a ramp sequence. We report the chi-square test statistic and P for each dataset to determine if, on average, variable ramp sequence presence within a gene corresponds with increased gene expression within the tissues that have ramp sequences.
Comparing ramp presence between tissues and cell types
We present an interactive, online comparison of the proportion of genes with ramp sequences across different tissues or cell types for each dataset (e.g. FANTOM5, GTEx, Human Protein Atlas, consensus, and Human Protein Atlas cell types), and we determined if the same genes always contained ramp sequences. We conducted a single chi-squared test for each dataset to determine the extent to which ramp sequences are distributed across tissue and cell types. Each tissue contained at least 2425 ramp sequences (i.e. 14% of sampled genes) and each cell type contained at least 1521 ramp sequences (i.e. 15% of sampled genes), which was a large enough sample size to adequately assess the distribution using the chi-squared test.
We followed these dataset-wide chi-squared tests with more specific pairwise, two-proportion z-tests between each tissue type to identify which tissues contained the most unique ramp sequences. Unlike the chi-squared test, the two-proportion z-test is a parametric test that assumes normality. However, z-tests are generally robust to small deviations from that assumption, especially when the sample size is large. Nevertheless, we tested each group for normality using the Shapiro-Wilk test and show the distributions of each group at https://github.com/ridgelab/rampAtlas/tree/main/histograms. We applied a Bonferroni correction for multiple testing (FANTOM5 alpha = 5.05 × 10–5, GTEx alpha = 8.91 × 10–5, Human Protein Atlas alpha = 5.54 × 10–5, consensus alpha = 2.64 × 10–5, cell type alpha = 2.33 × 10–5). Online Tableau charts are available for users to query bar charts depicting the total number of significant tests for each tissue/cell type with highlighted tissues/cell types containing more unique proportions of ramp sequences.
Data visualization
We used Tableau Software (www.tableau.com) to create The Ramp Atlas, which generates interactive graphics for users to query ramp sequence data across tissues and cell types. All Tableau charts are available at https://ramps.byu.edu/SearchDatabase. The bulk data files used to generate these Tableau charts are available for download at https://ramps.byu.edu/Downloads and the code used for all the analyses is publicly available on GitHub at https://github.com/ridgelab/rampAtlas.
SARS-CoV-2 ramp sequence analysis
We downloaded all SARS-CoV-2 nucleotide coding sequences from the National Center for Biotechnology Information (NCBI) SARS-CoV-2 Resources in November 2020, and we analyzed 406 882 available SARS-CoV-2 gene sequences and overlapping gene bodies. Since SARS-CoV-2 utilizes the host machinery for translation, the optimal codon usage for SARS-CoV-2 should be based on the relative synonymous codon usage of the host tissue. Therefore, we used ExtRamp coupled with the relative synonymous codon usage values calculated from 62 tissue-specific highly expressed genes in the Human Protein Atlas, GTEx, FANTOM5, and consensus datasets to determine the extent to which ramp sequences were present in SARS-CoV-2 genes for each dataset. We then determined the frequency of ramp sequences in SARS-CoV-2 genes across each tissue and identified the number of ramp sequences shared between any two tissues. We also identified tissue-specific ramp sequences in human genes that encode for SARS-CoV-2 entry factors, as reported in Singh et al. (90). We then ranked all tissues according to the total number of ramp sequences present in both SARS-CoV-2 and associated human entry factor genes. Using these data, we performed a one-tailed t-test to compare the mean number of SARS-CoV-2 and human entry factor genes with ramp sequences in tissues that show high SARS-CoV-2 proliferation (91,92) against those tissues that do not show high SARS-CoV-2 proliferation because ramp sequences are predicted to increase gene expression. All analyses are freely available and distributed using an ASP.NET web server hosted at Brigham Young University (https://ramps.byu.edu).
ExtRamp online
We developed an online web interface to facilitate tissue-specific ramp sequence calculations utilizing ExtRamp by using an ASP.NET Core 3.1 (https://dotnet.microsoft.com/apps/aspnet) framework on a Microsoft Windows server hosted at Brigham Young University. ExtRamp Online runs a modified version of ExtRamp within the web browser using Pyodide (https://pyodide.org/en/stable/). All parameters are customizable and preset to a default test case that allows users to see the expected output. Additionally, scroll-over hints teach users how to change parameters and effectively use those parameters in ramp sequence calculations. All associated scripts and information about the website are available at https://github.com/ridgelab/rampAtlas/tree/main/ExtRampOnline.
RESULTS
The Ramp Atlas (https://ramps.byu.edu) is a comprehensive online repository of tissue and cell type-specific ramp sequences spanning 18 388 human genes in 62 tissues and 66 cell types with additional interactive comparisons of SARS-CoV-2 ramp sequences and human entry factors. This resource provides a template for conducting ramp sequence analyses on viruses and identifying ramp sequences that are correlated with tissue or cell type-specific differential gene expression. Interactive graphics enable comprehensive user-specific analyses that can be integrated into a variety of downstream genetic analyses.
Table 1 shows that the presence of a ramp sequence significantly correlates with higher gene expression in each dataset: FANTOM5 (odds = 1.1152; P = 3.00 × 10–99), GTEx (odds = 1.1578; P = 9.48 × 10–155), Human Protein Atlas (odds = 1.1947; P = 1.27 × 10–306), consensus (odds = 1.1477; P = 1.00 × 10–254), and Human Protein Atlas tissue-stratified cell types (odds = 1.0407; P = 6.53 × 10–27). Table 2 shows that genes with tissue-specific ramp sequences (i.e. ramp sequences that were present in only some tissues) were also significantly more likely to increase gene expression in the consensus dataset (odds: 1.1937; P = 5.56 × 10–5), FANTOM5 (odds: 1.1314; P = 0.00589), and the Human Protein Atlas (odds: 1.1897; P = 2.74 × 10–5), although the difference was not significant in the GTEx dataset (odds: 0.9242; P = 0.15236). Notably, GTEx contains fewer tissue samples than all other datasets, with only 34 tissues compared to 43 in the Human Protein Atlas, 45 in FANTOM5, and 62 in the consensus dataset. Additionally, low-level basal contamination has previously been identified in approximately 40% of GTEx samples (93).
Table 1.
Odds of ramp sequence presence correlating with increased gene expression
| Dataset | Chi-square | P-value (P) | Odds ratio |
|---|---|---|---|
| FANTOM5 | 459.3981 | 3.00 × 10–99 | 1.1152 |
| GTEx | 715.4278 | 9.48 × 10–155 | 1.1578 |
| Human Protein Atlas | 1415.5104 | 1.27 × 10–306 | 1.1947 |
| Consensus | 1176.3328 | 1.00 × 10–254 | 1.1477 |
| Human Protein Atlas Tissue-stratified Cell Types | 115.3698 | 6.53 × 10–27 | 1.0407 |
For each dataset, the chi-squared value and P-value (P) for increased expression if a gene has a ramp sequence as well as the odds of having higher expression if ramp sequence present vs not present. The odds ratio shows a significant difference in gene expression if a gene has a ramp sequence.
Table 2.
Odds of ramp sequence presence correlating with tissue-specific gene expression
| Dataset | Genes with decreased tissue expression | Genes with increased tissue expression | Genes with no change in tissue expression | Chi-square | P-value (P) | Odds ratio |
|---|---|---|---|---|---|---|
| FANTOM5 | 1097 | 1282 | 32 | 14.3863 | 1.49 × 10–4 | 1.1686 |
| GTEx | 817 | 769 | 62 | 1.4527 | 0.2281 | 0.9412 |
| Human Protein Atlas | 1319 | 1550 | 41 | 18.5992 | 1.61 × 10–5 | 1.1751 |
| Consensus | 1134 | 1369 | 49 | 22.0635 | 2.64 × 10–6 | 1.2072 |
| Human Protein Atlas Tissue-stratified Cell Types | 265 | 366 | 39 | 16.1664 | 5.80 × 10–5 | 1.3811 |
For each dataset, the chi-squared statistic and P-value (P) for expression if a gene has a ramp sequence as well as the odds of having higher expression in a tissue where a ramp sequence is present. This odds ratio shows a significant difference in tissue-specific gene expression if a gene has a ramp sequence in that specific tissue.
A chi-squared test shows that ramp sequence prevalence differs significantly across all tissue types in every dataset (FANTOM5 P < 2.23 × 10–308, GTEx P = 8.52 × 10–81, Human Protein Atlas P < 2.23 × 10–308, consensus P < 2.23 × 10–308, cell type P = 6.06 × 10–20). Pairwise two-proportion z-tests that correct for multiple testing in each dataset showed that the proportion of genes with ramp sequences also differed significantly in many pairwise tissue comparisons: 43.99% of tissue comparisons in the consensus dataset, 50.51% of comparisons in FANTOM5, 35.65% of comparisons in GTEx, and 62.57% of comparisons in the Human Protein Atlas. Similarly, pairwise two-proportion z-tests in the Human Protein Atlas cell type dataset show that proportions of ramp sequences differed significantly in 4.34% of tissue-stratified cell type comparisons after correcting for multiple tests. The proportion of unique ramp sequences in the thymus, rectum, and total peripheral blood mononuclear cells (PBMC) were the highest compared to all other tissues (see Figure 1). We found that all 66 tissue-stratified cell types had at least one significant z-test result. Ramp sequence presence is most likely to differ in the seminiferous ducts in testis, cells in the white pulp in the spleen, and glandular cells in the parathyroid gland. Cells in the white pulp in the spleen use fewer ramp sequences than average, while the glandular and parathyroid gland cell types have more ramp sequences than average (see Figure 2). Figure 3 shows the percentage of ramp sequences shared between each tissue in the consensus dataset. The average percentage of ramp sequences shared between the urinary bladder and all other tissues is the highest (87.46%), while the rectum has is the fewest overlapping ramp sequences with other tissues (57.26%). Figure 4 shows ramp sequence overlap between different cell types from the Human Protein Atlas tissue-stratified cell types. Trophoblast cells in the placenta have the highest percentage of shared ramp sequences with other tissue-stratified cell types (94.96%), while cells in the seminiferous cells in the testes have the lowest (76.89%).
Figure 1.

Percentage of significant chi-square pairs in tissue types. We performed chi-square pair comparisons of ramp sequences across all tissues in each dataset and then pairwise two-proportion z-tests to identify which tissues have the most unique proportions of ramp sequences per gene. This figure shows, for each tissue, the percentage of pairwise z-tests that are significant, averaged across all datasets. Each tissue bar is colored based on the difference between the proportion of genes with ramp sequences and the average proportion of genes with ramp sequences across all tissues. Blue colored tissues have more ramp sequences than average while red colored tissues have fewer ramp sequences than average.
Figure 2.

Percentage of significant chi-square pairs in tissue-stratified cell types. We performed chi-square pair comparisons of ramp sequences across all tissue-stratified cell types in each dataset and then pairwise two-proportion z-tests to identify which cell types have the most unique proportions of ramp sequences per gene. This figure shows, for each cell type, the percentage of pairwise z-tests that are significant, averaged across all datasets. Each tissue-stratified cell type bar is colored based on the difference between the proportion of genes with ramp sequences in the cell type and the average proportion of genes with ramp sequences across all cell types. Blue colored tissue-stratified cell types have more ramp sequences than average while red colored cell types have fewer ramp sequences than average.
Figure 3.

Heat map of percent of ramp sequences shared between tissues. The percent of ramp sequences present in one tissue that are also present in another in the consensus dataset. Red indicates fewer shared ramp sequences and thus a more tissue-specific usage of ramp sequences. Tissue comparisons that are blue have more shared ramp sequences.
Figure 4.

Heat map of percent of ramp sequences shared between tissue-stratified cell types. The percent of ramp sequences present in one tissue-stratified cell type that are also present in another cell type. Red indicates fewer shared ramp sequences (i.e. more cell type-specific usage of ramp sequences). Tissue-stratified cell type comparisons that are blue have more shared ramp sequences.
The ‘Search Database’ page on The Ramp Atlas contains several interactive Tableau charts that allow users to query 18 388 genes for ramp sequences and explore differences between each dataset. For convenience, users can view ramp sequence results for multiple genes and datasets side-by-side. These interactive Tableau charts also display normalized gene expression levels across tissues, cell types, and datasets. Users can quickly view the extent to which ramp sequences differ between different tissues and visually determine if ramp sequences potentially affect gene expression. Interactive versions of Figures 1-4 (introduced above) are also available on the ‘Search Database’ page, which enable users to view tissue and cell type differences in ramp sequence presence in more detail. These ramp sequence results are also available for bulk download on the ‘Downloads’ page (https://ramps.byu.edu/Downloads).
SARS-CoV-2 ramp sequence analysis
We identified ramp sequences in seven SARS-CoV-2 genes: the 2′-O-RNA methyltransferase, the leader protein (non-structural protein 1), non-structural protein 6, the nucleocapsid phosphoprotein, the ORF3a protein, the ORF7a Nanoluciferase protein, and the surface glycoprotein. Ramp sequences in all but two of these genes (non-structural protein 6 and the surface glycoprotein) are tissue-specific (see Figure 5). Similarly, seven of the 26 analyzed human genes that encode SARS-CoV-2 entry factors (90) show tissue-specificity: Transmembrane protease, serine 2 (TMPRSS2); Membrane alanyl aminopeptidase (ANPEP); Charged Multivesicular Body Protein 2A (CHMP2A); Cathepsin L (CTSL); Furin, Paired Basic Amino Acid Cleaving Enzyme (FURIN); RAB1A, Member RAS Oncogene Family (RAB1A); and Transmembrane Anterior Posterior Transformation 1 (TAPT1) (see Figure 6). By combining these two analyses, we ranked tissues by ramp sequence prevalence in both SARS-CoV-2 and human entry factor genes (see Figure 7). We show that tissues with high SARS-CoV-2 proliferation have significantly more SARS-CoV-2 and human entry factor genes with ramp sequences than tissues that show less SARS-CoV-2 proliferation (P = 0.009918). Both the rectum and duodenum (tissues with high SARS-CoV-2 proliferation (91)) showed the most unique ramp sequence usage (see Figure 8).
Figure 5.

Percentage of tissues with a ramp sequence in SARS-CoV-2 genes. The percentage of 62 tissues in the consensus dataset in which seven SARS-CoV-2 genes have a ramp sequence. Error bars show the standard error between the four different datasets. In genes marked with an asterisk, less than one percent of all variants contained a ramp sequence. Notably, the other four genes had a ramp sequence in the one and only variant provided in the SARS-CoV-2 genome dataset. More exact data are available on The Ramp Atlas SARS-CoV-2 page: https://ramps.byu.edu/Covid.
Figure 6.
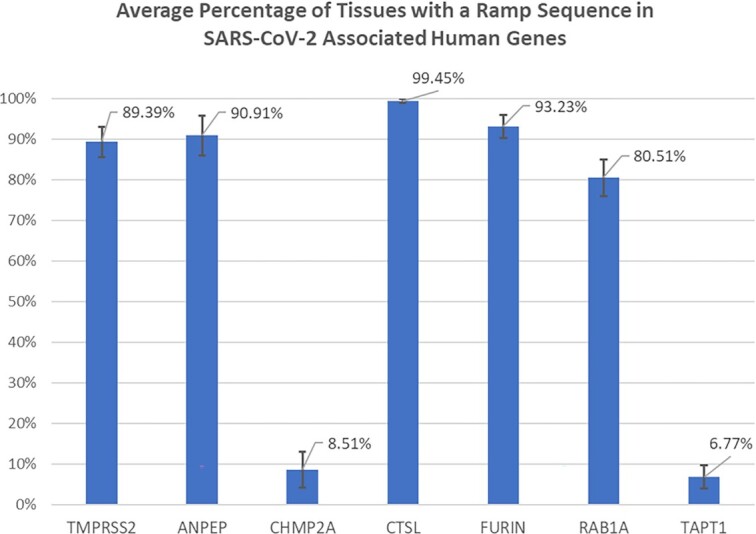
Percentage of tissues with a ramp sequence in human genes for SARS-CoV-2 entry factors. The percentage of the 62 tissues in the consensus dataset in which human genes that code for SARS-CoV-2 entry factors have a ramp sequence. Error bars show the standard error between the four different datasets.
Figure 7.

The total number of SARS-CoV-2 and associated human entry factor genes with a ramp sequence in 62 human tissues. An aggregation of the data presented in Figures 5 and 6, organized by tissue, and showing the total number of genes, both in the SARS-CoV-2 genome and associated human entry factors, with ramp sequences. Tissues where SARS-CoV-2 is found in higher amounts are marked in red boxes. These tissues had a significantly greater number of genes with ramp sequences (P= 0.0099).
Figure 8.

Heat map of percentage of SARS-CoV-2 ramp sequences shared between tissues. The percentage of ramp sequences in SARS-CoV-2 genes that are present in both tissues being compared. Calculated using the consensus dataset. The redder the tissue comparison, the smaller the percentage of ramp sequences were present in both tissues. The bluer the comparison, the higher percentage of shared ramp sequences.
Users can interactively query Tableau charts on the ‘SARS-CoV-2’ page of The Ramp Atlas to thoroughly explore ramp sequences within the SARS-CoV-2 genome as well as in seven human entry factors for SARS-CoV-2. An interactive heat map at the top of the page shows the percentage of SARS-CoV-2 ramp sequences that are shared between different tissues in the consensus dataset. Below, another interactive chart presents data on the presence of ramp sequences in different SARS-CoV-2 genes and across the 62 tissues in the consensus dataset. Under the ‘Ramp Sequences in Human Entry Factors’ section of the webpage, an interactive Tableau chart presents data for the presence of ramp sequences in SARS-CoV-2 human entry factors across all tissues in the consensus dataset. These interactive charts allow users to thoroughly explore ramp sequences in SARS-CoV-2.
ExtRamp online
We present an online implementation of the ExtRamp algorithm to facilitate wider adoption and application of ramp sequences for non-programmers, along with a significant improvement to the underlying algorithm to calculate tissue and cell type-specific ramp sequences (https://ramps.byu.edu/ExtRampOnline). To run ExtRamp Online, users can either paste a FASTA formatted gene header and sequence into a text box or upload a small FASTA file of coding sequences. The FASTA file must contain only coding sequences (i.e. non-translated regions are removed), and the coding sequences must contain all codons from the start codon to the stop codon, inclusively. Because ramp sequences are computed using tRNA abundances and codon usage biases, users can upload species-specific tRNA adaptation index values or use provided tissue or cell type-specific relative synonymous codon usage values provided in a simple drop-down menu. By default, relative synonymous codon usage values are calculated from the human reference genome GRCh38, but users can upload any reference genome. ExtRamp Online contains all options available in the original ExtRamp algorithm with additional pre-set default values to enable researchers to calculate human tissue-specific or genome-wide ramp sequences online. Hover-over hints provide users with detailed information on all available options, which will help facilitate future creative analyses using ramp sequences. ExtRamp Online generates a variety of output files, including a FASTA file containing the identified ramp sequences, a list or CSV of codon efficiency values, the headers of genes that did not contain a ramp sequence, the headers of genes that could not be processed by ExtRamp, and the segments of genes after identified ramp sequences. Examples of these files are available at ExtRamp Online, with checkboxes allowing users to select which files are downloaded. Additionally, The Ramp Atlas includes step-by-step instructions for running ExtRamp as well as descriptions of how to interpret output files. Because ExtRamp Online runs within the browser, the computer specifications of the user, Internet connection, and web browser may affect performance. For more intensive calculations (e.g. parallel comparisons or genome-wide analyses), we provide an automatic command generator that provides users with the command to run ExtRamp from the command line on their own machines in either a Linux, Mac, or Windows environment.
DISCUSSION
Unique and novel contributions of the ramp atlas
The Ramp Atlas allows researchers to query translational ramp sequences across human tissues and cell types through interactive tables and graphics online. These data are precomputed and available instantaneously for analysis. Tissue and cell type-specific ramp sequence data are presented alongside gene expression data, enabling users to further investigate the role of ramp sequences on expression. Additionally, we present analyses of ramp sequences in SARS-CoV-2 and associated human entry factors. This manuscript represents the first time that ramp sequences have been identified in SARS-CoV-2, and we show that the tissues with ramp sequences significantly intersect with tissues known to have higher rates of viral infection and proliferation. ExtRamp Online allows users to identify ramp sequences through a user-friendly interface. Additionally, precomputed tissue and cell type-specific input files are available to facilitate future single cell ramp analyses tailored toward unique user researcher questions. These novel contributions are unique to The Ramp Atlas and are an integral part of facilitating widespread ramp sequence research online and in single cells. We anticipate that The Ramp Atlas will allow future studies to identify how specific ramp sequences within single transcripts contribute to single cell gene expression. We also anticipate a wider adaption of ramp sequences in viral and disease research that are facilitated online through The Ramp Atlas.
Tissue-specific ramp sequence regulation
This study is the first comprehensive analysis of variable ramp sequences and presents a framework for conducting future tissue and cell type-specific ramp sequence analyses online. We recognize that the strength of the ramp sequence (i.e. the relative difference between translational efficiency at the 5′ end of the gene compared to the 3′ end of the gene) might play a significant role in determining transcript-specific affects in gene or protein levels. Although ramp sequences represent a true outlier region of decreased codon efficiency, some ramp sequences have a much higher difference between the harmonic mean of the codon efficiency within the ramp sequence and the harmonic mean of the codon efficiency following the ramp sequence. Those differences warrant future study to determine the extent to which transcript expression can be predicted based on the magnitude of the ramp sequence. The Ramp Atlas shows for the first time that ramp sequences within genes change based on the relative codon adaptiveness within a specific tissue or cell type, which often results in a ramp sequence being present in some tissues or cell types and not present in others even though the underlying genetic code does not change. Additionally, we show that tissue and cell type-specific ramp sequences are highly associated with increased gene expression when a ramp sequence is present. We propose that future ramp sequence calculations should consider ramp sequence variability that may occur within an organism based on tissue-specific codon optimality. We also propose that variable ramp sequences might be an additional mechanism for regulating tissue and cell type-specific differential gene expression that warrants further exploration.
Predicted SARS-CoV-2 tissue-specific ramp sequence optimization
Ramp sequences may play a significant role in determining tissue-specific SARS-CoV-2 infection and proliferation because tissues that experience higher rates of SARS-CoV-2 infection have significantly more ramp sequences in SARS-CoV-2 and associated human entry factor genes. These tissue-specific ramp sequences are predicted to increase expression of SARS-CoV-2 genes as well as the human genes that facilitate SARS-CoV-2 entry into host cells, resulting in tissue-specific increased rates of infection and proliferation. Specifically, tissues and tissue-stratified cell types with increased expression of human entry factors ACE2, TMPRSS2, and CTSL show increased SARS-CoV-2 infection (91,94–97). While both TMPRSS2 and CTSL contain ramp sequences, only ramp sequences in TMPRSS2 are tissue-specific and therefore may influence which tissues are most infected by SARS-CoV-2. Additionally, tissue-specific ramp sequences in the SARS-CoV-2 leader protein, which inhibits host gene expression (98,99) and helps the virus escape type-1 interferon cellular responses (98–100), may also influence tissue-specific infection and proliferation. Similar to how SARS-CoV-2 genes leverage tissue-specific codon usage biases to increase infection in lung tissue (67,70), we suggest that SARS-CoV-2 also utilizes tissue-specific ramp sequences to further improve its adaptiveness to the relative tRNA pool. Although we show a strong correlation between ramp sequence presence and SARS-CoV-2 proliferation, other biological mechanisms may also be responsible for this correlation, and further validation is required to determine if ramp sequences meaningfully affect SARS-CoV-2 viral proliferation.
In addition to potentially contributing to SARS-CoV-2 tissue-specific infection and proliferation, ramp sequences may also influence other fatal complications commonly found in SARS-CoV-2 patients. SARS-CoV-2 genes ORF3 and non-structural protein 1 (NSP1), respectively upregulate and downregulate APOB expression (101), whose expression is positively associated with atherosclerosis (102) that contributes to the severity of SARS-CoV-2 infection (101,103). We show that both ORF3 and NSP1 have ramp sequences specifically in the liver and small intestine, which is also where APOB expression is highest (86,104). We propose that these ramp sequences may affect ORF3 and NSP1 expression within the liver and small intestine, which likely alters APOB expression. While ORF3 and NSP1 have opposite effects on APOB expression, ramp sequences may play a role in determining the magnitude of those effects and the subsequent complications of atherosclerosis associated with increased APOB expression. These analyses constitute an important first step in characterizing variable, translational ramp sequences in SARS-CoV-2 genes and associated human entry factors as well as an exploration of their potential effects. Although these analyses provide only correlations between ramp sequences and SARS-CoV-2 proliferation, they indicate that future research into the effects of ramp sequences on viral proliferation might be warranted. The Ramp Atlas provides a framework to analyze ramp sequences in SARS-CoV-2 genes and perform similar analyses on novel viral strains to identify tissues where ramp sequences may aid viral proliferation.
CONCLUSION
The Ramp Atlas is the first comprehensive analysis of tissue and cell type-specific ramp sequences. We identified ramp sequences in SARS-CoV-2 that likely contribute to its widespread proliferation, and we show that those ramp sequences are most likely to occur in tissues known to be affected by SARS-CoV-2. Additionally, we provide a template for conducting similar ramp sequence analyses using a web resource to facilitate the widespread adaptation of ramp sequences in a variety of future applications.
The Ramp Atlas will help researchers better understand how ramp sequences affect gene expression, which may lead to more specific, targeted therapeutics. For example, tissue-specific gene and protein expression can affect tissue-specific responses to drug therapies (105,106), and protein expression profiles collected from tumor tissue samples can accurately predict patient responses to different cancer therapies (107–112). Because ramp sequence usage differs significantly between tissues and can be highly correlated with local gene expression, genetic variation affecting ramp sequences may also affect drug response and overall gene expression. Therefore, future ramp sequence analyses should consider local codon adaptation within different tissues and cell types before identifying ramp sequences. Since tissue-specific ramp sequences are highly correlated with increased gene expression, it is likely that future targeted genetic therapeutics that change ramp sequences may also have a tissue-specific effect on gene expression. The Ramp Atlas is a hypothesis-generating platform that provides a framework for researchers to conduct these sorts of analyses and creatively incorporate ramp sequences in their own research.
DATA AVAILABILITY
All scripts used to replicate this work are available at https://github.com/ridgelab/rampAtlas. The Ramp Atlas is publicly available at https://ramps.byu.edu.
ACKNOWLEDGEMENTS
We appreciate the contributions of Brigham Young University (BYU), the Office of Research Computing at BYU, Life Sciences IT at BYU and the Sanders-Brown Center on Aging at the University of Kentucky for supporting this research.
Contributor Information
Justin B Miller, Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40504, USA; Division of Biomedical Informatics, Department of Internal Medicine, University of Kentucky, Lexington, KY 40506, USA; Department of Pathology and Laboratory Medicine, University of Kentucky, Lexington, KY 40506, USA.
Taylor E Meurs, Department of Biology, Brigham Young University, Provo, UT 84602, USA.
Matthew W Hodgman, Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40504, USA.
Benjamin Song, Department of Biology, Brigham Young University, Provo, UT 84602, USA.
Kyle N Miller, Department of Computer Science, Utah Valley University, Orem, UT 84058, USA.
Mark T W Ebbert, Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40504, USA; Division of Biomedical Informatics, Department of Internal Medicine, University of Kentucky, Lexington, KY 40506, USA; Department of Neuroscience, University of Kentucky, Lexington, KY 40506, USA.
John S K Kauwe, Department of Biology, Brigham Young University, Provo, UT 84602, USA.
Perry G Ridge, Department of Biology, Brigham Young University, Provo, UT 84602, USA.
FUNDING
BrightFocus Foundation and its donors [A2020118F to Miller, A2020161S to Ebbert]; National Institutes of Health [1P30AG072946-01 to the University of Kentucky Alzheimer's Disease Research Center, AG068331 to Ebbert, GM138636 to Ebbert]; Alzheimer's Association [2019-AARG-644082 to Ebbert].
Conflict of interest statement. None declared.
REFERENCES
- 1. Kosti I., Jain N., Aran D., Butte A.J., Sirota M.. Cross-tissue analysis of gene and protein expression in normal and cancer tissues. Sci. Rep. 2016; 6:24799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pontén F., Gry M., Fagerberg L., Lundberg E., Asplund A., Berglund L., Oksvold P., Björling E., Hober S., Kampf C.et al.. A global view of protein expression in human cells, tissues, and organs. Mol. Syst. Biol. 2009; 5:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fagerberg L., Hallström B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K.et al.. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics. 2014; 13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Natarajan A., Yardimci G.G., Sheffield N.C., Crawford G.E., Ohler U.. Predicting cell-type-specific gene expression from regions of open chromatin. Genome Res. 2012; 22:1711–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kotliar D., Veres A., Nagy M.A., Tabrizi S., Hodis E., Melton D.A., Sabeti P.C.. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. Elife. 2019; 8:e43803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A.et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 7. Consortium GTEx Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lizio M., Abugessaisa I., Noguchi S., Kondo A., Hasegawa A., Hon C.C., de Hoon M., Severin J., Oki S., Hayashizaki Y.et al.. Update of the FANTOM web resource: expansion to provide additional transcriptome atlases. Nucleic Acids Res. 2019; 47:D752–D758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lizio M., Harshbarger J., Shimoji H., Severin J., Kasukawa T., Sahin S., Abugessaisa I., Fukuda S., Hori F., Ishikawa-Kato S.et al.. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015; 16:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prassas I., Chrystoja C.C., Makawita S., Diamandis E.P.. Bioinformatic identification of proteins with tissue-specific expression for biomarker discovery. BMC Med. 2012; 10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pontén F., Schwenk J.M., Asplund A., Edqvist P.H.. The human protein atlas as a proteomic resource for biomarker discovery. J. Intern. Med. 2011; 270:428–446. [DOI] [PubMed] [Google Scholar]
- 12. Uhlen M., Bjorling E., Agaton C., Szigyarto C.A., Amini B., Andersen E., Andersson A.C., Angelidou P., Asplund A., Asplund C.et al.. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics. 2005; 4:1920–1932. [DOI] [PubMed] [Google Scholar]
- 13. Mathivanan S., Lim J.W., Tauro B.J., Ji H., Moritz R.L., Simpson R.J.. Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Mol. Cell. Proteomics. 2010; 9:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kälin N., Claass A., Sommer M., Puchelle E., Tümmler B.. DeltaF508 CFTR protein expression in tissues from patients with cystic fibrosis. J. Clin. Invest. 1999; 103:1379–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Björling E., Lindskog C., Oksvold P., Linné J., Kampf C., Hober S., Uhlén M., Pontén F.. A web-based tool for in silico biomarker discovery based on tissue-specific protein profiles in normal and cancer tissues. Mol. Cell. Proteomics. 2008; 7:825–844. [DOI] [PubMed] [Google Scholar]
- 16. Arrell D.K., Neverova I., Van Eyk J.E.. Cardiovascular proteomics: evolution and potential. Circ. Res. 2001; 88:763–773. [DOI] [PubMed] [Google Scholar]
- 17. Korfali N., Wilkie G.S., Swanson S.K., Srsen V., de Las Heras J., Batrakou D.G., Malik P., Zuleger N., Kerr A.R., Florens L.et al.. The nuclear envelope proteome differs notably between tissues. Nucleus. 2012; 3:552–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson B.M., Zhang P., Schuetz J.D., Brouwer K.L.. Characterization of transport protein expression in multidrug resistance-associated protein (Mrp) 2-deficient rats. Drug Metab. Dispos. 2006; 34:556–562. [DOI] [PubMed] [Google Scholar]
- 19. Rho J.H., Roehrl M.H., Wang J.Y.. Tissue proteomics reveals differential and compartment-specific expression of the homologs transgelin and transgelin-2 in lung adenocarcinoma and its stroma. J. Proteome Res. 2009; 8:5610–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith M.Z., Nagy Z., Esiri M.M.. Cell cycle-related protein expression in vascular dementia and Alzheimer's disease. Neurosci. Lett. 1999; 271:45–48. [DOI] [PubMed] [Google Scholar]
- 21. Dittmar K.A., Goodenbour J.M., Pan T.. Tissue-specific differences in human transfer RNA expression. PLos Genet. 2006; 2:e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Payne B.L., Alvarez-Ponce D.. Codon usage differences among genes expressed in different tissues of drosophila melanogaster. Genome Biol Evol. 2019; 11:1054–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kames J., Alexaki A., Holcomb D.D., Santana-Quintero L.V., Athey J.C., Hamasaki-Katagiri N., Katneni U., Golikov A., Ibla J.C., Bar H.et al.. TissueCoCoPUTs: novel human tissue-specific codon and codon-pair usage tables based on differential tissue gene expression. J. Mol. Biol. 2020; 432:3369–3378. [DOI] [PubMed] [Google Scholar]
- 24. Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y.. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010; 141:344–354. [DOI] [PubMed] [Google Scholar]
- 25. Shao Z.Q., Zhang Y.M., Feng X.Y., Wang B., Chen J.Q.. Synonymous codon ordering: a subtle but prevalent strategy of bacteria to improve translational efficiency. PLoS One. 2012; 7:e33547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu Q. Mutational bias and translational selection shaping the codon usage pattern of tissue-specific genes in rice. PLoS One. 2012; 7:e48295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Camiolo S., Farina L., Porceddu A.. The relation of codon bias to tissue-specific gene expression in arabidopsis thaliana. Genetics. 2012; 192:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Plotkin J.B., Robins H., Levine A.J.. Tissue-specific codon usage and the expression of human genes. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:12588–12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sémon M., Lobry J.R., Duret L.. No evidence for tissue-specific adaptation of synonymous codon usage in humans. Mol. Biol. Evol. 2006; 23:523–529. [DOI] [PubMed] [Google Scholar]
- 30. Gingold H., Tehler D., Christoffersen N.R., Nielsen M.M., Asmar F., Kooistra S.M., Christophersen N.S., Christensen L.L., Borre M., Sorensen K.D.et al.. A dual program for translation regulation in cellular proliferation and differentiation. Cell. 2014; 158:1281–1292. [DOI] [PubMed] [Google Scholar]
- 31. Zhou H.-Q., Ning L.-W., Zhang H.-X., Guo F.-B.. Analysis of the relationship between genomic GC content and patterns of base usage, codon usage and amino acid usage in prokaryotes: similar GC content adopts similar compositional frequencies regardless of the phylogenetic lineages. PLoS One. 2014; 9:e107319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chamary J.V., Parmley J.L., Hurst L.D.. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 2006; 7:98–108. [DOI] [PubMed] [Google Scholar]
- 33. dos Reis M., Savva R., Wernisch L.. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 2004; 32:5036–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hia F., Yang S.F., Shichino Y., Yoshinaga M., Murakawa Y., Vandenbon A., Fukao A., Fujiwara T., Landthaler M., Natsume T.et al.. Codon bias confers stability to human mRNAs. EMBO Rep. 2019; 20:e48220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quax T.E., Claassens N.J., Soll D., van der Oost J.. Codon bias as a means to fine-tune gene expression. Mol. Cell. 2015; 59:149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao F., Yu C.H., Liu Y.. Codon usage regulates protein structure and function by affecting translation elongation speed in drosophila cells. Nucleic Acids Res. 2017; 45:8484–8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duret L. tRNA gene number and codon usage in the c. elegans genome are co-adapted for optimal translation of highly expressed genes. Trends Genet. 2000; 16:287–289. [DOI] [PubMed] [Google Scholar]
- 38. Duret L., Mouchiroud D.. Expression pattern and, surprisingly, gene length shape codon usage in caenorhabditis, drosophila, and arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:4482–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chamary J.V., Hurst L.D.. Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals. Genome Biol. 2005; 6:R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Doherty A., McInerney J.O.. Translational selection frequently overcomes genetic drift in shaping synonymous codon usage patterns in vertebrates. Mol. Biol. Evol. 2013; 30:2263–2267. [DOI] [PubMed] [Google Scholar]
- 41. Pouyet F., Mouchiroud D., Duret L., Sémon M.. Recombination, meiotic expression and human codon usage. Elife. 2017; 6:e27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Galtier N., Roux C., Rousselle M., Romiguier J., Figuet E., Glemin S., Bierne N., Duret L.. Codon usage bias in animals: disentangling the effects of natural selection, effective population size, and GC-Biased gene conversion. Mol. Biol. Evol. 2018; 35:1092–1103. [DOI] [PubMed] [Google Scholar]
- 43. Kirchner S., Cai Z., Rauscher R., Kastelic N., Anding M., Czech A., Kleizen B., Ostedgaard L.S., Braakman I., Sheppard D.N.et al.. Alteration of protein function by a silent polymorphism linked to tRNA abundance. PLoS Biol. 2017; 15:e2000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pavon-Eternod M., Gomes S., Geslain R., Dai Q., Rosner M.R., Pan T.. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009; 37:7268–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kirchner S., Ignatova Z.. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 2015; 16:98–112. [DOI] [PubMed] [Google Scholar]
- 46. Chittum H.S., Hill K.E., Carlson B.A., Lee B.J., Burk R.F., Hatfield D.L.. Replenishment of selenium deficient rats with selenium results in redistribution of the selenocysteine tRNA population in a tissue specific manner. Biochim. Biophys. Acta. 1997; 1359:25–34. [DOI] [PubMed] [Google Scholar]
- 47. Sagi D., Rak R., Gingold H., Adir I., Maayan G., Dahan O., Broday L., Pilpel Y., Rechavi O.. Tissue- and Time-Specific expression of otherwise identical tRNA genes. PLoS Genet. 2016; 12:e1006264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kondo K., Makovec B., Waterston R.H., Hodgkin J.. Genetic and molecular analysis of eight tRNA(Trp) amber suppressors in caenorhabditis elegans. J. Mol. Biol. 1990; 215:7–19. [DOI] [PubMed] [Google Scholar]
- 49. Waldman Y.Y., Tuller T., Shlomi T., Sharan R., Ruppin E.. Translation efficiency in humans: tissue specificity, global optimization and differences between developmental stages. Nucleic Acids Res. 2010; 38:2964–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smith D.W. Problems of translating heterologous genes in expression systems: the role of tRNA. Biotechnol Prog. 1996; 12:417–422. [DOI] [PubMed] [Google Scholar]
- 51. Gingold H., Dahan O., Pilpel Y.. Dynamic changes in translational efficiency are deduced from codon usage of the transcriptome. Nucleic Acids Res. 2012; 40:10053–10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Torrent M., Chalancon G., de Groot N.S., Wuster A., Madan Babu M.. Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci. Signal. 2018; 11:eaat6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pang Y.L., Abo R., Levine S.S., Dedon P.C.. Diverse cell stresses induce unique patterns of tRNA up- and down-regulation: tRNA-seq for quantifying changes in tRNA copy number. Nucleic Acids Res. 2014; 42:e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Puri P., Wetzel C., Saffert P., Gaston K.W., Russell S.P., Cordero Varela J.A., van der Vlies P., Zhang G., Limbach P.A., Ignatova Z.et al.. Systematic identification of tRNAome and its dynamics in lactococcus lactis. Mol. Microbiol. 2014; 93:944–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yona A.H., Bloom-Ackermann Z., Frumkin I., Hanson-Smith V., Charpak-Amikam Y., Feng Q., Boeke J.D., Dahan O., Pilpel Y.. tRNA genes rapidly change in evolution to meet novel translational demands. Elife. 2013; 2:e01339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Frenkel-Morgenstern M., Danon T., Christian T., Igarashi T., Cohen L., Hou Y.M., Jensen L.J.. Genes adopt non-optimal codon usage to generate cell cycle-dependent oscillations in protein levels. Mol. Syst. Biol. 2012; 8:572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goodarzi H., Nguyen H.C.B., Zhang S., Dill B.D., Molina H., Tavazoie S.F.. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell. 2016; 165:1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Netzer N., Goodenbour J.M., David A., Dittmar K.A., Jones R.B., Schneider J.R., Boone D., Eves E.M., Rosner M.R., Gibbs J.S.et al.. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature. 2009; 462:522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Polte C., Wedemeyer D., Oliver K.E., Wagner J., Bijvelds M.J.C., Mahoney J., de Jonge H.R., Sorscher E.J., Ignatova Z.. Assessing cell-specific effects of genetic variations using tRNA microarrays. BMC Genomics. 2019; 20:549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Torres A.G., Batlle E., Ribas de Pouplana L.. Role of tRNA modifications in human diseases. Trends Mol. Med. 2014; 20:306–314. [DOI] [PubMed] [Google Scholar]
- 61. Mahlab S., Tuller T., Linial M.. Conservation of the relative tRNA composition in healthy and cancerous tissues. RNA. 2012; 18:640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rudorf S. Efficiency of protein synthesis inhibition depends on tRNA and codon compositions. PLoS Comput. Biol. 2019; 15:e1006979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rudolph K.L., Schmitt B.M., Villar D., White R.J., Marioni J.C., Kutter C., Odom D.T.. Codon-Driven translational efficiency is stable across diverse mammalian cell states. PLoS Genet. 2016; 12:e1006024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pavon-Eternod M., Gomes S., Rosner M.R., Pan T.. Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA. 2013; 19:461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. van Weringh A., Ragonnet-Cronin M., Pranckeviciene E., Pavon-Eternod M., Kleiman L., Xia X.. HIV-1 modulates the tRNA pool to improve translation efficiency. Mol. Biol. Evol. 2011; 28:1827–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li M., Kao E., Gao X., Sandig H., Limmer K., Pavon-Eternod M., Jones T.E., Landry S., Pan T., Weitzman M.D.et al.. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature. 2012; 491:125–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Alonso A.M., Diambra L.. SARS-CoV-2 codon usage bias downregulates host expressed genes with similar codon usage. Front. Cell Dev. Biol. 2020; 8:831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen F., Wu P., Deng S., Zhang H., Hou Y., Hu Z., Zhang J., Chen X., Yang J.R.. Dissimilation of synonymous codon usage bias in virus-host coevolution due to translational selection. Nat. Ecol. Evol. 2020; 4:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Miller J.B., Hippen A.A., Wright S.M., Morris C., Ridge P.G.. Human viruses have codon usage biases that match highly expressed proteins in the tissues they infect. Biomed. Genet. Genomics. 2017; 2:1–5. [Google Scholar]
- 70. Li Y., Yang X., Wang N., Wang H., Yin B., Jiang W.. GC usage of SARS-CoV-2 genes might adapt to the environment of human lung expressed genes. Mol. Genet. Genomics. 2020; 295:1537–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Maldonado L.L., Bertelli A.M., Kamenetzky L.. Molecular features similarities between SARS-CoV-2, SARS, MERS and key human genes could favour the viral infections and trigger collateral effects. Sci. Rep. 2021; 11:4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhou J., Liu W.J., Peng S.W., Sun X.Y., Frazer I.. Papillomavirus capsid protein expression level depends on the match between codon usage and tRNA availability. J. Virol. 1999; 73:4972–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Miller J.B., Brase L.R., Ridge P.G.. ExtRamp: a novel algorithm for extracting the ramp sequence based on the tRNA adaptation index or relative codon adaptiveness. Nucleic Acids Res. 2019; 47:1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tuller T., Zur H.. Multiple roles of the coding sequence 5′ end in gene expression regulation. Nucleic Acids Res. 2015; 43:13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Verma M., Choi J., Cottrell K.A., Lavagnino Z., Thomas E.N., Pavlovic-Djuranovic S., Szczesny P., Piston D.W., Zaher H.S., Puglisi J.D.et al.. A short translational ramp determines the efficiency of protein synthesis. Nat. Commun. 2019; 10:5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tuller T., Veksler-Lublinsky I., Gazit N., Kupiec M., Ruppin E., Ziv-Ukelson M.. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011; 12:R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y.. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010; 141:344–354. [DOI] [PubMed] [Google Scholar]
- 78. Dana A., Tuller T.. The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res. 2014; 42:9171–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Park H., Subramaniam A.R.. Inverted translational control of eukaryotic gene expression by ribosome collisions. PLoS Biol. 2019; 17:e3000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. McKinnon L.M., Miller J.B., Whiting M.F., Kauwe J.S.K., Ridge P.G.. A comprehensive analysis of the phylogenetic signal in ramp sequences in 211 vertebrates. Sci. Rep. 2021; 11:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hodgman M.W., Miller J.B., Meurs T.E., Kauwe J.S.K.. CUBAP: an interactive web portal for analyzing codon usage biases across populations. Nucleic Acids Res. 2020; 48:11030–11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Goodman D.B., Church G.M., Kosuri S.. Causes and effects of N-terminal codon bias in bacterial genes. Science. 2013; 342:475–479. [DOI] [PubMed] [Google Scholar]
- 83. Villada J.C., Duran M.F., Lee P.K.. Interplay between position-dependent codon usage bias and hydrogen bonding at the 5′ end of ORFeomes. Msystems. 2020; 5:e00613–e00620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Quax T.E.F., Claassens N.J., Soll D., van der Oost J.. Codon bias as a means to fine-tune gene expression. Mol. Cell. 2015; 59:149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gorochowski T.E., Ignatova Z., Bovenberg R.A., Roubos J.A.. Trade-offs between tRNA abundance and mRNA secondary structure support smoothing of translation elongation rate. Nucleic Acids Res. 2015; 43:3022–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Thul P.J., Lindskog C.. The human protein atlas: a spatial map of the human proteome. Protein Sci. 2018; 27:233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A.et al.. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 88. Ponten F., Schwenk J.M., Asplund A., Edqvist P.H.D.. The human protein atlas as a proteomic resource for biomarker discovery. J. Intern. Med. 2011; 270:428–446. [DOI] [PubMed] [Google Scholar]
- 89. Thul P.J., Lindskog C.. The human protein atlas: a spatial map of the human proteome. Protein Sci. 2018; 27:233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Singh M., Bansal V., Feschotte C.. A single-cell RNA expression map of human coronavirus entry factors. Cell Rep. 2020; 32:108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Trypsteen W., Van Cleemput J., Snippenberg W.V., Gerlo S., Vandekerckhove L.. On the whereabouts of SARS-CoV-2 in the human body: a systematic review. PLoS Pathog. 2020; 16:e1009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Deinhardt-Emmer S., Wittschieber D., Sanft J., Kleemann S., Elschner S., Haupt K.F., Vau V., Häring C., Rödel J., Henke A.et al.. Early postmortem mapping of SARS-CoV-2 RNA in patients with COVID-19 and the correlation with tissue damage. Elife. 2021; 10:e60361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Nieuwenhuis T.O., Yang S.Y., Verma R.X., Pillalamarri V., Arking D.E., Rosenberg A.Z., McCall M.N., Halushka M.K.. Consistent RNA sequencing contamination in GTEx and other data sets. Nat. Commun. 2020; 11:1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Muus C., Luecken M.D., Eraslan G., Sikkema L., Waghray A., Heimberg G., Kobayashi Y., Vaishnav E.D., Subramanian A., Smillie C.et al.. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med. 2021; 27:546–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Matsuyama S., Nagata N., Shirato K., Kawase M., Takeda M., Taguchi F.. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010; 84:12658–12664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Glowacka I., Bertram S., Müller M.A., Allen P., Soilleux E., Pfefferle S., Steffen I., Tsegaye T.S., He Y., Gnirss K.et al.. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011; 85:4122–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A.et al.. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. 2020; 181:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Narayanan K., Huang C., Lokugamage K., Kamitani W., Ikegami T., Tseng C.T., Makino S.. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type i interferon, in infected cells. J. Virol. 2008; 82:4471–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kamitani W., Huang C., Narayanan K., Lokugamage K.G., Makino S.. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus nsp1 protein. Nat. Struct. Mol. Biol. 2009; 16:1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Xia H., Cao Z., Xie X., Zhang X., Chen J.Y., Wang H., Menachery V.D., Rajsbaum R., Shi P.Y.. Evasion of type i interferon by SARS-CoV-2. Cell Rep. 2020; 33:108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Stukalov A., Girault V., Grass V., Karayel O., Bergant V., Urban C., Haas D.A., Huang Y., Oubraham L., Wang A.et al.. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature. 2021; 594:246–252. [DOI] [PubMed] [Google Scholar]
- 102. Khalil M.F., Wagner W.D., Goldberg I.J.. Molecular interactions leading to lipoprotein retention and the initiation of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004; 24:2211–2218. [DOI] [PubMed] [Google Scholar]
- 103. Nicolai L., Leunig A., Brambs S., Kaiser R., Weinberger T., Weigand M., Muenchhoff M., Hellmuth J.C., Ledderose S., Schulz H.et al.. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation. 2020; 142:1176–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Demmer L.A., Levin M.S., Elovson J., Reuben M.A., Lusis A.J., Gordon J.I.. Tissue-specific expression and developmental regulation of the rat apolipoprotein b gene. Proc. Natl. Acad. Sci. U.S.A. 1986; 83:8102–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Duffy Á., Verbanck M., Dobbyn A., Won H.H., Rein J.L., Forrest I.S., Nadkarni G., Rocheleau G., Do R.. Tissue-specific genetic features inform prediction of drug side effects in clinical trials. Sci. Adv. 2020; 6:eabb6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hao Y., Quinnies K., Realubit R., Karan C., Tatonetti N.P.. Tissue-Specific analysis of pharmacological pathways. CPT Pharmacometrics Syst. Pharmacol. 2018; 7:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Nevins J.R., Huang E.S., Dressman H., Pittman J., Huang A.T., West M.. Towards integrated clinico-genomic models for personalized medicine: combining gene expression signatures and clinical factors in breast cancer outcomes prediction. Hum. Mol. Genet. 2003; 12:R153–R157. [DOI] [PubMed] [Google Scholar]
- 108. Gordon G.J., Jensen R.V., Hsiao L.L., Gullans S.R., Blumenstock J.E., Richards W.G., Jaklitsch M.T., Sugarbaker D.J., Bueno R.. Using gene expression ratios to predict outcome among patients with mesothelioma. J. Natl. Cancer Inst. 2003; 95:598–605. [DOI] [PubMed] [Google Scholar]
- 109. Nutt C.L., Mani D.R., Betensky R.A., Tamayo P., Cairncross J.G., Ladd C., Pohl U., Hartmann C., McLaughlin M.E., Batchelor T.T.et al.. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003; 63:1602–1607. [PubMed] [Google Scholar]
- 110. van de Vijver M.J., He Y.D., van’t Veer L.J., Dai H., Hart A.A., Voskuil D.W., Schreiber G.J., Peterse J.L., Roberts C., Marton M.J.et al.. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002; 347:1999–2009. [DOI] [PubMed] [Google Scholar]
- 111. van ’t Veer L.J., Dai H., van de Vijver M.J., He Y.D., Hart A.A., Mao M., Peterse H.L., van der Kooy K., Marton M.J., Witteveen A.T.et al.. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002; 415:530–536. [DOI] [PubMed] [Google Scholar]
- 112. Jacquemier J., Ginestier C., Rougemont J., Bardou V.J., Charafe-Jauffret E., Geneix J., Adélaïde J., Koki A., Houvenaeghel G., Hassoun J.et al.. Protein expression profiling identifies subclasses of breast cancer and predicts prognosis. Cancer Res. 2005; 65:767–779. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All scripts used to replicate this work are available at https://github.com/ridgelab/rampAtlas. The Ramp Atlas is publicly available at https://ramps.byu.edu.