

Abstract
The dark-eyed junco (Junco hyemalis) is one of the most common passerines of North America, and has served as a model organism in studies related to ecophysiology, behavior, and evolutionary biology for over a century. It is composed of at least 6 distinct, geographically structured forms of recent evolutionary origin, presenting remarkable variation in phenotypic traits, migratory behavior, and habitat. Here, we report a high-quality genome assembly and annotation of the dark-eyed junco generated using a combination of shotgun libraries and proximity ligation Chicago and Dovetail Hi-C libraries. The final assembly is ∼1.03 Gb in size, with 98.3% of the sequence located in 30 full or nearly full chromosome scaffolds, and with a N50/L50 of 71.3 Mb/5 scaffolds. We identified 19,026 functional genes combining gene prediction and similarity approaches, of which 15,967 were associated to GO terms. The genome assembly and the set of annotated genes yielded 95.4% and 96.2% completeness scores, respectively when compared with the BUSCO avian dataset. This new assembly for J. hyemalis provides a valuable resource for genome evolution analysis, and for identifying functional genes involved in adaptive processes and speciation.
Keywords: dark-eyed junco, Junco hyemalis, genome assembly, Hi-C
The dark-eyed junco (Junco hyemalis) is one of the most common passerines of North America, and has served as a model organism in different research disciplines for over a century. Here we report a high-quality genome assembly and annotation of the dark-eyed junco generated using a combination of shotgun and proximity ligation libraries. This new assembly for J. hyemalis provides a valuable resource for genome evolution analysis, and for identifying functional genes involved in adaptive processes and speciation.
Introduction
The dark-eyed junco (Junco hyemalis) is a common and widespread North American passerine that has been the subject of extensive research in multiple scientific disciplines for over 100 years (Miller 1941; Ketterson and Atwell 2016). The species present at least 6 distinct and geographically structured forms, showing marked levels of divergence in plumage coloration, habitat and life-history traits, including the timing of reproduction or migratory behavior (Fig. 1; Supplementary Table 1; Miller 1941; Nolan et al. 2002). These forms represent independent evolutionary lineages that radiated after the Last Glacial Maximum circa 18,000 years ago during a northward recolonization of the North American continent (Milá et al. 2007; Friis et al. 2022). Previous studies based on genome-wide data have documented a unique case of rapid diversification driven by the combined effects of sexual selection, ecological habitat features, and historical demographic factors (Friis et al. 2018; Friis and Milá 2020). The dark-eyed junco has also served as a model organism for the study of endocrine, neurological and behavioral aspects of migration, reproductive phenology (e.g. Ketterson and Nolan 1976; Cristol et al. 2003; Fudickar et al. 2017; Singh et al. 2021) and other life-history traits (Ketterson and Nolan 1999); ecophysiology (e.g. Stager et al. 2015, 2021); the hormonal basis of phenotypic variation, including traits related to sexual dimorphism, courtship behavior and mate choice (e.g. Ketterson et al. 1996, 2005, 2009); differential gene expression associated with pigmentation (Abolins-Abols et al. 2018); and phenotypic plasticity and adaptation to urban environments (e.g. Yeh and Price 2004; Atwell et al. 2014; Friis et al. 2022). For a review, see Ketterson and Atwell (2016). Despite the extensive research conducted on dark-eyed juncos, the molecular basis of the outstanding diversity found in the group remains poorly understood, in part because high-quality, full genome resources accounting for such variability have been sparse.
Fig. 1.

Geographic distribution and phenotypic diversity in the dark-eyed junco. a) Distribution map of the main dark-eyed junco forms. Colored areas correspond to the approximate breeding ranges of each form. b) Photographs of male individuals of the 6 main dark-eyed junco forms (Photos by BM).
A plethora of avian species has been fully sequenced and analyzed in the last decade (e.g. Warren et al. 2010; Zhang et al. 2012; Jarvis et al. 2014; Poelstra et al. 2014; Liu et al. 2019; Louha et al. 2020; Dussex et al. 2021; Recuerda et al. 2021). Recently, the Ten-Thousand Bird Genomes (B10K) consortium released over 300 avian genomes from 92.4% of all avian families, enabling genome-based comparative studies at phylogenetic scales never achieved before (Zhang et al. 2014; Feng et al. 2020). Yet within birds, the dark-eyed junco complex is of particular interest for the study of evolutionary processes. First, the system (which includes the closely related yellow-eyed junco Junco phaeonotus from Mexico and Central America), is composed of recently diversified yet phenotypically differentiated lineages, among which the signals of drift and selection at the molecular level are still recent and detectable. Second, the complex includes forms with broad geographic distributions encompassing heterogeneous habitats across ecological clines, but also spatially discontinuous habitats so that selective and neutral processes of divergence can be assessed in different spatial settings. Third, dark-eyed juncos show large variability in the degree of geographical isolation among phenotypically differentiated forms, from extensive gene flow to total isolation, which along with the first 2 points, makes them a suitable system for studying evolutionary processes related to dispersal, directional selection, and neutral evolution (Milá et al. 2006; Friis et al. 2018, 2022). Fourth, the dark-eyed juncos present highly divergent secondary sexual traits, enabling comparative analyses to test the role of sexual selection as a driver of genetic divergence (Friis and Milá 2020; Friis et al. 2022). And finally, as in most bird species, the dark-eyed junco genome is highly syntenic within the class Aves, relatively small, and structurally simple compared with other amniotes (Gregory 2002), which facilitates the identification of polymorphisms and structural variants.
Here, we report a high-quality, chromosome-level assembly obtained using shotgun and proximity ligation libraries as a resource for genome-based studies on J. hyemalis and related bird species. A structural and functional annotation using gene prediction and similarity approaches is also provided. To evaluate the performance of the assembly as a reference for genome-based studies, we conducted a demographic analysis using the pairwise sequentially Markovian coalescent method (PSMC) and the shotgun data generated for the assembly. Shotgun data are also used for the assembly of the mitochondrial genome.
Materials and methods
Genome sequencing and assembly
A high-quality genome was produced combining newly generated shotgun reads and sequence data from proximity ligation libraries. Preparation of proximity ligation libraries Chicago and Hi-C, as well as scaffolding with the software pipeline HiRise (Putnam et al. 2016; https://dovetailgenomics.com, last accesed on April 10, 2022) was conducted at Dovetail Genomics, LLC. The sequenced sample consisted of muscle tissue obtained from a female J. hyemalis carolinensis, collected at Mountain Lake Biological Station in Pembroke, Virginia, USA (37.3751°N, 80.5228°W), currently deposited at the Moore Laboratory of Zoology, Occidental College, Los Angeles, CA, USA (voucher number: MLZ: bird: 69236). Briefly, a de novo draft assembly was first built using shotgun, paired-end libraries (mean insert size ∼350 bp) and the Meraculous pipeline (Chapman et al. 2011). For the Chicago and the Dovetail Hi-C library preparation, chromatin was fixed with formaldehyde. Fixed chromatin was then digested with DpnII and free blunt ends were ligated. Crosslinks were reversed and the DNA was purified from protein. Resulting nucleic material was then sheared to ∼350 bp mean fragment size and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina-compatible adapters. Sequencing of the libraries was carried out on an Illumina HiSeq X platform. The shotgun reads, Chicago library reads, and Dovetail Hi-C library reads were then used as input data for HiRise, a software pipeline designed specifically for using proximity ligation data to scaffold genome assemblies (Supplementary Fig. 1, Putnam et al. 2016). More details on genome sequencing and assembly methods are available in the Supplementary Information.
A circularized assembly of the mitochondrial genome was generated using NOVOplasty2.7.2 (Dierckxsens et al. 2017) and the shotgun data. The NADH dehydrogenase subunit 2 (ND2) mitochondrial gene sequenced for a previous study (Friis et al. 2022) and available at NCBI (GenBank accession no. KX461682.1) was used for the input seed sequence.
Identification of repetitive regions
We first created a repeat library for the junco genome by modeling ab initio repeats using Repeat Modeler 1.0.11 (Smit and Hubley 2019) in scaffolds longer than 100 Kb with default options. The resulting repeat library was merged with known bird repeat libraries from the RepBase database (RepBase-20181026) (Bao et al. 2015), Dfam_Consensus-20181026 and repeats from the zebra finch. Then, we used Repeat Masker 4.0.7 (Smit et al. 2015) to identify and mask repeat regions in the whole-genome assembly and classified the repeat distribution by chromosome.
Gene prediction and functional annotation
Gene prediction was conducted using BRAKER v2.1.5 (Hoff et al. 2019) and GeMoMa v1.7.1 (Keilwagen et al. 2019). We used the repeat soft-masked genome assembly and we first trained Augustus with the conserved orthologous genes from BUSCO Aves_odb10 as proteins from short evolutionary distance (Gremme et al. 2005; Stanke et al. 2006; see Fig. 3b from Hoff et al. 2019). The predicted gene models obtained with Augustus were combined with homology-based annotations using the zebra finch (GCF_008822105.2; Warren et al. 2010) (GCF_008822105.2; Warren et al. 2010) and chicken (GCF_000002315.6; International Chicken Genome Sequencing Consortium 2004) annotated genes with the GeMoMa pipeline, obtaining the final reported gene models. We then applied a similarity-based search approach to conduct the functional annotation of the junco predicted proteins. We first used BLASTP against the UniProt SwissProt database and the annotated proteins from the zebra finch genome (Warren et al. 2010; UniProt Consortium 2019) (E-value 10−5). We only considered as positives those hits covering at least 2/3 of the query sequence length or 80% of the total subject sequence. We also used InterProScan v5.31 (Jones et al. 2014) to identify specific protein-domain signatures in the predicted genes. The functional annotation, including gene ontology (GO) terms, was integrated from all searches providing a curated set of junco coding genes (Supplementary Fig. 2). In addition, a visual summary of recovered GO categories for annotated genes was generated with WEGO 2.0 (Ye et al. 2018). We also used GenomeTools (Gremme et al. 2013) to calculate the number and mean length of genes, exons, introns, and CDS (coding sequence) from the annotation file in general feature format (GFF).
Fig. 3.

Changes in the ancestral effective population size of the dark-eyed junco. The dark red line represents the original effective population size through time, and light red lines represent 100 bootstrap estimates. The indices g and µ denote the generation time and the mutation rate, respectively.
Gene completeness assessment and genome synteny
We assessed gene completeness in the genome assembly and the gene annotation using BUSCO (Benchmarking Universal Single-Copy Orthologs) v4.0.5 (–auto-lineage-euk option; Waterhouse et al. 2018). BUSCO evaluations were conducted using the 255 and 8,338 single-copy orthologous genes in Eukaryota_odb10 and Aves_odb10 datasets, respectively. In addition, we used MUMmer (Delcher et al. 2003) to explore synteny with the zebra finch (Taeniopygia guttata) genome bTaeGut2.pri.v2 available at NCBI under the accession GCA_009859065.2.
Historic changes in effective population size
The pairwise sequentially Markovian coalescent (PSMC) (Li and Durbin 2011) was used to model historic changes in the effective population size of the dark-eyed junco. The shotgun data produced was mapped with bwa-mem (Li and Durbin 2009) against the fully assembled genome and a whole-genome diploid consensus sequence was generated using SAMtools v0.1.19 (Li et al. 2009) and bcftools (Danecek and McCarthy 2017). The default settings of PSMC were adopted, with a generation time of 1.5 years and a mutation rate of 4.6 × 10−8 per site per generation (Nam et al. 2010; Smeds et al. 2016). Confidence intervals were estimated based on bootstrapping (Li and Durbin 2011).
Results and discussion
Sequencing and genome assembly
We sequenced and assembled a reference genome of the dark-eyed junco. Shotgun library produced 465 million read pairs (2 × 150 bp). Chicago and Dovetail Hi-C libraries produced 218 million and 121 million read pairs (2 × 151 bp), respectively. Overall, 121 Gb were generated. Genome scaffolding with HiRise yielded an assembly of 4,684 scaffolds and 1.03 Gb, with a sequence coverage of 117x; an L50/N50 equal to 5 scaffolds/71.3 Mb and an L90/N90 of 19 scaffolds/14.1 Mb; and a relatively low number of ambiguous bases (i.e. N) inserted in the genome (3.13%; Table 1).
Table 1.
Summary statistics for the genome assembly of Junco hyemalis.
| Genome assembly | |
|---|---|
| Total length (bp) | 1,031,523,571 |
| Number of scaffolds | 4,684 |
| N50/L50 | 71,317,294 bp/5 scaffolds |
| N90/L90 | 14,099,349 bp/19 scaffolds |
| Chromosome scale | 1,013,712,310 bp/30 scaffolds |
| Longest scaffold (bp) | 152,011,357 |
| Missingness | 3.13% |
| GC content | 41.85% |
| BUSCO Eukaryota database | C: 92.9% [S: 92.5%, D: 0.4%], F: 3.9%, M: 3.2%, N: 255 |
| BUSCO Aves database | C: 95.4% [S: 95.2%, D: 0.2%], F: 1.6%, M: 3.0%, N: 8,338 |
CDS indicates protein-coding sequences. BUSCO parameters are C: Complete BUSCO; S: Complete and single-copy BUSCOs; D: Complete and duplicated BUSCOs; F: Fragmented BUSCOs; M: Missing BUSCOs; and N: Total BUSCO groups searched
The dark-eyed junco genome also showed high levels of synteny with the zebra finch Taeniopygia guttata (Fig. 2). The 30 longest scaffolds obtained based on the proximity ligation mate-paired libraries, ranging from 152.01 to 2.67 Mb (median = 18.90 Mb; Supplementary Fig. 4) and accounting for 98.27% of the whole assembled genome, were identified as full or nearly full chromosomes when compared to the zebra finch (Fig. 2). Both sexual chromosomes were successfully mapped, yet the chromosome W was highly fragmented (293 fragments), possibly due to difficulties assembling highly repetitive genomic regions (Dovetail, pers. comm.). In addition, a circularized assembly of the mitochondrial genome was 16,894 bp long with a 46.6% GC content.
Fig. 2.
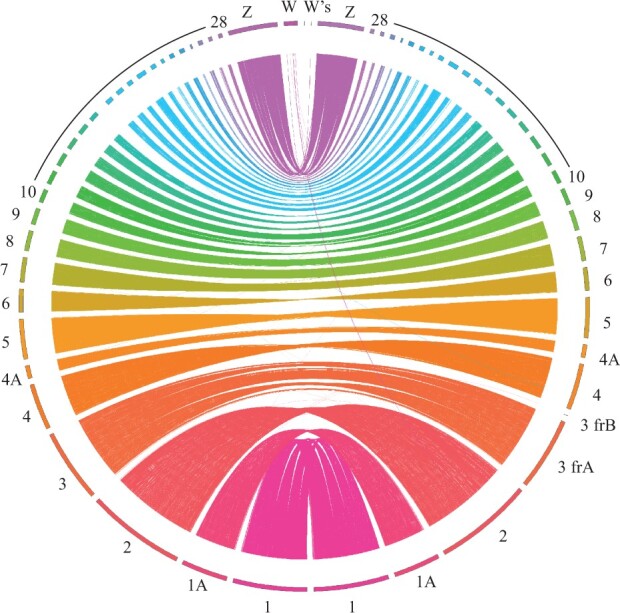
Circos plot showing synteny patterns between the zebra finch (left hemisphere) and the dark-eyed junco (right hemisphere) genome assemblies. Chromosome 3 is represented by 2 scaffolds (frA and frB). Only the 2 largest scaffolds of the 293 that mapped against the W chromosome are shown.
The integrity assessment of the J. hyemalis genome retrieved 92.9% and 95.4% of the tested BUSCO groups for the eukaryotes and aves databases, respectively, evidencing the completeness of our assembly (Table 1).
Genome annotation
We identified 19,026 protein-coding genes, from which 15,967 were associated to GO terms. The average gene length was 15.4 Kb, with a mean of 9.86 exons per gene. BUSCO integrity analysis reported 89.0% of recovered complete BUSCOs for the eukaryota database, and 96.2% in the case of Aves. Quantitatively, our genome annotation identified a considerably higher number of genes, coding sequences, exons and introns when compared with other passerines (i.e. Fringilla coelebs, Melospiza melodia, Taeniopygia guttata, Ficedula albicollis, Manacus vittelinus, and Geospiza fortis), while averaged lengths for these elements remained similar to other species annotations (Table 2). We also found that a total of 5.77% (59.5 Mb) of the J. hyemalis assembly consisted of repetitive elements, a value within the range expected for birds which is at 4%–10% of the genome (Zhang et al. 2014). Of the total of repetitive elements, the greatest proportions corresponded to long interspersed nuclear elements (LINEs, 52.51%) and to long terminal repeats (LTRs, 36.57%; Supplementary Table 2). In a WEGO analysis, the most represented GO terms where those associated to “cell,” “cell parts,” and “membrane part” in the cellular component category; “binding” in molecular functions; and “cellular processes” in the category of biological processes (Supplementary Fig. 4).
Table 2.
Summary statistics for the genome annotation of Junco hyemalis compared with other similarly sized avian species (Fringilla coelebs, Melospiza melodia, Taeniopygia guttata, Ficedula albicollis, Manacus vitellinus, and Geospiza fortis), modified from Recuerda et al. (2021).
| Genome annotation | J. hyemalis | F. coelebs | M. melodia | T. guttata | F. allbicollis | M. vittelinus | G. fortis |
|---|---|---|---|---|---|---|---|
| Number of genes | 19,026 | 17,703 | 15,086 | 17,561 | 16,763 | 18,976 | 14,399 |
| Average gene length (bp) | 15,402 | 15,818 | 14,457 | 26,458 | 31,394 | 27,847 | 30,164 |
| Number of CDS | 23,245 | 17,703 | 15,086 | 17,561 | 16,763 | 18,976 | 14,399 |
| Average CDS length (bp) | 1,647 | 1,679 | 1,325 | 1,677 | 1,942 | 1,929 | 1,766 |
| Number of exons | 229,210 | 221,872 | 131,940 | 171,767 | 189,043 | 190,390 | 164,721 |
| Average exon length (bp) | 167 | 165 | 153 | 255 | 253 | 264 | 195 |
| Average number of exons/gene | 9.86 | 10.16 | 8.67 | 10.25 | 12.22 | 11.51 | 11.41 |
| Number of introns | 205,965 | 200,041 | 116,724 | 153,909 | 171,236 | 171,089 | 149,563 |
| Average intron length (bp) | 1,945 | 1,902 | 1,695 | 2,930 | 3,257 | 3,294 | 2,813 |
| BUSCO Eukaryota database: C: 89.0% [S: 85.1%, D: 3.9%], F: 5.1%, M: 5.9%, N: 255 | |||||||
| BUSCO Aves database: C: 96.2% [S: 83.7%, D: 12.5%], F: 1.5%, M: 2.3%, N: 8,338 | |||||||
BUSCO parameters are C: Complete BUSCO; S: Complete and single-copy BUSCOs; D: Complete and duplicated BUSCOs; F: Fragmented BUSCOs; M: Missing BUSCOs; and N: Total BUSCO groups searched. CDS indicates protein-coding sequences.
Historic changes in effective population size
A PSMC analysis for the dark-eyed junco revealed an ancestral demographic expansion that started 500K years ago approximately. The trend was sustained till circa 100K years ago, followed by a decrease in the effective population size. This decrease roughly overlaps with the onset of the last glaciation period (Richmond and Fullerton 1986), which likely caused a demographic decline of Junco populations in the North American continent (Fig. 3).
Conclusion
We report here a high-quality assembly for the J. hyemalis genome along with a comprehensive functional annotation, a valuable resource to address a range of biological questions using genomic approaches. The combination of shotgun and proximity ligation libraries yielded an assembly 1.03 Gb long, with 98.3% of the sequence located in 30 full or nearly full chromosome scaffolds. The genome is highly contiguous and complete, enabling its use as a reference for variant calling and for the identification of candidate genes potentially involved in phenotypic variation. Improved scaffolding also enables the identification of regions putatively under selection, including structural variants such as chromosome rearrangements, repetitive elements or copy number variation, all relevant for investigating questions related to evolutionary biology and molecular ecology in the dark-eyed junco system and related species.
Data availability
Genome assembly, mitochondrial genome, genome annotation and related supporting resources have been deposited at DRYAD (doi: https://doi.org/10.5061/dryad.c59zw3r87). The genome assembly, including the raw shotgun sequencing data, Chicago and Hi-C libraries have been deposited at NCBI under accession QZWM00000000.2; BioProject PRJNA493001; BioSample: SAMN10120167.
Supplemental material is available at G3 online.
Supplementary Material
Acknowledgments
The authors thank María Recuerda for her assistance with synteny and PSMC analyses.
Funding
Funding was provided by grant CGL‐2011‐25866 from the Spanish Ministerio de Ciencia e Innovación to BM and by multiple awards from the National Science Foundation to EK.
Conflicts of interest
None declared.
Literature cited
- Abolins-Abols M, Kornobis E, Ribeca P, Wakamatsu K, Peterson MP, Ketterson ED, Milá B.. Differential gene regulation underlies variation in melanic plumage coloration in the dark-eyed junco (Junco hyemalis). Mol Ecol. 2018;27(22):4501–4515. [DOI] [PubMed] [Google Scholar]
- Atwell JW, Cardoso GC, Whittaker DJ, Price TD, Ketterson ED.. Hormonal, behavioral, and life-history traits exhibit correlated shifts in relation to population establishment in a novel environment. Am Nat. 2014;184(6):E147–E160. [DOI] [PubMed] [Google Scholar]
- Bao W, Kojima KK, Kohany O.. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 2015;6(1):11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JA, , HoI, , SunkaraS, , LuoS, , SchrothGP, , Rokhsar DS.. Meraculous: de novo genome assembly with short paired-end reads. PLoS One. 2011;6(8):e23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristol DA, Reynolds EB, Leclerc JE, Donner AH, Farabaugh CS, Ziegenfus CWS.. Migratory dark-eyed juncos, Junco hyemalis, have better spatial memory and denser hippocampal neurons than nonmigratory conspecifics. Anim Behav. 2003;66(2):317–328. [Google Scholar]
- Danecek P, McCarthy SA.. BCFtools/csq: haplotype-aware variant consequences. Bioinformatics. 2017;33(13):2037–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Salzberg SL, Phillippy AM.. Using MUMmer to identify similar regions in large sequence sets. Curr Protocols Bioinform. 2003;(1):10.13.18–11.13.18. [DOI] [PubMed] [Google Scholar]
- Dierckxsens N, Mardulyn P, Smits G.. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017;45(4):e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dussex N, van der Valk T, Morales HE, Wheat CW, Díez-del-Molino D, von Seth J, Foster Y, Kutschera VE, Guschanski K, Rhie A, et al. Population genomics of the critically endangered Kākāpō. Cell Genomics. 2021;1(1):100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Stiller J, Deng Y, Armstrong J, Fang Q, Reeve AH, Xie D, Chen G, Guo C, Faircloth BC, et al. Dense sampling of bird diversity increases power of comparative genomics. Nature. 2020;587(7833):252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friis G, Atwell JW, Fudickar AM, Greives TJ, Yeh PJ, Price TD, Ketterson ED, Milá B. Rapid evolutionary divergence of a population of the songbird Oregon junco (Junco hyemalis oreganus) following recent colonization of an urban area. Mol Ecol. 2022. [DOI] [PubMed] [Google Scholar]
- Friis G, Fandos G, Zellmer AJ, McCormack JE, Faircloth BC, Milá B.. Genome‐wide signals of drift and local adaptation during rapid lineage divergence in a songbird. Mol Ecol. 2018;27(24):5137–5153. [DOI] [PubMed] [Google Scholar]
- Friis G, Milá B.. Change in sexual signalling traits outruns morphological divergence across an ecological gradient in the post‐glacial radiation of the songbird genus Junco. J Evol Biol. 2020;33(9):1276–1293. [DOI] [PubMed] [Google Scholar]
- Fudickar AM, Greives TJ, Abolins-Abols M, Atwell JW, Meddle SL, Friis G, Stricker CA, Ketterson ED.. Mechanisms associated with an advance in the timing of seasonal reproduction in an urban songbird. Front Ecol Evol. 2017;5:85. [Google Scholar]
- Gregory TR. Animal genome size database. 2002. http://www.genomesize.com.
- Gremme G, Brendel V, Sparks ME, Kurtz S.. Engineering a software tool for gene structure prediction in higher organisms. Inf Softw Technol. 2005;47(15):965–978. [Google Scholar]
- Gremme G, Steinbiss S, Kurtz S.. GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinform. 2013;10(3):645–656. [DOI] [PubMed] [Google Scholar]
- Hoff KJ, Lomsadze A, Borodovsky M, Stanke M.. Whole-genome annotation with BRAKER. In: Gene Prediction. Humana, New York, NY: Springer; 2019. p. 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432(7018):695–716. [DOI] [PubMed] [Google Scholar]
- Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P, Li C, Ho SYW, Faircloth BC, Nabholz B, Howard JT, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346(6215):1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilwagen J, Hartung F, Grau J.. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol Biol. 2019;1962:161–177. [DOI] [PubMed] [Google Scholar]
- Ketterson E, Nolan V Jr, Sandell M.. Testosterone in females: mediator of adaptive traits, constraint on sexual dimorphism, or both? Am Nat. 2005;166(S4):S85–S98. [DOI] [PubMed] [Google Scholar]
- Ketterson ED, Atwell JW.. Snowbird: Integrative Biology and Evolutionary Diversity in the Junco. Chicago: University of Chicago Press; 2016. [Google Scholar]
- Ketterson ED, Atwell JW, McGlothlin JW.. Phenotypic integration and independence: hormones, performance, and response to environmental change. Integr Comp Biol. 2009;49(4):365–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketterson ED, Nolan V Jr.. Adaptation, exaptation, and constraint: a hormonal perspective. Am Nat. 1999;154(S1):S4–S25. [DOI] [PubMed] [Google Scholar]
- Ketterson ED, Nolan V Jr. Geographic variation and its climatic correlates in the sex ratio of eastern‐wintering dark‐eyed juncos (Junco hyemalis hyemalis). Ecology. 1976;57(4):679–693. [Google Scholar]
- Ketterson ED, Nolan V Jr, Cawthorn MJ, Parker PG, Ziegenfus C.. Phenotypic engineering: using hormones to explore the mechanistic and functional bases of phenotypic variation in nature. Ibis. 1996;138(1):70–86. [Google Scholar]
- Li H, Durbin R.. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. Inference of human population history from individual whole-genome sequences. Nature. 2011;475(7357):493–U484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu S, Zhang N, Chen D, Que P, Liu N, Höglund J, Zhang Z, Wang B.. Genome assembly of the common pheasant Phasianus colchicus: a model for speciation and ecological genomics. Genome Biol Evol. 2019;11(12):3326–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louha S, Ray DA, Winker K, Glenn TC.. A high-quality genome assembly of the North American Song Sparrow, Melospiza melodia. G3 (Bethesda). 2020;10(4):1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milá B, McCormack JE, Castaneda G, Wayne RK, Smith TB.. Recent postglacial range expansion drives the rapid diversification of a songbird lineage in the genus Junco. Proc Biol Sci. 2007;274(1626):2653–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milá B, Smith TB, Wayne RK.. Postglacial population expansion drives the evolution of long-distance migration in a songbird. Evolution. 2006;60(11):2403–2409. [PubMed] [Google Scholar]
- Miller A. Speciation in the avian genus Junco. Univ Californ Public Zool. 1941;44(3):173–434. [Google Scholar]
- Nam K, Mugal C, Nabholz B, Schielzeth H, Wolf JB, Backström N, Künstner A, Balakrishnan CN, Heger A, Ponting CP, et al. Molecular evolution of genes in avian genomes. Genome Biol. 2010;11(6):R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan VJ, Ketterson ED, Cristol DA, Rogers CM, Clotfelter ED, et al. Dark-eyed Junco (Junco hyemalis). In: Poole A, Gill F, editors. The Birds of North America. Philadelphia, Pennsylvania: The Birds of North America, Inc; 2002. pp. 1–44. [Google Scholar]
- Poelstra JW, Vijay N, Bossu CM, Lantz H, Ryll B, Müller I, Baglione V, Unneberg P, Wikelski M, Grabherr MG, et al. The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science. 2014;344(6190):1410–1414. [DOI] [PubMed] [Google Scholar]
- Putnam NH, O'Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, Troll CJ, Fields A, Hartley PD, Sugnet CW, et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016;26(3):342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recuerda M, Vizueta J, Cuevas-Caballé C, Blanco G, Rozas J, Milá B.. Chromosome-level genome assembly of the common chaffinch (Aves: Fringilla coelebs): a valuable resource for evolutionary biology. Genome Biol Evol. 2021;13(4):evab034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond GM, Fullerton DS.. Summation of Quaternary glaciations in the United States of America. Quat Sci Rev. 1986;5:183–196. [Google Scholar]
- Singh D, Reed SM, Kimmitt AA, Alford KA, Stricker CA, Polly PD, Ketterson ED.. Breeding at higher latitude is associated with higher photoperiodic threshold and delayed reproductive development in a songbird. Horm Behav. 2021;128:104907. [DOI] [PubMed] [Google Scholar]
- Smeds L, Qvarnström A, Ellegren H.. Direct estimate of the rate of germline mutation in a bird. Genome Res. 2016;26(9):1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R. RepeatModeler-1.0. 11. Institute for Systems Biology. 2019. http://www.repeatmasker.org/RepeatModeler/. Accessed 15.
- Smit A, Hubley R, Green P. RepeatMasker Open-4.0. 2013–2015. 2015.
- Stager M, Senner NR, Swanson DL, Carling MD, Eddy DK, Greives TJ, Cheviron ZA.. Temperature heterogeneity correlates with intraspecific variation in physiological flexibility in a small endotherm. Nat Commun. 2021;12(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stager M, Swanson DL, Cheviron ZA.. Regulatory mechanisms of metabolic flexibility in the dark-eyed junco (Junco hyemalis). J Exp Biol. 2015;218(Pt 5):767–777. [DOI] [PubMed] [Google Scholar]
- Stanke M, Schöffmann O, Morgenstern B, Waack S.. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 2006;7(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, Künstner A, Searle S, White S, Vilella AJ, Fairley S, et al. The genome of a songbird. Nature. 2010;464(7289):757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, Kriventseva EV, Zdobnov EM.. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 2018;35(3):543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Zhang Y, Cui H, Liu J, Wu Y, Cheng Y, Xu H, Huang X, Li S, Zhou A, et al. WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018;46(W1):W71–W75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh PJ, Price TD.. Adaptive phenotypic plasticity and the successful colonization of a novel environment. Am Nat. 2004;164(4):531–542. [DOI] [PubMed] [Google Scholar]
- Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C, Storz JF, Antunes A, Greenwold MJ, Meredith RW, et al. ; Avian Genome Consortium. Comparative genomics reveals insights into avian genome evolution and adaptation. Science. 2014;346(6215):1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Parker P, Li B, Li H, Wang J, et al. The genome of Darwin’s Finch (Geospiza fortis). Gigascience. 2012;1:13.23587407 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genome assembly, mitochondrial genome, genome annotation and related supporting resources have been deposited at DRYAD (doi: https://doi.org/10.5061/dryad.c59zw3r87). The genome assembly, including the raw shotgun sequencing data, Chicago and Hi-C libraries have been deposited at NCBI under accession QZWM00000000.2; BioProject PRJNA493001; BioSample: SAMN10120167.
Supplemental material is available at G3 online.