

Abstract
Objective.
Hypoxia occurs in tumors, infections, and inflammatory sites such as joints of patients with rheumatoid arthritis (RA). It alleviates inflammatory responses and increases bone resorption in inflammatory arthritis by enhancing osteoclastogenesis. The mechanism by which hypoxia response is linked to osteoclastogenesis and inflammatory bone resorption is unclear. We showed that LSD1 metabolically integrates inflammatory osteoclastogenesis and bone resorption.
Methods.
LSD1 specific inhibitors and siRNAs were used to inhibit LSD1. Using human osteoclast precursors derived from CD14+ cells, RNA-sequencing and subsequent analysis were performed. Accelerated bone loss and inflammatory osteolysis models were tested as mice model. After cis-expression quantitative trait locus (cis-eQTL) analysis for HIF-1α, the association between HIF-1α allelic variants bone erosion was evaluated in RA patients.
Results.
RANKL induces LSD1 expression in an mTOR-dependent manner. The expression of LSD1 was higher in RA synovium than that from osteoarthritis. LSD1 inhibition suppresses osteoclast differentiation. Transcriptome analysis identified LSD1-mediated hypoxia and cell-cycle pathways during human osteoclastogenesis. Furthermore, HIF-1α protein, which is rapidly degraded by the proteasome in normoxia, was expressed in RANKL-stimulated osteoclast precursor cells. RANKL-induced LSD1 stabilized HIF-1α protein, thereby promoted glycolysis, along with E2F1, which was also upregulated by LSD1. cis-eQTL analysis revealed that higher HIF-1α expression was associated with increased bone erosion in RA. LSD1 inhibition decreased pathological bone resorption in an accelerated osteoporosis mouse model and arthritis and inflammatory osteolysis mouse models.
Conclusion.
LSD1 metabolically regulates osteoclastogenesis in an inflammatory energy-demanding environment and provides new therapeutic strategies targeting osteoclasts against inflammatory arthritis including RA.
Keywords: rheumatoid arthritis, osteoclast, LSD1, hypoxia
INTRODUCTION
Bone destruction is a hallmark and serious consequence of various skeletal diseases, including osteoporosis and inflammatory arthritis. It affects the quality of life of patients and increases the risk of disability and mortality. Osteoclasts are multinucleated bone-resorbing cells derived from myeloid lineage cells. Excessive generation of osteoclasts and/or increased osteoclast activity in these patients results in pathological bone resorption (1, 2). Antiresorptive therapies, including bisphosphonates and anti-receptor activator of nuclear factor-κB ligand (RANKL) monoclonal antibody (denosumab), have become the first-line treatments for osteoporosis, and denosumab is the currently available drug for treating rheumatoid arthritis (RA) with the aim to suppress progressive bone erosion (3, 4). These agents effectively reduce bone resorption and limit bone loss. However, these agents still face problems associated with long-term use and drug discontinuation (5).
Macrophage colony stimulating factor (M-CSF) and RANKL are key cytokines for osteoclast differentiation and survival. During osteoclast differentiation, the binding of RANKL to its receptor, receptor activator of NF-κB (RANK), activates various transcription factors including MYC, NF-κB, AP-1, CREB, and NFATc1, which work in a complex network to promote osteoclast differentiation (6). Effective osteoclastogenesis requires both glycolysis and oxidative phosphorylation (OXPHOS) for energy generation (7–13). We previously showed that E2F1, a transcription factor that mainly controls the cell cycle, is induced by RANKL stimulation in non-proliferative human osteoclast precursors (OCPs) and promotes glycolysis during the early phases of osteoclastogenesis (14). Hypoxia inducible factor 1 (HIF-1) appears to be another factor responsible for the induction of glycolytic genes during osteoclastogenesis because GLUT1, VEGF, and glycolytic enzymes, the well-known target genes of HIF-1, are upregulated during osteoclastogenesis, even in normoxia (15). However, HIF-1α, a subunit of HIF-1, is unstable in normoxia because of its rapid degradation by O2-dependent prolyl hydroxylation (PHD) and von Hippel-Lindau (VHL) protein-dependent proteasomal degradation (15). However, the association between RANKL signaling and HIF-1 has not been completely elucidated.
Lysine-specific demethylase 1 (LSD1, also known as KDM1A, BHC110, and AOF2) catalyzes the demethylation of mono- and di-methylated histone 3 lysine 4 (H3K4me1, H3K4me2) for transcriptional repression and mono- and di-methylated H3K9 (H3K9me1, H3K9me2) for gene activation. LSD1 regulates not only histone substrates but also the methylation dynamics of non-histone proteins, including E2F1, DNMT1, HIF-1α, and STAT3, which play a crucial role in a wide range of cellular processes, including stem cell pluripotent regulation, metabolism, cell proliferation, embryonic development, and human cancer development (16). However, how LSD1 affects inflammatory responses and osteoclastogenesis remains unclear.
In the present study, we showed that LSD1 is essential for osteoclastogenesis in vivo and in vitro. LSD1 was induced by RANKL in an mTOR-dependent manner. RANKL-induced LSD1 stabilized HIF-1α protein even in normoxia and mediated metabolic pathways along with E2F1. LSD1 was upregulated in RA synovium and was induced by TNF-α stimulation. Expression quantitative trait locus analysis revealed that higher HIF-1α expression was associated with increased bone erosion in RA. Inhibition of LSD1 suppressed pathological bone resorption in an accelerated osteoporosis mouse model and in arthritis and inflammatory osteolysis mouse models. These results suggest that targeting LSD1 may be an attractive therapeutic target to prevent pathological bone resorption including RA.
MATERIALS AND METHODS
Details in RNA interference, RNA sequencing, glycolytic rate assay, immunofluorescence staining, immunohistochemistry, rheumatoid arthritis patient analysis and micro-computed tomography (μCT) analysis are described in Supplementary Methods.
Reagents.
Human and murine M-CSF, sRANKL, and TNF were purchased from Peprotech (Rocky Hill, NJ, USA). Inhibitors, SP2509, RN-1, ORY-1001, and Torin1, were purchased from Cayman Chemical Company (Ann Arbor, MI). The antibodies used were as follows: LSD1, phospho-NFkB p65, Phospho-4E-BP1, HIF1a, E2F1, PHD-2 (Cell Signaling Technologies, Danvers, MA), p38, c-Myc (BioLegend, San Diego, California) and Swine Anti-Rabbit Immunoglobulins/HRP (DAKO, Hovedstaden, Denmark).
Cell culture.
Peripheral blood mononuclear cells (PBMCs) were obtained with lymphocyte separation solution (d=1.077)(Nacalai Tesque, Kyoto, Japan) from leukoreduction filters purchased from Japanese Red Cross Society, using a protocol approved by the ethics committee of Kyoto University Graduate School and Faculty of Medicine. CD14 monocytes were obtained from PBMCs with anti-CD14 magnetic beads (Miltenyi Biotec, Auburn CA). CD14-positive monocytes were cultured in alpha modified essential medium (SIGMA, St. Louis, MO) with 10% FBS (MP Biomedicals, Irvine, CA), penicillin, streptomycin (Nacalai Tesque), and L-glutamine (Nacalai Tesque) supplemented with 20 ng/ml M-CSF (Peprotech, Rocky Hill, NJ) overnight to obtain OCPs expressing RANK. For osteoclast differentiation, OCPs were cultured with 20 ng/mL M-CSF and 40 ng/mL RANKL. Cells were stained for TRAP using the TRAP Kit (SIGMA). For the hypoxia experiment, cells were placed in a hypoxic incubator (4% oxygen, WakenBtech, Kyoto, Japan). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Cayman Chemical, MI) was performed, according to the manufacturer’s protocol. In vitro resorption activity of osteoclasts was measured using bone resorption assay plate (PG research, Tokyo, Japan). Cells were seeded in bone resorption assay plates and cultured in the presence of M-CSF and RANKL. After 5 days of culture with RANKL, cells were removed, according to the manufacturer’s protocol.
In vivo mouse bone phenotype.
All animal studies were conducted in accordance with the principles of the Kyoto University Committee of Animal Resources, which are based on the International Guiding Principles for Biomedical Research Involving Animals. The experimental sample size was determined by our previous studies (14). The mice at a similar age were purchased and randomly assigned to each group. No randomization allocation sequence was assigned. For the experiment involving arthritis, 8-week-old SKG female mice were purchased from Clea Japan (Fujinomiya, Japan). Arthritis was induced by intraperitoneal injection of 20 mg mannan (Sigma). SP2509 (25 mg/kg) was administered intraperitoneally twice per week for 3 weeks. The severity of arthritis was scored in a blinded fashion by three investigators for each paw on a 3-point scale, as previously described (14). To evaluate osteoclast formation as a primary outcome measure, TRAP staining of histological sections from calcaneocuboid and tarsometatarsal joints were evaluated.
For inflammatory osteolysis experiments, we used an established mouse model, which is the TNF-induced supracalvarial osteolysis model (17) with minor modifications. TNF (Peprotech) was administered on days 0, 2, and 4 to 6-week-old female C57BL/6N mice (Clea-Japan). SP2509 (25 mg/kg) was administered intraperitoneally on days 0 and 3. On day 4, mice were sacrificed 6 h after TNF-α injection, and calvarial bones were collected for sectioning.
For the enhanced osteoporosis experiment, 14–16-week-old C57BL/6N male mice were purchased from Clea-Japan and were fed a low-Ca diet (less than 0.01%, Clea Japan) or a control diet (Clea Japan). SP2509 was administered intraperitoneally at 25 mg/kg twice per week for 3 weeks for μCT analysis and 2 weeks for sectioning.
Statistical analysis.
Statistical analysis was performed using JMP Pro version 13.0.0 (SAS, Institute Inc., Cary, NC, USA). In two conditions, we used the two-tailed unpaired t test, and in more than 2 conditions, we used Tukey’s multiple comparison test. A p value of less than 0.05 was considered statistically significant. For all experiments, * p < 0.05 and ** p < 0.01.
RESULTS
LSD1 is required for osteoclast differentiation.
First, we tested the effects of LSD inhibition on osteoclast differentiation using human blood-derived OCPs, which provides the following advantages: osteoclastogenesis can be studied separately from the effects on proliferation, and cells directly relevant for human diseases can be used (18). SP2509, a specific inhibitor of LSD1, inhibited the differentiation of OCPs into multinucleated tartrate-resistant acid phosphate (TRAP)-positive cells in a dose-dependent manner (Figure 1A, Supplementary Figure 1). Accordingly, SP2509 suppressed RANKL-induced expression of osteoclast-related genes, including cathepsin K (CTSK) and β3 integrin (ITGB3), in a dose-dependent manner (Figure 1B). RANKL-induction of NFATc1, which is essential for osteoclast differentiation and the expression of many osteoclast genes, was also suppressed by SP2509. We directly tested the role of LSD1 in osteoclastogenesis by silencing LSD1 using siRNA. LSD1 silencing suppressed RANKL-induced osteoclast differentiation and the expression of osteoclast-related genes, ITGB3 and CTSK (Figures 1C, D, E, Supplementary Figure 2). siRNA-mediated knockdown of LSD1 expression also suppressed RANKL-induced expression of NFATc1. These results indicate that LSD1 plays a positive role in osteoclast differentiation.
Figure 1. LSD1 is required for osteoclast differentiation.
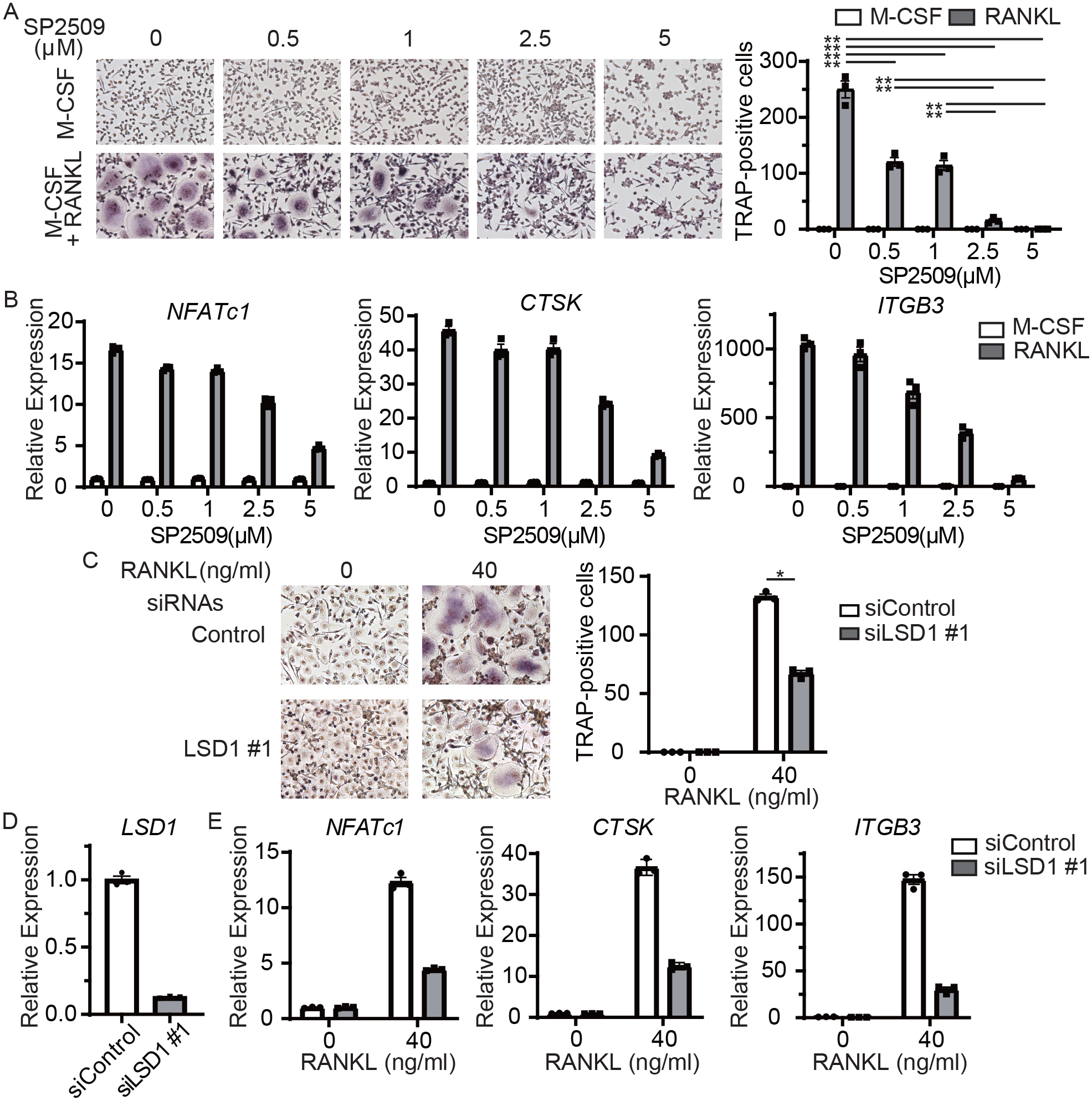
A, Osteoclast precursors (OCPs) derived from human CD14-positive monocytes were treated with 40 ng/ml RANKL. TRAP-positive, multinucleated cells were counted in triplicate. Left panel, representative results from one of three independent donors. Right panel, number of TRAP-positive, multinucleated cells. Data are shown as mean ± SEM from aggregate data from three independent donors. B, RT-qPCR analysis of osteoclast-related genes (normalized relative to TBP mRNA). OCPs were treated with RANKL for 48 h. Data are mean ± SEM of triplicates from one representative experiment of three independent donors. C, Human CD14-positive monocytes were nucleofected with LSD1-specific or scrambled control siRNAs, and osteoclast were generated. TRAP-positive, multinucleated cells were counted in triplicate. Left panel, representative results from three independent experiments. Right panel, number of TRAP-positive, multinucleated cells. Data are shown as mean ± SEM from aggregate data from three independent donors. D, RT-qPCR analysis of osteoclast-related gene expression. Data are mean ± SEM of triplicates from one representative experiment of three independent experiments.
* p < 0.05, using the Tukey-Kramer test.
LSD1 expression is induced by RANKL and TNF-α in OCPs, and high in RA synovium.
To determine how LSD1 expression is regulated in inflammatory bone resorption, OCPs were stimulated with RANKL. RANKL stimulation slightly upregulated mRNA levels of LSD1 (Figure 2A). Immunochemistry revealed that the protein levels of LSD1 were significantly upregulated in the nucleus 48 h after RANKL stimulation (Figure 2B). These results suggest that LSD1 expression is induced by RANKL at the protein level rather than at the mRNA level. Because we previously showed that RANKL stimulation upregulated mTORC1, a key positive regulator of translation (14), we tested whether mammalian target of rapamycin complex 1 (mTORC1) activity has a role in LSD1 protein expression. RANKL stimulation upregulated the phosphorylation of eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) and LSD1 (Figure 2C). Torin 1, an mTORC1 inhibitor, suppressed both RANKL-induced LSD1 and phospho-4E-BP1, suggesting that LSD1 protein expression is mTOR-dependent. Next, we investigated the level of LSD1 expression in RA synovium. Immunohistochemistry revealed that LSD1 expression was higher in RA synovium than in osteoarthritis synovium (Figure 2D). We also stimulated OCPs with proinflammatory cytokines, TNF-α, IL-1β, and IL-6. The expression of LSD1 protein was significantly upregulated with TNF-α stimulation, but not with IL-1β or IL-6 treatment (Figure 2E). These data suggested that, LSD1 expression was induced by RANKL and TNF-α stimulation in OCPs and was elevated in inflammatory RA synovial tissue.
Figure 2. LSD1 expression is induced by RANKL and TNF-α in OCPs and is elevated in RA synovium.
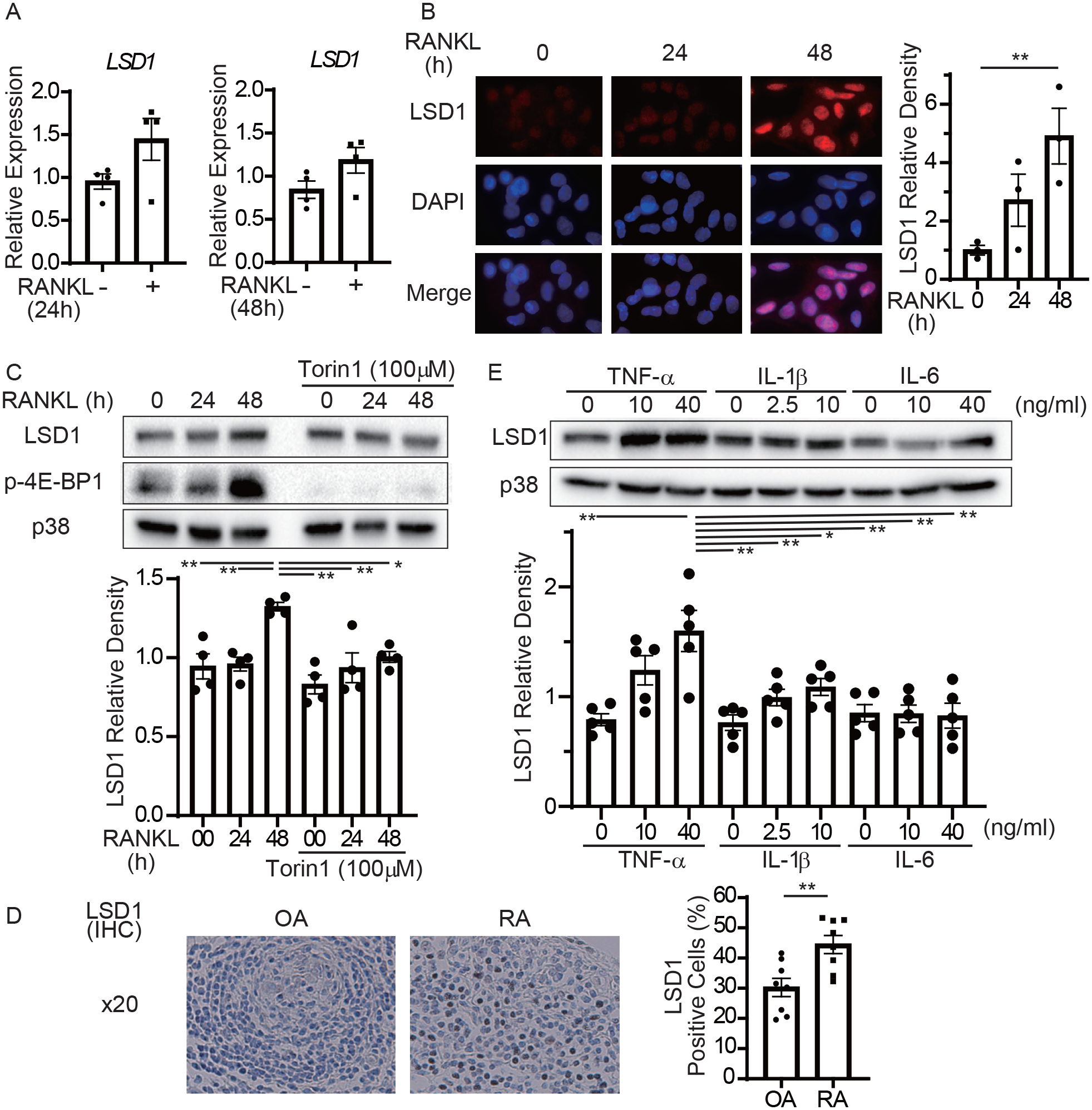
A, RT-qPCR of LSD1 mRNA normalized relative to TBP mRNA. B, Immunofluorescence microscopy showing the subcellular localization of LSD1. Human OCPs were treated with RANKL for the indicated hours. Left panel, representative images from one of the three different donors. LSD1 (red) and nuclear (DAPI, blue) staining images are merged in the lowest panels. Right panel, relative fluorescence intensity of LSD1 amounts from three different donors. C, Immunoblot of whole-cell lysates using LSD1, p-4E-BP1, and p38 antibodies. p38 served as a loading control. Blots are representative (n = 4). D, Immunohistochemistry of synovium of RA or OA patients using LSD1 antibody. Light panel, representative images from eight patients with RA and OA. Right panel, the ratio of LSD1 positive cells.
E, Immunoblot of whole-cell lysates using LSD1 and p38 antibodies. OCPs were treated with TNF-α, IL-1β, and IL-6. Blots show representative data (n = 5).
Data are shown as mean ± SEM from aggregate data. * p < 0.05, ** p < 0.01 from a 2-tailed, unpaired t-test (D) and Tukey-Kramer test (B, C, E)
LSD1 regulates metabolic pathways in RANKL-stimulated OCPs.
The known non-histone substrates of LSD1 demethylation are p53, DNMT1, E2F1, MYC, MYPT1, STAT3, ERα, HIF-1α, MTA, and AGO2 (16). We previously showed that E2F1 and MYC are important factors for osteoclastogenesis linked to metabolism (7, 14). LSD1 inhibition by SP2509 and LSD1 silencing inhibited RANKL-induced MYC expression and translocation to the nucleus (Supplementary Figures 3A–C). SP2509 also suppressed RANKL-induced E2F1 expression and E2F1 target gene expression (Supplementary Figures 3D–G).
We performed RNA-seq and bioinformatic analysis to identify genes regulated by LSD1 in RANKL-stimulated OCPs (Supplementary Figure 4). Pathways enriched by RANKL-inducible genes were compared between LSD1-specific siRNAs and scrambled controls. The cell cycle (which is associated with anabolic metabolism), metabolic, inflammatory-NF-κB, and hypoxia pathways were altered (Figure 3A). Consistently, gene set enrichment analysis (GSEA) revealed that LSD1 regulated metabolic pathways including MYC, oxidative phosphorylation, mTORC1, and glycolysis, as well as the inflammatory pathway (Figure 3B). Furthermore, LSD1 mediated hypoxia and COMMD1 (linked to inflammation, metabolism, and hypoxia) pathways (Figures 3B, C). Analysis of transcription factors regulated by LSD1 showed that E2F1, E2F3, E2F4, and MYC (linked to cell cycle and metabolism) were highly enriched (Figure 3D). It also revealed that RELA, NFKB1 (linked to inflammation), and VHL (linked to hypoxia) were highly enriched.
Figure 3. LSD1 regulates metabolic pathways in RANKL-stimulated OCPs.
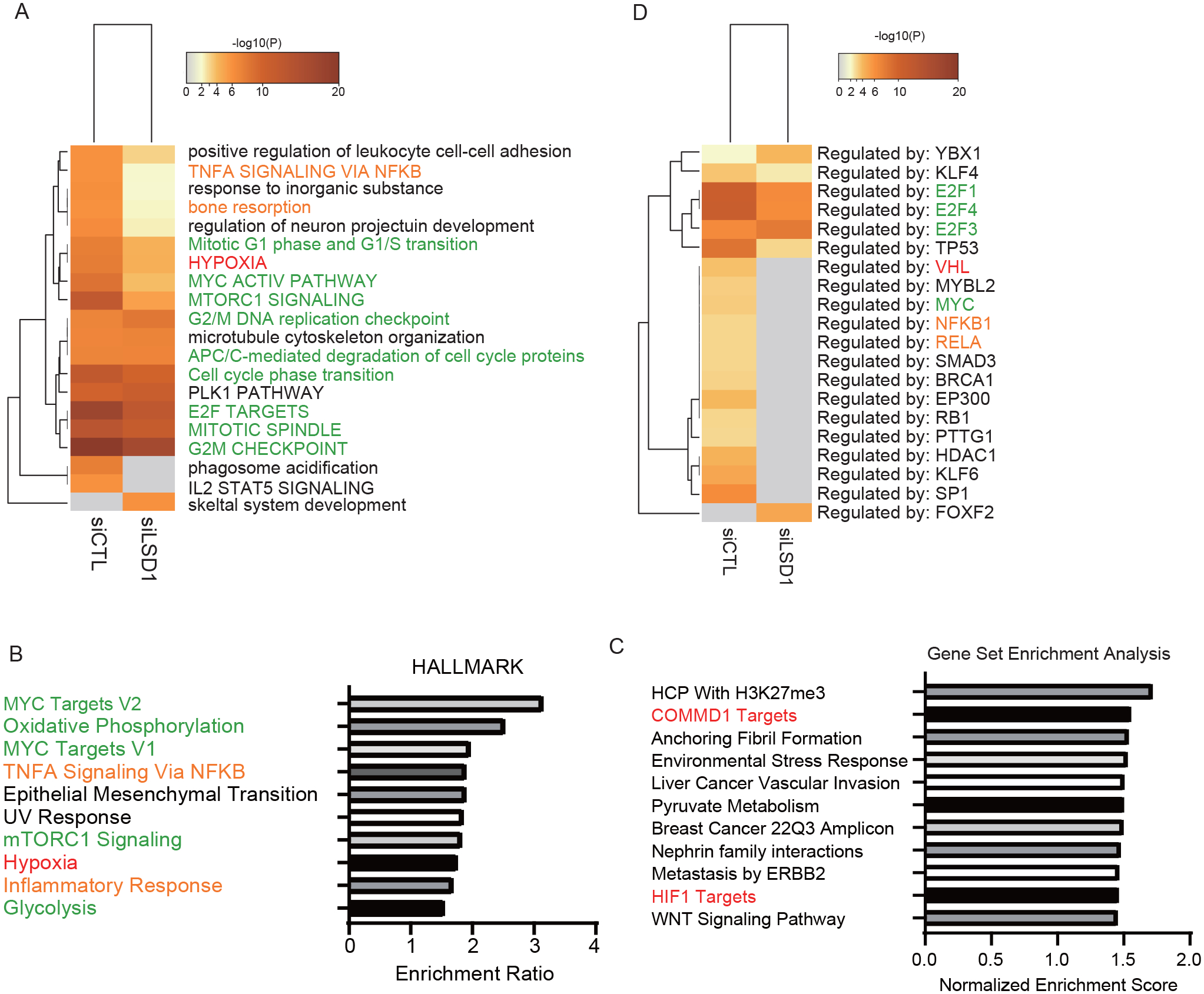
A, Human OCPs transfected with LSD1 siRNA or scrambled control were stimulated with RANKL for 0, 24, or 48 h. The top 20 pathways that were differentially enriched in siRNA-mediated LSD1 knockdown and scrambled control-treated human OCPs stimulated with RANKL among genes with more than 2-fold increase at 48 h compared with that at 0 h after RANKL stimulation. B, C, Gene set enrichment analysis (GSEA), comparing RANKL-inducible genes between human OCPs transfected with LSD1-specific siRNA and scrambled control. Genes upregulated by RANKL (> 1.5 folds) and suppressed by LSD1 siRNA (< 1.5 folds) after 48 h of RANKL stimulation and not upregulated by LSD1 siRNA without RANKL treatment were analyzed. D, The top 20 transcription factors that were differentially enriched in OCPs treated with RANKL. Genes described in A were used for analysis.
Font colors of labels correspond to the metabolic and cell cycle gene set (green), inflammation-NF-κB signaling gene set (orange), and hypoxia gene set (red).
The cell cycle and hypoxia pathways are associated with anabolic metabolism. We previously showed that COMMD1 is an important factor for hypoxia-enhancement of osteoclastogenesis through the HIF, E2F, and NF-kB/metabolism-mediated axis through E3 ubiquitination of HIF-1α and NF-κB (14). RNA-seq analysis suggested that LSD1 also has an integrative role in inflammation, hypoxia, and metabolism.
HIF-1α regulates osteoclast differentiation in normoxia.
Osteoclast formation and bone resorption are regarded as energy-demanding processes that require active metabolic reprogramming, although these have not been extensively studied (12). During osteoclastogenesis, the expression of glucose transporters (GLUT)—GLUT1 (SLC2A1) and GLUT3 (SLC2A3)—is known to be upregulated (Figure 4A), and glycolysis is increased (11, 15). HIF-1α is a strong inducer of glycolysis, which targets these genes, and is suspected to have a critical role in osteoclastogenesis; however, the association between RANKL signaling and HIF-1α remains unclear. The HIF-1α protein is unstable because of the hydroxylation by PHD in normoxia.
Figure 4. HIF-1α regulates osteoclast differentiation in normoxia.
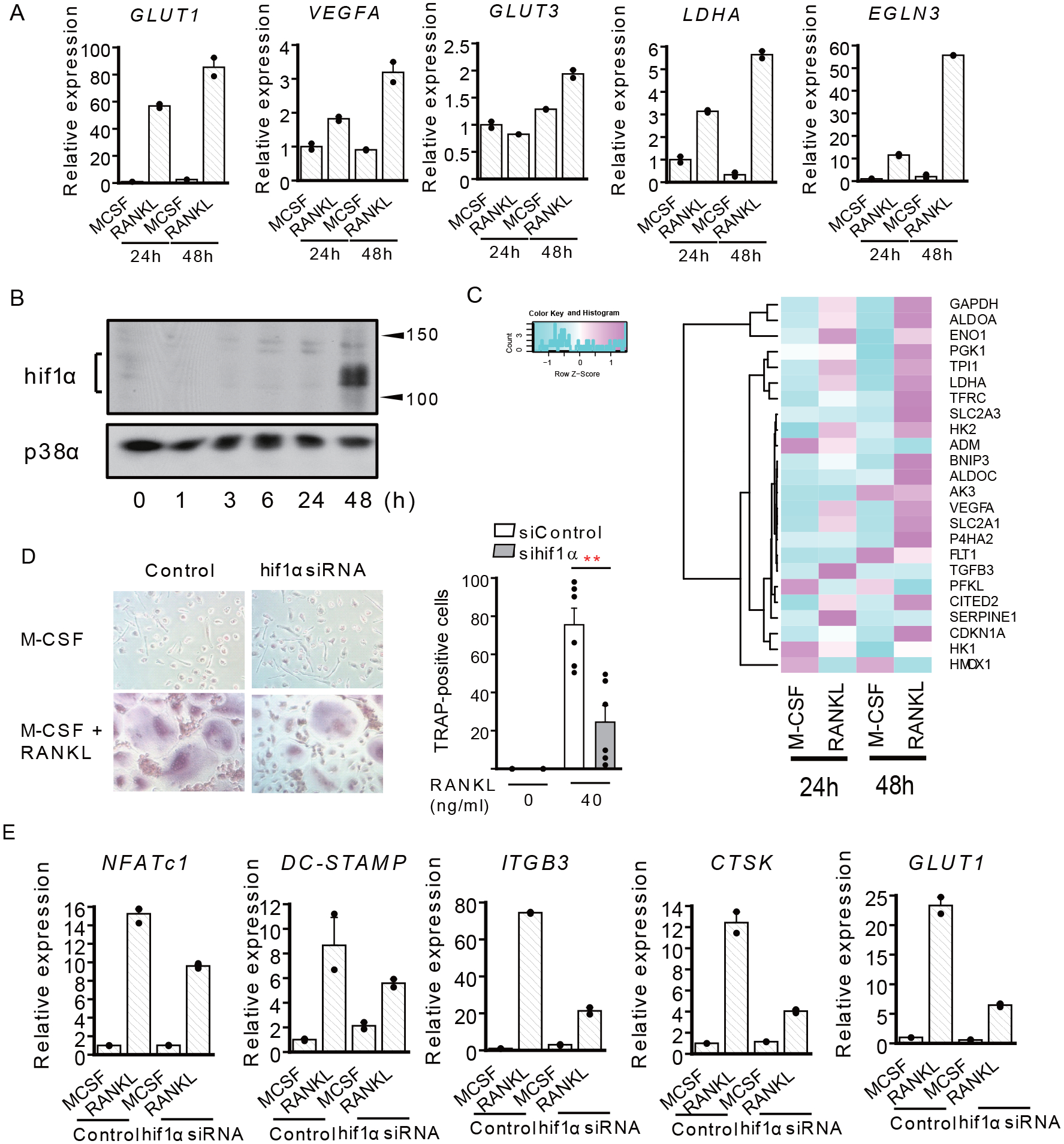
A, RT-qPCR of HIF-1α target gene expression relative to TBP. Data show the mean ± SEM of duplicates from one representative experiment from three different donors. B, Immunoblot of whole-cell lysates using HIF-1α and p38 antibodies. OCPs were stimulated with RANKL for the indicated hours. Representative blot from five experiments with different donors is shown. C, Heatmap of RNA-seq FPKM values for HIF-1α target genes. RNA-seq data deposited at GEO database (GSE99987) were used for the analysis. D, Human CD14-positive monocytes were nucleofected with the control or HIF-1α-specific siRNA and osteoclasts were generated. TRAP-positive, multinucleated cells were counted in triplicate. Left panel, representative results from six independent experiments. Right panel, data are shown as mean ± SEM from aggregate data from six independent donors. E, RT-qPCR of osteoclast-related genes expression, relative to TBP. Data are shown as mean ± SEM of duplicates of one representative experiment from three different donors.
** p < 0.01 by Tukey-Kramer test.
HIF-1α mRNA levels remained unchanged at 24 h of RANKL stimulation and increased slightly at 48 h (Supplementary Figure 5). Surprisingly, however, immunoblotting showed that the expression of HIF-1α protein could be detected at 48 h after RANKL stimulation in normoxia, although it could not be observed within 24 h of RANKL stimulation (Figure 4B). Concomitantly, expression of HIF-1α target genes, including VEGFA, PHD3, and LDHA, as well as GLUT1 and GLUT3, was induced 48 h after RANKL treatment (Figures 4A, C). Furthermore, siRNA-mediated knockdown of HIF-1α suppressed osteoclastogenesis and the expression of osteoclast-related genes including TRAP, CTSK, ITGB3, and DC-STAMP in normoxia (Figures 4D, E). These results suggest that RANKL-induced osteoclastogenesis is metabolically regulated by HIF-1α, even in normoxia.
LSD1 promotes glycolysis via HIF-1α.
To gain additional insights into the mechanism by which LSD1 regulates metabolism through the HIF-1α pathway, we analyzed the expression of RANKL-induced HIF-1α, whose protein expression is known to be stabilized by demethylation activity of LSD1 in other cell systems (19, 20). LSD1 inhibitors, SP2509 and RN1, significantly suppressed RANKL-induced HIF-1α protein expression (Figure 5A), while SP2509 and silencing of LSD1 minimally decreased HIF-1α mRNA (Supplementary Figures 6A, B). Concordantly, HIF-1α target genes, which were upregulated by RANKL, were suppressed by SP2509 and siRNA-mediated silencing of LSD1 (Figures 5B, C). RANKL induced the expression of PHD2, which induced HIF-1α degradation by mediating HIF-1α hydroxylation; however, SP2509 did not inhibit RANKL-induced expression of PHD2 (Supplementary Figure 6C). Finally, we evaluated the level of glycolysis after RANKL-stimulation. SP2509 significantly suppressed glycolysis and glycolytic capacity (Figures 5D, E). These results suggest that LSD1 regulates osteoclast differentiation by controlling glycolysis via stabilizing HIF-1α in addition to E2F1 (Figure 5F).
Figure 5. LSD1 promotes glycolysis via HIF-1α.
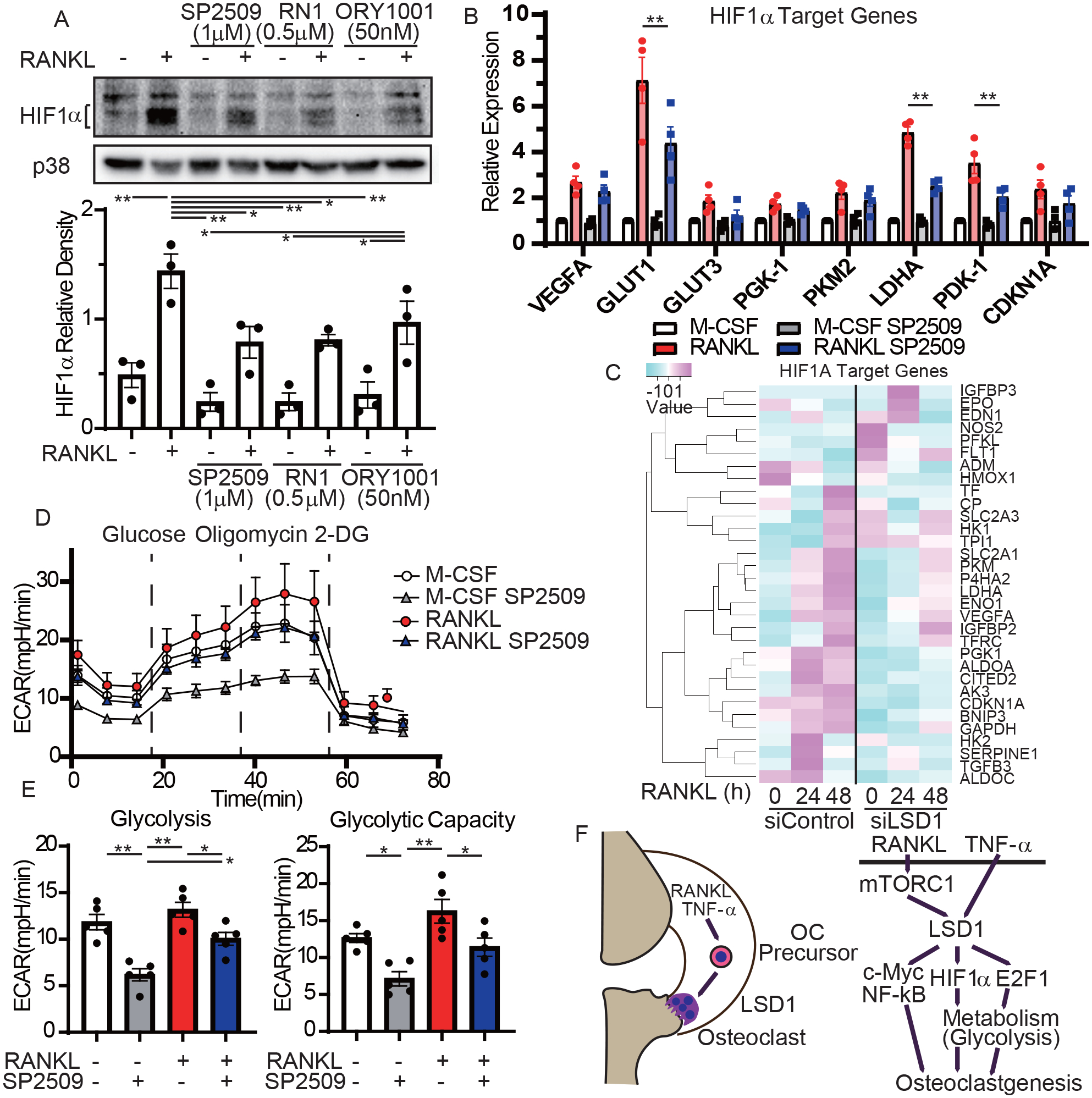
A, Immunoblot of whole-cell lysates using HIF-1α and p38 antibodies. Human CD14-positive monocytes were cultured with M-CSF and LSD1 inhibitors, SP-2509, RN1, or ORY 1001. Cells were treated with RANKL for 48 h. Representative blots (upper) from three different donors and densitometric quantitation are shown. B, RT-qPCR of HIF-1α target gene expression relative to TBP. Data are shown as mean ± SEM from aggregate data of four independent donors. C, The heatmap shows RNA-seq gene expressions of human OCPs transfected with siRNA of LSD1 or scrambled control treated with RANKL for the indicated hours. D, E, The extracellular acidification rate (ECAR) was recorded. The ECAR was normalized to the relative cell counts. Representative time course data (left panel) and assessment of glycolysis and glycolytic capacity (n = 5). F, Schematic diagram of LSD1-mediated pathways during osteoclastogenesis. All data are shown as mean ± SEM. * p < 0.05 and ** p < 0.01 by Tukey-Kramer test.
LSD1 inhibition suppressed osteoclastogenesis in hypoxia.
In addition to osteoclastogenesis in normoxia, we confirmed that LSD1 inhibition by SP2509 suppressed osteoclast differentiation in hypoxia (Supplementary Figure 7A). Suppression of LSD1 by SP2509 (1μM) did not strongly suppress the mRNA level of NFATc1 (Figure 1B, Supplementary Figure 3B); however, RANKL and hypoxia induction of the HIF-1α target genes was effectively suppressed by SP2509 (Supplementary Figure 7C). These results suggest that LSD1 plays a metabolically important role in hypoxic enhancement of osteoclastogenesis. Furthermore, HIF-1α is known to promote bone resorption even under normoxia (11). We confirmed that siRNA-mediated knockdown of LSD1 inhibited bone resorption and HIF-1α downregulation (Supplementary Figure 7D), suggesting that LSD1 could also promote bone resorption.
LSD1 is a potential therapeutic target for pathological bone resorption.
We investigated whether LSD1 could be a potential therapeutic target for pathological bone resorption. Next, we tested the relationship between LSD1 allelic variants that affect gene expression and bone loss in RA patients. We obtained eQTL associations showing p values less than 0.05 with HIF-1α and LSD1 expression from the data based on a previously published large eQTL study using a healthy donor (21). A total of four SNPs (rs646193, rs2092933, rs12137132, and rs10917219) were associated with LSD1 expression; the first two were observed in CD4-positive cells, while the latter were observed in monocytes. Three single nucleotide polymorphisms (SNPs) (rs912619, rs2252267, and rs4902069) were associated with HIF-1α expression: the first two in B cells and last in peripheral blood. We could not find a significant relationship between LSD1 allelic variants and bone erosion in RA patients. However, among the SNPs with cis eQTL association with HIF-1α, allelic variant rs4902069 (G) was significantly associated with higher expression of HIF-1α in healthy controls (p = 0.0017, Figure 6A). It was also associated with increased bone erosion of the hand joints in RA patients (p = 0.01, Figure 6B).
Figure 6. HIF-1α and LSD1 positively affect bone erosion in inflammatory arthritis.

A, Cis-expression quantitative trait locus (eQTL) association of rs4902069 (AA, AG, GG) and HIF-1α expression in Japanese healthy donor. B, vdH-modified total Sharp score (mTSS) of hand joints of RA patients with the three genotypes for rs4902069. C, D, C57BL/6N male mice were fed a control diet or a low-Ca (less than 0.01%) diet and were treated with DMSO or SP2509. C, μCT analysis of distal femurs. n = 10 from three independent experiments. D, TRAP staining of histological sections from distal femur and histomorphometric analysis. n = 8 from two experiments. E, F, Arthritis was induced in SKG mice with mannan, and the mice were treated with DMSO or SP2509. E, Time course of arthritis score and joint swelling. F, TRAP staining of histological sections from calcaneocuboid and tarsometatarsal joints. n = 8 from two experiments. G, H, TNF-induced supracalvarial osteolysis model. TRAP staining of histological sections of calvaria. n =10 from two experiments. All data are shown as mean ± SEM. * p < 0.05 and ** p < 0.01 by a 2-tailed, unpaired t-test (F, H) and Tukey-Kramer test (C, D).
Next, we examined the effect of LSD1 on accelerated bone loss using a low-calcium diet. Microcomputed tomography (μCT) analysis revealed that a low-calcium diet decreased bone mass and that LSD1 inhibition by SP2509 significantly suppressed the low calcium-induced bone loss relative to that in mice treated with dimethyl sulfoxide (DMSO) (Figure 6C). Histomorphometry showed that SP2509 decreased osteoclast number and surface area, supporting that LSD1 is crucial for osteoclastogenesis and pathological bone resorption (Figure 6D).
Finally, we tested the effect of LSD1 on inflammatory bone resorption. Mannan-induced arthritis in ZAP70-mutated SKG mice, which developed T cell-mediated autoimmune arthritis, clinically and immunologically resembled RA in humans (22). LSD1 inhibition by SP2509 treatment resulted in a minor decrease in arthritis score that did not reach statistical significance (Figure 6E). LSD1 inhibition significantly suppressed osteoclast number in periarticular bone (Figure 6F), and inhibited the decrease in the bone volume of the calcaneus in an SKG arthritis model without affecting LSD1 expression (Supplementary Figures 8A–D).
In the TNF-induced supracalvarial osteolysis model, SP2509 also strongly inhibited osteoclastogenesis and associated bone erosion in vivo (Figure 6G, Supplementary Figure 8E). Histomorphometric analysis showed decreased osteoclast numbers and surface area (osteoclast surface area per bone surface (OcS/BS) and osteoclast numbers per bone surface (NOc/BS), respectively (Figure 6H).
These results suggest that LSD1 is a potential therapeutic target for metabolically energy demanding pathological bone resorption.
DISCUSSION
The importance of functions and drivers of metabolic reprogramming, especially glycolysis, during osteoclastogenesis has been highlighted; however, the process of metabolic reprogramming in osteoclastogenesis is still not completed understood (12, 13). We investigated the mechanisms by which LSD1 mediates osteoclastogenesis. In this study, we show that RANKL-induced LSD1 stabilizes the HIF-1α protein even in normoxia. LSD1 mediates glycolysis along with HIF-1α and E2F1. LSD1 inhibition suppressed energy-demanding pathological bone resorption.
In RA synovium, synovial proliferation and leukocyte extravasation outstrip the oxygen supply and create a hypoxic microenvironment (15). Abnormal cellular metabolism and mitochondrial dysfunction increase production of reactive oxygen species and worsen the inflammation. Hypoxia is also known to enhance osteoclast differentiation (14). It is noteworthy hypoxic response is involved in osteoclast differentiation even under normoxic condition and LSD1 integrates its response.
HIF-1α, which increases glycolysis and decreases mitochondrial function in response to hypoxia stimuli, is unstable in normoxia. However, stimulation with cytokines, growth factors, and vascular hormones can also lead to the induction and activation of HIF-1α in different cell types (23–26). These factors mostly regulate HIF-1α at the transcriptional, post-transcriptional, or translational levels (27, 28). We showed that RANKL stabilized HIF-1α protein under normoxia and HIF-1α was important for human osteoclastogenesis even under normoxia. Because of the complexity and difficulty in detecting the protein expression of HIF-1α, HIF-1α depleted cells tend to be used in normoxia without showing protein expression and the mechanism of stabilization of HIF-1α. A recent study also demonstrated that metabolic processes are regulated by HIF-1α even in normoxia using HIF-1α-depleted neuroblastoma (NB) cells (29), without confirming the expression of HIF-1α protein in normoxia. Although we showed that the stabilization of HIF-1α protein under normoxia during osteoclastogenesis was partly due to its modification by LSD1, HIF-1α protein expression in normoxia was also shown to be regulated by the increased expression of HIF-1α mRNA and mTOR activity (27). RANKL could also induce the expression of HIF-1α mRNA and increase mTOR activity in our model. Although it was not investigated in this study, it has been reported that a non-hypoxic simulator regulates HIF-1α expression through microRNAs (28). Whether the RANKL-LSD1 axis stabilizes HIF-1α via microRNAs needs to be investigated in further study.
In this study, LSD1 was induced via the RANKL-mTOR pathway, resulting in the induction of the HIF-1α and E2F1 pathways. We previously demonstrated the importance of RANKL-mediated induction of anabolic, cell-cycle, and E2F-mediated pathways using non-proliferating human OCPs: the effects of RANKL on cell proliferation and energy production using proliferating mouse OCPs are difficult to distinguish (14). E2F target genes regulate many anabolic pathway genes. Human OCPs use the cell cycle pathway to prepare for the high energy demand for terminal differentiation, just as proliferating cells utilize the cell cycle pathway to prepare the energy required for cell division. Recent analysis of stepwise events during mouse osteoclastogenesis using single-cell RNA-seq supports this idea (30). This study provides new insights into how RANKL stimulates metabolic reprogramming mediating HIF-1α and E2F1.
LSD1 targets both histone and non-histone proteins with demethylase enzymatic activity (16). In HIF-1α, Set9 histone methyltransferase induces HIF-1α methylation at lysine (K) 32 or 391, and LSD1 reverses this process, which protects HIF-1α against ubiquitin-mediated protein degradation (19, 31). LSD1 also inhibits HIF-1α hydroxylation by PHD2 in cancer cell lines. SP2509 blocks LSD1 methylase function allosterically, that is, SP2509 interrupts the binding of the partner protein by binding with the H3 pocket within LSD1. RN1 and ORY1001 are trans-2phenylcyclopropylamine (2-PCPA) derivatives that inhibit LSD1 methylase activity as covalent adducts through a flavin loop, which is a cofactor during catalysis of demethylation (32, 33). Through these processes, RANKL-induced HIF-1α protein stabilization is considered to be inhibited by these inhibitors (Figure 5A).
The inhibition of LSD1 successfully suppressed bone loss via a low-calcium diet. We used SP2509 at a dose that was used in a previous study (34). However, we still need to study whether SP2509 at this dose could effectively suppress pathological bone resorption in vivo or might have some toxic effects. Furthermore, inhibition of LSD1 was shown to increase BMP2, WNT7B, and RUNX2 expression in osteoblasts, and mice lacking LSD1 in mesenchymal cells exhibit increased bone mass (35, 36). Treatment with an LSD1 inhibitor for bone loss may have dual effects on the bone via an increase in bone formation and reduction in bone resorption.
SP2509 did not effectively suppress inflammatory arthritis. Elevated LSD1 expression has been shown in various cancers and is closely related to many cellular effects, including malignant transformation, epithelial-mesenchymal transition, and cell proliferation and differentiation (33). Thus, it is considered a promising therapeutic target for malignant tumors. It is reasonable to think that SP2509, an LSD1 inhibitor, suppresses inflammatory arthritis because it suppresses cell cycle and metabolic pathways, which are essential for the proliferation of inflammatory synovial cells (37). In fact, disease severity was alleviated in collagen-induced arthritis model mice when LSD1 was knocked down (38). However, this effect was not confirmed in this study. The following are the possible reasons: 1. inflammation induced in SKG mice was fulminant or 2. SP2509 was insufficient to control arthritis. However, bone erosion was suppressed in SKG mice, and LSD1 remains a promising target that can alleviate both arthritis and bone erosion by changing the type and dose of inhibitors.
One of the limitations of this study is the lack of epigenetic analysis. Because LSD1 mediates transcriptional repression by demethylation of H3K4me1/2 and transcriptional activation by demethylation of H3K9me1/2, determining which genes are epigenetically regulated in RANKL-induced osteoclast differentiation may be a challenging task by histone modification analysis, and it is reasonable to investigate the LSD1-regulated pathway by siRNA silencing. Another limitation is that the specificity and toxicity of SP2509 are not shown. It has been shown to exhibit a low activity toward D-lactate dehydrogenase, glucose oxidase, cytochrome P450s (CYPs), and human ether-a-go-go-related gene (hERG) (39). A transcriptomic analysis using Ewing sarcoma cell line also showed that the SP2509-regulated genes mimicked those regulated by LSD1 knockdown (40). However, further studies are still needed before the application of SP2509 for the treatment of human diseases.
In summary, we identified the integrative role of LSD1 in osteoclastogenesis. LSD1 stabilizes RANKL-induced HIF-1α protein even in normoxia, which promotes osteoclast differentiation via an anabolic pathway. During osteoclastogenesis, LSD1 is induced via the RANKL-induced mTOR pathway. TNF-α also induces LSD1 expression. Inhibition of LSD1 effectively suppresses osteoclastogenesis and bone loss in an accelerated osteoporosis model and in inflammatory osteolysis and arthritis models. These findings indicate that LSD1 plays a crucial role in osteoclastogenesis under inflammatory energy-demanding challenging conditions and will provide new therapeutic strategies that target osteoclasts against inflammatory arthritis including RA.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Editage (www.editage.com) for English language editing.
FUNDINGS
This work was supported by the KANAE Foundation for the Promotion of Medical Science, Takeda Science Foundation, Inamori Foundation, JSPS KAKENHI (Grant Number 17H06816, 18H02926) to KM. Research conducted in the USA at Hospital for Special Surgery was supported by NIH grant DE019420.
CONFLICTS OF INTERESTS
The Department of Advanced Medicine for Rheumatic Diseases is supported by Nagahama City, Shiga, Japan, Toyooka City, Hyogo, Japan and five pharmaceutical companies (Mitsubishi Tanabe Pharma Co., Chugai Pharmaceutical Co. Ltd, UCB Japan Co. Ltd, Asahi Kasei Pharma Corp., and AYUMI Pharmaceutical Co.). The KURAMA cohort study was supported by a grant from Daiichi Sankyo Co. Ltd. This study was conducted as an investigator initiated the study. These companies had no role in the design of the study, the collection or analysis of data, the writing of the manuscript, or the decision to submit the manuscript for publication.
All authors have declared no conflicts of interests.
DATA AVAILABILITY
RNA-seq data for this project have been deposited at NCBI’s Gene Expression Omnibus (GEO) with GSE number GSE166559.
REFERENCES
- 1.Mbalaviele G, Novack DV, Schett G, Teitelbaum SL. Inflammatory osteolysis: a conspiracy against bone. The Journal of clinical investigation. 2017;127(6):2030–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh MC, Takegahara N, Kim H, Choi Y. Updating osteoimmunology: regulation of bone cells by innate and adaptive immunity. Nature reviews Rheumatology. 2018;14(3):146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black DM, Rosen CJ. Clinical Practice. Postmenopausal Osteoporosis. The New England journal of medicine. 2016;374(3):254–62. [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi T, Tanaka Y, Soen S, Yamanaka H, Yoneda T, Tanaka S, et al. Effects of the anti-RANKL antibody denosumab on joint structural damage in patients with rheumatoid arthritis treated with conventional synthetic disease-modifying antirheumatic drugs (DESIRABLE study): a randomised, double-blind, placebo-controlled phase 3 trial. Ann Rheum Dis. 2019;78(7):899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsourdi E, Zillikens MC, Meier C, Body JJ, Gonzalez Rodriguez E, Anastasilakis AD, et al. Fracture risk and management of discontinuation of denosumab therapy: a systematic review and position statement by ECTS. The Journal of clinical endocrinology and metabolism. 2020. [DOI] [PubMed] [Google Scholar]
- 6.Tsukasaki M, Takayanagi H. Osteoimmunology: evolving concepts in bone-immune interactions in health and disease. Nature reviews Immunology. 2019;19(10):626–42. [DOI] [PubMed] [Google Scholar]
- 7.Bae S, Lee MJ, Mun SH, Giannopoulou EG, Yong-Gonzalez V, Cross JR, et al. MYC-dependent oxidative metabolism regulates osteoclastogenesis via nuclear receptor ERRalpha. The Journal of clinical investigation. 2017;127(7):2555–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishikawa K, Iwamoto Y, Kobayashi Y, Katsuoka F, Kawaguchi S, Tsujita T, et al. DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S-adenosylmethionine-producing metabolic pathway. Nature medicine. 2015;21(3):281–7. [DOI] [PubMed] [Google Scholar]
- 9.Lemma S, Sboarina M, Porporato PE, Zini N, Sonveaux P, Di Pompo G, et al. Energy metabolism in osteoclast formation and activity. The international journal of biochemistry & cell biology. 2016;79:168–80. [DOI] [PubMed] [Google Scholar]
- 10.Jin Z, Wei W, Yang M, Du Y, Wan Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell metabolism. 2014;20(3):483–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Indo Y, Takeshita S, Ishii KA, Hoshii T, Aburatani H, Hirao A, et al. Metabolic regulation of osteoclast differentiation and function. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2013;28(11):2392–9. [DOI] [PubMed] [Google Scholar]
- 12.Park-Min KH. Metabolic reprogramming in osteoclasts. Seminars in immunopathology. 2019;41(5):565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivashkiv LB. Metabolic-epigenetic coupling in osteoclast differentiation. Nature medicine. 2015;21(3):212–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murata K, Fang C, Terao C, Giannopoulou EG, Lee YJ, Lee MJ, et al. Hypoxia-Sensitive COMMD1 Integrates Signaling and Cellular Metabolism in Human Macrophages and Suppresses Osteoclastogenesis. Immunity. 2017;47(1):66–79 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41(4):518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu F, Lin Y, Wang Z, Wu X, Ye Z, Wang Y, et al. Biological roles of LSD1 beyond its demethylase activity. Cellular and molecular life sciences : CMLS. 2020;77(17):3341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitaura H, Zhou P, Kim HJ, Novack DV, Ross FP, Teitelbaum SL. M-CSF mediates TNF-induced inflammatory osteolysis. The Journal of clinical investigation. 2005;115(12):3418–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sorensen MG, Henriksen K, Schaller S, Henriksen DB, Nielsen FC, Dziegiel MH, et al. Characterization of osteoclasts derived from CD14+ monocytes isolated from peripheral blood. Journal of bone and mineral metabolism. 2007;25(1):36–45. [DOI] [PubMed] [Google Scholar]
- 19.Lee JY, Park JH, Choi HJ, Won HY, Joo HS, Shin DH, et al. LSD1 demethylates HIF1alpha to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene. 2017;36(39):5512–21. [DOI] [PubMed] [Google Scholar]
- 20.Sacca CD, Gorini F, Ambrosio S, Amente S, Faicchia D, Matarese G, et al. Inhibition of lysine-specific demethylase LSD1 induces senescence in Glioblastoma cells through a HIF-1alpha-dependent pathway. Biochimica et biophysica acta Gene regulatory mechanisms. 2019;1862(5):535–46. [DOI] [PubMed] [Google Scholar]
- 21.Ishigaki K, Kochi Y, Suzuki A, Tsuchida Y, Tsuchiya H, Sumitomo S, et al. Polygenic burdens on cell-specific pathways underlie the risk of rheumatoid arthritis. Nature genetics. 2017;49(7):1120–5. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi Y, Hirota K, Sakaguchi S. Impaired T cell receptor signaling and development of T cell-mediated autoimmune arthritis. Immunological reviews. 2020;294(1):164–76. [DOI] [PubMed] [Google Scholar]
- 23.Dery MA, Michaud MD, Richard DE. Hypoxia-inducible factor 1: regulation by hypoxic and non-hypoxic activators. The international journal of biochemistry & cell biology. 2005;37(3):535–40. [DOI] [PubMed] [Google Scholar]
- 24.McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, et al. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 alpha. Blood. 2012;120(15):3118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. The Journal of experimental medicine. 2012;209(13):2441–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453(7196):807–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayashi Y, Yokota A, Harada H, Huang G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1alpha in cancer. Cancer science. 2019;110(5):1510–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuschel A, Simon P, Tug S. Functional regulation of HIF-1alpha under normoxia--is there more than post-translational regulation? Journal of cellular physiology. 2012;227(2):514–24. [DOI] [PubMed] [Google Scholar]
- 29.Cimmino F, Avitabile M, Lasorsa VA, Montella A, Pezone L, Cantalupo S, et al. HIF-1 transcription activity: HIF1A driven response in normoxia and in hypoxia. BMC medical genetics. 2019;20(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsukasaki M, Huynh NC, Okamoto K, Muro R, Terashima A, Kurikawa Y, et al. Stepwise cell fate decision pathways during osteoclastogenesis at single-cell resolution. Nature metabolism. 2020;2(12):1382–90. [DOI] [PubMed] [Google Scholar]
- 31.Kim Y, Nam HJ, Lee J, Park DY, Kim C, Yu YS, et al. Methylation-dependent regulation of HIF-1alpha stability restricts retinal and tumour angiogenesis. Nature communications. 2016;7:10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng YC, Ma J, Wang Z, Li J, Jiang B, Zhou W, et al. A Systematic Review of Histone Lysine-Specific Demethylase 1 and Its Inhibitors. Medicinal research reviews. 2015;35(5):1032–71. [DOI] [PubMed] [Google Scholar]
- 33.Fang Y, Liao G, Yu B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. Journal of hematology & oncology. 2019;12(1):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiskus W, Sharma S, Shah B, Portier BP, Devaraj SG, Liu K, et al. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia. 2014;28(11):2155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munehira Y, Yang Z, Gozani O. Systematic Analysis of Known and Candidate Lysine Demethylases in the Regulation of Myoblast Differentiation. Journal of molecular biology. 2017;429(13):2055–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun J, Ermann J, Niu N, Yan G, Yang Y, Shi Y, et al. Histone demethylase LSD1 regulates bone mass by controlling WNT7B and BMP2 signaling in osteoblasts. Bone research. 2018;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falconer J, Murphy AN, Young SP, Clark AR, Tiziani S, Guma M, et al. Review: Synovial Cell Metabolism and Chronic Inflammation in Rheumatoid Arthritis. Arthritis & rheumatology (Hoboken, NJ). 2018;70(7):984–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu W, Fan JB, Xu DW, Zhu XH, Yi H, Cui SY, et al. Knockdown of LSD1 ameliorates the severity of rheumatoid arthritis and decreases the function of CD4 T cells in mouse models. International journal of clinical and experimental pathology. 2018;11(1):333–41. [PMC free article] [PubMed] [Google Scholar]
- 39.Sorna V, Theisen ER, Stephens B, Warner SL, Bearss DJ, Vankayalapati H, et al. High-throughput virtual screening identifies novel N’-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. Journal of medicinal chemistry. 2013;56(23):9496–508. [DOI] [PubMed] [Google Scholar]
- 40.Pishas KI, Drenberg CD, Taslim C, Theisen ER, Johnson KM, Saund RS, et al. Therapeutic Targeting of KDM1A/LSD1 in Ewing Sarcoma with SP-2509 Engages the Endoplasmic Reticulum Stress Response. Molecular cancer therapeutics. 2018;17(9):1902–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data for this project have been deposited at NCBI’s Gene Expression Omnibus (GEO) with GSE number GSE166559.