

Abstract
In neuroinflammatory and neurodegenerative disorders such as multiple sclerosis, mitochondrial damage caused by oxidative stress is believed to contribute to neuroaxonal damage. Previously, we demonstrated that exposure to hydrogen peroxide (H2O2) alters mitochondrial morphology and motility in myelinated axons and that these changes initiate at the nodes of Ranvier, where numerous sodium channels are located. Therefore, we suggested that mitochondrial damage may lead to ATP deficit, thereby affecting the efficiency of the sodium-potassium ATPase and eventually leading to sodium overload in axons. The increased intra-axonal sodium may revert the axonal sodium-calcium exchangers and thus may lead to a pathological calcium overload in the axoplasm and mitochondria. Here, we used the explanted murine ventral spinal roots to investigate whether modulation of sodium or calcium influx may prevent mitochondrial alterations in myelinated axons during exogenous application of H2O2 inducing oxidative stress. For that, tetrodotoxin, an inhibitor of voltage-gated sodium ion channels, and ruthenium 360, an inhibitor of the mitochondrial calcium uniporter, were applied simultaneously with hydrogen peroxide to axons. Mitochondrial shape and motility were analyzed. We showed that inhibition of axonal sodium influx prevented oxidative stress-induced morphological changes (i.e., increase in circularity and area and decrease in length) and preserved mitochondrial membrane potential, which is crucial for ATP production. Blocking mitochondrial calcium uptake prevented decrease in mitochondrial motility and also preserved membrane potential. Our findings indicate that alterations of both mitochondrial morphology and motility in the contexts of oxidative stress can be counterbalanced by modulating intramitochondrial ion concentrations pharmacologically. Moreover, motile mitochondria show preserved membrane potentials, pointing to a close association between mitochondrial motility and functionality.
1. Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) that affects approximately 2.5 million people worldwide [1]. The pathological hallmarks of MS include inflammation, demyelination, and neurodegeneration; however, its pathogenesis and the relationship between those three aspects are not completely understood [1].
In this context, mitochondria have emerged as one of the key players that are affected by inflammation and contribute to neuroaxonal loss [2–4]. During neuroinflammatory events in MS, activated CNS-invading leukocytes, as well as microglia, are potential sources of reactive oxygen species (ROS), mainly via increased activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases [5–8]. It is assumed that excessive ROS production may lead to oxidative stress and consequently to the inhibition of adenosine triphosphate (ATP) production. Activation of oxidative phosphorylation under pathological stress conditions may also lead to increased chances of electron slippage to oxygen and the formation of additional detrimental ROS [9–11]. In a physiological state, cells have mechanisms to cope with increased ROS production [12, 13]. However, sustained inflammation and oxidative stress may lead to irreversible damage in mitochondria and affect the survivability of the cells [14].
To investigate the impact of oxidative stress on neuroaxonal mitochondria, we have developed an ex vivo model to monitor mitochondrial alterations in murine spinal roots [15, 16]. We focused on ventral spinal roots because they consist predominantly of efferent motor axons and are thicker than dorsal roots making them easier to handle. Using this model, we previously showed that oxidative stress alters both mitochondrial morphology (increases mitochondrial circularity and decreases mitochondrial area and length) and mitochondrial motility (reduces the percentage of moving mitochondria, length of their trajectories and their velocity) [17]. We also observed that, following an oxidative insult, all these alterations consistently initiate at the nodes of Ranvier [17].
In axons, voltage-gated sodium channels (NaV) are mainly located near the nodes of Ranvier [18]. In the presence of oxidative stress, mitochondrial damage may lead to reduced ATP generation [19] and the consequent failure of the sodium-potassium-ATPase (Na+/K+-ATPase), leading to sodium (Na+) accumulation inside the axons [20]. Moreover, in a degeneration paradigm using dorsal root ganglion cells, it has been demonstrated that influx of Na+ via NaV contributes to intraneuronal Na+ accumulation [13]. To compensate for the excess of intracellular Na+ in the presence of a dysfunctional Na+/K+-ATPase, the axonal sodium-calcium exchanger (NCX) may start acting in a reverse mode, causing axonal calcium (Ca2+) overload [5, 13, 20].
High cytosolic Ca2+ concentration directly impacts mitochondria, which in turn are part of the Ca2+ buffering system of cells [13, 21, 22]. Tightly regulated intracellular Ca2+ homeostasis is crucial because an excessive mitochondrial Ca2+ uptake may lead to the opening of the permeability transition pore (PTP), resulting in apoptosis [9, 14]. A mitochondrial Ca2+ uniporter (MCU) transports Ca2+ into the mitochondrial matrix [19]. It has been shown that overexpression of MCU and subsequent mitochondrial Ca2+ overload results in neuronal death, both in vitro and in vivo [23]. Moreover, mitochondria are linked to motor proteins via Miro-1/2, which have Ca2+-sensing structures, suggesting that mitochondrial motility is also Ca2+-sensitive [18]. Although during physiological state, a slight increase in mitochondrial Ca2+ appears to directly stimulate mitochondrial ATP production by activating Ca2+-sensitive enzymes of Krebs' Cycle [21], high levels of Ca2+ may lead to the suppression of mitochondrial movement [20].
In neuroinflammation, the assumption that alteration of ion concentrations and neuronal damage are connected is supported by the beneficial effects of ion channel blockers reported in experimental autoimmune encephalitis (EAE), where blocking NaV or voltage-gated Ca2+ channels attenuates the disease course [13, 24]. Hence, we hypothesized that the abnormal activity of ion channels at the nodes of Ranvier following oxidative stress may cause the observed mitochondrial alterations [5, 13, 24].
Thus, we investigated here if preventing Na+ overload within axons and Ca2+ overload within mitochondria using the NaV blocker tetrodotoxin (TTX) and the MCU inhibitor ruthenium 360 (Ru360), respectively, would protect both mitochondria and axons from oxidative-stress mediated damage.
2. Material and Methods
2.1. Ethics Statement
All experimental procedures were approved by the regional animal study committee of Berlin (Landesamt für Gesundheit und Soziales Berlin). Animal experiments were conducted in strict accordance with Directive 2010/63/EU of the European Parliament and of the European Council of 22 September 2010. Female and male mice (8-10 weeks old) were used for the experiments. The mice were housed and maintained in a temperature-controlled environment on a 12 h light-dark cycle.
2.2. Preparation and Maintenance of Ventral Spinal Roots
Ventral spinal roots were prepared as described previously [15]. Briefly, C57BL/6 mice were deeply anesthetized with isoflurane before cervical dislocation. After separating the connective tissue, the dorsal side of the spinal cord was exposed, and the vertebrae were cut laterally from rostral to caudal. The spinal cord was sectioned at the thoracic level and the ventral spinal roots were cut distal to the spinal cord. Together with the attached spinal roots, the explanted spinal cord was then placed into artificial cerebrospinal fluid (aCSF), saturated with carbogen (95% O2 and 5% CO2), and adjusted to a pH of 7.3-7.4. Under a dissecting microscope, the lumbar ventral roots were finally selected and separated from the spinal cord. Explanted ventral roots were maintained in aCSF, containing the following solutions: Solution I – 124 mM NaCl, 1.25 mM NaH2PO4, 10.0 mM Glucose, 1.8 mM MgSO4, 1.6 mM CaCl2, 3.00 mM KCl; Solution II – 26.0 mM NaHCO3. Both solutions were mixed immediately before use.
2.3. Induction of Oxidative Stress and Treatment Groups
All experiments were conducted in a submerged incubation chamber (Brain Slice Keeper-BSK 6 Scientific Systems Design Inc., Ontario, Canada), allowing up to five different treatment conditions and continuous carbogen perfusion of each submersion well throughout the entire process. Although the BSK 6 has 6 individual tubes to supply gas to each of the six wells, one tube had to be used to carbogenate the aCSF stock and therefore only 5 wells were available for the experiments.
To assess the effect of TTX and Ru360 on mitochondrial alterations induced by oxidative stress, we assigned spinal roots randomly to the following experimental groups: a) Negative controls of TTX experiments consisted of axons incubated with aCSF for 30 min at room temperature (RT). Negative controls of Ru360 experiments consisted of axons incubated with the corresponding solvent dimethyl sulfoxide (DMSO) at 1 μl/ml (0,001%) for 30 min at RT. This concentration corresponded to the one used to solve Ru360. DMSO does not exert an effect on investigated mitochondrial parameters (data not shown). We also refer to the negative groups as “untreated groups”. b) In the oxidatively-stressed control group, ventral spinal roots were incubated with 100 μM H2O2 for 30 min at RT along with the corresponding vehicle (aCSF for TTX experiments, DMSO for Ru360 experiments). We also refer to this group as “positive control”. c) Effects of blocking NaV channels on spinal roots were investigated by incubating the spinal roots with 100 nM or 1 μM TTX along with 100 μM H2O2. d) Effects of blocking mitochondrial Ca2+ influx were determined by incubation with 5, 10, or 20 μM Ru360 along with 100 μM H2O2.
2.4. Labeling of Mitochondria, Microscopy, and Analysis of Mitochondrial Dynamics (Morphology and Motility)
After incubation with the treatments, transected ventral spinal roots were washed and transferred into aCSF containing 100 nM MitoTracker® Orange CMTMRos (Life Technologies, Darmstadt, Germany) dissolved in DMSO for 30 min at RT and then washed again with fresh aCSF.
Microscopy and imaging analysis of the ventral spinal roots were performed as previously described [15]. For microscopy, spinal roots were placed on a glass coverslip and transferred to an imaging chamber containing carbogenated aCSF. A custom-built nylon net was placed on top of the spinal roots to prevent them from moving during image acquisition. For all experiments, an inverted laser-scanning confocal microscope adapted for live-cell imaging was used. Experiments with Na+ channel blockade were imaged with an LSM 710 (Carl Zeiss, Jena, Germany). Experiments with Ca2+ channel blockade were conducted using a Nikon Scanning Confocal A1Rsi+. MitoTracker® Orange was excited at 561 nm with a diode-pumped solid-state (DPSS) laser. Visualization of mitochondria was performed through a 100x (LSM 710, Carl Zeiss) or 60x (Nikon Scanning Confocal A1Rsi+) oil immersion objective. Regions of interest (ROI) were chosen based on the following criteria: 1) clearly visible node of Ranvier 2) well-labeled mitochondria 3) axon with intact myelin sheath and no signs of membrane disruption in regions adjacent to the selected ROI 4) areas at least 2 mm away from the end of the roots. Scrutinizing the spinal roots from the proximal to the distal end, three separate ROI were chosen. For each ROI, a time-lapse (60-second duration, 2 s/frame) with a resolution of 512x512 pixels was recorded. Exposure time and laser power were reduced to minimize photobleaching and phototoxicity.
The first frame of every time-lapse video was used to assess mitochondrial morphology with an automated analysis tool of the Volocity®6.3 software (Perkin Elmer, Rodgau, Germany). To determine the changes in mitochondrial morphology, the following parameters were analyzed: shape factor (4πX [Area/Perimeter2]), a measure of circularity ranging from 0 to 1, in which “1” indicates a perfect circle, length (μm) and area (μm2) of an individual mitochondrion. To assess motility, mitochondria were tracked manually using Volocity®6.3 software (Perkin Elmer, Rodgau, Germany). Any mitochondrion with a displacement of ≥1 μm was considered “mobile”. For experiments with Ru360, mobile mitochondria were further analyzed for track length (μm), the measure of the real distance traveled by a mitochondrion, and velocity (μm/s).
Under physiological and pathological conditions, mitochondrial populations display high heterogeneity within one cell due to their adaption to different energetic states. Thus, to minimize selection bias, large amounts of mitochondria in different axons of several experiments were analyzed and matched.
2.5. Assessment of Mitochondrial Membrane Potential
To determine mitochondrial membrane potential, spinal roots were stained with 20 μg/ml 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1; Life Technologies, Darmstadt, Germany) in aCSF at RT for 1 h. JC-1 accumulates in mitochondria with intact membrane potential and negative charge. Sufficient accumulation due to unaltered mitochondrial membrane potential leads to the formation of J aggregates and a shift in emitted fluorescence from green (529 nm) to red (590 nm) [25]. To minimize background noise, roots were washed with fresh aCSF before imaging. JC-1 was excited with dual illumination with argon (514 nm) and DPSS (561 nm) lasers.
Red/green fluorescence ratio of JC-1 stained mitochondria determined at a Nikon Scanning Confocal A1Rsi+microscope was used for the analysis of mitochondrial membrane potential. Results of the red/green fluorescence ratio of individual mitochondrion were normalized to the average red/green fluorescence ratio of the untreated group as established by others [26].
2.6. Statistical Analysis
Acquired data were analyzed with Prism 8 Software (GraphPad, CA, USA). All datasets were first subjected to D'Agostino and Pearson omnibus K2 normality test and Shapiro-Wilk normality test for Gaussian distribution. Data fitting the criteria for normal distribution were subsequently analyzed using a one-way ANOVA with Bonferroni's post hoc test. Data following a non-parametric distribution were analyzed using a Kruskal-Wallis test followed by a Dunn's post hoc multiple comparisons test. p values ≤ 0.05 were considered significant. The significance of the data was further depicted as ∗ implying p ≤ 0.05, ∗∗ implying p ≤ 0.01, ∗∗∗ implying p ≤ 0.001, and ∗∗∗∗ implying p ≤ 0.0001. All data are shown in mean ± SEM.
3. Results
3.1. Blocking Axonal Na+ Influx Prevents Oxidative Stress-Induced Morphological Changes in Mitochondria
To investigate the effect of Na+ channel blockade on mitochondrial morphology, the explanted ventral spinal roots were treated with 100 μM H2O2 alone, or 100 μM H2O2 along with different concentrations of TTX (100 nM or 1 μM). Explants were then imaged using a confocal microscope (Figure 1(a)). Shape factor (Figure 1(b)), mitochondrial length (Figure 1(c)), and mitochondrial area (Figure 1(d)) were analyzed.
Figure 1.
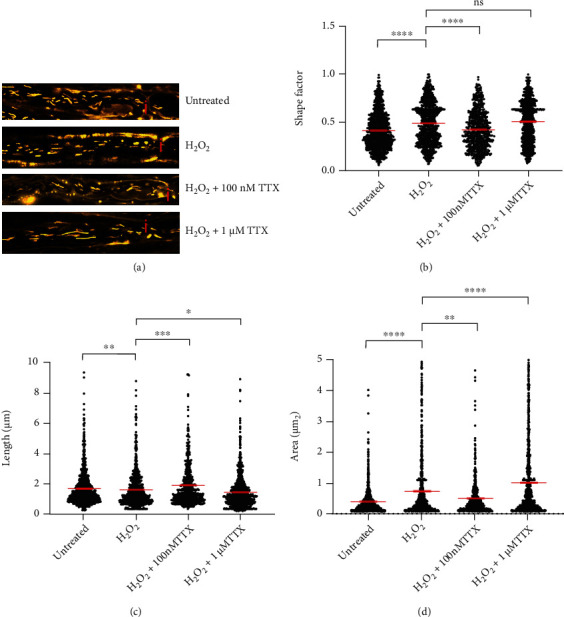
Blocking axonal Na+ influx with tetrodotoxin (TTX) prevents oxidative stress-induced mitochondrial morphology alterations. (a) Representative original images of all different experimental conditions; axons incubated with aCSF alone contained elongated mitochondria; incubation with 100 μM H2O2 led to the generation of smaller and rounder mitochondria, and some diffuse MitoTracker® distribution; axon simultaneously incubated with 100 μM H2O2 and 100 nM TTX contained elongated mitochondria; axon simultaneously incubated with 100 μM H2O2 and 1 μM TTX contained short mitochondria but with increased area. (b–d) Shape factor (b), length (c), and area (d) of mitochondria located near the nodes of Ranvier in axons incubated with the above-mentioned treatments. Nodes of Ranvier are marked with a red “i”. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001. The error bars represent the standard error of mean; n = 6 animals and 22 roots; untreated 7 roots, H2O2 6 roots, H2O2+100 nM TTX 4 roots, and H2O2+1 μM TTX 5 roots.
During oxidative stress, mitochondrial shape factor (untreated: 0.4148 ± 0.0060; H2O2: 0.4854 ± 0.0074) and area (untreated: 0.4043 ± 0.0124 μm2; H2O2: 0.7557 ± 0.0335 μm2) increased while mitochondrial length decreased (untreated: 1.684 ± 0.0375 μm; H2O2: 1.5800 ± 0.0431 μm; Figures 1(b) and 1(c)). All observed morphological changes induced by oxidative stress were prevented with 100 nM of TTX (shape factor = 0.4202 ± 0.0079; length = 1.8990 ± 0.0702 μm; area = 0.5247 ± 0.0268 μm2; Figures 1(b)–1(d)). In contrast, 1 μM of TTX did not affect the H2O2-induced increase in shape factor (0.5030 ± 0.0072; Figure 1(b)), but significantly reduced length (1.4400 ± 0.0362 μm, Figure 1(c)) and increased mitochondrial area in comparison to oxidative stress conditions (1.0150 ± 0.0377 μm2; Figure 1(d)).
3.2. Blocking Axonal Na+ Influx Prevents Oxidative Stress-Induced Changes of Mitochondria Motility
Next, we performed time-lapse imaging and analyzed mitochondrial motility parameters under the above-mentioned experimental conditions (Figure 2(a)). We analyzed the percentage of manually tracked motile mitochondria (Figure 2(b)). The untreated group with aCSF alone showed an average percentage of motile mitochondria of about 16% (15.890 ± 1.395%), while in the presence of 100 μM H2O2 only around 5% (5.044 ± 1.228%) of mitochondria were motile (Figure 2(b)). Blocking Na+ influx with 1 μM TTX prevented the oxidative stress-induced reduction of motile mitochondria (11.460 ± 1.826%; Figure 2(b)). The effect of 100 nM TTX was not significant compared to the H2O2-treated group (Figure 2(b)).
Figure 2.
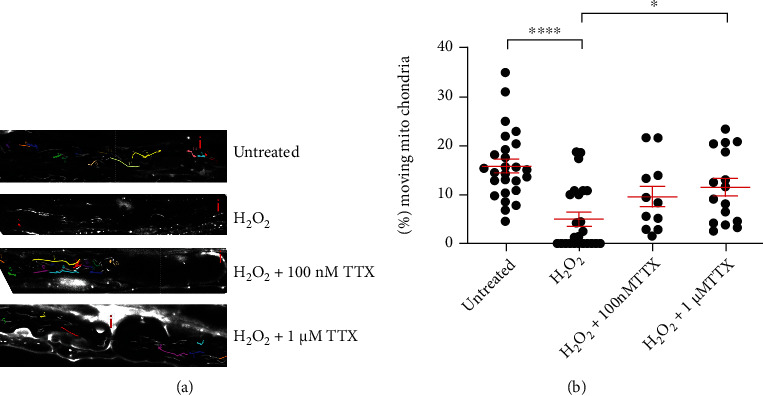
Blocking axonal Na+ influx with TTX barely affects mitochondrial motility parameters altered due to oxidative stress. (a) Representative original images of all different experimental conditions: axon incubated with aCSF alone contained multiple moving mitochondria that c5over larger distances in the axon; 100 μM H2O2 induced a strong reduction of motile mitochondria; axons simultaneously incubated with 100 μM H2O2 and 100 nM TTX or 1 μM TTX contained more motile mitochondria, covering longer distances. (b) Percentage of moving mitochondria per axon. Nodes of Ranvier are marked with a red “i”. ∗p ≤ 0.05, ∗∗∗∗p ≤ 0.0001. The error bars represent the standard error of mean; n = 6 animals and 22 roots; untreated 7 roots, H2O2 6 roots, H2O2+100 nM TTX 4 roots, and H2O2+1 μM TTX 5 roots.
3.3. Blocking Mitochondrial Ca2+ Uptake Prevents Oxidative Stress-Induced Alterations of Mitochondrial Length
Then, we examined the influence of mitochondrial Ca2+ on mitochondrial morphology. Oxidative stress was induced again with 100 μM H2O2. Blocking mitochondrial Ca2+ influx via mitochondrial Ca2+ uniporter channels was performed by simultaneous incubation of mitochondria with H2O2 and 5, 10, or 20 μM Ru360. We observed that H2O2 led to a decrease in mitochondrial length (untreated: 1.9260 ± 0.0343 μm; H2O2: 1.6920 ± 0.0302 μm) and area (untreated: 1.0890 ± 0.0292 μm2; H2O2: 0.9756 ± 0.0268 μm2) compared to the untreated group (Figures 3(c) and 3(d)). However, shape factor did not increase under H2O2-treatment (untreated: 0.4703 ± 0.0072; H2O2: 0.4841 ± 0.0071) when compared to the untreated group (Figure 3(b)). Blocking mitochondrial Ca2+ influx with 5 μM Ru360 prevented changes in shape factor (0.4474 ± 0.0090, Figure 3(b)). A similar trend was observed in roots treated with 10 μM Ru360 (0.4741 ± 0.0083; Figure 3(b)). However, at 20 μM, Ru360 induced an even more pronounced increase in shape factor values (0.5214 ± 0.0095) when compared to the H2O2-treated group (Figure 3(b)). Incubation with 5 μM Ru360 did not increase mitochondrial length compared to the H2O2-treated group (1.881 ± 0.0426 μm, Figure 3(c)). In the presence of 10 μM Ru360, mitochondrial length increased (1.7780 ± 0.0390 μm; Figure 3(c)), while at 20 μM Ru360 promoted decrease in mitochondrial length compared to treatment with oxidative stress alone (Figure 3(c)). Regarding area, we did not observe significant alterations in either of the treatment groups (Figure 3(d)).
Figure 3.
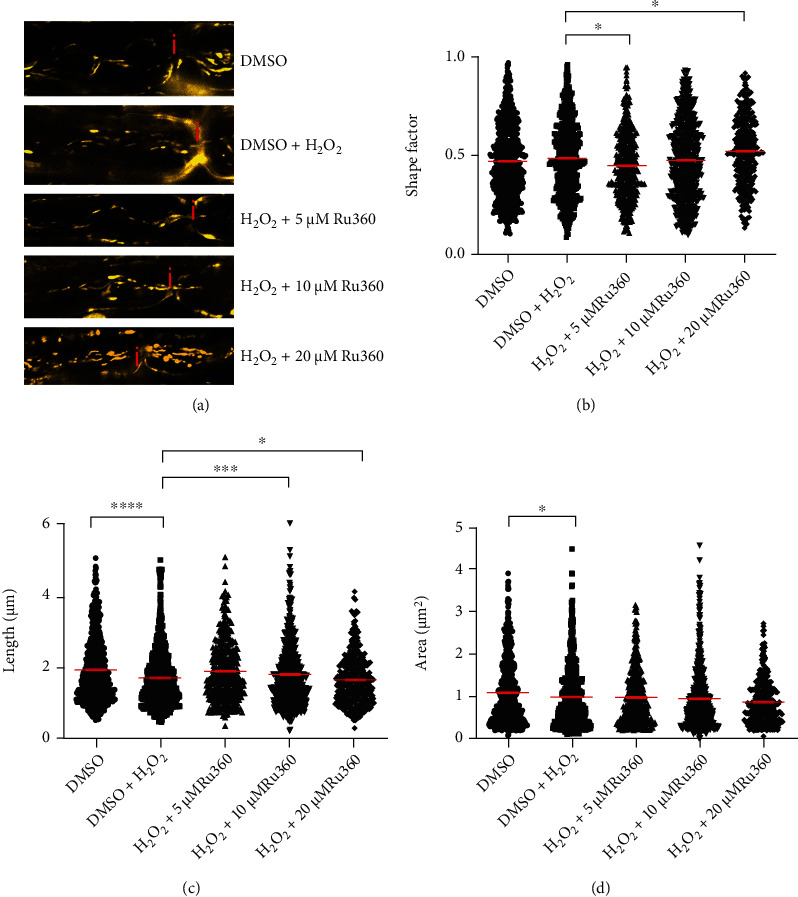
Blocking mitochondrial Ca2+ influx with Ru360 alters mitochondrial morphology. (a) Representative original images of all different experimental conditions; axon incubated with DMSO alone showed elongated mitochondria; 100 μM H2O2 induced shorter and smaller mitochondria; axonal mitochondria exposed to 100 μM H2O2 and 5 μM Ru360 showed slightly longer and less round morphology than mitochondria exposed to H2O2 alone; incubation with 100 μM H2O2 and 10 μM Ru360 led to the formation of longer mitochondria compared with H2O2 control; axon incubated with 100 μM H2O2 and 20 μM Ru360 showed shorter mitochondria than oxidative stress control. (b–d) Mitochondrial shape factor (b), length (c), and area (d) of single mitochondria. Nodes of Ranvier are marked with a red “i”. ∗p ≤ 0.05, ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001. The error bars represent the standard error of mean; n = 7 animals and 29 roots; DMSO 7 roots, H2O2 7 roots, H2O2+5 μM Rui360 4 roots, H2O2+10 μM Ru360 7 roots, and H2O2+20 μM Ru360 4 roots.
3.4. Blocking Mitochondrial Ca2+ Uptake Prevents Reduction of Mitochondrial Motility in Stressed Axons
To investigate the effect of blocking MCU on oxidative stress-induced alterations in mitochondrial motility, we incubated explanted ventral spinal roots with DMSO alone, DMSO plus 100 μM H2O2, or with 100 μM H2O2 along with three different concentrations (5, 10 or 20 μM) of Ru360 (Figure 4(a)).
Figure 4.
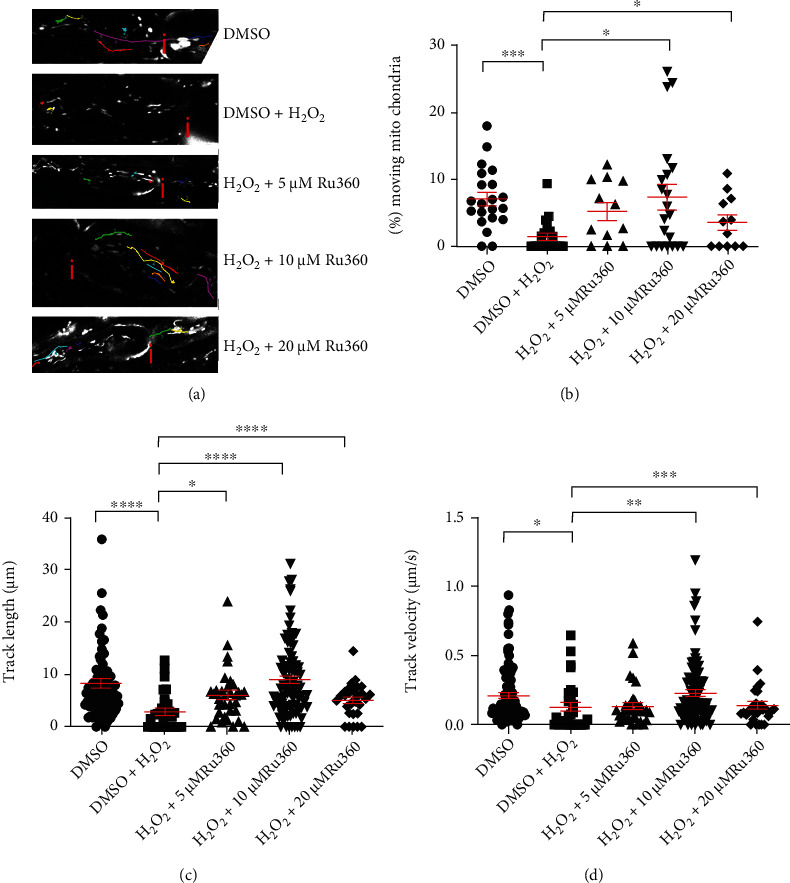
Blocking mitochondrial Ca2+ uptake with Ru360 prevents oxidative stress-induced loss of mitochondrial motility. (a) Representative original images of all different experimental conditions: axon incubated with DMSO alone contained multiple moving mitochondria that cover larger distances in the axon.; axon incubated with 100 μM H2O2, showed few motile mitochondria with short track length; simultaneous incubation with 100 μM H2O2 and 5 or 20 μM Ru360 led to more motile mitochondria that move longer distances; axon simultaneously incubated with 100 μM H2O2 and 10 μM Ru360 showed nearly normal mitochondrial motility. (b–d) Quantification of the percentage of motile mitochondria (a), track length (b), and track velocity (c). Nodes of Ranvier are marked with a red “i”. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001. The error bars represent the standard error of mean; n = 7 animals and 29 roots; DMSO 7 roots, H2O2 7 roots, H2O2+5 μM Rui360 4 roots, H2O2+10 μM Ru360 7 roots, and H2O2+20 μM Ru360 4 roots.
In the untreated group, we observed an average of 7% (7.103% ± 0.997) of moving mitochondria (Figure 4(b)). H2O2 at 100 μM caused a significant reduction in motile mitochondria (1.447% ± 0.507) as well as a decrease in track length (untreated: 8.2722 ± 0.8433 μm; H2O2-treated: 2.8750 ± 0.6442 μm) and track velocity (untreated: 0.2094 ± 0.0210 μm/s; H2O2-treated: 0.1265 ± 0.0320 μm/s) (Figures 4(a)–4(c)). H2O2-induced decrease in percentage of motile mitochondria, mitochondrial track length, and track velocity was prevented with 10 μM Ru360 (% of moving mitochondria: 7.393 ± 1.861%; track length: 8.9410 ± 0.7597 μm; track velocity: 0.2293 ± 0.0243 μm/s; Figures 4(a)–4(c)) and 20 μM Ru360 (% of moving mitochondria: 3.549 ± 1.124%; track length: 4.989 ± 0.6025 μm; track velocity: 0.1384 ± 0.0280 μm/s; Figures 4(a)–4(c)). However, in spinal roots treated with 5 μM Ru360, only H2O2-induced changes for track length (μm, Figure 4(c)) were prevented. No effects were observed on percentage of moving mitochondria or track velocity ((% of moving mitochondria: 5.205 ± 1.325%; track velocity: 0.1331 ± 0.0235 μm/s, Figures 4(a) and 4(d)).
3.5. Blocking Axonal Na+ Influx Prevents Oxidative Stress-Induced Reduction of Mitochondrial Membrane Potential
Next, we investigated whether inhibition of axonal Na+ influx may preserve mitochondrial functionality altered by H2O2. Four groups of spinal roots were treated for 30 min with either aCSF alone (vehicle control group), 100 μM H2O2, 100 μM H2O2+1 μM TTX or 1 μM TTX alone, respectively. Since the incubation chamber permitted the simultaneous assessment of maximally 5 conditions, only the 1 μM TTX concentration, which showed best protecting effects in Figure 2(a), was tested in these experiments. Treated spinal roots were then incubated for 30 min with the ratiometric indicator JC-1. The red/green fluorescence ratio is an indication of the mitochondrial membrane potential and thereby mitochondrial ability to produce ATP (Figure 5(a)).
Figure 5.
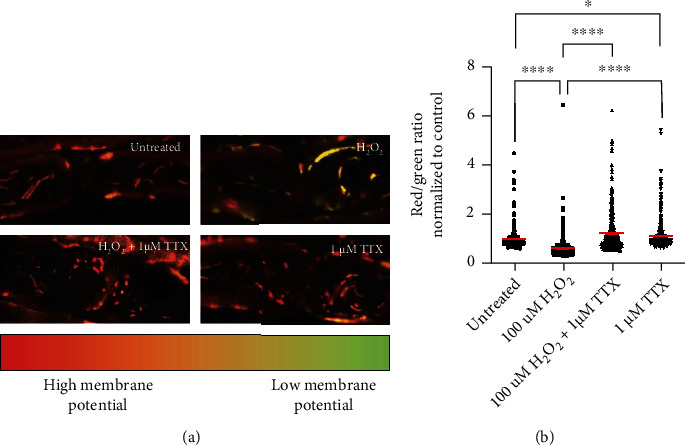
Blocking axonal Na+ influx with TTX prevents loss of mitochondrial membrane potential. (a) Representative images of axons in the different treatment groups. The upper left image shows mitochondrial membrane potential in untreated condition. Oxidative stress led to loss of mitochondrial membrane potential (upper right image) and a shift to green fluorescence. TTX prevented the H2O2 effects (lower left image). The lower right image shows that the application of TTX alone led to preserved mitochondrial membrane potential. (b) Data represent normalized values of individual mitochondria to the mean of the control group (red/green ratio = 1 ± 0.0383). ∗∗∗∗p ≤ 0.0001. The error bars represent the standard error of mean; n = 3 animals and 12 roots; untreated 3 roots, H2O2 3 roots, H2O2+1 μM TTX 3 roots, and 1 μM TTX 3 roots.
The application of 100 μM H2O2 resulted in a shift to green fluorescence (0.6374 ± 0.0291; Figures 5(a) and 5(b)), as a sign of a loss of mitochondrial membrane potential. 1 μM TTX applied simultaneously with 100 μM H2O2 prevented the loss of mitochondrial membrane potential (untreated: 1.0000 ± 0.0297; 1 μM TTX: 1.2410 ± 0.0432; Figures 5(a) and 5(b)). TTX alone led to higher mitochondrial membrane potential than in the untreated group (TTX: 1.1270 ± 0.0309; Figures 5(a) and 5(b)).
3.6. Blocking Mitochondrial Ca2+ Uptake Prevents Oxidative Stress-Induced Reduction of Mitochondrial Membrane Potential
To assess effects of Ca2+ uptake on mitochondrial functionality, four groups of spinal roots were treated for 30 min with either DMSO (vehicle control group), DMSO +100 μM H2O2, 100 μM H2O2+10 μM Ru360 or 10 μM Ru360 alone, respectively. We selected 10 μM Ru360 for these experiments because it was the concentration that showed the best protection against oxidative stress-induced loss of motility (Figure 4(a)). Treated spinal roots were then incubated for 30 min with the ratiometric indicator JC-1. The red/green fluorescence ratio is an indication of the mitochondrial membrane potential and thereby mitochondrial ability to produce ATP (Figure 6(a)).
Figure 6.
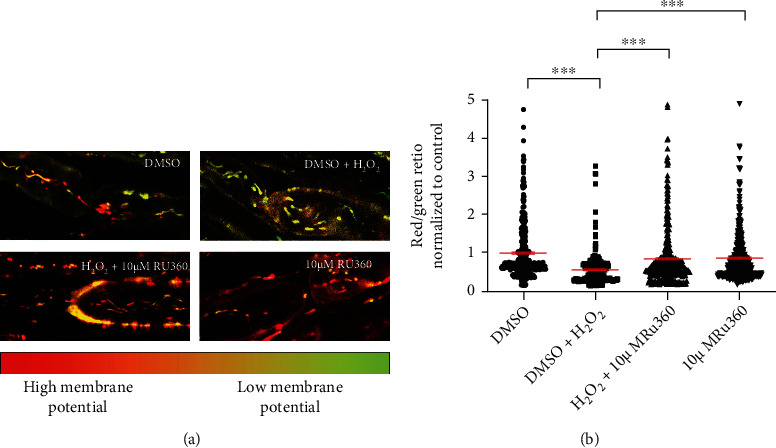
Blocking mitochondrial Ca2+ uptake with Ru360 prevents loss of mitochondrial membrane potential. (a) Representative images of axons in the different treatment groups. The upper left image shows mitochondrial membrane potential under negative control conditions containing mitochondria with high (red) and low (green) mitochondrial membrane potential. Oxidative stress led to loss of mitochondrial membrane potential (upper right image) and a shift to green fluorescence. Ru360 prevented the H2O2 effects (lower left image). The lower right image shows that the application of the Ru360 alone had no effects on mitochondrial functionality compared to control group. (b) Data represent normalized values of individual mitochondria to the mean of the control group (red/green ratio = 1 ± 0.0383). ∗∗∗p ≤ 0.001. The error bars represent the standard error of mean; n = 5 animals and 20 roots; DMSO 5 roots, H2O2 5 roots, H2O2+10 μM Ru360 5 roots, and 10 μM Ru360 5 roots.
The application of 100 μM H2O2 resulted in a shift to green fluorescence (0.5638 ± 0.0250; Figures 6(a) and 6(b)), as a sign of a severe loss of mitochondrial membrane potential. 10 μM Ru360 applied simultaneously with 100 μM H2O2 prevented the loss of mitochondrial membrane potential and restored it to values close to the untreated group (untreated: 1.0000 ± 0.0383; 10 μM Ru360: 0.8507 ± 0.0395; Figures 6(a) and 6(b)). With Ru360 alone, we did not observe any effects on mitochondrial membrane potential in comparison to DMSO treated condition (Figures 6(a) and 6(b)).
4. Discussion
Mitochondrial alterations linked to oxidative stress [9] are reported to occur in the early stages of MS [4, 12] and are believed to contribute to neurodegenerative processes observed in MS patients [2, 27–29]. Therefore, mitochondria have emerged as potential therapeutic targets to limit disease progression [30, 31]. In this study, we investigated using an ex vivo model of peripheral axons [17] whether the effects of oxidative stress on mitochondria can be prevented by targeting pathological ion alterations affecting, in particular, the levels of axonal Na+ and mitochondrial Ca2+.
In this model, oxidative stress was induced by a 30-minute incubation with 100 μM H2O2, a concentration that led to reversible structural and functional alterations in mitochondria [32]. We observed oxidative stress-induced decrease in mitochondrial length (Figures 1(c) and 3(c)) as well as a decrease in the number of motile mitochondria (Figures 2(b) and 4(b)). Additionally, consistent with our previous reports [16, 33] and those of others describing inhibition of axonal transport by oxidative stress [34–36], we observed a decrease in both track length and track velocity of mitochondria exposed to 100 μM H2O2 (Figures 4(c) and 4(d)). The observed reduction in mitochondrial length supports previous findings of our group [17] and may be the consequence of an increase in the fission process, which is induced in stressed and damaged mitochondria to get rid of the damaged portion [37].
We also expected that oxidative stress would damage mitochondria and reduce their functionality in our model causing ATP depletion as it has been reported for highly energy-dependent neuronal cells [9, 38]. We showed a decrease in mitochondrial membrane potential under oxidative stress conditions (Figures 5(a), 5(b), 6(a), and 6(b)). As an intact mitochondrial membrane potential is an important determinant for mitochondrial ATP production via oxidative phosphorylation [39], we assumed ATP depletion in oxidatively injured mitochondria. In a novel CNS model established in our lab, we were indeed able to show decreased ATP levels upon oxidative stress induced by 100 μM H2O2 [33]. Thus, our paradigm of stressed mitochondria in explanted roots may serve in the future to examine effects of antioxidative interventions on ATP levels.
We previously reported that alterations of mitochondria during oxidative stress initiate at the nodes of Ranvier [17]. NaVs are abundantly present at the nodes of Ranvier and are important for saltatory conduction [40, 41]. In MS lesions, the expression of these channels is reported to be altered [42–44]. In this line, during exposure to H2O2, blocking NaV with 100 nM TTX prevented the decrease in length and increase in shape factor and area (Figures 1(a)–1(c)). In contrast, 1 μM TTX along with H2O2 led to the generation of short mitochondria that display however large areas (Figures 1(b) and 1(c)). A large mitochondrial area could reflect either detrimental swelling [45, 46] or fusion [1, 35]. We speculate that in the group treated with H2O2 and 1 μM TTX, transient mitochondrial fusion followed by fission as reported by Liu et al. [45] may occur. Transient fusion seems to be central for maintaining metabolism and motility [45]. In this line, we observed that 1 μM TTX could prevent the motility decrease and the loss of membrane potential observed in mitochondria exposed to H2O2 (Figures 2(a), 5(a), and 5(b)).
Interestingly, 1 μM TTX alone induced an elevation of the mitochondrial membrane potential when compared to the untreated group. This may reflect a state defined as mitochondrial hyperpolarization [46, 47]. We hypothesize that the presence of TTX and the consequent reduce Na+ influx may lead to a diminished activity of the ATP-dependent Na+/K+-ATPase and induce an increase of ATP. Thus, in our setup, hyperpolarization may be generated by the ATP-consuming reverse action mode of complex V [46]. The exact mechanism underlying the elevation of the mitochondrial membrane potential with TTX alone will be part of future investigations.
Subsequently to Na+ overload, intra-axonal Ca2+ accumulation occurs via reverse action mode of NCX, as described in other studies [13, 48]. During axonal Ca2+ overload, mitochondria may uptake Ca2+ and function as an intracellular Ca2+ buffering system [49]. However, excessive intramitochondrial Ca2+ may affect mitochondrial function and motility. It has been shown that dynamin-related protein 1 (Drp1), responsible for mitochondrial fission, as well as Miro, connecting mitochondria via other proteins to motor proteins, are directly or indirectly controlled by Ca2+ [29, 50–52]. Moreover, mitochondrial swelling seems to be Ca2+-related, too [53]. In this case, we demonstrated that inhibition of Ca2+ influx into mitochondria with 10 μM Ru360 completely prevents oxidative stress-induced reduction of mitochondrial length and all motility parameters (Figures 3(b) and 4(a)–4(c)). Further, with 10 μM Ru360, we observed preserved mitochondrial membrane potential (Figures 5(a) and 5(b)). Thus, a rise in intramitochondrial Ca2+ concentration appears to contribute to mitochondrial alterations during oxidative stress in our model. In the motility experiments, we observed a biphasic effect of Ru360 with similar absolute values for 5 and 20 μM and a clearly different response for 10 μM Ru360. This biphasic effect was observed in all investigated mitochondrial motility parameters, i.e., percentage of motile mitochondria, mitochondrial track length, and track velocity (Figures 3(a)–3(c)).
Our data confirm previous studies that indicated that ion concentrations show no linear correlation with mitochondrial morphology, motility, or membrane potential [54, 55]. While a slight increase in mitochondrial Ca2+ concentration may increase mitochondrial ATP production and be beneficial [55], elevated levels of mitochondrial Ca2+ may lead to the opening of the PTP with possible detrimental effects [56]. In addition, PTP opening does not only depend on ion concentrations but also on ATP/ADP levels, mitochondrial ROS, fatty acids, and magnesium levels [57–59]. ROS function as signaling molecules, reversibly oxidizing defined structures and thereby regulating transcription or enzyme activity [6, 31, 60–62]. ROS regulates among others the activity of MCU [63], as well as of voltage-gated sodium channels, including NaV1.7 [64]. These potential cellular mechanisms to cope with increased ROS should be kept in mind when dealing with oxidative stress and ion alterations.
Ru360 is a specific inhibitor of the MCU [65, 66]. However, blocking MCU may not result in a complete inhibition of mitochondrial Ca2+ influx. As described in metabolically inhibited cells [47], a reverse action mode of mitochondrial Na+/Ca2+-exchanger may enhance intramitochondrial Ca2+ in stressed axons. Additionally, mitochondria closely interact with the endoplasmic reticulum (ER), forming mitochondria-associated membranes (MAMs) [67]. MAMs play a role in the exchange of Ca2+ or metabolites [68, 69], mitochondrial fusion and fission processes, and induction of apoptosis [70].
Mitochondria possess different mechanisms of Ca2+ influx [71], but also of Ca2+ efflux. The two most important mechanisms are via mitochondrial Na+/Ca2+-exchanger and via 2H+/Ca2+-exchanger [71, 72]. Mitochondrial Ca2+ uptake is therefore most likely directly influenced by intra-axonal Na+ concentration because this affects mitochondrial Ca2+ efflux mechanisms via mitochondrial Na+/Ca2+-exchanger. Interestingly, a reverse action mode is also described for mitochondrial Na+/Ca2+-exchanger in metabolically inhibited cells [48]. Thus, blocking either axonal Na+ influx or mitochondrial Ca2+ uptake may likely indirectly interfere with other pathways, for example via mitochondria-associated membranes (MAMs) or mitochondrial Na+/Ca2+-exchanger of a tightly regulated and interconnected Na+- Ca2+-homeostasis.
5. Limitations of the Study
One technical limitation of our setup was the restricted number of experimental conditions that could be conducted simultaneously within one experiment. The size of the incubation chamber and the narrow time-window, in which transplants could be imaged ex vivo, permitted only the comparison of maximally five different culture conditions. Therefore, using this setup, we were unable to compare effects on mitochondria of different concentrations of inhibitors both in the absence and the presence of the oxidative insult.
Therefore, using this setup, we were able to show only effects on mitochondria of different concentrations of inhibitors in the oxidative stress paradigm and not in the absence of H2O2.
Moreover, although our data indicate that modulation of Ca2+ influx with Ru360 protects mitochondria from oxidative stress-induced damage, we could not define which Ca2+ concentrations are protective and which concentrations are detrimental for mitochondria. Basically, we attested that the explanted root model was not suitable for intra-axonal Ca2+quantification using, for instance, Ca2+-sensitive dyes or roots from Ca2+ reporter mice.
6. Conclusion
In conclusion, explanted murine spinal roots appear to be a suitable model to investigate oxidative stress-induced ion alterations affecting axonal mitochondria, in particular, Na+ and Ca2+ overload. Using the model, we demonstrated that inhibition of axonal Na+ influx prevented oxidative stress-induced alterations of mitochondrial morphology. On the other hand, blocking mitochondrial Ca2+ uptake prevented the oxidative stress-induced reduction of both mitochondrial motility and mitochondrial membrane potential, which is crucial for ATP production.
The fact that H2O2-induced alterations in mitochondria morphology and motility were prevented by pharmacologic inhibitors of NaV and MCU indicates a direct participation of Na+ and Ca2+ on oxidative stress-mediated mitochondrial changes. Further investigations in this direction are needed to explore the therapeutic potential of the modulation of Na+ and Ca2+ ion channel for mitochondrial protection during oxidative stress.
Acknowledgments
We thank the Core Facility Advanced Medical Bioimaging (AMBIO) at Charité Universitätsmedizin Berlin for imaging infrastructure and technical support. The study was supported by the Charité Universitätsmedizin and by a fellowship from Berlin Institute of Health (BIH) to R. Ulshöfer. The work of C. Infante-Duarte was supported by the DFG (SFB1340-1, Project B05), and the work of R. Niesner and A. E. Hauser was supported by the DFG (SFB1444, Project 14).
Data Availability
The main data supporting the findings of this study are listed in Tables 1–6 of the Supplementary Materials.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Bimala Malla and Carmen Infante-Duarte contributed equally to this work.
Supplementary Materials
Table 1: summary of morphology parameters of untreated mitochondria, mitochondria under H2O2 treatment alone, and mitochondria treated with H2O2 in the presence of 100 nM and 1 μM TTX.
Table 2: summary of motility parameters of untreated mitochondria, mitochondria under H2O2 treatment alone, and mitochondria treated with H2O2 in the presence of 100 nM and 1 μM TTX.
Table 3: summary of morphology parameters of untreated mitochondria, mitochondria under H2O2 treatment alone, and mitochondria treated with H2O2 in the presence of 5 μM, 10 μM, and 20 μM Ru360.
Table 4: summary of motility parameters of untreated mitochondria, mitochondria under H2O2 treatment alone, and mitochondria treated with H2O2 in the presence of 5 μM, 10 μM, and 20 μM Ru360.
Table 5: summary of the red-green ratio normalized to untreated mitochondria, mitochondria treated with 100 μM H2O2 alone or in the presence of 1 μM TTX, and mitochondria treated with 1 μM TTX alone.
Table 6: summary of the red-green ratio normalized to untreated mitochondria, mitochondria treated with 100 μM H2O2 alone or in the presence of 10 μM Ru360, and mitochondria treated with 10 μM Ru360 alone.
References
- 1.Thompson A. J., Baranzini S. E., Geurts J., Hemmer B., Ciccarelli O. Multiple sclerosis. The Lancet . 2018;391(10130):1622–1636. doi: 10.1016/S0140-6736(18)30481-1. [DOI] [PubMed] [Google Scholar]
- 2.Su K. G., Banker G., Bourdette D., Forte M. Axonal degeneration in multiple sclerosis: the mitochondrial hypothesis. Current Neurology and Neuroscience Reports . 2009;9(5):411–417. doi: 10.1007/s11910-009-0060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haider L. Inflammation, iron, energy failure, and oxidative stress in the pathogenesis of multiple sclerosis. Oxidative Medicine and Cellular Longevity . 2015;2015:10. doi: 10.1155/2015/725370.725370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patergnani S., Fossati V., Bonora M., et al. Mitochondria in multiple sclerosis: molecular mechanisms of pathogenesis. International Review of Cell and Molecular Biology . 2017;328:49–103. doi: 10.1016/bs.ircmb.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Waxman S. G. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nature Reviews Neuroscience . 2006;7(12):932–941. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- 6.Ohl K., Tenbrock K., Kipp M. Oxidative stress in multiple sclerosis: central and peripheral mode of action. Experimental Neurology . 2016;277:58–67. doi: 10.1016/j.expneurol.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mossakowski A. A., Pohlan J., Bremer D., et al. Tracking CNS and systemic sources of oxidative stress during the course of chronic neuroinflammation. Acta Neuropathologica . 2015;130(6):799–814. doi: 10.1007/s00401-015-1497-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radbruch H., Bremer D., Guenther R., et al. Ongoing oxidative stress causes subclinical neuronal dysfunction in the recovery phase of EAE. Frontiers in Immunology . 2016;7(92) doi: 10.3389/fimmu.2016.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Federico A., Cardaioli E., Da Pozzo P., Formichi P., Gallus G. N., Radi E. Mitochondria, oxidative stress and neurodegeneration. Journal of the Neurological Sciences . 2012;322(1-2):254–262. doi: 10.1016/j.jns.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 10.Kadenbach B. Introduction to mitochondrial oxidative phosphorylation. Advances in Experimental Medicine and Biology . 2012;748:1–11. doi: 10.1007/978-1-4614-3573-0_1. [DOI] [PubMed] [Google Scholar]
- 11.Wilson D. F. Oxidative phosphorylation: regulation and role in cellular and tissue metabolism. The Journal of Physiology . 2017;595(23):7023–7038. doi: 10.1113/JP273839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikić I., Merkler D., Sorbara C., et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nature Medicine . 2011;17(4):495–499. doi: 10.1038/nm.2324. [DOI] [PubMed] [Google Scholar]
- 13.Persson A. K., Kim I., Zhao P., Estacion M., Black J. A., Waxman S. G. Sodium channels contribute to degeneration of dorsal root ganglion neurites induced by mitochondrial dysfunction in an in vitro model of axonal injury. The Journal of Neuroscience . 2013;33(49):19250–19261. doi: 10.1523/JNEUROSCI.2148-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pegoretti V., Swanson K. A., Bethea J. R., Probert L., Eisel U. L. M., Fischer R. Inflammation and oxidative stress in multiple sclerosis: consequences for therapy development. Oxidative Medicine and Cellular Longevity . 2020;2020:19. doi: 10.1155/2020/7191080.7191080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bros H., Niesner R., Infante-Duarte C. An ex vivo model for studying mitochondrial trafficking in neurons. Methods in Molecular Biology . 2015;1264:465–472. doi: 10.1007/978-1-4939-2257-4_38. [DOI] [PubMed] [Google Scholar]
- 16.Malla B., Cotten S., Ulshoefer R., et al. Teriflunomide preserves peripheral nerve mitochondria from oxidative stress-mediated alterations. Therapeutic Advances in Chronic Disease . 2020;11 doi: 10.1177/2040622320944773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bros H., Millward J. M., Paul F., Niesner R., Infante-Duarte C. Oxidative damage to mitochondria at the nodes of Ranvier precedes axon degeneration in ex vivo transected axons. Experimental Neurology . 2014;261:127–135. doi: 10.1016/j.expneurol.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 18.Wang J., Ou S.-W., Wang Y.-J. Distribution and function of voltage-gated sodium channels in the nervous system. Channels . 2017;11(6):534–554. doi: 10.1080/19336950.2017.1380758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X., Wang W., Li L., Perry G., Lee H.-g., Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease . 2014;1842(8):1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Criste G., Trapp B., Dutta R. Axonal loss in multiple sclerosis: causes and mechanisms. Handbook of Clinical Neurology . 2014;122:101–113. doi: 10.1016/B978-0-444-52001-2.00005-4. [DOI] [PubMed] [Google Scholar]
- 21.Lin Y., Li L.-L., Nie W., et al. Brain activity regulates loose coupling between mitochondrial and cytosolic Ca2+ transients. Nature Communications . 2019;10(1):p. 5277. doi: 10.1038/s41467-019-13142-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Csordás G., Várnai P., Golenár T., et al. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Molecular Cell . 2010;39(1):121–132. doi: 10.1016/j.molcel.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Granatiero V., Pacifici M., Raffaello A., De Stefani D., Rizzuto R. Overexpression of mitochondrial calcium uniporter causes neuronal death. Oxidative Medicine and Cellular Longevity . 2019;2019:15. doi: 10.1155/2019/1681254.1681254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schattling B., Eggert B., Friese M. A. Acquired channelopathies as contributors to development and progression of multiple sclerosis. Experimental Neurology . 2014;262:28–36. doi: 10.1016/j.expneurol.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 25.Sivandzade F., Bhalerao A., Cucullo L. Analysis of the mitochondrial membrane potential using the cationic JC-1 dye as a sensitive fluorescent probe. Bio-Protocol . 2019;9(1, article e3128) doi: 10.21769/BioProtoc.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kilbride S. M., Telford J. E., Davey G. P. Complex I controls mitochondrial and plasma membrane potentials in nerve terminals. Neurochemical Research . 2021;46(1):100–107. doi: 10.1007/s11064-020-02990-8. [DOI] [PubMed] [Google Scholar]
- 27.Witte M. E., Mahad D. J., Lassmann H., van Horssen J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends in Molecular Medicine . 2014;20(3):179–187. doi: 10.1016/j.molmed.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Mahad D., Lassmann H., Turnbull D. Review: mitochondria and disease progression in multiple sclerosis. Neuropathology and Applied Neurobiology . 2008;34(6):577–589. doi: 10.1111/j.1365-2990.2008.00987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer C. S., Osellame L. D., Stojanovski D., Ryan M. T. The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cellular Signalling . 2011;23(10):1534–1545. doi: 10.1016/j.cellsig.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 30.Reddy P. H., Reddy T. P. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Current Alzheimer Research . 2011;8(4):393–409. doi: 10.2174/156720511795745401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Camara A. K., Lesnefsky E. J., Stowe D. F. Potential therapeutic benefits of strategies directed to mitochondria. Antioxidants & Redox Signaling . 2010;13(3):279–347. doi: 10.1089/ars.2009.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gülden M., Jess A., Kammann J., Maser E., Seibert H. Cytotoxic potency of H2O2 in cell cultures: impact of cell concentration and exposure time. Free Radical Biology and Medicine . 2010;49(8):1298–1305. doi: 10.1016/j.freeradbiomed.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 33.Malla B., Liotta A., Bros H., et al. Teriflunomide preserves neuronal activity and protects mitochondria in brain slices exposed to oxidative stress. International Journal of Molecular Sciences . 2022;23(3):p. 1538. doi: 10.3390/ijms23031538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fang C., Bourdette D., Banker G. Oxidative stress inhibits axonal transport: implications for neurodegenerative diseases. Molecular Neurodegeneration . 2012;7(1):p. 29. doi: 10.1186/1750-1326-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Errea O., Moreno B., Gonzalez-Franquesa A., Garcia-Roves P. M., Villoslada P. The disruption of mitochondrial axonal transport is an early event in neuroinflammation. Journal of Neuroinflammation . 2015;12(1):p. 152. doi: 10.1186/s12974-015-0375-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Isonaka R., Hiruma H., Kawakami T. Inhibition of axonal transport caused by tert-butyl hydroperoxide in cultured mouse dorsal root ganglion neurons. Journal of Molecular Neuroscience . 2011;45(2):194–201. doi: 10.1007/s12031-010-9457-3. [DOI] [PubMed] [Google Scholar]
- 37.Carinci M., Vezzani B., Patergnani S., et al. Different roles of mitochondria in cell death and inflammation: focusing on mitochondrial quality control in ischemic stroke and reperfusion. Biomedicine . 2021;9(2):p. 169. doi: 10.3390/biomedicines9020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Islam M. T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurological Research . 2017;39(1):73–82. doi: 10.1080/01616412.2016.1251711. [DOI] [PubMed] [Google Scholar]
- 39.Brand M. D. The efficiency and plasticity of mitochondrial energy transduction. Biochemical Society Transactions . 2005;33(5):897–904. doi: 10.1042/BST0330897. [DOI] [PubMed] [Google Scholar]
- 40.Freeman S. A., Desmazières A., Fricker D., Lubetzki C., Sol-Foulon N. Mechanisms of sodium channel clustering and its influence on axonal impulse conduction. Cellular and Molecular Life Sciences . 2016;73(4):723–735. doi: 10.1007/s00018-015-2081-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seidl A. H. Regulation of conduction time along axons. Neuroscience . 2014;276:126–134. doi: 10.1016/j.neuroscience.2013.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Black J. A., Newcombe J., Trapp B. D., Waxman S. G. Sodium channel expression within chronic multiple sclerosis plaques. Journal of Neuropathology and Experimental Neurology . 2007;66(9):828–837. doi: 10.1097/nen.0b013e3181462841. [DOI] [PubMed] [Google Scholar]
- 43.Craner M. J., Newcombe J., Black J. A., Hartle C., Cuzner M. L., Waxman S. G. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proceedings of the National Academy of Sciences of the United States of America . 2004;101(21):8168–8173. doi: 10.1073/pnas.0402765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bouafia A., Golmard J. L., Thuries V., et al. Axonal expression of sodium channels and neuropathology of the plaques in multiple sclerosis. Neuropathology and Applied Neurobiology . 2014;40(5):579–590. doi: 10.1111/nan.12059. [DOI] [PubMed] [Google Scholar]
- 45.Liu X., Weaver D., Shirihai O., Hajnóczky G. Mitochondrial 'kiss-and-run': interplay between mitochondrial motility and fusion-fission dynamics. The EMBO Journal . 2009;28(20):3074–3089. doi: 10.1038/emboj.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forkink M., Manjeri G. R., Liemburg-Apers D. C., et al. Mitochondrial hyperpolarization during chronic complex I inhibition is sustained by low activity of complex II, III, IV and V. Bioenergetics . 2014;1837(8):1247–1256. doi: 10.1016/j.bbabio.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 47.Abramov A. Y., Smulders-Srinivasan T. K., Kirby D. M., et al. Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations. Brain: a Journal of Neurology . 2010;133(3):797–807. doi: 10.1093/brain/awq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smets I., Caplanusi A., Despa S., et al. Ca2+ uptake in mitochondria occurs via the reverse action of the Na+/Ca2+ exchanger in metabolically inhibited MDCK cells. American Journal of Physiology Renal Physiology . 2004;286(4):F784–F794. doi: 10.1152/ajprenal.00284.2003. [DOI] [PubMed] [Google Scholar]
- 49.Nicholls D. G. Mitochondrial calcium function and dysfunction in the central nervous system. Biochimica et Biophysica Acta (BBA)-Bioenergetics . 2009;1787(11):1416–1424. doi: 10.1016/j.bbabio.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mishra P., Chan D. C. Metabolic regulation of mitochondrial dynamics. The Journal of Cell Biology . 2016;212(4):379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cereghetti G. M., Stangherlin A., de Brito O. M., et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proceedings of the National Academy of Sciences of the United States of America . 2008;105(41):15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y., Sheng Z. H. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. The Journal of Cell Biology . 2013;202(2):351–364. doi: 10.1083/jcb.201302040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Courtes A. A., de Carvalho N. R., Gonçalves D. F., et al. Guanosine protects against Ca2+-induced mitochondrial dysfunction in rats. Biomedicine & Pharmacotherapy . 2019;111:1438–1446. doi: 10.1016/j.biopha.2019.01.040. [DOI] [PubMed] [Google Scholar]
- 54.Bagur R., Hajnóczky G. Intracellular Ca2+ sensing: its role in calcium homeostasis and signaling. Molecular Cell . 2017;66(6):780–788. doi: 10.1016/j.molcel.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffiths E. J., Rutter G. A. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochimica et Biophysica Acta (BBA)-Bioenergetics . 2009;1787(11):1324–1333. doi: 10.1016/j.bbabio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 56.Peng T. I., Jou M. J. Oxidative stress caused by mitochondrial calcium overload. Annals of the New York Academy of Sciences . 2010;1201(1):183–188. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- 57.Naumova N., Šachl R. Regulation of cell death by mitochondrial transport systems of calcium and Bcl-2 proteins. Membranes . 2020;10(10):p. 299. doi: 10.3390/membranes10100299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Belosludtsev K. N., Dubinin M. V., Belosludtseva N. V., Mironova G. D. Mitochondrial Ca2+ transport: mechanisms, molecular structures, and role in cells. Biochemistry Biokhimiia . 2019;84(6):593–607. doi: 10.1134/S0006297919060026. [DOI] [PubMed] [Google Scholar]
- 59.Golshani-Hebroni S. Mg++ requirement for MtHK binding, and Mg++ stabilization of mitochondrial membranes via activation of MtHK & MtCK and promotion of mitochondrial permeability transition pore closure: a hypothesis on mechanisms underlying Mg++'s antioxidant and cytoprotective effects. Gene . 2016;581(1):1–13. doi: 10.1016/j.gene.2015.12.046. [DOI] [PubMed] [Google Scholar]
- 60.Kumar A., Ratan R. R. Oxidative stress and Huntington's disease: the good, the bad, and the ugly. Journal of Huntington's Disease . 2016;5(3):217–237. doi: 10.3233/JHD-160205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stone J. R., Yang S. Hydrogen peroxide: a signaling messenger. Antioxidants & Redox Signaling . 2006;8(3-4):243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 62.Stowe D. F., Camara A. K. Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxidants & Redox Signaling . 2009;11(6):1373–1414. doi: 10.1089/ars.2008.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nemani N., Shanmughapriya S., Madesh M. Molecular regulation of MCU: implications in physiology and disease. Cell Calcium . 2018;74:86–93. doi: 10.1016/j.ceca.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel R., Sesti F. Oxidation of ion channels in the aging nervous system. Brain Research . 2016;1639:174–185. doi: 10.1016/j.brainres.2016.02.046. [DOI] [PubMed] [Google Scholar]
- 65.Reed K. C., Bygrave F. L. A low molecular weight ruthenium complex inhibitory to mitochondrial Ca2+ transport. FEBS Letters . 1974;46(1-2):109–114. doi: 10.1016/0014-5793(74)80346-7. [DOI] [PubMed] [Google Scholar]
- 66.Moore C. L. Specific inhibition of mitochondrial Ca++ transport by ruthenium red. Biochemical and Biophysical Research Communications . 1971;42(2):298–305. doi: 10.1016/0006-291X(71)90102-1. [DOI] [PubMed] [Google Scholar]
- 67.Bernard-Marissal N., Chrast R., Schneider B. L. Endoplasmic reticulum and mitochondria in diseases of motor and sensory neurons: a broken relationship? Cell Death & Disease . 2018;9(3):p. 333. doi: 10.1038/s41419-017-0125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giorgi C., Missiroli S., Patergnani S., Duszynski J., Wieckowski M. R., Pinton P. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxidants & Redox Signaling . 2015;22(12):995–1019. doi: 10.1089/ars.2014.6223. [DOI] [PubMed] [Google Scholar]
- 69.Rizzuto R., Marchi S., Bonora M., et al. Ca2+ transfer from the ER to mitochondria: when, how and why. Biochimica et Biophysica Acta (BBA) - Bioenergetics . 2009;1787(11):1342–1351. doi: 10.1016/j.bbabio.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krols M., van Isterdael G., Asselbergh B., et al. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathologica . 2016;131(4):505–523. doi: 10.1007/s00401-015-1528-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feissner R. F., Skalska J., Gaum W. E., Sheu S.-S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Frontiers in Bioscience . 2009;14(4):1197–1218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pizzo P., Drago I., Filadi R., Pozzan T. Mitochondrial Ca2+ homeostasis: mechanism, role, and tissue specificities. Pflügers Archiv - European Journal of Physiology . 2012;464(1):3–17. doi: 10.1007/s00424-012-1122-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table 1: summary of morphology parameters of untreated mitochondria, mitochondria under H2O2 treatment alone, and mitochondria treated with H2O2 in the presence of 100 nM and 1 μM TTX.
Table 2: summary of motility parameters of untreated mitochondria, mitochondria under H2O2 treatment alone, and mitochondria treated with H2O2 in the presence of 100 nM and 1 μM TTX.
Table 3: summary of morphology parameters of untreated mitochondria, mitochondria under H2O2 treatment alone, and mitochondria treated with H2O2 in the presence of 5 μM, 10 μM, and 20 μM Ru360.
Table 4: summary of motility parameters of untreated mitochondria, mitochondria under H2O2 treatment alone, and mitochondria treated with H2O2 in the presence of 5 μM, 10 μM, and 20 μM Ru360.
Table 5: summary of the red-green ratio normalized to untreated mitochondria, mitochondria treated with 100 μM H2O2 alone or in the presence of 1 μM TTX, and mitochondria treated with 1 μM TTX alone.
Table 6: summary of the red-green ratio normalized to untreated mitochondria, mitochondria treated with 100 μM H2O2 alone or in the presence of 10 μM Ru360, and mitochondria treated with 10 μM Ru360 alone.
Data Availability Statement
The main data supporting the findings of this study are listed in Tables 1–6 of the Supplementary Materials.