

Abstract
Identification of adaptive targets in experimental evolution typically relies on extensive replication and genetic reconstruction. An alternative approach is to directly assay all mutations in an evolved clone by generating pools of segregants that contain random combinations of evolved mutations. Here, we apply this method to 6 Saccharomyces cerevisiae clones isolated from 4 diploid populations that were clonally evolved for 2,000 generations in rich glucose medium. Each clone contains 17–26 mutations relative to the ancestor. We derived intermediate genotypes between the founder and the evolved clones by bulk mating sporulated cultures of the evolved clones to a barcoded haploid version of the ancestor. We competed the resulting barcoded diploids en masse and quantified fitness in the experimental and alternative environments by barcode sequencing. We estimated average fitness effects of evolved mutations using barcode-based fitness assays and whole-genome sequencing for a subset of segregants. In contrast to our previous work with haploid evolved clones, we find that diploids carry fewer beneficial mutations, with modest fitness effects (up to 5.4%) in the environment in which they arose. In agreement with theoretical expectations, reconstruction experiments show that all mutations with a detectable fitness effect manifest some degree of dominance over the ancestral allele, and most are overdominant. Genotypes with lower fitness effects in alternative environments allowed us to identify conditions that drive adaptation in our system.
Keywords: experimental evolution, adaptation, dominance, overdominance, fitness
Introduction
Over the course of adaptation of a clonal population, selection acts on genetic variation that is generated within the first few hundred generations (Lang et al. 2011; Blundell et al. 2019). The fates of these mutations are not independent, and beneficial mutations can be lost due to genetic drift and clonal interference (Desai and Fisher 2007; Kao and Sherlock 2008; Lang et al. 2011; Maddamsetti et al. 2015; Good et al. 2017; Lenski 2017). In addition, genetic hitchhiking may result in the fixation of many neutral or deleterious mutations (Desai et al. 2007; Lang et al. 2013; Payen et al. 2016). Collectively, these effects can hinder efforts to determine adaptive mutational spectra. Common targets of selection can be identified through molecular characterization of many independently evolved lineages (Lang et al. 2013; Levy et al. 2015; Venkataram et al. 2016; Fisher et al. 2018; Marad et al. 2018; Blundell et al. 2019). However, relying on recurring targets is prone to miss beneficial mutations with weak effects, low mutation rates, or genetic interactions (Buskirk et al. 2017). Unambiguously distinguishing beneficial mutations from hitchhikers requires measuring the fitness effect of all mutations within an individual evolved clone via reconstructions (Chou et al. 2011; Khan et al. 2011) or bulk-segregant approaches (Brauer et al. 2006; Ehrenreich et al. 2010; Wenger et al. 2010; Magwene et al. 2011b; Cubillos et al. 2013; Sigwalt et al. 2016; Buskirk et al. 2017).
Increased ploidy state can amplify the problem for identification of adaptive variation. Specifically diploidy appears to be the converging ploidy in yeast under many conditions (Gerstein et al. 2006; Oud et al. 2013; Selmecki et al. 2015; Venkataram et al. 2016; Gorter et al. 2017; Fisher et al. 2018), and although there are conditions in which other ploidies may be advantageous (Zörgö et al. 2013; Hope and Dunham 2014; Zhu et al. 2016) meiosis and a functional mating-type switching system among natural isolates (Hanson and Wolfe 2017; Peter et al. 2018) suggest that diploidy is the default. Ploidy state is an important determinant of yeast physiology, affecting gene expression levels (Galitski et al. 1999; de Godoy et al. 2008) and responses across a range of environments (Zörgö et al. 2013). Under laboratory conditions, adaptation differences between the 2 ploidies mainly stem from gene copy number differences, with the following important implications. Unlike haploids, both fitness and dominance effects determine selection in diploids (Haldane 1924; Orr and Otto 1994; Gerstein et al. 2011, 2014; Sellis et al. 2016; Sharp et al. 2018), and dominance effects, much like fitness (Wenger et al. 2011; Gerstein et al. 2012; Kvitek and Sherlock 2013; Jerison et al. 2017), are environment and genetic background dependent (Gerstein et al. 2014; Matsui et al. 2022). According to Haldane’s sieve, adaptive mutations are expected to manifest at least some degree of dominance, in order to be visible by selection in diploids (Haldane 1924; Orr and Otto 1994; Sellis et al. 2016).
Fully dominant mutations are rare relative to recessive mutations (Deutschbauer et al. 2005; Zörgö et al. 2012). Therefore, diploids should have access to fewer large-effect adaptive mutations, which contributes to their comparatively slower adaptation rate relative to haploids (Orr and Otto 1994; Gerstein et al. 2011; Gerstein and Otto 2011; McDonald et al. 2016; Marad et al. 2018; Johnson et al. 2021). Similarly, recessive maladaptive mutations are not expected to be purged from clonally evolving diploids (Nishant et al. 2010; Sharp et al. 2018), contributing to the hitchhiking mutational load, their neutral evolution, and eventually to genetic diversity with adaptive and innovation potentials (Wideman et al. 2019). In addition, aneuploidies and other structural variants are more common in diploids (Sellis et al. 2016; Fisher et al. 2018) and can further constrain clonal adaptation (Yona et al. 2012; Assaf et al. 2015; Sellis et al. 2016; Fisher et al. 2021).
Previously, we sequenced 2 clones each of 24 evolving diploid yeast populations and identified adaptive mutations based on recurrence (Marad et al. 2018). To quantify the fitness effects of all mutations in 4 of these populations, we combined a bulk segregant approach that we have successfully applied in adapted haploids (Buskirk et al. 2017) with DNA barcoding (Levy et al. 2015; Liu et al. 2019; Nguyen Ba et al. 2019; Matsui et al. 2022). We find that while diploid clones carry on average 22 mutations each, only 1 or 2 per clone (6% total) are beneficial with effects ranging between 1% and 5.4%. For comparison, beneficial mutations in haploids have larger effect sizes (1–10%) and are accompanied by half-as-many hitchhiking mutations (Buskirk et al. 2017). Environmental perturbations change the fitness effects of the evolved mutations, suggesting pleiotropy and pinpointing to a selective pressure driving adaptation in our environment. Reconstructions of evolved alleles show that adaptive mutations that were in heterozygous state in the evolved clone are frequently overdominant, that the overdominance effect depends on the background, and that adaptive mutations that were in homozygous state in the evolved clone are either partially or fully dominant, consistent with Haldane’s sieve. These data indicate that both smaller fitness effects and reduced availability of beneficial mutations contribute to relatively slower adaptation of diploids. The present study corroborates prior findings that compare adaptation rates of clonally evolving yeast haploids and diploids (Gerstein et al. 2011; Johnson et al. 2021).
Materials and methods
Yeast strains and strain construction
The strains used in this experiment are derived from diploid yGIL672 (W303 background), with genotype MATa/α, and homozygous for ade2-1, CAN1, his3-11,15, leu2-3,112, trp1-1, bar1Δ::ADE2, hmlαΔ::LEU2, GPA1::NatMX, ura3Δ::pFUS1-yEVenus.
Evolved clones of yGIL672 were isolated from generation 2,000 of a 5,000-generation experiment described previously (Marad et al. 2018). Killing ability and sensitivity to killer toxin were assayed as described previously (Buskirk et al. 2020) using a modified version of the standard halo assay. Killing ability was assayed against a hypersensitive tester strain (yGIL1097) and killer toxin sensitivity was assayed against yGIL672.
Gene deletions were introduced by amplifying the KanMX cassette from the hemizygous or MATa deletion collections (Euroscarf) and transforming using the standard lithium acetate protocol (Gietz and Schiestl 2007). Lethality of the essential gene deletions was verified via tetrad dissection. Evolved mutations were introduced into the ancestral background (yGIL432, yGIL646, and yGIL672; MATa, MATα, and MATa/α, respectively) using CRISPR/Cas9 allele swaps as described previously (Fisher et al. 2019). Briefly, oligonucleotides specifying the gRNA were hybridized and introduced into the SWA1 and BCL1 restriction sites of pML104 (Addgene #67638). Repair templates containing evolved mutations were generated by amplifying ∼500 bp fragments centered around the mutation of interest or by gBlock synthesis (IDT) containing the mutation or interest along with synonymous PAM site changes. For driving loss-of-heterozygosity (LOH) in diploids, no repair template was provided. All plasmids and strain constructions were validated by Sanger sequencing (Genscript).
Barcode library transformation
Introduction of the barcoding system includes 2 sequential transformations. The elements introduced in each reaction and the final locus are shown in Supplementary Fig. 1. Briefly, the first transformation introduces a 26-mer barcode, gal-induced Cre recombinase, a lox site, half of URA3 next to an artificial intron, and a kanamycin selection marker. The second transformation introduces a second 26-mer barcode and the rest of the URA3 gene next to an artificial intron. Second round transformants selection in medium lacking uracil relies on reconstruction of the split URA3 and artificial intron splicing. The first barcode was amplified out of a yeast library [strain XLY092 (Liu et al. 2019)] in 3 overlapping amplicons 1–2 kb long and the amplicons were introduced into yGIL432 via a lithium acetate transformation (Gietz and Schiestl 2007). The amplicons were targeted upstream of the GPA1 locus (primers in Supplementary Table 1), replacing the NatMX cassette that marks our ancestors and all derivatives. The choice of GPA1 locus has the advantage of being located close to chromosome VIII centromere, thus reducing the chance of gene conversion and LOH. Successful transformants were selected on YPD supplemented with kanamycin. Integration at the intended locus was screened via lack of growth on media supplemented with ClonNAT and amplification of the expected junctions and/or receptiveness to the high complexity barcode [pBAR7-L1 plasmid library (Liu et al. 2019)]. The second barcode library was introduced to individual transformants with 1 barcode. High efficiency in genomic integration of the high complexity barcode was driven by overnight galactose induction of Cre recombination. Transformants were selected on synthetic complete media lacking uracil on 150 mm x 15 mm petri dishes and harvested via pooling after 2 days. The number of unique barcodes or integration events was estimated by plating dilutions of the transformation reaction on 100 mm × 15 mm petri dishes. The number of transformants after induction was estimated by plating 1:10 and 1:100 dilutions on selective media and was corrected for growth by plating 1:106 and 1:107 dilutions on YPD before and after gal-induction.
Construction of barcoded bulk segregant pools
Sporulating cultures of evolved diploid clones were mated each with a different pool of the barcoded strain with an estimated diversity of 400,000 barcodes, as follows. Four milliliters of sporulating cultures (∼2 × 107 tetrads/ml and ∼30% sporulation efficiency) were resuspended in 60 μl water and 2,000 U zymolase (USBiological) and the samples were incubated at 30°C for 1 h. Then 20 μl glass beads and 100 μl Triton X-100 were added, the samples were vortexed for 2 min, incubated at 30°C for 40 min and vortexed for an additional 2 min; 1.8 ml water was added, and the samples were sonicated at full power for 4 s. The solution was mixed with the haploid mating partner on nylon membranes (GVS, Sanford NE) at an excess of 1:100 (100 barcoded cells for each MATα spore) using a vacuum manifold. The membranes were incubated on a YPD plate at room temperature overnight and subsequently the cells were harvested from the membranes with PBS and were plated on synthetic complete medium lacking uracil and supplemented with ClonNAT (selection for mated diploids with a barcode) at different densities. Small pools were generated by picking 192 individual colonies off of each low-density CSM-ura + ClonNAT plate, propagating them in 96-well plates and mixing them at equal volumes. Large pools were generated by scraping ∼60,000 colonies off of high-density CSM-ura + ClonNAT plates. Control barcoded parental strains were derived from yGIL672. At each step of the barcoding process, parental clones were isolated and assayed for fitness, killing ability and sensitivity to killer toxin. Barcoded diploid segregants were also assayed for killing ability and sensitivity to killer toxin. With the exception of parental clones from population F04, phenotypes of the parental clones remained consistent throughout the barcoding process (Supplementary Fig. 2a).
Quantifying the recovery of recessive lethal mutations
To estimate mating efficiency of query haplotypes, sporulating cultures of diploid lab W303 derivatives, engineered with the respective gene deletions, were digested with zymolase (USBiological), the asci were broken, and the resulting spores were mass mated to a haploid mating partner, as follows: 3 ml sporulating cultures were resuspended in 60 μl water and 2,000 U zymolase and the samples were incubated at 30°C for 1 h. Then 20 μl glass beads and 20 μl Triton X-100 were added, the samples were vortexed for 2 min, incubated at 30°C for 40 min and vortexed for an additional 2 min; 1.2 ml water was added, and the samples were sonicated at power 4 for 4 s. The broken asci mix was mixed with the haploid mating partner either in patches on YPD plates or in membranes using a vacuum manifold. The resulting query diploids were quantified by plating to YPD supplemented with G418 and clonNAT. Percent recovery was estimated as the fraction of colony-forming units (cfu) on YPD media supplemented with G418 and clonNAT over the cfu on YPD supplemented with G418 (marker of the limiting mating partner) and expressed as a percentage of the nonessential control (yur1Δ) mating efficiency (Supplementary Fig. 3). For the mating efficiency calculation, it was assumed that sporulation efficiency was the same for all genotypes. Euploidy of the resulting query diploids was verified via tetrad dissection.
Backcrossing of evolved clones
A backcrossing approach was used to introgress putative recessive lethal mutations into the ancestral background. We transformed evolved clones with plasmids carrying ancestral versions of genes harboring putative recessive lethal mutations [MoBY collection (Ho et al. 2009)]. We sporulated each evolved clone and screened a single 4-spore tetrad for the allele of interest. An appropriate spore was selected and backcrossed to either yGIL432 (MATa) or yGIL646 (MATα). For each clone, we performed at least 5 backcrosses to replace the rest of the evolved background with the ancestral. Haploid progeny of the final tetrad dissection and haploid ancestral strains were used to generate a heterozygous backcrossed evolved mutation (cross of a segregant with the evolved allele and the ancestor), a heterozygous intratetrad mating (cross of segregants with and without the evolved allele), a homozygous ancestral backcross (cross of a segregant without the evolved allele and the ancestor), a homozygous ancestral intratetrad mating (cross between 2 segregants with the ancestral allele), and a homozygous mutant intratetrad mating (cross between 2 segregants with the evolved allele). Each of these 5 types of crosses were performed in at least triplicate and the final strains were plated on 5-FOA to select for loss of the MoBY plasmid.
Fitness assays
Unless otherwise stated, all fitness assays were performed under the same conditions as the evolution experiment. The populations were diluted daily 1:210 using a Biomek FX liquid handler into 128 ml of YPD plus 100 mg/ml ampicillin and 25 mg/ml tetracycline to prevent bacterial contamination. The cultures were incubated at 30°C in an unshaken 96-well plate.
The fluorescence-based fitness assays were previously described (Buskirk et al. 2017). Briefly, saturated cultures of the query strain and a ymCitrine-labeled version of the ancestor were mixed isovolumetrically at Generation 0. The assays were performed for 30–50 generations and sampled every 10 generations (4–6 timepoints total). Following each transfer, 4 μl of the saturated culture was diluted in 60 μl PBS and the samples were stored at 4°C for 1–2 days before being assayed by flow cytometry (BD FACSCanto II). Data were analyzed in FlowJo. Fitness was calculated as the linear regression of the log ratio of experimental-to-reference frequencies over time in generations.
For the barcode-based fitness assays, the small pools of 192 barcoded diploid segregants were mixed with 4 independently barcoded ancestral derivatives and between 7 and 11 independently barcoded evolved derivatives (with appropriate volumes adjustments to achieve equal strain representation). Each pool was used to seed 2 columns of a 96-well plate (16 wells) and the populations were propagated for 110 generations in the same way as the original evolution experiment, with sampling every 10 generations. For each timepoint, each column (8 wells) was pooled, the cells were spun down and stored at −20°C for genomic DNA preparations. To monitor changes in allele frequencies, fitness assays were performed in the same way, using the large pools of ∼60,000 segregants.
Library preparations and sequencing
Barcode determination of individual segregants
To identify the barcodes of the isolated segregants in 96-well plates, we employed a 3- (column, row, plate) dimensional pooling strategy (Evans and Lewis 1989; Baym et al. 2016). Briefly, we inoculated 13 96-deep well plates with YPD from our frozen stocks (12 plates with segregants, 2 corresponding to each evolved parent, and 1 with all the barcoded parents). The cultures grew for 2 days at 30°C and then the contents of wells were pooled by column, row, and plate, resulting in 12, 8, and 13 pools, respectively. Genomic DNA was isolated and barcode libraries were prepared from the pools as described below.
gDNA preparation
Cells from ∼1.5 to 2 ml saturated culture were resuspended in 100 µl lysis buffer (0.9 M sorbitol, 50 mM sodium phosphate pH 7.5, 240 µg/ml zymolase, 14 mM β-mercaptoethanol) and incubated at 37°C for 30 min. Ten microliters of 0.5M EDTA and 10 µl 10% SDS were added consecutively, with brief vortexing after each addition, and the samples were incubated at 65°C for 30 min and then on ice for 5 min. 50 µl of 5M potassium acetate were added, the samples were mixed, incubated on ice for 30 min and spun down at full speed in a microcentrifuge for 10 min. The supernatant was transferred to a new tube with 200 µl isopropanol and was incubated on ice for 5 min. The nucleic acid was spun down full speed in a microcentrifuge for 10 min, washed twice with 70% ethanol, was let to dry completely, and then resuspended in 20 μl 10 mM Tris pH 7.5. Overnight incubation at room temperature or short incubation at 65°C was sometimes necessary for complete resuspension. RNA was digested with the addition of 0.5 µl 20 mg/ml RNase A (ThermoFisher Scientific, Waltham, MA) and incubation at 37°C for 1 h or at room temperature overnight.
Barcode sequencing libraries
A 2-step PCR protocol was used to amplify the barcoded locus (primers in Supplementary Table 1). For the first amplification a maximum of 200 ng genomic DNA (corresponding to 7.5 × 106 diploid Saccharomyces cerevisiae genomes) was used as template in a 20-μl reaction with the following composition: 20 nM each forward and reverse primer (PU1 and PU2; Supplementary Table 1), 10 ng/µl gDNA, 1 mM dNTPs, 0.2 μl Herculase II fusion DNA polymerase (Agilent, Santa Clara, CA), 1× Herculase buffer, in the following conditions: hot start, initial denaturation at 98°C for 2 min, 2 cycles of 98°C for 10 s, 61°C for 20 s and 72°C for 30 s, and final extension at 72°C for 1 min. Primers PU1 and PU2 introduce unique molecular identifiers (UMI) and this first-step reaction is used as is as a template for the second step reaction, which introduces library-specific indexes for multiplex sequencing (Supplementary Table 1). To the first-step reaction, 30 μl with the following composition are added: 0.4 µl Herculase, 1× Herculase buffer, 1 mM dNTPs, and 417 nM each of BC_i5 and BC_i7 (Supplementary Table 1), and amplification happened in the following conditions: hot start, initial denaturation at 98°C for 2 min, 22 cycles of 98°C for 10 s, 61°C for 20 s, and 72°C for 30 s, and final extension at 72°C for 1 min. DNA from all libraries was pooled isostoichiometrically, based on DNA concentrations estimated by Nanodrop. A 350-bp band was gel-purified of the final pool with the QIAGEN gel extraction kit (QIAGEN, Germantown, MD).
Whole genome and whole genome whole population sequencing protocol
Genomic DNA was prepared for each of the segregants, after they were grown to saturation on YPD for whole-genome sequencing (WGS). Whole-genome whole population time–course sequencing of the fitness assays was performed to monitor changes in allele frequencies. Samples were thawed from −20°C and sequencing libraries were prepared according to Baym et al. (2015) with the modifications described in Buskirk et al. (2017). Individual libraries were quantified by Nanodrop and pooled. Gel extraction included fragments in the 350–650 bp range.
Library QC and sequencing
Final sample quantification was done by Qubit. Final pools were analyzed by BioAnalyzer on a High-Sensitivity DNA Chip (BioAnalyzer 2100, Agilent), before sequencing on an Illumina HiSeq 2500 sequencer with 250-bp single-end reads or on a NovaSeq with 2× 150 bp paired-end reads at the Sequencing Core Facility within the Lewis-Sigler Institute for Integrative Genomics at Princeton University.
Data analysis
Raw sequencing data were split by index using a dual-index barcode splitter (barcode_splitter.py) from L. Parsons (Princeton University).
Fitness estimation from barcode sequencing
Lineage fitness estimation from barcode sequencing data was performed as in Venkataram et al. (2016). Note that the fitness algorithm calculates fitness per transfer and that is how it is reported in the extended tables (Supplementary File 1). In figures and supplementary tables, fitness is expressed per generation, assuming 10 generations per transfer. Raw barcode counts were prepared from barcode sequencing reads with use of existing software and a custom python script (https://github.com/Dangeli/Barcode-counting). Briefly, reads derived from paired-end sequencing were merged with pear (v0.9.11) (Zhang et al. 2014). Merged reads (or reads derived from single-end sequencing) were aligned against the expected barcoded locus sequence with bowtie2 (v2.3.4.1) (Langmead and Salzberg 2012). Barcodes and UMI were extracted from the aligned reads and clustered using bartender (v1.1) (Zhao et al. 2018). Barcodes from reads were updated using the cluster centers derived from bartender and Levenshtein distance with threshold 2. The updated reads and the UMI were used to derive raw barcode counts, which were used as input to the lineage fitness algorithm.
WGS analysis
Data from libraries from genomic DNA were subsequently trimmed from adaptor sequences using fastx_clipper from the FASTX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/download.html), version 0.0.14 if they originated from a single-end or trimmomatic, version 0.36 (Bolger et al. 2014) with option PE if they originated from a paired-end sequencing run. Each sample was aligned to the complete and annotated W303 genome (Buskirk et al. 2017) using Burrows–Wheeler Aligner (v.0.7.15) (Li and Durbin 2009), option mem. BAM files were generated from SAM files, sorted and indexed with samtools, version 1.4 (Li et al. 2009). BAM files from libraries originating from the same sample were merged prior to sorting and indexing. Variants were called using freebayes, version 1.1.0 (https://github.com/ekg/freebayes), with option pooled-continuous for population data or option pooled-discrete with ploidy 2 for clonal data. Variant call format (VCF) files were annotated using SnpEff, version 4.3 (Cingolani et al. 2012).
Variant discovery in evolved clones
We generated a consensus evolved variant list for each of the evolved parents, considering segregant, parental, and population WGS data. First, we merged BAM files derived from segregant libraries by common descend and from population libraries from the same initial pool. Merged segregant datasets consisted of 21 sequenced derivatives from clone A05-C, 28 from clone A05-D, 26 from clone A07-C, 24 from clone H06-C, and 35 each from clones F04-C and F04-D. Merged population datasets consisted of 3 fitness assays each for initial evolved clones A05-C and F04-D and 2 assays each for initial evolved clones A05-D, A07-C, and F04-C. Each assay is made up of 6 timepoints. Downstream analysis was performed as described for WGS analysis. Variants were called with freebayes setting the ploidy option at 8 (considering that heterozygous mutations will appear in half the diploid derivatives in heterozygosities and trying to correct for small sample sizes). Mutations from the population merged datasets were called with parameters -F 0.01 -C 5 and –pooled-continuous. Parental variants parameters are as described for clonal data in WGS analysis. Each of these call sets were filtered as follows: calls that mapped on 2-µm plasmid or mitochondria, calls with low quality score (<19.99) and calls with more than 1 alternative allele were excluded. Additionally, total coverage of variant, fraction of alternative calls, as well as forward and reverse fraction of alternative calls were considered. In particular, we included calls with coverage z-score between −0.5 and 3 and forward to reverse alternative allele ratio between 0.4 and 2.5. The filtered variant list was then manually curated by visual inspection of the alignments on IGV (Robinson et al. 2011). We also computationally filtered the list using the following criteria for inclusion: The variant is called in the evolved parent and at least 1 more dataset. The variant is called in a single population and not in the ancestor. The list that resulted after application of these criteria overlapped with the manually curated list that resulted after visual inspection of alignments. To specifically discover homozygous mutations, we applied the following 2 criteria for consideration in each of the 3 datasets: total coverage >29 and forward and reverse representation of the alternative allele. Subsequently, we categorized mutations as heterozygous in the dataset (if alternative allele to total coverage was between 0.3 and 0.7) or homozygous (if alternative allele to total coverage was >0.7). Mutations that passed this filtering had to be called “homozygous” in the parental dataset and “heterozygous” in at least one of the merged segregants or merged populations datasets in order to be categorized as homozygous in the evolved parent.
Assign fitness values to mutations from WGS and time–course barcode sequencing of segregants
Freebayes parameters for variant calling from clonal data were as described in WGS analysis. Evolved mutations in the consensus list were scored for presence/absence in each segregant. Fitness values were attached to each genotype by using barcode and well coordinate information. For mutations represented by at least 3 fitness values in each of the presence, absence groups we estimated their fitness effect as the difference between the averages of the presence and absence groups. Significance was initially assessed by t-test and rank sum test and was Bonferroni-adjusted. Additionally, we performed ANOVA with input all mutations that appeared significant in at least one test before Bonferroni correction in at least one assay (including assays in the evolutionary condition and in conditions that deviate from the evolutionary when available).
Assign fitness values to mutations from time–course whole population WGS data
Freebayes parameters for variant calling from individual timepoints data were -F 0.05 -C 3 and –pooled-continuous. For each of the mutations in the consensus lists, we calculated the natural logarithm of 2*evo/(anc-evo) per replicate assay and timepoint, where evo represents the evolved variant coverage and anc represents the ancestral variant coverage. We also used directly the FASTQ files from population sequencing to estimate the evolved variant fitness over the ancestral as follows. For each variant in the consensus list, we generated 12 search terms. The 12 terms were 20-base strings containing either the ancestral or the evolved allele, in the forward or reverse orientation and for three 5-base sliding windows centered around the variant position. For all search terms, aggregate counts were generated per allele and timepoint. In both cases, since we are mainly interested in heterozygous mutations, we assume that when anc > evo the population is a mix of individuals with the ancestral allele in homozygosity and heterozygosity with the evolved allele only. Mutations for which anc = evo or anc < evo in at least 1 timepoint were suggestive of being represented by at least a fraction of individuals homozygous for the evolved allele. Nevertheless, they were still included in the analysis as far as they resulted in at least 3 timepoints for which anc > evo, since that could be an artifact because of low locus coverage. We used linear regression to model the natural logarithm of heterozygotes over the ancestor [2*evo/(anc− evo)] over time, where the slope represents the fitness coefficient of the variant in heterozygosity. Linearity was assessed with the Durbin–Watson statistic.
Results
Diploid populations harbor recessive deleterious and lethal mutations
According to Haldane’s sieve, recessive beneficial mutations fail to fix in asexual diploid populations because selection cannot act on the heterozygote (Haldane 1924; Connallon and Hall 2018). By similar logic, selection should not prevent the accumulation of recessive deleterious mutations in asexual diploid populations either. To test whether diploid populations contain recessive deleterious mutations we subjected 17 diploid populations from Generation 4,000 and 9 clones from diploid populations from Generation 2,000 of a previously performed evolution experiment to a meiotic cycle (Marad et al. 2018). Although sporulation efficiency was uniformly high, segregant viability was not. At one extreme, Population C04 failed to produce a single viable spore across 10 tetrads, indicating a severe meiotic defect, while 5 of the 17 populations and 2 of the 9 clones had spore viability at least as high as the ancestor (Supplementary Table 2). Overall germination and colony size segregation patterns suggest that there are multiple recessive deleterious mutations.
Our strategy to quantify fitness effects of evolved mutations in diploids relies on reshuffling of the evolved mutations on the ancestral background, achieved via bulk-sporulation of evolved clones followed by bulk-mating of the spores to a haploid version of their ancestor (Fig. 1). Nevertheless, haplotypes are briefly exposed to haploidy, which may limit the recovery of haplotypes with recessive lethal mutations. Prior studies have shown that lethal haplotypes can be recovered as long as mating with a complementing strain is introduced soon after meiosis (Haarer et al. 2011). To show that we can recover recessive deleterious and recessive lethal mutations in our pools, we performed a pilot experiment, using six strains. Five of them were hemizygous for an essential gene, each implicated in one of the following biological processes: protein folding (CNS1), actin turnover (COF1), cytokinesis (IQG1), karyogamy and spindle pole body formation (KAR1), and secretion (SEC27), and one for a nonessential gene involved in N-glycosylation (YUR1) as a control (Supplementary Fig. 3). In all cases, we recovered the recessive-lethal mutation, though below the expected 1/2 rate (recovery ranged from 1/1,000 for sec27Δ to 1/8 for kar1Δ; Supplementary Fig. 3).
Fig. 1.
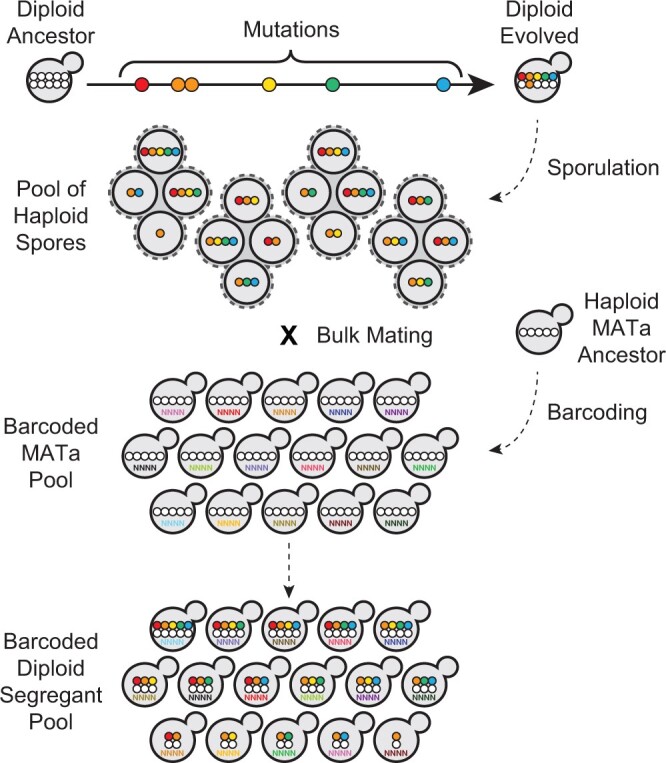
Strategy used to generate diploid segregant pools. Diploid evolved clones isolated from Generation 2000 were sporulated and mated en masse to barcoded MATα version of the ancestor. Each unique barcode represents a single mating event. The resulting diploids have genotypes intermediate to their ancestor and evolved parents and carry a barcode. Mutations homozygous in the evolved parent (in orange) end up in heterozygosity in all segregants. Supplementary Fig. 1 shows the barcoded locus.
Diploid evolved clones carry homozygous and heterozygous adaptive mutations
To identify beneficial mutations in diploid evolved populations, we performed bulk-sporulation of six parental clones from Generation 2,000, followed by bulk-matings to their ancestor, and created large pools of barcoded diploid segregants, each with a random combination of heterozygous mutations (Fig. 1). The six parental clones derived from Populations A05, A07, F04, and H06 were chosen on the basis of high fitness, high spore viability, and absence of aneuploidies in clones from the same generation (Marad et al. 2018). Because our strain background harbors the killer virus [a cytoplasmic dsRNA virus that can impact fitness (Buskirk et al. 2020)], we assayed killing ability and toxin sensitivity for each parental clone and monitored killing ability throughout pool construction (data for parental clones are shown in Table 1 and Supplementary Fig. 2a). We deemed that necessary to at least be aware of phenotypic changes that may be associated with the killer virus.
Table 1.
Population and clone phenotypes.
| Population | Estimated population fitness (%) a | Population killing activity b | Clone | Meiotic progeny viability (%) | Clone killing activity b |
|---|---|---|---|---|---|
| A05 | 5.47 | 0.67 | A05-C | 49.50 | 0.67 |
| A05 | 5.47 | 0.67 | A05-D | 50 | 0.33 |
| A07 | 5.60 | 0.67 | A07-C | 17.50 | 0.67 |
| H06 | 6.14 | 1 | H06-C | 51 | 1 |
| F04 | 6.41 | 1 | F04-C | 45 | 0.67 |
| F04 | 6.41 | 1 | F04-D | 92.50 | 1 |
Killing activity is scored as 0 (none), 0.33 (intermediate low), 0.67 (intermediate high), and 1 (same as in the ancestor).
Fitness values are from Marad et al. (2018).
Sensitivity assay showed that all clones were equally resistant to the killer toxin, and same as the ancestor
For each evolved parental clone, we generated two segregant pools: a “small pool” of ∼200 individuals and a “large pool” of >60,000 individuals. We used the small pools to quantify the segregant fitness via pooled competition assays and barcode sequencing (Venkataram et al. 2016) and the large pool to perform whole genome whole population sequencing. Barcoded derivatives of the ancestor and evolved parental clones were spiked into the segregant pools, which were then propagated under conditions identical to the evolution experiment for 110 generations in 2 replicates. In addition, we measured the fitness of the ancestor and evolved barcoded derivatives using a fluorescence-based fitness assay (Supplementary Fig. 2a). Fitness values from these independent assays are highly correlated (Supplementary Fig. 2b, R = 0.92).
Independently barcoded derivatives of evolved clones that originated from Population F04 displayed variability in fitness, while technical replicates were far more reproducible (Fig. 2; Supplementary Fig. 2). The strain background we employ harbors killer and helper viruses that can contribute to interesting evolutionary outcomes (Buskirk et al. 2020), but we are largely unaware of how transformation and meiosis affect inheritance of these elements. Motivated by the fitness variations of individually barcoded clones F04-C and F04-D, we checked the killer phenotypes of all barcoded control strains. Barcoded derivatives of evolved parental clones from Population F04 displayed variability in their killer phenotypes as well (Supplementary Fig. 2a includes killing activity data for Population F04 clones and barcoded derivatives origin can be traced). Clones from Population F04 carry mutations in KRE6, whose loss of function has been associated with increased resistance to killer toxins (Brown et al. 1993; Kasahara et al. 1994). Mutations in KRE6 and inconsistencies in the fitness and killer phenotypes on barcoded clones were not observed in other populations. Barcoding of diploid segregants was achieved via meiosis and mating to a barcoded haploid version of the ancestor, and that could also affect killer phenotypes and fitness. However, killer phenotypes were more uniform across diploid barcoded derivatives from clones of the F04 population. Based on that we assumed that meiosis contributed less instability in the killer-associated phenotypes and fitness.
Fig. 2.

Fitness distributions of diploids derived from mating between a haploid version of the ancestor and the meiotic progeny of evolved diploid clones. The assays were barcode-based and the fitness was estimated with the algorithm published in Venkataram et al. (2016). Fitness correlations are shown for 2 replicates per clone. Ancestor and evolved parents are annotated in cyan and orange, respectively. Derived diploids are annotated in gray. Clones for which there are WGS data available are annotated with a closed circle. Outliers were identified via boxplot and Rosner tests performed in R (Supplementary Table 3; Tables E1–E6 in Supplementary File 1). Pearson’s correlations for each replicate pair are shown at the top left of each panel.
Homozygous and heterozygous beneficial mutations affect the segregant fitness distribution in different ways. Homozygous mutations in the evolved parent end up as heterozygous mutations in all segregants (Fig. 1, orange mutation). In the case of a single homozygous beneficial mutation as the only mutation with a fitness effect, we expect a unimodal distribution of fitness in the segregants. This pattern is observed in the segregants from clones A05-C, A05-D, and A07-C (Fig. 2; Supplementary Table 3 and Tables E1–E6 in Supplementary File 1). We estimated the fitness of the beneficial mutation in homozygous state from the fitness difference between evolved and ancestor and in heterozygous state from the difference between the average segregant fitness and the ancestor fitness.
In contrast, the fitness distributions of segregants from the F04-C, F04-D, and H06-C evolved parental clones suggest fitness gains due to heterozygous adaptive mutations (Fig. 2). The evolved and ancestral parents are not detected as outliers in these populations, but as expected for a nontransgressive segregation, they are at the extremes of the segregant distributions (Wilcoxon P-values: ancestral—segregants comparisons <0.1 for individual replicates and segregants—evolved comparisons <0.04 for averaged replicates).
To identify candidate homozygous beneficial mutations in A05-C, A05-D, and A07-C and candidate heterozygous beneficial mutations in F04-C, F04-D, and H06-C, we sequenced the ancestral and evolved parental clones as well as a total of 160 segregants across the six small pools. We genotyped each segregant and identified the corresponding barcode sequence. The evolved parental clones carry on average 1 homozygous and 20 heterozygous mutations (all mutations shown in Supplementary Table 4). Both parental clones from population A05 have a single homozygous mutation in a previously identified common target of selection, ACE2 (Oud et al. 2013; Ratcliff et al. 2015; Fisher et al. 2018). Parental clone A07-C has 3 homozygous mutations, including in WHI3 and CTS1. CTS1 is the most common mutational target across diploid populations and CTS1 mutations are always observed in homozygosity due to their proximity to the highly recombinogenic rDNA locus (Marad et al. 2018). WHI3 is not a known target of selection. The third homozygous mutation in A07-C is at an intergenic position on chromosome II. Parental clone from population H06 carries heterozygous mutations in common targets of selection PDR5 and PTR2. Population F04 carries heterozygous nonsynonymous mutations in the common targets of selection ACE2, and KRE6 (Marad et al. 2018). Interestingly, the two parental clones from population F04 show subtly different behavior in all assays. Segregants from F04-C show more of a continuum of fitness values between ancestral and evolved parents (Fig. 2), with the evolved parental clone appearing as an outlier in one replicate. Furthermore, the genetic data suggest that F04-C, but not F04-D, carries at least one recessive deleterious mutation (Supplementary Table 2).
Most genome evolution in clonally evolved diploids is nonadaptive
In addition to identifying candidate beneficial mutations, we measured the fitness effects of all heterozygous mutations in each evolved clone using two methods. First, we combined the genotype information of a subset of segregants from WGS and the fitness from barcode sequencing. The fitness effect of each variant was estimated as the difference in mean fitness of segregants with and without the variant (Fig. 3; Supplementary Table 5). To assess significance of the effect we used Bonferroni-corrected t-tests and Wilcoxon rank-sum tests (Tables E7–E12 in Supplementary File 1). Few heterozygous mutations appeared significant in the evolved clones that harbored beneficial homozygous mutations, and none were significant after the Bonferroni correction (Tables E7–E9 in Supplementary File 1). In populations with heterozygous beneficial mutations, we initially identified up to four putatively significant mutations (Fig. 3). However, after correcting for cosegregation of mutations in our sequenced segregants (ANOVA, Tables E10–E12 in Supplementary File 1), we show that there are two adaptive mutations in each clone (Table 2). Clones from population F04 carry heterozygous beneficial mutations in ACE2 and KRE6 with average fitness effects 1.33% and 1.89%, respectively, for clone F04-C and 1.76% and 2.42%, respectively, for clone F04-D. Clone H06-C carries adaptive mutations in PDR5 and PTR2 with average fitness effects 3.57% and 2.03%, respectively.
Fig. 3.

Quantification of heterozygous variant fitness effects using time–course barcode sequencing and segregant WGS. Annotations reflect population and clone. Only mutations with data in both replicates are included. Mutations are ranked by mean average fitness for both replicates, where mean fitness was calculated as the difference evolved-ancestral allele mean fitness. Error bars represent 95% confidence intervals for replicate 1 on the left and replicate 2 on the right, of the t-test comparing evolved and ancestral alleles mean fitness. Mutations with neutral effect are considered those whose t-test confidence interval encompasses 0 of at least 1 replicate and are annotated in light blue. Of the rest, mutations with positive or negative effects are annotated in red and purple, respectively. Variant fitness data reported in Supplementary Table 5 and available genotyping information associated with segregant fitness are reported in Tables E7–E12 in Supplementary File 1.
Table 2.
Adaptive mutations in diploids.
| Data from segregant fitness and genotyping |
Data from overdominance analysis |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | Population | Evolved state | Heterozygous clone C | Homozygous clone C | Heterozygous clone D | Homozygous clone D | Evolved background heterozygous | Evolved background homozygous | Ancestral background heterozygous | Ancestral background homozygous |
| ace2-R617G | A05 | Homozygous | 1.65% | 4.48% | 0.97% | 4.22% | ND | ND | ND | ND |
| cts1-G109D | A07 | Homozygous | 2.15% | 5.36% | n/a | n/a | ND | ND | ND | ND |
| ace2-R669* | F04 | Heterozygous | 1.33% | n/a | 1.76% | n/a | 1.84% | 4.25% | ND | ND |
| kre6-S453L | F04 | Heterozygous | 1.89% | n/a | 2.42% | n/a | 5.06% | 4.30% | 7.25% | −11.28% |
| pdr5-F562I | H06 | Heterozygous | 3.57% | n/a | n/a | n/a | ND | ND | ND | ND |
| ptr2-V243fs | H06 | Heterozygous | 2.03% | n/a | n/a | n/a | ND | ND | ND | ND |
| pdr3-Y276S | F05 | Heterozygous | n/a | n/a | n/a | n/a | ND | ND | 4.81% | −9.36% |
| pse1-Q651* | G06 | Heterozygous | n/a | n/a | n/a | n/a | 2.27% | −6.10% | ND | ND |
| kre6-A521T | G06 | Heterozygous | n/a | n/a | n/a | n/a | 4.45% | ND | ND | ND |
To corroborate these results, we also quantified the fitness effects of heterozygous mutations by propagating the large segregant pools (∼60,000 segregants each for clones A05-C, A05-D, A07-C, F04-C, and F04-D) for ∼110 generations. We estimated the fitness effect by tracking the frequency of each mutation by WGS every 20 generations with a minimum of 60× coverage. These data were noisy and contained a high rate of both false-positives and false-negatives (Supplementary Tables 6 and 7). Nevertheless, the KRE6 mutation was identified in both clones from population F04, but in one replicate each and significance after correction holds only for the one clone. ACE2 appeared significant in a single replicate with both methodologies but was not significant after correction.
Overall, we find that the majority of heterozygous mutations (∼96%) in our evolved clones do not have a detectable effect on fitness, with the remaining ∼4% having modest fitness benefits ranging from 1.3% to 3.6% (Tables 2 and 3). Among homozygous mutations, which make up ∼10% of the total evolved, the proportion of beneficial mutations is much higher (38%) and their fitness effects are larger (4.2–5.4%). Considering both heterozygous and homozygous mutations, ∼6% have a detectable fitness effect in our evolved diploid parental clones.
Table 3.
Adaptive genome evolution in diploid and haploid populations.
| Background | Mutations a | Beneficial mutations a | Adaptive evolution (%) | Nonadaptive evolution (%) |
|---|---|---|---|---|
| Diploid | ||||
| Heterozygous | 139 | 6 | 4 | 96 |
| Homozygous | 8 | 3 | 38 | 62 |
| Total | 147 | 9 | 6 | 94 |
| Haploid | ||||
| Total | 116 | 24 | 21 | 79 |
We considered each clone as an independent genotype, therefore mutations that appear in both clones from the same population are counted twice. Otherwise, the number of hitchhikers would be artificially inflated relative to beneficial mutations.
Heterozygous beneficial mutations are frequently overdominant
Short-term evolution in diploids is predicted to favor selection for overdominant mutations (Sellis et al. 2011, 2016). We determined the dominance of candidate beneficial mutations using three approaches: forcing LOH in the evolved strains, backcrossing the evolved strains, and/or reconstructing the evolved mutations in the ancestral background. We selected putative beneficial mutations that satisfy the following three criteria based on our previous work (Marad et al. 2018). First, we chose mutations in common targets of selection. Second, we considered mutations in euploid populations. Third, we excluded populations with linked heterozygous mutations to avoid losing heterozygosity at multiple loci (Fisher et al. 2018).
We used CRISPR/Cas9 to force gene conversion of individual heterozygous evolved alleles within evolved strains toward homozygosity for either the ancestral or evolved allele. We performed competitive fitness assays on these strains and the evolved heterozygote to ascertain the fitness effects of the mutations in the context of the evolved background (Fig. 4a and Table 2). Three mutations (bck1-S945S, bst1-S740R, and ubp12-V279L) have no detectable fitness effect on the evolved background upon LOH in either direction (t-test, P > 0.3). Three mutations (pse1-Q6562*, kre6-S453L, and kre6-A521T) are overdominant, with highest fitness while in heterozygosity (t-test, P < 0.05 for the pse1-Q6562* and kre6-A521T alleles and P = 0.057 for the kre6-S453L allele). PSE1 had been previously identified as an adaptive target, exclusively in a heterozygous form (Fisher et al. 2018). Of those, we were unable to recover the homozygous mutant for kre6-A521T suggesting lethality. The last mutation (ace2-R669*) showed partial dominance. ACE2 is a common target that is observed in both heterozygous and homozygous forms (Marad et al. 2018).
Fig. 4.

Overdominance of diploid-evolved heterozygous mutations. Average fitness of LOH of putative beneficial mutations was determined by driving LOH in the evolved background (a) or by introgressing the evolved mutations into the ancestral background (b). Small markers represent individual fitness values from independently constructed strains and large markers represent the respective averages. The homozygous ancestral alleles are annotated by two open circles, heterozygous by one open and one white and homozygous evolved by two white circles. Open big circles in (b) annotate final segregant backcrossed to haploid version of the ancestor and filled circles annotate intra-ascus matings of the final cross.
Independently, we introgressed pdr3-Y276S and kre6-S453L in the ancestral background to generate heterozygotes and homozygotes. We chose pdr3-Y276S because this mutation cosegregated with a small colony phenotype (Supplementary Table 2). For each mutation, we performed five rounds of backcrossing to replace most of the evolved background with the ancestral, while selecting for the allele of interest. Haploid segregants from the fifth backcross were used to construct diploids heterozygous and homozygous for the query allele, whose fitness was determined against a fluorescent version of the diploid ancestor (Fig. 4b and Table 2). We found that both the pdr3-Y276S and the kre6-S453L mutants are beneficial (4.0% and 3.3%, respectively) when heterozygous, but strongly deleterious (−10.2% and −15.0%, respectively) when homozygous (Fig. 4b). Though both the forced LOH experiment and the backcrossing experiment show that the kre6-S453L mutation is overdominant, the deleterious effect of the homozygous mutation is far less severe in the evolved background (compare Fig. 4a with b).
Adaptive mutations are pleiotropic
Evolution experiments in yeast that favor selection of fast-settling mutants have targeted genes implicated in cell wall metabolism (Koschwanez et al. 2013; Ratcliff et al. 2015). Spatiotemporal heterogeneity arising from the lack of agitation during growth, leads to nutrient, and oxygen gradients, providing the selective forces driving adaptation (Lang et al. 2011; Frenkel et al. 2015). We observed that the genetic targets of selection in our system impinge on similar biological processes across ploidies, such as cell wall metabolism, drug transport and nutrient sensing, and signaling. Here, we sought to identify these common selective pressures, by modifying the evolutionary environment.
We measured the fitness of evolved clones H06-C and F04-D, as well as a reconstructed heterozygous kre6-S453L strain in the ancestral background, in environments that prohibit cell settling in order to disrupt spatial heterogeneity by agitating the 96-well plates during growth (Fig. 5a). We also experimented by removing the antibiotics, which were included to prevent bacterial contamination (Lang et al. 2011). Although we know that ampicillin and tetracyclin do not affect yeast growth, we are unaware on how their presence may affect adaptation. Clone H06-C displayed a strong tradeoff on the well-mixed environments, in which it had an almost 5% fitness deficit compared with its ancestor, while the presence of antibiotics in the growth medium did not further affect its fitness in either the well-mixed or the static environment. Clone F04-D is always more fit than its ancestor, but the fitness advantage was decreased without antibiotics and/or with agitation. Interestingly, the heterozygous kre6-S453L strain and the F04-D clone (which contains this mutation) had the lowest fitness in the well-mixed environment with antibiotics, suggesting that antibiotics contributed a relative fitness deficit in the well-mixed environment.
Fig. 5.

Fitness of evolved mutations changes upon environmental perturbation. Relative fitness was estimated against a fluorescent version of the respective ancestor. a) Fitness of evolved clones, H06-C and F04-D, and of KRE6 allele from population F04 engineered into the ancestral background were assayed in the different conditions as annotated on the first panel. T-test significance, annotated in black on top of the graphs, is with respect to the experimental condition (ns, not significant; *P < 0.05; **P < 0.001; ***P < 0.0001). T-test significance annotated in green is between the well-mixed (with and without antibiotics) environments. b) Mutations that emerged during haploid evolution (left panel) or loss-of-function mutations of genes mutated during haploid evolution (right panel) were engineered in the ancestral background and fitness was assayed in a static (evolutionary) and a well-mixed environment, shown on the x- and y-axis, respectively. Error bars represent SE from at least three replicate assays.
To determine if this was a general effect of cell wall mutations, we measured the fitness effects of cell wall mutants that had emerged during haploid evolutions, as well as null alleles of adaptive targets (Lang et al. 2013; Buskirk et al. 2017), in static and well-mixed environments (Fig. 5b, only in the presence of antibiotics). KRE6 and KRE5 mutations showed a range of tradeoffs, with KRE5 being the most compromising in the well-mixed environment (Fig. 5b, left panel). We also tested the fitness effects of null alleles for CNE1, ROT2, STE11, and YUR1, of which only YUR1 is involved in the cell wall metabolism. CNE1 and ROT2 deletions did not phenocopy the evolved alleles (Buskirk et al. 2017), as they displayed no fitness effect in the evolutionary condition. The STE11 and YUR1 deletions displayed a fitness advantage in the static environment, but only YUR1 manifested a strong tradeoff in the well-mixed environment. Collectively, these data suggest that adaptation in our environment that relies on modifications of the cell wall is typically lost under well-mixed environments.
To identify mutations with pleiotropic effects, we assayed fitness of the small pools of barcoded segregants under the different environments (Supplementary Fig. 4 and Tables 8–11; Tables E11–E14 in Supplementary File 1). We found that removing antibiotics causes a wider spread in the fitness distributions of both pools (Supplementary Fig. 4), observation for which we do not have an explanation. As expected, the evolved parent H06-C displayed strong trade-offs in the well-mixed environments, but only a few segregants appeared to approach the fitness deficits of the evolved parent, suggesting a multiallelic effect. Interestingly, PTR2 is adaptive in all environments, but PDR5 is adaptive only in static environments. In addition, a mutation in FLO9 appears to have a small fitness deficit in the well-mixed environments (−0.7% in three out of the four assays) and a mutation in ASK10 a small fitness advantage in the mixed environment without antibiotics. Assays of the segregant pool from clone F04-D supported the fitness advantage of KRE6 and ACE2 mutations, but in only one replicate of the evolutionary condition, presumably because of the scarcity of information in this round. Unexpectedly, ACE2 had a small positive effect (∼1%) in the well-mixed environments. KRE6 did not appear as consequential in any other environment. Three more mutations (a missense in SNU114, a missense in VPS41 and an intergenic mutation) had a significant deficit in a single environment each (in well-mixed no antibiotic, in well-mixed with antibiotic, and in the evolutionary condition, respectively) but only in one of the two replicates. In conclusion, it seems that the highest fitness mutations confer smaller effects in alternative environments, without necessarily being maladaptive, whereas fitness deficits may originate from molecular changes with no detectable fitness effect under the evolutionary environment.
Discussion
Recessive beneficial mutations are unlikely to become fixed in diploid populations, a phenomenon known as Haldane’s Sieve (Haldane 1924; Connallon and Hall 2018). Diploids, therefore, adapt more slowly than haploids (Zeyl et al. 2003; Gerstein et al. 2011) and have a different spectrum of genetic changes (Fisher et al. 2018; Marad et al. 2018; Johnson et al. 2021). Here, we quantified the fitness effects and the degree of dominance of all mutations that emerged in six clones during diploid evolution using bulk-segregant fitness assays, genetic reconstructions, and backcrossing. Collectively, four principles emerge from this study. First, diploid populations accumulate many recessive deleterious or lethal mutations that reduce spore viability. Second, all beneficial mutations in diploids display some degree of dominance with respect to fitness. Third, most heterozygous beneficial mutations are overdominant. Fourth, adaptation in our environment is predominately driven by spatial heterogeneity.
Our results support theoretical predictions and prior experimental results suggesting that heterozygous beneficial mutations are likely to be overdominant (Haldane 1924; Sellis et al. 2011, 2016; Johnson et al. 2021). Even those mutations that are not overdominant (like ace2 in Population F04) are at least partially dominant. We previously observed the inverse pattern for beneficial mutations that evolved in haploid populations: they are either recessive or underdominant (Buskirk et al. 2017; Fisher et al. 2018; Marad et al. 2018). Using data from our diploid, autodiploid and haploid datasets (Lang et al. 2013; Buskirk et al. 2017; Fisher et al. 2018; Marad et al. 2018), we summarize the patterns of dominance for haploid and diploid laboratory evolution experiments (Fig. 6). Beneficial mutations that arose on the haploid and diploid backgrounds occupy separate but overlapping regions of this space. Note that the same genes often underlie adaptation in both haploids and diploids, but their dominance effects depend on the background in which they arose.
Fig. 6.

Dominance of beneficial mutations can be predicted by the ploidy of the background in which they were selected. Stepwise fitness gains of beneficial mutations from homozygous ancestral to homozygous mutant are shown. The mutations were identified as adaptive in diploid, autodiploid, and haploid datasets (Lang et al. 2013; Buskirk et al. 2017; Fisher et al. 2018; Marad et al. 2018). The ploidy of the background and the zygosity of the mutation at selection are annotated, and selected genes are identified by name. Transparency has been applied at overlapping points for clarity. Overdominant mutations are in the orange-shaded region and arise exclusively as heterozygous mutations in diploids. Only diploids have access to these mutations. Partially dominant mutations are in the green-shaded region. Both haploids and diploids have access to these mutations. Recessive mutations and underdominant mutations are in the brown-shaded and purple-shaded regions, respectively, and arise exclusively in haploid backgrounds.
We show that a bulk-segregant approach can be used to identify and quantify the fitness effects of all heterozygous mutations in evolved diploid clones. By mating immediately after breaking the tetrad asci, our method can recover recessive lethal mutations in the final pools, an essential feature for the recovery of strong effect overdominant alleles. Tetrad dissection data show that our evolved clones harbor recessive deleterious and/or recessive lethal mutations (Supplementary Table 2). According to our proof-of-principle experiment with known recessive lethal mutations (Supplementary Fig. 3), alleles with strong deleterious effects may be under-represented in our pools. We used the WGS information from the sequenced segregants and the first sequenced timepoint from the population data to search for under-represented nonsynonymous mutations in coding sequences (Supplementary Fig. 5 and Table 12). Mutations in four genes (HIS6, CEP3, GAS5, and POR1) are under-represented among the segregants as estimated by both sequencing of the segregant pool and individual segregants. Of those the frameshift mutation in CEP3 is expected to be deleterious, as the gene encodes an essential kinetochore protein. The rest of the mutations are missense, making it difficult to interpret potential deleterious effects. HIS6 and GAS5 are involved in histidine and cell wall biosynthesis, respectively, and POR1 encodes for a mitochondrial porin.
By including the ancestor and evolved diploid parents into the segregant pools, we can identify homozygous beneficial mutations in the evolved parent and quantify their combined fitness effects. This is a useful feature of our assay because, although homozygous mutations contributed only ∼5% of the total genetic variation, they contributed ∼33% of the adaptive variation (Table 3). Additionally, the method can identify adaptive variation that is missed by approaches that identify genes mutated more often than expected by chance across replicate populations, such as PDR5 and PTR2.
Time–course sequencing of segregant pools assays were less reproducible in diploid segregant pools with 60× to 360× whole-genome coverage compared with haploid segregant pools sequenced with 100× coverage (Buskirk et al. 2017). We attributed this to the higher precision requirements for accurate fitness prediction of heterozygous mutations. For example, an evolved mutation in a haploid segregant pool starts at 50% and fixes at 100%, whereas an evolved heterozygous mutation in a diploid segregant pool starts at 25% and fixes at 50%. In addition, LOH rates are high and more variable across the genome, compared with point mutations (Lee et al. 2009; Peter et al. 2018), which would add noise to this approach. Despite the noise in our bulk-fitness assays, both the fitness distributions of all segregants and the genotyping of individual segregants are consistent with a small number of adaptive mutations in our diploid populations.
The differences in adaptation rates as suggested by the fitness increases during the diploid and haploid evolutions are striking: diploid fitness increased by an average of 5.8% over 4,000 generations (or ∼1.5% per 1,000 generations; fitness increase was linear), whereas haploid fitness increased by 8.5% over 1,000 generations (of which ∼6% was gained within the first 500 generations) (Marad et al. 2018). Because diploids are more fit compared with haploids (Venkataram et al. 2016; Fisher et al. 2018) some of this difference in adaptation rate may be attributable to diminishing returns (Kryazhimskiy et al. 2014; Johnson et al. 2019, 2021). Nevertheless, we find that most of the variation that emerges during short-term clonal evolution in diploids does not contribute to adaptation, as only 6% of the identified mutations have a detectable effect on fitness. We identify both heterozygous and homozygous beneficial mutations. Consistent with our work in haploids we did not identify any beneficial synonymous or intergenic mutations in diploids. Diploid populations also carry a load of recessive-deleterious and recessive-lethal mutations. In contrast, 20% of mutations in haploid populations were shown to be beneficial and <1% deleterious (Buskirk et al. 2017).
By altering specific environmental parameters, we found that cell settling at the bottom of the vessel exerts a dominant selective pressure, driving adaptation in our system. Populations growing statically in liquid media are subjected to nutrient and oxygen gradients, as well as the spatial structure imposed by the shape of the well, which has been previously shown to drive evolutionary dynamics and adaptations in that and similar systems (Ratcliff et al. 2012; Frenkel et al. 2015). When adapted clones were exposed to a well-mixed environment, they lost their competitive advantage over their ancestor. However, the specific patterns of the evolved parents and their segregants’ fitness effects across environments suggest complex origins of these trade-offs, due only in part to the adaptive mutations. Trade-offs have been observed before upon reversion of the selective pressures that drive adaptation in a system. Specific examples include adaptations driven by carbon limitation resulting in fitness costs in carbon excess (Wenger et al. 2011), loss of signaling/regulatory pathways driven by environmental constancy (chemostat) but becoming maladaptive in oscillating environments (batch culture) (Kvitek and Sherlock 2013) and adaptive responses to high salinity (Hietpas et al. 2013).
Maintenance of heterozygosity in overdominant loci is affected by the rate of mitotic recombination during asexual propagation and the frequency of sexual reproduction (Sellis et al. 2011, 2016; Fisher et al. 2021). In fully asexual diploid populations, LOH depends on mitotic recombination rates in combination with the distribution of fitness and dominance effects of linked heterozygous loci (Fisher et al. 2021). In sexual populations, the maintenance of heterozygosity is affected by the frequency of sexual reproduction and the degree of outcrossing. During evolution experiments, frequent sexual cycles (1 every 30–120 generations) purge deleterious variants (including recessive and overdominant) (Burke et al. 2014; Kosheleva and Desai 2018). However, in nature, Saccharomyces yeasts undergo infrequent sexual cycles (in the order of 1 every 1,000 mitotic divisions) (Tsai et al. 2008; Lee et al. 2022), and while most mating events result from inbreeding (Ruderfer et al. 2006; Tsai et al. 2008), the exact rate of outcrossing depends on the environment (Magwene et al. 2011a), In fact, both theory and simulation have shown that even in obligate sexual populations, overdominant mutations can be maintained by balancing selection for thousands of generations (Sellis et al. 2011). All these suggest that overdominant mutations are likely to contribute disproportionately to short-term adaptation in diploid populations.
Data availability
The raw short-read sequencing data reported in this paper are deposited under accession no. PRJNA775967 in the NCBI BioProject database. Custom code for barcode sequencing data processing https://github.com/Dangeli/Barcode-counting.
Supplemental material is available at GENETICS online.
Supplementary Material
Acknowledgments
We wish to thank Ryan Vignogna for help with the design of CRISPR/Cas9 replacement constructs, all members of the Lang Lab for feedback on the manuscript and David Amberg and Brian Haarer from SUNY Upstate Medical University for providing hemizygote strains from the Euroscarf collection.
Funding
Portions of this research were conducted on Lehigh University’s Research Computing infrastructure partially supported by National Science Foundation Award 2019035. This study was supported by grants from the National Institutes of Health R01GM127420 (GIL) and R01AI164530 (SFL), and by the National Institute of Standards and Technology (SFL) and the Department of Energy (SFL).
Conflicts of interest
None declared.
Contributor Information
Dimitra Aggeli, Department of Biological Sciences, Lehigh University, Bethlehem, PA 18015, USA.
Daniel A Marad, Department of Biological Sciences, Lehigh University, Bethlehem, PA 18015, USA.
Xianan Liu, Joint Initiative for Metrology in Biology, Stanford, CA 94025, USA; SLAC National Accelerator Laboratory, Menlo Park, CA, 94025, USA.
Sean W Buskirk, Department of Biological Sciences, Lehigh University, Bethlehem, PA 18015, USA; Department of Biology, West Chester University, West Chester, PA 19383, USA.
Sasha F Levy, Joint Initiative for Metrology in Biology, Stanford, CA 94025, USA; SLAC National Accelerator Laboratory, Menlo Park, CA, 94025, USA.
Gregory I Lang, Department of Biological Sciences, Lehigh University, Bethlehem, PA 18015, USA.
Literature cited
- Assaf ZJ, Petrov DA, Blundell JR. Obstruction of adaptation in diploids by recessive, strongly deleterious alleles. Proc Natl Acad Sci U S A. 2015;112(20):E2658–E2666. doi: 10.1073/pnas.1424949112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One. 2015;10(5):e0128036. doi: 10.1371/journal.pone.0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M, Shaket L, Anzai IA, Adesina O, Barstow B. Rapid construction of a whole-genome transposon insertion collection for Shewanella oneidensis by knockout Sudoku. Nat Commun. 2016;7:13270. doi: 10.1038/ncomms13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell JR, Schwartz K, Francois D, Fisher DS, Sherlock G, Levy SF. The dynamics of adaptive genetic diversity during the early stages of clonal evolution. Nat Ecol Evol. 2019;3(2):293–301. doi: 10.1038/s41559-018-0758-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer MJ, Christianson CM, Pai DA, Dunham MJ. Mapping novel traits by array-assisted bulk segregant analysis in Saccharomyces cerevisiae. Genetics. 2006;173(3):1813–1816. doi: 10.1534/genetics.106.057927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Kossaczka Z, Jiang B, Bussey H. A mutational analysis of killer toxin resistance in Saccharomyces cerevisiae identifies new genes involved in cell wall (1–>6)-beta-glucan synthesis. Genetics. 1993;133(4):837–849. doi: 10.1093/genetics/133.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke MK, Liti G, Long AD. Standing genetic variation drives repeatable experimental evolution in outcrossing populations of Saccharomyces cerevisiae. Mol Biol Evol. 2014;31(12):3228–3239. doi: 10.1093/molbev/msu256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buskirk SW, Peace RE, Lang GI. Hitchhiking and epistasis give rise to cohort dynamics in adapting populations. Proc Natl Acad Sci U S A. 2017;114(31):8330–8335. doi: 10.1073/pnas.1702314114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buskirk SW, Rokes AB, Lang GI. Adaptive evolution of nontransitive fitness in yeast. eLife. 2020;9:e62238.doi: 10.7554/eLife.62238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou H-H, Chiu H-C, Delaney NF, Segrè D, Marx CJ. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science. 2011;332(6034):1190–1192. doi: 10.1126/science.1203799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connallon T, Hall MD. Genetic constraints on adaptation: a theoretical primer for the genomics era. Ann N Y Acad Sci. 2018;1422(1):65–87. doi: 10.1111/nyas.13536. [DOI] [PubMed] [Google Scholar]
- Cubillos FA, Parts L, Salinas F, Bergström A, Scovacricchi E, Zia A, Illingworth CJR, Mustonen V, Ibstedt S, Warringer J, et al. High-resolution mapping of complex traits with a four-parent advanced intercross yeast population. Genetics. 2013;195(3):1141–1155. doi: 10.1534/genetics.113.155515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai MM, Fisher DS, Murray AW. The speed of evolution and maintenance of variation in asexual populations. Curr Biol. 2007;17(5):385–394. doi: 10.1016/j.cub.2007.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai MM, Fisher DS. Beneficial mutation–selection balance and the effect of linkage on positive selection. Genetics. 2007;176(3):1759–1798. doi: 10.1534/genetics.106.067678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschbauer AM, Jaramillo DF, Proctor M, Kumm J, Hillenmeyer ME, Davis RW, Nislow C, Giaever G. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169(4):1915–1925. doi: 10.1534/genetics.104.036871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenreich IM, Torabi N, Jia Y, Kent J, Martis S, Shapiro JA, Gresham D, Caudy AA, Kruglyak L. Dissection of genetically complex traits with extremely large pools of yeast segregants. Nature. 2010;464(7291):1039–1042. doi: 10.1038/nature08923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans GA, Lewis KA. Physical mapping of complex genomes by cosmid multiplex analysis. Proc Natl Acad Sci U S A. 1989;86(13):5030–5034. doi: 10.1073/pnas.86.13.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher KJ, Buskirk SW, Vignogna RC, Marad DA, Lang GI. Adaptive genome duplication affects patterns of molecular evolution in Saccharomyces cerevisiae. PLoS Genet. 2018;14(5):e1007396. doi: 10.1371/journal.pgen.1007396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher KJ, Kryazhimskiy S, Lang GI. Detecting genetic interactions using parallel evolution in experimental populations. Philos Trans R Soc Lond B Biol Sci. 2019;374(1777):20180237.doi: 10.1098/rstb.2018.0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher KJ, Vignogna RC, Lang GI. Overdominant mutations restrict adaptive loss of heterozygosity at linked loci. Genome Biol Evol. 2021;13(8):evab181.doi: 10.1093/gbe/evab181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel EM, McDonald MJ, Van Dyken JD, Kosheleva K, Lang GI, Desai MM. Crowded growth leads to the spontaneous evolution of semistable coexistence in laboratory yeast populations. Proc Natl Acad Sci U S A. 2015;112(36):11306–11311. doi:10.1073/pnas.1506184112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galitski T, Saldanha AJ, Styles CA, Lander ES, Fink GR. Ploidy regulation of gene expression. Science. 1999;285(5425):251–254. doi: 10.1126/science.285.5425.251. [DOI] [PubMed] [Google Scholar]
- Gerstein AC, Chun H-JE, Grant A, Otto SP. Genomic convergence toward diploidy in Saccharomyces cerevisiae. PLoS Genet. 2006;2(9):e145.doi: 10.1371/journal.pgen.0020145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, Cleathero LA, Mandegar MA, Otto SP. Haploids adapt faster than diploids across a range of environments: haploids adapt faster than diploids. J Evol Biol. 2011;24(3):531–540. doi: 10.1111/j.1420-9101.2010.02188.x. [DOI] [PubMed] [Google Scholar]
- Gerstein AC, Otto SP. Cryptic fitness advantage: diploids invade haploid populations despite lacking any apparent advantage as measured by standard fitness assays. PLoS One. 2011;6(12):e26599. doi: 10.1371/journal.pone.0026599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, Lo DS, Otto SP. Parallel genetic changes and nonparallel gene–environment interactions characterize the evolution of drug resistance in yeast. Genetics. 2012;192(1):241–252. doi: 10.1534/genetics.112.142620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, Kuzmin A, Otto SP. Loss-of-heterozygosity facilitates passage through Haldane’s sieve for Saccharomyces cerevisiae undergoing adaptation. Nat Commun. 2014;5:3819.doi:10.1038/ncomms4819. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2(1):31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- de Godoy LMF, Olsen JV, Cox J, Nielsen ML, Hubner NC, Fröhlich F, Walther TC, Mann M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455(7217):1251–1254. doi: 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- Good BH, McDonald MJ, Barrick JE, Lenski RE, Desai MM. The dynamics of molecular evolution over 60,000 generations. Nature. 2017;551(7678):45–50. doi: 10.1038/nature24287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter FA, Derks MFL, van den Heuvel J, Aarts MGM, Zwaan BJ, de Ridder D, de Visser JAGM. Genomics of adaptation depends on the rate of environmental change in experimental yeast populations. Mol Biol Evol. 2017;34(10):2613–2626. doi: 10.1093/molbev/msx185. [DOI] [PubMed] [Google Scholar]
- Haarer B, Aggeli D, Viggiano S, Burke DJ, Amberg DC. Novel interactions between actin and the proteasome revealed by complex haploinsufficiency. PLoS Genet. 2011;7(9):e1002288. doi: 10.1371/journal.pgen.1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS. A mathematical theory of natural and artificial selection. Part II. The influence of partial self-fertilisation, inbreeding, assortative mating, and selective fertilisation on the composition of mendelian populations, and on natural selection. Biol Rev. 1924;1(3):158–163. doi: 10.1111/j.1469-185X.1924.tb00546.x. [DOI] [Google Scholar]
- Hanson SJ, Wolfe KH. An evolutionary perspective on yeast mating-type switching. Genetics. 2017;206(1):9–32. doi: 10.1534/genetics.117.202036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietpas RT, Bank C, Jensen JD, Bolon DNA. Shifting fitness landscapes in response to altered environments: fitness landscapes in altered environments. Evolution. 2013;67(12):3512–3522. doi: 10.1111/evo.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CH, Magtanong L, Barker SL, Gresham D, Nishimura S, Natarajan P, Koh JLY, Porter J, Gray CA, Andersen RJ, et al. A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat Biotechnol. 2009;27(4):369–377. doi: 10.1038/nbt.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope EA, Dunham MJ. Ploidy-regulated variation in biofilm-related phenotypes in natural isolates of Saccharomyces cerevisiae. G3 (Bethesda). 2014;4(9):1773–1786. doi: 10.1534/g3.114.013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerison ER, Kryazhimskiy S, Mitchell JK, Bloom JS, Kruglyak L, Desai MM. Genetic variation in adaptability and pleiotropy in budding yeast. eLife. 2017;6:e27167.doi: 10.7554/eLife.27167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MS, Martsul A, Kryazhimskiy S, Desai MM. Higher-fitness yeast genotypes are less robust to deleterious mutations. Science. 2019;366(6464):490–493. doi: 10.1126/science.aay4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MS, Gopalakrishnan S, Goyal J, Dillingham ME, Bakerlee CW, Humphrey PT, Jagdish T, Jerison ER, Kosheleva K, Lawrence KR, et al. Phenotypic and molecular evolution across 10,000 generations in laboratory budding yeast populations. eLife. 2021;10:e63910. doi: 10.7554/eLife.63910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao KC, Sherlock G. Molecular characterization of clonal interference during adaptive evolution in asexual populations of Saccharomyces cerevisiae. Nat Genet. 2008;40(12):1499–1504. doi: 10.1038/ng.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara S, Ben Inoue S, Mio T, Yamada T, Nakajima T, Ichishima E, Furuichi Y, Yamada H. Involvement of cell wall β-glucan in the action of HM-1 killer toxin. FEBS Lett. 1994;348(1):27–32. doi: 10.1016/0014-5793(94)00575-3. [DOI] [PubMed] [Google Scholar]
- Khan AI, Dinh DM, Schneider D, Lenski RE, Cooper TF. Negative epistasis between beneficial mutations in an evolving bacterial population. Science. 2011;332(6034):1193–1196. doi:10.1126/science.1203801. [DOI] [PubMed] [Google Scholar]
- Koschwanez JH, Foster KR, Murray AW. Improved use of a public good selects for the evolution of undifferentiated multicellularity. Elife. 2013;2:e00367.doi: 10.7554/eLife.00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosheleva K, Desai MM. Recombination alters the dynamics of adaptation on standing variation in laboratory yeast populations. Mol Biol Evol. 2018;35(1):180–201. doi: 10.1093/molbev/msx278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryazhimskiy S, Rice DP, Jerison ER, Desai MM. Global epistasis makes adaptation predictable despite sequence-level stochasticity. Science. 2014;344(6191):1519–1522. doi: 10.1126/science.1250939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitek DJ, Sherlock G. Whole genome, whole population sequencing reveals that loss of signaling networks is the major adaptive strategy in a constant environment. PLoS Genet. 2013;9(11):e1003972. doi: 10.1371/journal.pgen.1003972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Botstein D, Desai MM. Genetic variation and the fate of beneficial mutations in asexual populations. Genetics. 2011;188(3):647–661. doi: 10.1534/genetics.111.128942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Rice DP, Hickman MJ, Sodergren E, Weinstock GM, Botstein D, Desai MM. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature. 2013;500(7464):571–574. doi: 10.1038/nature12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TJ, , LiuY, , LiuW-A, , LinY-F, , LeeH-H, , KeH-M, , HuangJ-P, , LuJ, , HsiehC-L, , Chung K-F, et al. Extensive sampling of Saccharomyces cerevisiae in Taiwan reveals ecology and evolution of predomesticated lineages. Genome Res. 2022; 10.1101/gr.276286.121 35361625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PS, Greenwell PW, Dominska M, Gawel M, Hamilton M, Petes TD. A fine-structure map of spontaneous mitotic crossovers in the yeast Saccharomyces cerevisiae. PLoS Genet. 2009;5(3):e1000410. doi: 10.1371/journal.pgen.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenski RE. Experimental evolution and the dynamics of adaptation and genome evolution in microbial populations. ISME J. 2017;11(10):2181–2194. doi: 10.1038/ismej.2017.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SF, Blundell JR, Venkataram S, Petrov DA, Fisher DS, Sherlock G. Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature. 2015;519(7542):181–186. doi: 10.1038/nature14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Liu Z, Dziulko AK, Li F, Miller D, Morabito RD, Francois D, Levy SF. iSeq 2.0: a modular and interchangeable toolkit for interaction screening in yeast. Cell Syst. 2019;8(4):338–344.e8. doi: 10.1016/j.cels.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddamsetti R, Lenski RE, Barrick JE. Adaptation, clonal interference, and frequency-dependent interactions in a long-term evolution experiment with Escherichia coli. Genetics. 2015;200(2):619–631. doi: 10.1534/genetics.115.176677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magwene PM, Kayıkçı Ö, Granek JA, Reininga JM, Scholl Z, Murray D. Outcrossing, mitotic recombination, and life-history trade-offs shape genome evolution in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2011a;108(5):1987–1992. doi: 10.1073/pnas.1012544108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magwene PM, Willis JH, Kelly JK. The statistics of bulk segregant analysis using next generation sequencing. PLoS Comput Biol. 2011b;7(11):e1002255.doi: 10.1371/journal.pcbi.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marad DA, Buskirk SW, Lang GI. Altered access to beneficial mutations slows adaptation and biases fixed mutations in diploids. Nat Ecol Evol. 2018;2(5):882–889. doi: 10.1038/s41559-018-0503-9. [DOI] [PubMed] [Google Scholar]
- Matsui T, Mullis MN, Roy KR, Hale JJ, Schell R, Levy SF, Ehrenreich IM. The interplay of additivity, dominance, and epistasis on fitness in a diploid yeast cross. Nat Commun. 2022;13(1):1463.doi: 10.1038/s41467-022-29111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald MJ, Rice DP, Desai MM. Sex speeds adaptation by altering the dynamics of molecular evolution. Nature. 2016;531(7593):233–236. doi: 10.1038/nature17143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Ba AN, Cvijović I, Rojas Echenique JI, Lawrence KR, Rego-Costa A, Liu X, Levy SF, Desai MM. High-resolution lineage tracking reveals travelling wave of adaptation in laboratory yeast. Nature. 2019;575(7783):494–499. doi: 10.1038/s41586-019-1749-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishant KT, Wei W, Mancera E, Argueso JL, Schlattl A, Delhomme N, Ma X, Bustamante CD, Korbel JO, Gu Z, et al. The Baker’s yeast diploid genome is remarkably stable in vegetative growth and meiosis. PLoS Genet. 2010;6(9):e1001109. doi: 10.1371/journal.pgen.1001109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA, Otto SP. Does diploidy increase the rate of adaptation? Genetics. 1994;136(4):1475–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oud B, Guadalupe-Medina V, Nijkamp JF, de Ridder D, Pronk JT, van Maris AJA, Daran J-M. Genome duplication and mutations in ACE2 cause multicellular, fast-sedimenting phenotypes in evolved Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2013;110(45):E4223–E4231. doi: 10.1073/pnas.1305949110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen C, Sunshine AB, Ong GT, Pogachar JL, Zhao W, Dunham MJ. High-throughput identification of adaptive mutations in experimentally evolved yeast populations. PLoS Genet. 2016;12(10):e1006339. doi: 10.1371/journal.pgen.1006339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J, De Chiara M, Friedrich A, Yue J-X, Pflieger D, Bergström A, Sigwalt A, Barre B, Freel K, Llored A, et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature. 2018;556(7701):339–344. doi: 10.1038/s41586-018-0030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliff WC, Denison RF, Borrello M, Travisano M. Experimental evolution of multicellularity. Proc Natl Acad Sci U S A. 2012;109(5):1595–1600. doi: 10.1073/pnas.1115323109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliff WC, Fankhauser JD, Rogers DW, Greig D, Travisano M. Origins of multicellular evolvability in snowflake yeast. Nat Commun. 2015;6:6102.doi: 10.1038/ncomms7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderfer DM, Pratt SC, Seidel HS, Kruglyak L. Population genomic analysis of outcrossing and recombination in yeast. Nat Genet. 2006;38(9):1077–1081. doi: 10.1038/ng1859. [DOI] [PubMed] [Google Scholar]
- Sellis D, Callahan BJ, Petrov DA, Messer PW. Heterozygote advantage as a natural consequence of adaptation in diploids. Proc Natl Acad Sci U S A. 2011;108(51):20666–20671. doi: 10.1073/pnas.1114573108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellis D, Kvitek DJ, Dunn B, Sherlock G, Petrov DA. Heterozygote advantage is a common outcome of adaptation in Saccharomyces cerevisiae. Genetics. 2016;203(3):1401–1413. doi: 10.1534/genetics.115.185165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki AM, Maruvka YE, Richmond PA, Guillet M, Shoresh N, Sorenson AL, De S, Kishony R, Michor F, Dowell R, et al. Polyploidy can drive rapid adaptation in yeast. Nature. 2015;519(7543):349–352. doi: 10.1038/nature14187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp NP, Sandell L, James CG, Otto SP. The genome-wide rate and spectrum of spontaneous mutations differ between haploid and diploid yeast. Proc Natl Acad Sci U S A. 2018;115(22):E5046–E5055. doi: 10.1073/pnas.1801040115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigwalt A, Caradec C, Brion C, Hou J, de Montigny J, Jung P, Fischer G, Llorente B, Friedrich A, Schacherer J, et al. Dissection of quantitative traits by bulk segregant mapping in a protoploid yeast species FEMS Yeast Res. 2016;16(5):fow056. doi: 10.1093/femsyr/fow056. [DOI] [PubMed] [Google Scholar]
- Tsai IJ, Bensasson D, Burt A, Koufopanou V. Population genomics of the wild yeast Saccharomyces paradoxus: quantifying the life cycle. Proc Natl Acad Sci U S A. 2008;105(12):4957–4962. doi: 10.1073/pnas.0707314105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataram S, Dunn B, Li Y, Agarwala A, Chang J, Ebel ER, Geiler-Samerotte K, Hérissant L, Blundell JR, Levy SF, et al. Development of a comprehensive genotype-to-fitness map of adaptation-driving mutations in yeast. Cell. 2016;166(6):1585–1596.e22. doi: 10.1016/j.cell.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger JW, Schwartz K, Sherlock G. Bulk segregant analysis by high-throughput sequencing reveals a novel xylose utilization gene from Saccharomyces cerevisiae. PLoS Genet. 2010;6(5):e1000942.doi: 10.1371/journal.pgen.1000942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger JW, Piotrowski J, Nagarajan S, Chiotti K, Sherlock G, Rosenzweig F. Hunger artists: yeast adapted to carbon limitation show trade-offs under carbon sufficiency. PLoS Genet. 2011;7(8):e1002202. doi: 10.1371/journal.pgen.1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wideman JG, Novick A, Muñoz-Gómez SA, Doolittle WF. Neutral evolution of cellular phenotypes. Curr Opin Genet Dev. 2019;58–59:87–94. doi: 10.1016/j.gde.2019.09.004. [DOI] [PubMed] [Google Scholar]
- Yona AH, Manor YS, Herbst RH, Romano GH, Mitchell A, Kupiec M, Pilpel Y, Dahan O. Chromosomal duplication is a transient evolutionary solution to stress. Proc Natl Acad Sci U S A. 2012;109(51):21010–21015. doi: 10.1073/pnas.1211150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeyl C, Vanderford T, Carter M. An evolutionary advantage of haploidy in large yeast populations. Science. 2003;299(5606):555–558. doi: 10.1126/science.1078417. [DOI] [PubMed] [Google Scholar]
- Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30(5):614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Liu Z, Levy SF, Wu S. Bartender: a fast and accurate clustering algorithm to count barcode reads. Bioinformatics. 2018;34(5):739–747. doi: 10.1093/bioinformatics/btx655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YO, Sherlock G, Petrov DA. Whole genome analysis of 132 clinical Saccharomyces cerevisiae strains reveals extensive ploidy variation. G3 (Bethesda). 2016;6(8):2421–2434. doi:10.1534/g3.116.029397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zörgö E, Gjuvsland A, Cubillos FA, Louis EJ, Liti G, Blomberg A, Omholt SW, Warringer J. Life history shapes trait heredity by accumulation of loss-of-function alleles in yeast. Mol Biol Evol. 2012;29(7):1781–1789. doi: 10.1093/molbev/mss019. [DOI] [PubMed] [Google Scholar]
- Zörgö E, Chwialkowska K, Gjuvsland AB, Garré E, Sunnerhagen P, Liti G, Blomberg A, Omholt SW, Warringer J. Ancient evolutionary trade-offs between yeast ploidy states. PLoS Genet. 2013;9(3):e1003388. doi: 10.1371/journal.pgen.1003388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw short-read sequencing data reported in this paper are deposited under accession no. PRJNA775967 in the NCBI BioProject database. Custom code for barcode sequencing data processing https://github.com/Dangeli/Barcode-counting.
Supplemental material is available at GENETICS online.