

Abstract
Histone modifications play a crucial role in the integration of environmental signals to mediate gene expression outcomes. However, genetic and pharmacological interference often causes pleiotropic effects, creating the urgent need for methods that allow locus-specific manipulation of histone modifications, preferably in an inducible manner. Here, we report an inducible system for epigenome editing in Arabidopsis (Arabidopsis thaliana) using a heat-inducible dCas9 to target a JUMONJI (JMJ) histone H3 lysine 4 (H3K4) demethylase domain to a locus of interest. As a model locus, we target the ASCORBATE PEROXIDASE2 (APX2) gene that shows transcriptional memory after heat stress (HS), correlating with H3K4 hyper-methylation. We show that dCas9–JMJ is targeted in a HS-dependent manner to APX2 and that the HS-induced overaccumulation of H3K4 trimethylation (H3K4me3) decreases when dCas9–JMJ binds to the locus. This results in reduced HS-mediated transcriptional memory at the APX2 locus. Targeting an enzymatically inactive JMJ protein in an analogous manner affected transcriptional memory less than the active JMJ protein; however, we still observed a decrease in H3K4 methylation levels. Thus, the inducible targeting of dCas9–JMJ to APX2 was effective in reducing H3K4 methylation levels. As the effect was not fully dependent on enzyme activity of the eraser domain, the dCas9–JMJ fusion protein may act in part independently of its demethylase activity. This underlines the need for caution in the design and interpretation of epigenome editing studies. We expect our versatile inducible epigenome editing system to be especially useful for studying temporal dynamics of chromatin modifications.
A temperature-inducible epigenome editing system to knock down histone methylation can be used to study the role of histone H3K4 methylation during heat stress memory in Arabidopsis.
Introduction
Recent years have seen great progress in our knowledge of the distribution and regulation of histone modifications and their involvement in various plant processes including development, as well as short- and long-term adaptation to changing environments (Lämke and Bäurle, 2017; Xiao et al., 2017; Cheng et al., 2020; Ueda and Seki, 2020; Samo et al., 2021). While the dynamics of histone modifications are well characterized, it remains challenging to establish causal relationships between histone modifications and changes in gene expression or epigenetic states. Such analyses require the knockout of a specific histone modification, ideally in a locus-specific manner. In plants, this is often hampered by functional redundancy of writer/eraser enzymes and/or their lack of specificity (Cheng et al., 2020; Ueda and Seki, 2020). The known writer/eraser mutants affect many genes, causing pleiotropic effects that may be difficult to disentangle. For example, at least eight histone H3 lysine 4 (H3K4) methyltransferases (SET DOMAIN GROUP [SDG] 4, 8, 25; ARABIDOPSIS TRITHORAX [ATX] 1, 2, 3, 4, 5) have been described in Arabidopsis (Arabidopsis thaliana) (Cheng et al., 2020; Ueda and Seki, 2020). In addition, these enzymes are not necessarily specific toward histone H3K4, making genetic analysis and its interpretation difficult. Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9) provides a powerful method to selectively manipulate individual loci by targeting them with the help of specific single-guide RNAs (sgRNAs) (Doudna and Charpentier, 2014; Kumlehn et al., 2018). Targeting of heterologous proteins can be achieved by fusing an enzymatically inactive version of Cas9 (dCas9) with an effector protein of interest (Hilton et al., 2015; Kearns et al., 2015; Kwon et al., 2017; Lee et al., 2019; Papikian et al., 2019).
In plants, the CRISPR/dCas9-editing system has been previously used to manipulate DNA methylation at individual loci including the imprinted FLOWERING WAGENINGEN (FWA) locus by expressing DNA methyltransferases (Ghoshal et al., 2021; Liu et al., 2021) or the TEN–ELEVEN TRANSLOCATION1 (TET1) DNA demethylase (Gallego-Bartolomé et al., 2018). For histone modifications, it has been used to target the HISTONE ACETYLTRANSFERASE1 and the H3 lysine 9 methyltransferase KRYPTONITE to an abscisic acid-response gene (AREB1) and the flowering time gene FLOWERING LOCUS T, respectively (Lee et al., 2019; Roca Paixão et al., 2019). While moderate effects on gene expression have been observed in both cases, neither study provided evidence that the effect on gene expression was due to modification of histones at the locus of interest following the binding of the dCas9-effector fusion protein.
Moderate heat stress (HS) primes a plant to acquire thermotolerance and better withstand a subsequent HS (Bäurle, 2016; Ohama et al., 2017; Oberkofler et al., 2021). A short HS pulse induces the acquisition of thermotolerance, but also activates a lasting stress memory that is maintained for several days after the HS has subsided (referred to as recovery phase). At the physiological level, this HS memory mediates enhanced survival upon a second HS that would be lethal to a naïve plant (Charng et al., 2007; Stief et al., 2014). At the molecular level, HS induces two types of transcriptional memory at partially overlapping sets of genes (Lämke et al., 2016; Liu et al., 2018); first, it induces sustained induction of gene expression that is actively maintained during several days of recovery at normal growth temperatures (Type I). Second, HS induces enhanced transcriptional re-activation upon a recurrent HS in certain genes, after their expression has returned to baseline levels during the recovery phase (Type II). In our experimental setup to analyze Type II memory, we refer to the first HS as priming HS (P) and to the second HS as triggering HS (T). Notably, P and T consist of an identical HS treatment (1 h at 37°C). Types I and II transcriptional memory require the HEAT SHOCK FACTOR A2 (HSFA2) transcription factor and correlate with enhanced H3K4 methylation (Charng et al., 2007; Lämke et al., 2016; Liu et al., 2018). Notably, the H3K4 hyper-methylation is maintained at elevated levels even after active transcription has subsided. The ASCORBATE PEROXIDASE2 (APX2) gene displays Type II transcriptional memory (Lämke et al., 2016). While the current model posits that HSFA2 recruits a histone methyltransferase that sets and maintains H3K4 hyper-methylation, the identity of this methyltransferase is still elusive. Although SDG25 and ATX1 have been implicated in HS-induced H3K4 trimethylation (H3K4me3), it is unclear, whether they target APX2 or other memory genes (Song et al., 2021). Thus, it remains an open question whether H3K4 methylation mediates the transcriptional memory or whether it is only correlated with the state of activated memory in Arabidopsis. In yeast and mammalian cells, H3K4 hyper-methylation marks recent transcriptional activity of a locus and may cause modified re-activation after a second stimulus (Ng et al., 2003; D’Urso et al., 2016). In plants, H3K4 hyper-methylation was also associated with somatic stress memory in response to dehydration stress and salt stress (Ding et al., 2012; Sani et al., 2013; Feng et al., 2016). Thus, we were curious to investigate the effects of perturbing H3K4 methylation at APX2 after HS.
Histone demethylation is an enzymatically mediated process, and two families of histone demethylases are present in eukaryotes (Dimitrova et al., 2015; Cheng et al., 2020). While LYSINE-SPECIFIC DEMETHYLASE1 (LSD1)-like amine oxidases only demethylate dimethylated and monomethylated lysines, JUMONJI (JMJ) demethylases are active on trimethylated, dimethylated, and monomethylated lysines (Dimitrova et al., 2015). In Arabidopsis, there are four LSD1-like genes that all act on H3K4me2/1 (LSD1-LIKE1 [LDL]1, LDL2, LDL3, FLOWERING LOCUSD) (Jiang et al., 2007; Liu et al., 2007; Ishihara et al., 2019), while the JMJ family consists of 21 members (Liu et al., 2010; Chen et al., 2011; Cheng et al., 2020). Typically, JMJ proteins show a high specificity toward the lysine that they demethylate (Ueda and Seki, 2020). Based on experimental and phylogenetic analysis, at least five JMJ proteins act on H3K4, namely JMJ14, JMJ15, JMJ16, JMJ17, and JMJ18 (Lu et al., 2010; Yang et al., 2012b, 2012a; Huang et al., 2019; Liu et al., 2019). These proteins are at least partially functionally redundant, rendering genetic analyses cumbersome. We selected the well-characterized JMJ18 as a demethylase for use in targeted epigenome editing (Yang et al., 2012a). To our knowledge, a histone demethylase has not been used in an epigenome editing experiment in plants. In animals, this has been done with an LSD family demethylase (Kearns et al., 2015), and with the H3K9 demethylase JMJD2 (Baumann et al., 2019). Targeting dCas9–JMJD2 together with a transcriptional activator was evaluated for its ability to enhance gene activation and found to have no substantial effect on target gene expression (Baumann et al., 2019). In addition, when targeted by a customized Zinc finger protein the H3K4me3 demethylase KDM5A/RBP2/JARID1A was able to reduce transcript levels of the targeted gene in a cancer cell line (Horton et al., 2016; Cano-Rodriguez et al., 2017). To create an inducible system for H3K4 demethylation, we expressed the dCas9–JMJ18 fusion under the control of a HS-inducible promoter.
In summary, we report epigenome editing by expressing a histone H3K4-specific demethylase in Arabidopsis to evaluate the role of H3K4 methylation in HS memory. We show that the effector construct binds to the targeted APX2 locus, where it decreases HS-induced transcriptional memory and H3K4 methylation. We highlight a need for careful design of controls, including enzyme-dead versions of the constructs, to be able to ascribe effects to the enzymatic activity of the targeted protein. Our HS-inducible epigenome editing system is widely applicable to address the role of epigenetic modifications for various processes such as stress responses and cell differentiation.
Results
Design and construction of inducible locus-specific epigenome editing for H3K4me demethylation
We have previously shown that HS-induced transcriptional memory is tightly correlated with H3K4me3 hyper-methylation at several loci including APX2 (Lämke et al., 2016; Liu et al., 2018). To establish whether H3K4 methylation is required for Type II transcriptional memory (enhanced re-induction), we sought to target an H3K4 demethylase specifically after HS to APX2. We first created a construct where the HS-inducible pHSP21 promoter drives expression of a fusion protein consisting of an inactive Cas9 (dCas9) and the catalytically active domain of JMJ18, referred to as JMJ in the following (Figure 1, A–C; Yang et al., 2012a; Stief et al., 2014; Hilton et al., 2015; Kearns et al., 2015). This domain of JMJ18 was previously shown to be sufficient to specifically demethylate histone H3K4me3/me2 (Yang et al., 2012a). To be able to test whether the catalytic activity of JMJ18 is necessary for any potential effects, we also generated a construct with a catalytically inactive JMJ18, dJMJ, where we mutated the highly conserved histidine 395 to alanine. In JMJ14, the corresponding point mutation (H397A) abolishes binding of the essential Fe(II)-cofactor and renders the enzyme catalytically inactive (Lu et al., 2010).
Figure 1.

Principle and design of JMJ epigenome editing constructs targeting APX2. Schematic representation of generated constructs for validation of sgRNA design by transient transactivation in N. benthamiana (A) and for generation of stable lines (B) in the pHEE401E vector backbone. Both types of constructs contain an sgRNA cassette, which is consisting of three repeats of a U6 promoter variant (light gray box) driving expression of three different sgRNA + scaffold sequence (purple arrow) followed by a U6 terminator variant (dark gray box). To validate sgRNA targeting efficiency, constitutively expressed 3xFlag-dCas9 was translationally fused to VP64 (A, light purple arrow), followed by the rbcS-E9 terminator sequence. To generate stable lines, 3xFlag-dCas9–(d)JMJ (B, orange arrow) was expressed under the HS-inducible pHSP21 promoter, followed by the rbcS-E9 terminator sequence. C, sgRNA (purple line)-mediated targeting of 3xFlag-dCas9–(d)JMJ (orange) to the promoter region of APX2 is expected to affect histone H3 lysine K4 demethylation (dashed arrow) and type II transcriptional memory of APX2 endogenous and pAPX2::LUC genes. Both genes show enhanced re-induction of transcription after repeated HS. Purple box, sgRNA-targeted region; blue insets, HSEs; gray box, untranslated region; black box; coding region exon; black line, DNA.
Three sgRNA cassettes consisting of three individual sgRNAs each were designed and evaluated (Figure 1, A and B; Table 1). SgRNA cassette a binds just upstream of the heat shock elements (HSEs) (Nover et al., 2001; Schramm et al., 2006) between –113- and –205-bp relative to the transcriptional start site (TSS) of APX2 (Table 1). Previous analyses indicated that HSFA2 and other HSFs bind to these HSEs (Schramm et al., 2006; Friedrich et al., 2021). Cassette b binds the promoter further upstream in a region that overlaps with the 5′-end of H3K4 hyper-methylation after HS (between –238- and –387-bp relative to the TSS). We chose this location because a previous epigenome editing study reported that effector complexes were most efficient when targeted to the boundaries of histone modification domains (Kwon et al., 2017). The third cassette (c) was designed by scrambling the cassette a sequences, resulting in no predicted target sequences in the genome.
Table 1.
Sequences and positions of sgRNAs relative to TSS of APX2
| Cassette | sgRNA sequences (5′–3′) | Position rel. to TSS (bp) |
|---|---|---|
| a | GGACGGTCATGATTTGTAGA | −113 |
| GACTATGGATGGTTTCTGGA | −160 | |
| GACACGTGGTGTGTATCTGT | −205 | |
| b | GAGTTGGTACAACATACTCG | −238 |
| GATCGAGCACAAAAACGTTA | −326 | |
| GAACCCTTAAGTCTTTTTGG | −387 | |
| c | GGTATAGGCATTAAGTGGTC | NA (Scrambled sequences of cassette a) |
| GGTAGGTCGTTAGGTATACT | ||
| GGTTGTATACTGTCGGCGTA |
Selection and validation of sgRNAs in a transient assay
To evaluate the efficiency of sgRNA cassettes a–c in targeting dCas9 to APX2, we took advantage of a transient transformation system, where we combined the pAPX2::LUCIFERASE (LUC) reporter gene, the respective sgRNA cassette, and a 35S::dCas9–VP64 effector construct (Figure 1A). VP64 is a transcriptional activator consisting of four tandem repeats of herpes simplex virus early transcriptional activator VP16 that mediates transcriptional activation if bound to the promoter of a gene (Sadowski et al., 1988; Beerli et al., 1998). SgRNA-targeted dCas9–VP64 has been successfully used for transcriptional activation of protein-coding and noncoding genes in Arabidopsis and Nicotiana benthamiana (Lowder et al., 2015). Targeting of the dCas9–VP64 activator to the APX2 promoter via the sgRNAs can be monitored by LUC-dependent bioluminescence. Three days after co-infiltrating the constructs into N. benthamiana leaves, LUC activity was analyzed (Figure 2). Cassette a promoted high LUC activity upon co-infiltration with the effector and reporter constructs. In contrast, co-infiltration of cassettes b (distal promoter) and c (scrambled), respectively, did not induce LUC activity above baseline levels (Figure 2), suggesting that these sgRNAs are not efficient in targeting dCas9–VP64 for transactivation. Thus, we selected cassette a for further studies.
Figure 2.
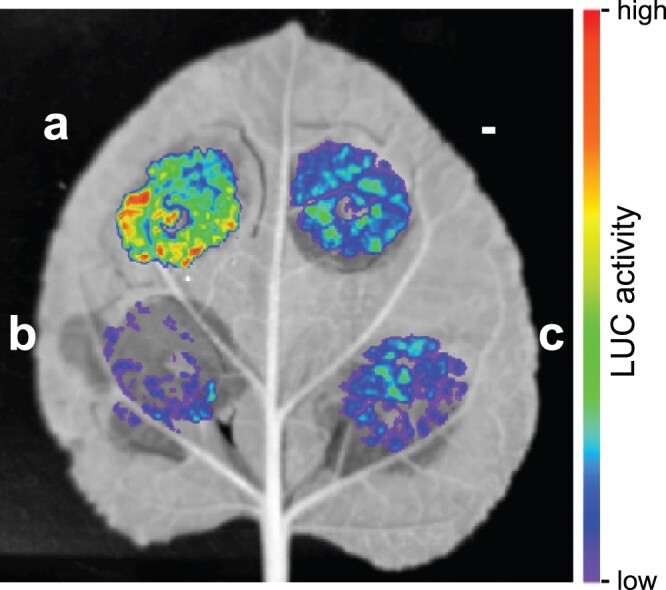
Validation of sgRNA targeting of pAPX2 in a transient transactivation assay in N. benthamiana. LUC-based assay to validate targeting efficiency of sgRNA cassettes to APX2600 bp promoter fragment. Luciferin-infiltrated N. benthamiana leaf 3 d after co-infiltration with pAPX2600 bp::LUC and p2x35S::3xFlag-dCas9-VP64 in combination with sgRNA cassette a (a), cassette b (b), or scrambled cassette (c), or without any cassette (–). High LUC activity in presence of cassette a indicates binding of 3xFlag-dCas9–VP64 to pAPX2600bp and transactivation of LUC expression by transcriptional activator VP64. The image is representative of three independent experiments.
dCas9–JMJ affects pAPX2::LUC expression after recurrent HS
We next generated transgenic plants that carried cassette a, pAPX2::LUC, and pHSP21::dCas9–JMJ or pHSP21::dCas9–dJMJ, respectively. We selected two representative lines from 20 primary transformants for detailed analysis of LUC expression after HS. In the absence of the dCas9–JMJ effector, expression of LUC after a priming HS (P d4, 1 h 37°C) was low and was slightly increased after 2 d of recovery (P d6, Figure 3, A and B). Similarly, expression of LUC after only a triggering HS (T, 1 h 37°C) was low. However, LUC expression was strongly increased in seedlings that received both the P and T treatments (P + T, Figure 3). For plants expressing pHSP21::dCas9–JMJ, LUC expression was similar to plants without the effector in either P or T conditions (P d4, T, Figure 3), and it was lower in seedlings that received P treatment and 2 d of recovery before imaging (P d6). The strongest difference, however, was observed after P + T treatment, where the dCas9–JMJ seedlings had similar LUC signal to plants that only received the T treatment, while plants lacking this construct showed a much stronger LUC activity (Figure 3B). LUC expression after a two-step acclimation (ACC) treatment, which induces more persistent type I memory than the P treatment described above, was reduced throughout the recovery phase (Supplemental Figure S1). Together, these findings suggest that dCas9–JMJ expression interferes with both, Types I and II transcriptional memory after HS.
Figure 3.

Effect of dCas9–(d)JMJ on type II transcriptional memory of pAPX2::LUC. A, Treatment scheme for LUC-based type II transcriptional memory assay. Four-day-old seedlings were either exposed to priming HS (P) on Day 4 and imaged on Day 4 (P d4) or on Day 6 (P d6), exposed to a triggering HS (T) on Day 6 and imaged on Day 6 (T), exposed to priming HS on Day 4 and a triggering HS on Day 6 (P + T) and imaged on Day 6 (P + T), or not exposed to any HS and imaged on Day 4 (N d4) or on Day 6 (N d6). B, LUC-based type II transcriptional memory assay shows reduced transcriptional activity of the pAPX2600bp::LUC reporter in dCas9-JMJ lines (top panel, JMJ #1 and JMJ #2) compared to the parent, as well as different behavior of two dCas9–dJMJ lines (middle and bottom, dJMJ #1 and dJMJ #2). For images within one panel the indicated genotypes were grown and treated side-by-side on the same set of plates. Images are representative of three independent experiments.
To investigate the function of the JMJ domain, we analyzed transgenic plants expressing the presumably inactive dCas9–dJMJ fusion. In contrast to the active JMJ, where all transformants showed a very similar LUC expression phenotype, the dJMJ transformants fell into two phenotypic classes with respect to this phenotype. One class was indistinguishable from the plants that did not carry any dCas9 construct (dJMJ#2), while the second class was more similar to dCas9–JMJ plants (dJMJ#1) (Figure 3B; Supplemental Figures S1 and S2A). Both dJMJ phenotypic classes were equally frequent among independent transformants (n = 8; four lines dJMJ#1-like, four lines dJMJ#2-like).
dCas9–JMJ affects APX2 expression after recurrent HS
We next sought to confirm and extend the findings of the bioluminescence assay at the transcript level of pAPX2::LUC and the endogenous APX2 gene by reverse transcription-quantitative PCR (RT-qPCR, Figure 4). Compared to plants without a dCas9 construct, LUC transcript levels after T treatment of naïve plants (Figure 4A) were slightly lower in dCas9–JMJ plants, and similar to the control in dCas9–dJMJ (Figure 4B). However, if the T treatment was preceded by a priming treatment (P) 2 d earlier (P + T), LUC transcript levels were strongly and significantly reduced in dCas9–JMJ. For dCas9–dJMJ plants, in contrast, LUC transcript levels were reduced in the dJMJ#1-like class, but not in the dJMJ#2-like class (Figure 4B; Supplemental Figure S2B). We next analyzed transcript levels of the endogenous APX2 gene (Figure 4B; Supplemental Figure S2B). All genotypes showed similar induction after T treatment; however, the dCas9–JMJ lines showed a strongly reduced expression after P + T treatment, suggesting that transcriptional memory is impaired in these plants. For the dCas9–dJMJ lines, the results were heterogeneous, but in full agreement with the LUC bioluminescence assay, with dCas9–dJMJ #1-class showing a reduced and #2-class showing parent-like APX2 expression levels. For the non-memory HS-inducible HSP101 gene, expression was similar in all lines. Thus, transcript analyses confirm that the dCas9–JMJ lines show decreased expression after P + T treatment, consistent with the notion that these lines are impaired in HS-induced transcriptional memory. We selected two dCas9–JMJ lines (JMJ#1, #2) and one representative lines of each dJMJ class (dJMJ#1, #2) for further analyses. The four selected dCas9–JMJ and dCas9–dJMJ lines had similar protein expression levels, as confirmed by immunoblotting protein extracts from HS-treated plants (Supplemental Figure S3).
Figure 4.
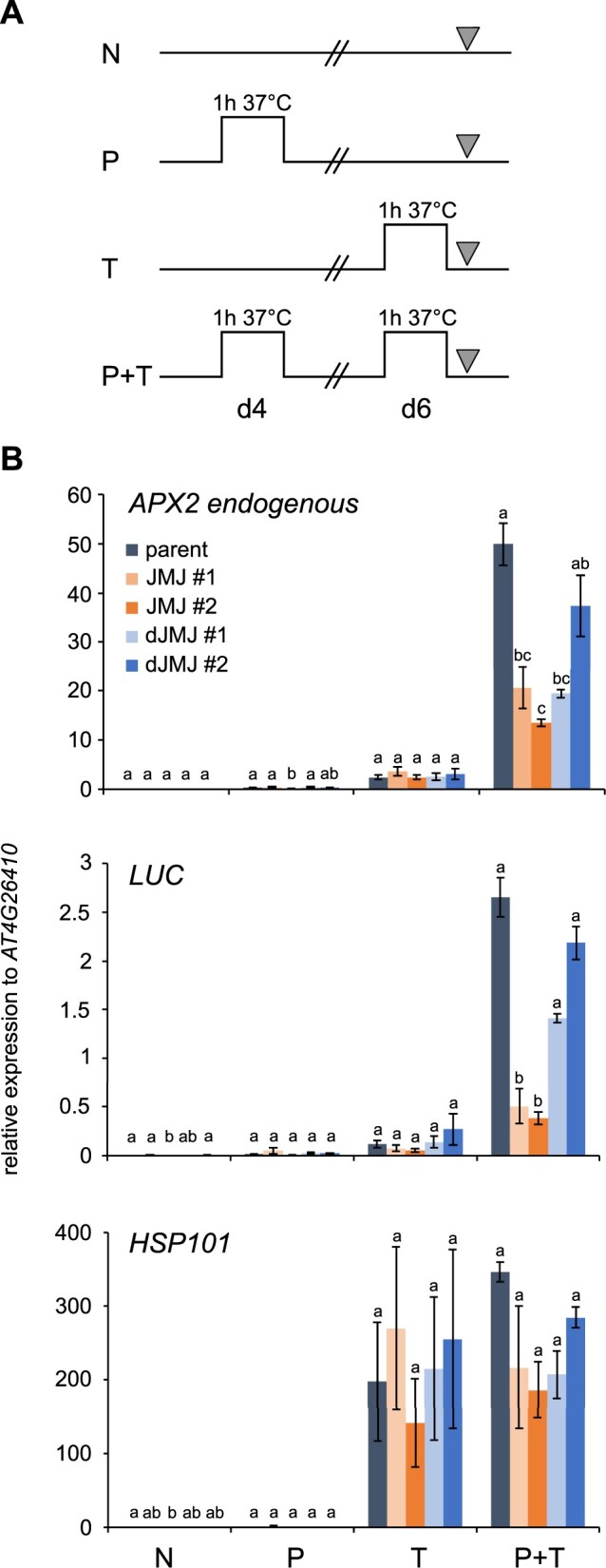
Type II transcriptional memory of endogenous APX2 and pAPX2::LUC after recurrent HS is reduced in dCas9–JMJ lines. A, Treatment scheme for RT-qPCR-based type II transcriptional memory assay. Four-day-old seedlings were either exposed to priming HS (P) on Day 4, exposed to a triggering HS (T) on Day 6, exposed to a priming and a triggering HS (P + T), or not exposed to any HS (N). All samples were taken on d6 corresponding to the end of the HS treatment. B, Transcript levels of endogenous APX2 (At3g09640), LUC (pAPX2600 bp::LUC), and HSP101 (At1g74310) in parent, two dCas9–JMJ lines (JMJ #1 and JMJ #2), and two dCas9-dJMJ lines (dJMJ #1 and dJMJ #2) as measured by RT-qPCR. Expression values are relative to At4g26410. Data are mean ± sem of three independent experiments. Transcript levels were statistically evaluated for all genotypes within each time point by Tukey’s HSD (P < 0.05). Genotypes are assigned one or more letters based on their statistical group. Genotypes sharing one letter are not significantly different.
The dCas9–(d)JMJ fusion proteins bind to the APX2 promoter
To test whether the reduced transcriptional memory is mediated by direct binding of dCas9–JMJ to the APX2 promoter, we performed chromatin immunoprecipitation (ChIP) using the Flag epitope in the dCas9–(d)JMJ fusion protein (Figure 1). We found enrichment of Flag-dCas9–(d)JMJ relative to the control amplicon outside the APX2 locus (amplicon 2) after heat treatment (P, T, P + T, Figure 5, A and B). However, dJMJ line #2 had a weaker signal compared to the others. All lines showed the strongest enrichment after P + T treatment. The enrichment peaked at the TSS-160 amplicon (4, Figure 5, A and B), overlapping with the location of the sgRNAs of cassette a. The signal decreased shortly after the TSS, consistent with the idea that dCas9–JMJ does not spread far beyond the targeted binding site. Very slight but significant binding was also found in N conditions, indicating a basal activity of pHSP21 under no-HS conditions. We did not find any enrichment at the ACTIN locus, which we included as a negative control (Figure 5B). Thus, dCas9–(d)JMJ is expressed and targeted to pAPX2.
Figure 5.

dCas9–(d)JMJ proteins bind APX2 after HS. A, Schematic representation of positions of analyzed amplicons by ChIP-qPCR. For the APX2 endogenous locus and the pAPX2600 bp::LUC transgene, the TSS is shown with a black arrow, UTRs are shown as gray boxes, exons are shown as black boxes, and the introns of the endogenous APX2 are shown as black lines. The sgRNA cassette a binding area is shown as purple box. Amplicons are shown as colored lines below the gene models. Pink amplicons are specific for APX2 endogenous, yellow amplicons are specific for pAPX2600 bp::LUC, and gray amplicons are not specific, that is, amplicons 3 and 4 amplify both the endogenous APX2 and the transgene. 2, APX2 – (3 kb upstream of At3g09640); 3, TSS −300 bp; 4, TSS −160 bp; 5, pAPX2-APX2; 6, APX2 CDS; 7, pAPX2-LUC; 8, LUC CDS. B, Occupancy of dCas9–JMJ and dCas9–dJMJ as determined by ChIP-qPCR. Sampling time points as described in Figure 4. Amplicons shown on the x-axis as indicated in (A), an amplicon of the ACTIN locus is shown as control. Enrichment normalized to Input. Data are mean ± sem of three independent experiments. Asterisks mark significant differences to amplicon 2 (APX2−) of the same genotype (unpaired two-sided t test, *P < 0.05, **P < 0.01).
dCas9–JMJ reduces H3K4me3 at APX2
We next studied the effect of dCas9–(d)JMJ expression on H3K4 methylation at APX2 using ChIP. H3K4me3 is induced after HS at APX2, and is maintained for at least 3 d, thus corresponding with the duration of the transcriptional memory (Lämke et al., 2016; Liu et al., 2018). Fully consistent with previous reports, H3K4me3 was enriched in P, T, and P + T conditions relative to N in the control line without the dCas9 fusion protein (Figures 4A and 6; e.g. amplicon 6, P versus N, P = 0.025, unpaired two-sided t test). The enrichment of H3K4me3 was strongest around the TSS and in the coding region (amplicons 5, 6 in Figure 6). As expected, after P (HS + 2 d recovery) the signal was stronger than after T (45 min after HS). The strongest enrichment, was observed in P + T conditions, which was significantly higher than the enrichment in T (amplicon 6, P + T versus T, P = 0.003, unpaired two-sided t test). Here, a reduction in H3K4me3 levels was observed in both dCas9–JMJ lines (#1, P = 0.004, unpaired two-sided t test). A slight reduction in H3K4me3 levels was also observed in the P condition, but not in the T condition (Figure 6). Thus, dCas9–JMJ is effective in reducing H3K4me3 levels at APX2 after HS. However, due to the use of a HS-inducible system, the demethylation function may not yet be active at the time when T was sampled. In contrast, we did not find any consistent changes at the ACTIN locus. For pAPX2::LUC, H3K4me3 was overall less enriched, however, we nevertheless observed a significant decrease in the enrichment around the TSS in dCas9–JMJ #1. For dCas9–dJMJ, the overall pattern was similar to dCas9–JMJ, but with nonsignificant reductions after P and P + T relative to the parent at the same treatment.
Figure 6.

Epigenome editing reduces H3K4me3 enrichment after HS. Enrichment of H3K4me3 as determined by ChIP-qPCR. Sampling time points as described in Figure 4A. Amplicons shown on the x-axis as indicated in Figure 5A, an amplicon of the ACTIN locus is shown as control. Enrichment is normalized to histone H3 and Input. Data are mean ± sem of three independent experiments. Asterisks mark significant differences to parent (unpaired two-sided t test, *P < 0.05, **P < 0.01).
Thus, we found that dCas9–JMJ reduced H3K4 methylation at APX2, strongly suggesting that the demethylase activity of JMJ18 causes reduced transcriptional memory. However, we cannot exclude that this is mediated by nonenzymatic effects of the JMJ fusion protein since similar phenotypes were observed with the catalytically inactive dJMJ18 domain.
Discussion
We have developed an inducible epigenome editing system for H3K4 methylation that can be easily adapted to other chromatin modifiers. We provide a framework of how the functional relevance of histone methylation may be tested in a locus-specific manner, thus avoiding pleiotropic effects of other approaches. The system may not only be useful for studying environmentally mediated chromatin modifications, but also for studying the role of histone methylation in the determination of cell fates during cell differentiation. For example, our system may be easily adapted to study the effect of removing the Polycomb mark H3K27me3 in flowering time or flower development (Xiao et al., 2017; Pelayo et al., 2021).
Our results are consistent with the notion that H3K4 hyper-methylation marks the chromatin of APX2 and mediates enhanced transcriptional re-induction upon a recurrent HS. Histone demethylation was detected in a region close to the binding site of cassette a. H3K4me3 typically peaks at the 5′-end of genes and we found the strongest enrichment just downstream of the TSS. Dynamic H3K4me3 has been observed in many contexts, however, its biological function is still not fully clear. The transient removal of this mark may shed light onto this function in an unprecedented manner.
We evaluated the efficiency of the sgRNA cassettes using a rapid transient N. benthamiana assay. Only one of the two sgRNA cassettes that we tested was efficient in targeting dCas9–VP64 to APX2. Thus, we focused further analysis on this cassette. However, we cannot exclude that the nonfunctional cassette was located simply too far from the TSS to provide efficient transactivation. Thus, this cassette might have been efficient in targeting dCas9–JMJ as the methylation is enriched also in the promoter (Figure 6). A previous study in mammalian cells found that epigenome editing of the p300 acetyl transferase was most efficient if the sgRNAs were located just outside the acetylated region (Kwon et al., 2017). Further studies will be needed to test whether this requirement is generally applicable also for histone (de-)methylation in plants.
The use of a HS-inducible promoter allowed us to minimize effects in the noninduced condition. The HSP21 promoter was previously characterized to have very low baseline expression and very high induction levels (Stief et al., 2014). It also shows sustained induction during the recovery phase, thus boosting protein levels during this phase. We did not precisely determine the lag time until functional dCas9–(d)JMJ protein is generated. However, the observed H3K4me3 dynamics (Figure 6) suggest that 45 min of recovery time may be too short to detect effects of the editing if plants were only triggered (T). On the other hand, this recovery time was sufficient if plants were primed before the triggering HS (P + T).
We included an inactive JMJ domain in our study to be able to assess whether the effects are linked to the demethylase activity of the JMJ domain. The mutated residue is required for binding of the Fe(II)-cofactor and was previously shown to be essential for enzymatic activity of JMJ14 (Lu et al., 2010). The inactive dJMJ construct reduced the transcriptional memory to some extent and caused a reduction in H3K4me3 levels. This may be explained by at least two different scenarios. First, it is possible that the dJMJ domain has a scaffolding function that is able to recruit endogenous H3K4 demethylases. While demethylase activity was shown for JMJ18 (Yang et al., 2012a), it is possible that it also fulfills structural functions for which the demethylase activity is dispensable. Second, the effect may be caused by steric hindrance or interference with the binding and functioning of gene-specific and general components of the transcriptional machinery. The size of the dCas9–JMJ fusion protein (dCas9 + (d)JMJ = 180 kDa) is roughly the same as that of the presumed trimeric HSF complex (Friedrich et al., 2021). It may be helpful for future studies to include a fusion of dCas9 to an unrelated protein of a similar size as the effector protein (here: (d)JMJ) to disentangle steric effects from effects caused by the active or inactive JMJ protein. Protein size is a highly relevant consideration for construct design, as several available epigenome editing systems (such as SunTag) target multiple effector proteins to one target site (Tanenbaum et al., 2014; Morita et al., 2016; Papikian et al., 2019). Steric effects may be minimal when using a system that mediates binding of only one effector protein per target site. Thus, our data indicate the need to carefully design epigenome editing studies, at least those in which mechanistic insights are desired in addition to effects on expression levels of the modified gene.
In summary, we developed an inducible epigenome editing system to modify H3K4 methylation, which can be easily adapted to other histone modifications. We report an efficient system to evaluate sgRNA cassettes and provide guidelines for study design. Our results demonstrate that H3K4 hyper-methylation is tightly connected with transcriptional memory of APX2. This versatile system is widely applicable to modify histone modification levels in both basic research questions and applied settings.
Materials and methods
Plant materials, growth conditions, and HS treatments
All Arabidopsis (A. thaliana) lines generated and used in this study harbor the pAPX2600bp::LUC transgene (Liu et al., 2018) in Col-0 background; hence, we refer to this line as parent. Seedlings were grown on GM medium (1% (w/v) glucose) under 16-h/8-h light/dark cycle at 23°C/21°C. For the analysis of Type II transcriptional memory, seedlings were exposed to HS at 37°C for 1 h either on Day 4 (priming, P), or on Day 6 (triggering, T), or on Days 4 and 6 (priming and triggering, P + T), or not at all (no HS, NHS). For the analysis of type I transcriptional memory by LUC imaging, 4-d-old seedlings were exposed to a two-step acclimation (ACC) protocol as follows: 37°C for 1 h, 23°C for 1.5 h, 44°C for 45 min.
Construction of transgenic lines
To generate dCas9–JMJ constructs, pHEE401E (Wang et al., 2015) was used as backbone. 3xFLAG-dCas9 was amplified from pHSN6A01 (Wang et al., 2015) with primer 3,197 containing XbaI restriction site and primer 3,198 containing AscI and SacI restriction sites. pHSP21 was amplified from genomic DNA with primers 3,033/34 containing NcoI and XbaI restriction sites. The JmjC domain of JMJ18 (JMJ) was amplified from cDNA with primers 3,203/04 containing AscI and SacI restriction sites and stop codon. To generate the catalytically inactive JMJ, site-directed mutagenesis was performed on the JMJ fragment, corresponding to H395A substitution of the full-length JMJ18 protein. sgRNA cassettes flanked by HindIII and NcoI restriction sites were obtained from BioCat (Heidelberg, Germany) in pUC57 and subcloned into pHEE401E vector. To generate dCas9–VP64 constructs, dCas9–VP64 and 2x35S promoter were amplified from pHSN6A01. dCas9–VP64 was amplified using primers 3,459/60 containing XbaI and SacI restriction sites, 2x35S was amplified using primers 3,035/36 containing NcoI and XbaI restriction sites. Both fragments were subcloned into pHEE401E containing the sgRNA cassette. All constructs were introduced into Agrobacterium tumefaciens GV3,101 for plant transformation. For generation of stable transgenic lines, pAPX2::LUC plants were transformed using the floral dip method (Clough and Bent, 1998). Primer sequences are listed in Supplemental Table S1.
LUC imaging and immunoblotting
For LUC imaging in the transient transactivation assay, 4-week-old leaves of N. benthamiana were co-infiltrated (Sparkes et al., 2006) with the following A. tumefaciens GV3101 suspensions: pGreenII binary vector containing pAPX2::LUC (kindly provided by Y. Pan) and pHEE401E binary vector containing p2x35S::dCas9–VP64 and either one of the three sgRNA cassettes, or no sgRNA cassette. After 3 d, leaves were infiltrated with 2-mM firefly luciferin (Promega, Madison, WI, USA) and immediately imaged with NightOWL In Vivo Imaging System (Berthold Technologies) using standard settings. For LUC imaging of seedlings, they were sprayed with 2-mM luciferin and imaged as above. For immunoblotting, total protein was extracted from frozen tissue (Smaczniak et al., 2012). An amount of 40 µg of total protein was separated by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS–PAGE) on a 4% (v/v) gel and immunoblotted with anti-Flag or anti-histone H3 antibodies (F1804, Sigma, ab1791, Abcam, Cambridge, UK) and secondary anti-mouse antibody (926-32210) on an Odyssey imaging system (both LI-COR Biosciences Lincoln, NE, USA).
RT-qPCR
To examine enhanced re-induction of gene expression, seedlings were exposed to 37°C for 1 h on Day 4 and/or Day 6 after germination. All samples, including nontreated controls, were snap-frozen 6 d after germination at a time corresponding to the end of the Day 6 HS treatment. To extract RNA frozen tissue was ground to a fine powder and resuspended in 500-μL RNA extraction buffer (100-mM Tris–HCl pH 8.5, 5-mM EDTA, 100-mM NaCl, 0.5% (w/v) SDS), 250-μL phenol, and 5-μL β-mercaptoethanol. Samples were incubated at 60°C for 15 min. An aliquot of 250-μL chloroform was added and samples were incubated at RT for 15 min, followed by centrifugation at RT for 10 min at 16,000 g. An aliquot of 550 μL of the aqueous phase were recovered and mixed with 550-μL phenol:chloroform:isoamyl alcohol (25:24:1), incubated and centrifuged as above. Then 500 μL of the aqueous phase was recovered and mixed with 400-μL isopropanol and 50-μL 3-M sodium acetate and precipitated at –80°C for 15 min, followed by centrifugation at 4°C for 30 min at 20,500 g. RNA was precipitated overnight at 4°C in 2 M LiCl. RNA was pelleted by centrifugation at 4°C for 30 min at 20,500g, washed in 80% (v/v) EtOH, dried, and resuspended in 40-μL H2O. For RT-qPCR, total RNA was treated with TURBO DNA-free (Ambion, Austin, TX, USA) and reverse transcribed with SuperScript III (Invitrogen, Waltham, MA, USA) according to manufacturer’s instructions. For each 13.33-μL RT-qPCR reaction, 0.167-μL cDNA was used with GoTaq qPCR Master Mix (Promega Madison, WI, USA) on a LightCycler480 II system (Roche, Basel, Switzerland). All analyzed transcripts were normalized to the reference gene At4g26410 (Czechowski et al., 2005) using the comparative Cq method. Primer sequences are listed in Supplemental Table S1. To perform multiple comparison of gene expression results between transgenic lines, one-way ANOVA with post-hoc Tukey’s HSD analysis (confidence level = 0.95) were performed using the TukeyHSD() function of the R package multcompView (https://cran.r-project.org/web/packages/multcompView/index.html).
ChIP
For dCas9–JMJ and histone modification ChIP, seedlings were exposed to 37°C for 1 h on Day 4 and/or Day 6 after germination. All samples, including nontreated controls, were harvested 6 d after germination at a time corresponding to 45 min after the d6 HS treatment. Cross-linking of samples was done under vacuum in ice-cold MC buffer with 1% (v/v) formaldehyde for 2 × 10 min. Chromatin was extracted as follows (Kaufmann et al., 2010): frozen tissue was ground to a fine powder, resuspended in 25-mL M1 buffer, filtered through Miracloth mesh (Merck Kenilworth, NJ, USA), washed five times in 5-mL M2 buffer, and once in 5-mL M3 buffer, with a centrifugation step at 4°C, 10 min, 1,000g in between each wash. The resulting chromatin pellet was resuspended in 1-mL Sonication buffer and sheared with a Bioruptor Pico (Diagenode Denville, NJ, USA) for 17 cycles (30 s on/30 s off) on low-intensity settings. Equal chromatin amounts of chromatin from each sample were immunoprecipitated over night at 4°C using antibodies against Flag (F1804, Sigma-Aldrich), H3 (ab1791, Abcam), and H3K4me3 (ab8580, Abcam). Immunoprecipitated DNA was quantified by qPCR on a LightCycler480 II system (Roche). Primer sequences are listed in Supplemental Table S1. To statistically evaluate dCas9–(d)JMJ and H3K4me3 enrichment, independent t tests were performed (https://www.graphpad.com/quickcalcs/ttest1.cfm).
Accession numbers
Accession numbers for genes mentioned in this work are as follows: APX2 (AT3G09640), JMJ18 (AT1G30810), and HSP101 (AT1G74310).
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Effect of (d)JMJ on type I transcriptional memory of pAPX2::LUC.
Supplemental Figure S2. Type II transcriptional memory of additional dCas9–dJMJ lines.
Supplemental Figure S3. Immunoblot detection of dCas9– (d)JMJ proteins.
Supplemental Table S1. Oligonucleotides used in this study.
Supplementary Material
Acknowledgments
We thank Y.Y. Charng (Taipei, Taiwan) for pAPX2::LUC seeds. We thank S. Hirsch and J. Baltzer for excellent technical assistance and R. Urrea Castellanos, T. Friedrich and M. Lenhard for helpful comments. We thank D. Mäker and C. Schmidt for excellent plant care.
Funding
Funding was provided by the European Research Council (ERC CoG 725295) and the Deutsche Forschungsgemeinschaft (CRC973, Project A2) to I.B.
Conflict of interest statement. None declared.
V.O. and I.B. conceived and designed research, V.O. performed experiments, and V.O. and I.B. analyzed experiments and wrote the manuscript.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plphys/pages/general-instructions) is: Isabel Bäurle (isabel.baeurle@uni-potsdam.de).
References
- Baumann V, Wiesbeck M, Breunig CT, Braun JM, Köferle A, Ninkovic J, Götz M, Stricker SH (2019) Targeted removal of epigenetic barriers during transcriptional reprogramming. Nat Commun 10: 2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäurle I (2016) Plant heat adaptation: priming in response to heat stress. F1000Res 5: 694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli RR, Segal DJ, Dreier B, Barbas CF (1998) Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci USA 95: 14628–14633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano-Rodriguez D, Campagnoli S, Grandi A, Parri M, Camilli ED, Song C, Jin B, Lacombe A, Pierleoni A, Bombaci M, et al. (2017) TCTN2: a novel tumor marker with oncogenic properties. Oncotarget 8: 95256–95269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charng YY, Liu HC, Liu NY, Chi WT, Wang CN, Chang SH, Wang TT (2007) A heat-inducible transcription factor, HsfA2, is required for extension of acquired thermotolerance in Arabidopsis. Plant Physiol 143: 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Hu Y, Zhou DX (2011) Epigenetic gene regulation by plant Jumonji group of histone demethylase. Biochim Biophys Acta 1809: 421–426 [DOI] [PubMed] [Google Scholar]
- Cheng K, Xu Y, Yang C, Ouellette L, Niu L, Zhou X, Chu L, Zhuang F, Liu J, Wu H, et al. (2020) Histone tales: lysine methylation, a protagonist in Arabidopsis development. J Exp Bot 71: 793–807 [DOI] [PubMed] [Google Scholar]
- Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16: 735–743 [DOI] [PubMed] [Google Scholar]
- Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139: 5–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova E, Turberfield AH, Klose RJ (2015) Histone demethylases in chromatin biology and beyond. EMBO Rep 16: 1620–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Fromm M, Avramova Z (2012) Multiple exposures to drought “train” transcriptional responses in Arabidopsis. Nat Commun 3: 740. [DOI] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E (2014) The new frontier of genome engineering with CRISPR-Cas9. Science 346: 1258096. [DOI] [PubMed] [Google Scholar]
- D’Urso A, Takahashi YH, Xiong B, Marone J, Coukos R, Randise-Hinchliff C, Wang JP, Shilatifard A, Brickner JH (2016) Set1/COMPASS and Mediator are repurposed to promote epigenetic transcriptional memory. eLife 5: e16691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XJ, Li JR, Qi SL, Lin QF, Jin JB, Hua XJ (2016) Light affects salt stress-induced transcriptional memory of P5CS1 in Arabidopsis. Proc Natl Acad Sci USA 113: E8335–E8343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich T, Oberkofler V, Trindade I, Altmann S, Brzezinka K, Lämke J, Gorka M, Kappel C, Sokolowska E, Skirycz A, et al. (2021) Heteromeric HSFA2/HSFA3 complexes drive transcriptional memory after heat stress in Arabidopsis. Nat Commun 12: 3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego-Bartolomé J, Gardiner J, Liu W, Papikian A, Ghoshal B, Kuo HY, Zhao JM-C, Segal DJ, Jacobsen SE (2018) Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc Natl Acad Sci USA 115: E2125–E2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal B, Picard CL, Vong B, Feng S, Jacobsen SE (2021) CRISPR-based targeting of DNA methylation in Arabidopsis thaliana by a bacterial CG-specific DNA methyltransferase. Proc Natl Acad Sci USA 118: e2125016118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA (2015) Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33: 510–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JR, Engstrom A, Zoeller EL, Liu X, Shanks JR, Zhang X, Johns MA, Vertino PM, Fu H, Cheng X (2016) Characterization of a linked jumonji domain of the KDM5/JARID1 family of histone H3 lysine 4 demethylases. J Biol Chem 291: 2631–2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Zhang A, Jin JB, Zhao B, Wang TJ, Wu Y, Wang S, Liu Y, Wang J, Guo P, et al. (2019) Arabidopsis histone H3K4 demethylase JMJ17 functions in dehydration stress response. New Phytol 223: 1372–1387 [DOI] [PubMed] [Google Scholar]
- Ishihara H, Sugimoto K, Tarr PT, Temman H, Kadokura S, Inui Y, Sakamoto T, Sasaki T, Aida M, Suzuki T, et al. (2019) Primed histone demethylation regulates shoot regenerative competency. Nat Commun 10: 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Yang W, He Y, Amasino RM (2007) Arabidopsis relatives of the human lysine-specific Demethylase1 repress the expression of FWA and FLOWERING LOCUS C and thus promote the floral transition. Plant Cell 19: 2975–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann K, Muino JM, Osteras M, Farinelli L, Krajewski P, Angenent GC (2010) Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP). Nat Protocol 5: 457–472 [DOI] [PubMed] [Google Scholar]
- Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, Maehr R (2015) Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods 12: 401–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumlehn J, Pietralla J, Hensel G, Pacher M, Puchta H (2018) The CRISPR/Cas revolution continues: From efficient gene editing for crop breeding to plant synthetic biology. J Integr Plant Biol 60: 1127–1153 [DOI] [PubMed] [Google Scholar]
- Kwon DY, Zhao YT, Lamonica JM, Zhou Z (2017) Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat Commun 8: 15315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämke J, Bäurle I (2017) Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol 18: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämke J, Brzezinka K, Altmann S, Bäurle I (2016) A hit-and-run heat shock factor governs sustained histone methylation and transcriptional stress memory. EMBO J 35: 162–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Neumann M, Duro DI, Schmid M (2019) CRISPR-based tools for targeted transcriptional and epigenetic regulation in plants. PLoS One 14: e0222778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Lu F, Cui X, Cao X (2010) Histone methylation in higher plants. Ann Rev Plant Biol 61: 395–420 [DOI] [PubMed] [Google Scholar]
- Liu F, Quesada V, Crevillen P, Bäurle I, Swiezewski S, Dean C (2007) The Arabidopsis RNA-binding protein FCA requires a lysine-specific demethylase 1 homolog to downregulate FLC. Mol Cell 28: 398–407 [DOI] [PubMed] [Google Scholar]
- Liu HC, Lamke J, Lin SY, Hung MJ, Liu KM, Charng YY, Baurle I (2018) Distinct heat shock factors and chromatin modifications mediate the organ-autonomous transcriptional memory of heat stress. Plant J 95: 401–413 [DOI] [PubMed] [Google Scholar]
- Liu P, Zhang S, Zhou B, Luo X, Zhou XF, Cai B, Jin YH, Niu D, Lin J, Cao X, et al. (2019) The histone H3K4 demethylase JMJ16 represses leaf senescence in Arabidopsis. Plant Cell 31: 430–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Gallego-Bartolomé J, Zhou Y, Zhong Z, Wang M, Wongpalee SP, Gardiner J, Feng S, Kuo PH, Jacobsen SE (2021) Ectopic targeting of CG DNA methylation in Arabidopsis with the bacterial SssI methyltransferase. Nat Commun 12: 3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowder LG, Zhang D, Baltes NJ, Paul JW, Tang X, Zheng X, Voytas DF, Hsieh T-F, Zhang Y, Qi Y (2015) A CRISPR/Cas9 Toolbox for Multiplexed Plant Genome Editing and Transcriptional Regulation. Plant Physiol 169: 971–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Cui X, Zhang S, Liu C, Cao X (2010) JMJ14 is an H3K4 demethylase regulating flowering time in Arabidopsis. Cell Res 20: 387–390 [DOI] [PubMed] [Google Scholar]
- Morita S, Noguchi H, Horii T, Nakabayashi K, Kimura M, Okamura K, Sakai A, Nakashima H, Hata K, Nakashima K, et al. (2016) Targeted DNA demethylation in vivo using dCas9–peptide repeat and scFv–TET1 catalytic domain fusions. Nat Biotechnol 34: 1060–1065 [DOI] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K (2003) Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell 11: 709–719 [DOI] [PubMed] [Google Scholar]
- Nover L, Bharti K, Doring P, Mishra SK, Ganguli A, Scharf KD (2001) Arabidopsis and the heat stress transcription factor world: how many heat stress transcription factors do we need? Cell Stress Chaperones 6: 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberkofler V, Pratx L, Bäurle I (2021) Epigenetic regulation of abiotic stress memory: maintaining the good things while they last. Curr Opin Plant Biol 61: 102007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohama N, Sato H, Shinozaki K, Yamaguchi-Shinozaki K (2017) Transcriptional regulatory network of plant heat stress response. Trends Plant Sci 22: 53–65 [DOI] [PubMed] [Google Scholar]
- Papikian A, Liu W, Gallego-Bartolomé J, Jacobsen SE (2019) Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat Commun 10: 729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelayo MA, Yamaguchi N, Ito T (2021) One factor, many systems: the floral homeotic protein AGAMOUS and its epigenetic regulatory mechanisms. Curr Opin Plant Biol 61: 102009. [DOI] [PubMed] [Google Scholar]
- Roca Paixão JF, Gillet FX, Ribeiro TP, Bournaud C, Lourenço-Tessutti IT, Noriega DD, de Melo BP, de Almeida-Engler J, Grossi-de-Sa MF (2019) Improved drought stress tolerance in Arabidopsis by CRISPR/dCas9 fusion with a Histone AcetylTransferase. Sci Rep 9: 8080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski I, Ma J, Triezenberg S, Ptashne M (1988) GAL4-VP16 is an unusually potent transcriptional activator. Nature 335: 563–564 [DOI] [PubMed] [Google Scholar]
- Samo N, Ebert A, Kopka J, Mozgová I (2021) Plant chromatin, metabolism and development-an intricate crosstalk. Curr Opin Plant Biol 61: 102002. [DOI] [PubMed] [Google Scholar]
- Sani E, Herzyk P, Perrella G, Colot V, Amtmann A (2013) Hyperosmotic priming of Arabidopsis seedlings establishes a long-term somatic memory accompanied by specific changes of the epigenome. Genome Biol 14: R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm F, Ganguli A, Kiehlmann E, Englich G, Walch D, von Koskull-Doring P (2006) The heat stress transcription factor HsfA2 serves as a regulatory amplifier of a subset of genes in the heat stress response in Arabidopsis. Plant Mole Biol 60: 759–772 [DOI] [PubMed] [Google Scholar]
- Smaczniak C, Immink RG, Muino JM, Blanvillain R, Busscher M, Busscher-Lange J, Dinh QD, Liu S, Westphal AH, Boeren S, et al. (2012) Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc Natl Acad Sci USA 109: 1560–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Zhang L, Han J, Zhou M, Liu J (2021) Histone H3K4 methyltransferases SDG25 and ATX1 maintain heat‐stress gene expression during recovery in Arabidopsis. Plant J 105: 1326–1338 [DOI] [PubMed] [Google Scholar]
- Sparkes IA, Runions J, Kearns A, Hawes C (2006) Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat Protocol 1: 2019–2025 [DOI] [PubMed] [Google Scholar]
- Stief A, Altmann S, Hoffmann K, Pant BD, Scheible W-R, Bäurle I (2014) Arabidopsis miR156 Regulates Tolerance to Recurring Environmental Stress through SPL Transcription Factors. The Plant Cell 26: 1792–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD (2014) A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159: 635–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda M, Seki M (2020) Histone modifications form epigenetic regulatory networks to regulate abiotic stress response. Plant Physiol 182: 15–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZP, Xing HL, Dong L, Zhang HY, Han CY, Wang XC, Chen QJ (2015) Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation. Genome Biol 16: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Jin R, Wagner D (2017) Developmental transitions: integrating environmental cues with hormonal signaling in the chromatin landscape in plants. Genome Biol 18: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Han Z, Cao Y, Fan D, Li H, Mo H, Feng Y, Liu L, Wang Z, Yue Y, et al. (2012a) A companion cell-dominant and developmentally regulated H3K4 demethylase controls flowering time in Arabidopsis via the repression of FLC expression. PLoS Genet 8: e1002664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Mo H, Fan D, Cao Y, Cui S, Ma L (2012b) Overexpression of a histone H3K4 demethylase, JMJ15, accelerates flowering time in Arabidopsis. Plant Cell Rep 31: 1297–308 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.