

Abstract
The new national guidelines for clinical research, the Ethical Guidelines for Medical and Biological Research Involving Human Subjects, were implemented in Japan in June 2021. The guidelines were developed by integrating two ethical guidelines: Ethical Guidelines for Medical and Health Research Involving Human Subjects and Ethical Guidelines for Human Genome/Gene Analysis Research. The Ethical Guidelines for Clinical Research were originally developed as three separate guidelines: Ethical Guidelines for Human Genome/Gene Analysis Research formulated in 2001, Ethical Guidelines for Epidemiological Research in 2002 and Ethical Guidelines for Clinical Research in 2003. They have undergone several amendments and integration in response to the government’s policy changes, such as the protection of personal information, conflicts of interest and reliability of clinical research. The three major changes introduced in the New Integrated Guidelines in 2021 are centralized review, electromagnetic informed consent and research cooperating organization. These are expected to be used as tools to facilitate the conduct of research. This review discusses the regulations of academic clinical research in Japan, the history of ethical guidelines and the three major changes introduced in the New Integrated Guidelines.
Keywords: Japanese regulations, ethical guidelines, clinical trial, clinical research
The new ethical guidelines were implemented in 2021. This review discusses the academic clinical research regulations in Japan, the history of ethical guidelines and major changes in the new guidelines.
Introduction
In June 2021, the new ethical guidelines for clinical research, the Ethical Guidelines for Medical and Biological Research Involving Human Subjects (hereinafter referred to as the ‘New Integrated Guidelines’), were implemented in Japan (1). The new guidelines were developed by integrating the existing Ethical Guidelines for Medical and Health Research Involving Human Subjects and the Ethical Guidelines for Human Genome/Gene Analysis Research. Both guidelines were to be reviewed entirely and amended as necessary ~5 years after they came into force. Additionally, with the enactment of the Clinical Trials Act (CTA) in April 2018, some provisions of the ethical guidelines must be aligned with those of the CTA. Revisions to the Ethical Guidelines for Human Genome/Gene Analysis Research were also needed since some parts of the ethical guidelines no longer fit the current clinical practice.
This review article discusses the regulations of academic clinical research in Japan, the history of the establishment and integration of ethical guidelines and the three major changes introduced in the New Integrated Guidelines.
Regulation of academic clinical research in Japan
Before describing the details of the new ethical guidelines, this article begins with an explanation of the regulatory system of academic research in Japan and where the research regulated by the ethical guidelines is positioned within that system.
With the enactment of the CTA in April 2018, which will be discussed in detail later, the classification of academic research has changed from a regulatory perspective. Until April 2018, research was roughly classified into two categories: (i) investigator-initiated registration-directed trial (IIRDT) under the Pharmaceuticals and Medical Devices Act (PMD Act) and (ii) clinical research, including interventional studies (which is synonymous with ‘clinical trial’), and observational studies under the ethical guidelines (Fig. 1A) (2). After April 2018, academic research was classified into four categories: (i) IIRDT under the PMD Act; (ii) clinical trials wherein researchers must comply with the CTA, called ‘specified clinical trials’; (iii) clinical trials wherein researchers are required to make an effort to comply with CTA, called ‘nonspecified clinical trials’; and (iv) clinical research under the ethical guidelines (Fig. 1B) (2,3). Clinical trials using unapproved or off-label regenerative medical products under another law, the Act to Ensure the Safety of Regenerative Medicine, were omitted from this review because of their relatively small number.
Figure 1.
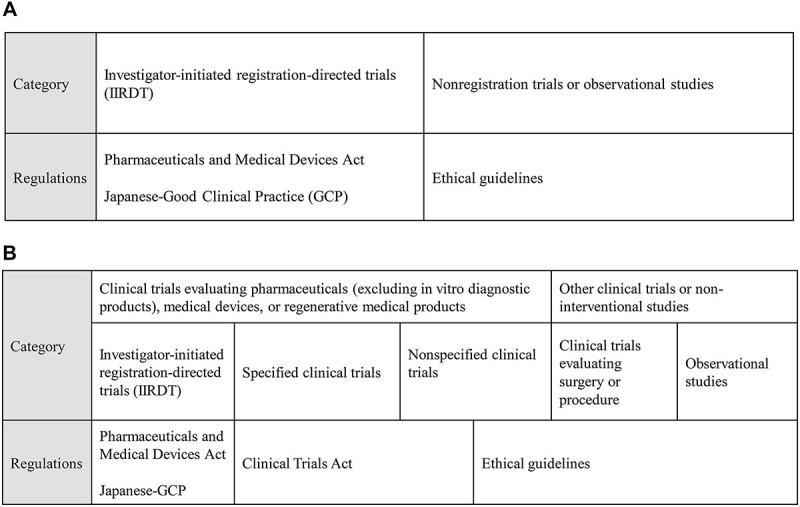
(A) Previous classification and applicable regulations for Japanese academic clinical research applied before April 2018. (B) The present classification and applicable regulations of Japanese academic clinical research applied from April 2018.
Researchers determine which of these four categories apply to their study purpose based on the definitions that are described subsequently. Among clinical trials evaluating the efficacy or safety of pharmaceuticals (excluding in vitro diagnostic products), medical devices or regenerative medical products (‘pharmaceuticals’ hereafter), a clinical trial is classified as IIRDT under the PMD Act when its purpose is to file a new pharmaceutical application or expand an indication. The rest of the trials that do not fall under the above category are classified either as a specified clinical trial or a nonspecified clinical trial under the CTA. The difference between a specified clinical trial and a nonspecified clinical trial is the use of unapproved/off-label pharmaceuticals in the trial and/or the funding by a manufacturer that has marketing approval for the pharmaceuticals. When either of these applies, a study is called a specified clinical trial, and researchers must comply with the CTA and file a clinical trial application to the Ministry of Health, Labour and Welfare (MHLW). When neither of these applies, the study is called a nonspecified clinical trial, and researchers do not need to file a clinical trial application to the MHLW. Nevertheless, they are required to ‘make an effort’ to comply with parts of the provisions specified in the CTA. ‘Make an effort’ means the applicability of this act is limited when the researchers could not comply with the CTA for unavoidable reasons, such as a lack of research funding that could pay the review fee of an ethics review committee or the cost of hiring the personnel necessary to prepare documentation. In fact, some nonspecified clinical trials are conducted under the ethical guidelines for these reasons. When investigators do not intend to evaluate the efficacy or safety of pharmaceuticals, clinical trials, such as surgical trials or radiotherapy trials using radiation therapy equipment within the scope of approval, are conducted under the ethical guidelines. Non-interventional studies are also subjected to the ethical guidelines (Fig. 1B) (3).
Ethical guidelines
History of ethical guidelines
The ethical guidelines for clinical research were originally developed as three separate guidelines: The Ethical Guidelines for Human Genome/Gene Analysis Research formulated in 2001, the Ethical Guidelines for Epidemiological Research in 2002 and the Ethical Guidelines for Clinical Research in 2003. They have been amended several times and integrated because of changes in government policies on the protection of personal information, conflicts of interest and reliability of clinical research, among others (Fig. 2) (4).
Figure 2.
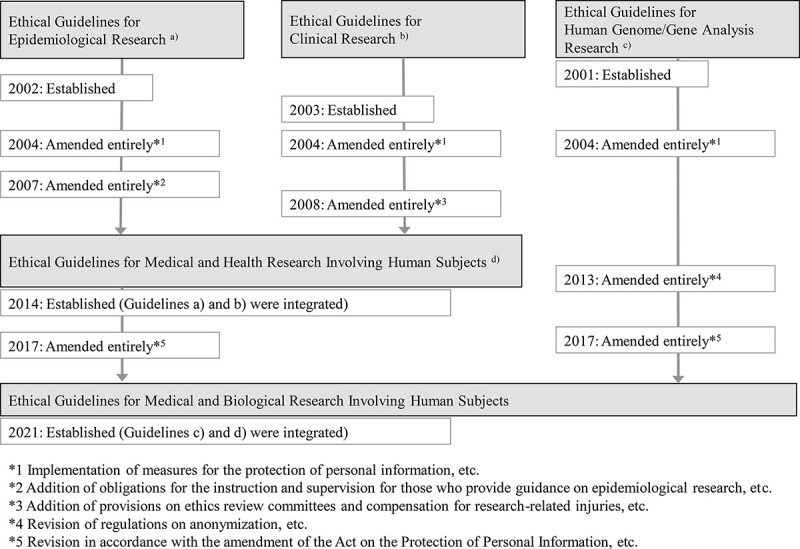
History of the Japanese ethical guidelines.
In 2012, a series of research misconducts, including the Valsartan case, came to light (5,6), and the investigative commission established under the Minister of Health, Labor and Welfare concluded that the following issues observed in the Valsartan case must be resolved to recover trust in clinical research in Japan: (1) the purpose of the research was not directed towards the clarification of the medical research problem; (2) inappropriate data management led to erroneous research conclusions; (3) the principal investigator did not adequately fulfil his/her responsibilities; (4) the ethical review committee failed to prevent research misconduct; (5) the research materials had already been discarded and could not be verified retrospectively; and (6) insufficient consideration was given to the protection of the research subjects. The investigative commission presented the following recommendations to prevent recurrence of research misconducts: (a) strengthening the functions and ensuring the transparency of ethical review committees; (b) clarification of the responsibilities of principal and associate investigators and providing thorough education and training for them; (c) establishment of a system of preventing data falsification, such as introducing a third-party organization that is exclusively responsible for data management, and consideration of the appropriateness of a research-conducting organization not only for conflict of interest management but also for ensuring research quality by the ethics review committee; and (d) obligation to store clinical research-related materials.
Additionally, the diversification of research types at that time made it unclear which guidelines (i.e. Ethical Guidelines for Epidemiological Research or Ethical Guidelines for Clinical Research) applied to the research. Due to these circumstances, the two ethical guidelines were amended and integrated into the Ethical Guidelines for Medical and Health Research Involving Human Subjects in 2014 (Fig. 2). In response to the investigative commission’s recommendations, several new requirements were introduced in the new ethical guidelines, such as clarifying researcher responsibilities, requiring education and training for ethical review committee members, disclosing the rules and regulations of the ethics review committee and the list of its members to the public, storing specimens and information, managing conflicts of interest, monitoring and auditing.
Although the regulation of clinical research conducted under the ethical guidelines was strengthened, the investigative commission under the Minister of Health, Labor and Welfare concluded that a certain range of clinical research should be regulated by law, considering both the risk to the research subjects participating in the clinical research and the social risk, such as the degree of impact of the research results on treatment policy in clinical practice. This was because the ethical guidelines were previously just administrative guidance jointly issued by the Ministry of Education, Culture, Sports, Science and Technology, the MHLW and the Ministry of Economy, Trade and Industry. They were not legal regulations adopted by the National Diet and thus not legally binding, that is, no penalties were imposed for violations. Thus, compliance with the ethical guidelines alone was not regarded as sufficient to recover trust in clinical research in Japan that was lost due to a series of research misconduct cases.
Consequently, the CTA was adopted by the National Diet in 2017 and enacted in April 2018. Specified clinical trials using unapproved/off-label pharmaceuticals or funded by a manufacturer that has marketing approval for the pharmaceuticals were defined as research corresponding to two scopes determined on the basis of the risks mentioned above by the investigative commission.
New ethical guidelines in 2021
The New Integrated Guidelines, which integrated two existing ethical guidelines, was implemented in June 2021 (Fig. 2). Before the execution of the new guidelines, some studies fell under both the Ethical Guidelines for Medical and Health Research Involving Human Subjects and Ethical Guidelines for Human Genome/Gene Analysis Research (hereinafter referred to as the ‘Human Genome Guidelines’), which have many similarities in terms of what they specify.
The other reason for the integration of the two ethical guidelines is that the Human Genome Guidelines no longer fit the current clinical practice. The Human Genome Guidelines were formulated on 29 March 2001, more than 20 years ago, when human genome research was literally exploratory ‘research’, and the results were rarely disclosed to the patients. At that time, research dealing with germline mutations was subject to the Human Genome Guidelines and was treated as a separate category of research that deals with sensitive information of genes and requires special care in handling. However, in recent years, to detect acquired somatic mutations in cancer cells, for example, a method of comparing the DNA sequence of cancer tissue with that of a blood-derived germline is frequently used. In this case, although the researcher is in fact reading the germline, the purpose of the research is to detect somatic mutations, and the question arises as to whether the applicability of the Human Genome Guidelines should be judged by ‘action’ or ‘purpose.’
Furthermore, a controversy has arisen in treating only germline mutations with special care and requiring complicated procedures. In other words, other types of information in medical information should also be handled as sensitively as germline mutations. Additionally, the results of germline mutation testing are now being returned to patients as part of usual clinical practice. However, if test results were to be returned to patients in research under the Human Genome Guidelines, the procedure would have been extremely complicated and would have involved anonymization and deanonymization by a ‘privacy officer’ who would be defined in the Human Genome Guidelines and would be independent of the researcher during genome analysis.
With the integration of the two ethical guidelines, the following provisions unique to the Human Genome Guidelines have been basically abolished: (1) the requirement of assigning a privacy officer independent of the researcher; (2) the requirement of an ethical review when an existing specimen and/or information is provided to an external institution, although only notification to the chief executive of the investigating institution was required in the Ethical Guidelines for Medical and Health Research Involving Human Subjects; (3) the requirement of an on-site investigation by outside experts once a year; and (4) the requirement of a withdrawal of consent to be done in writing. Although the procedure has been simplified in this way, it is important to note that the individual’s germline information is protected by the concise procedures following the New Ethical Guidelines.
Practical changes in the New Integrated Guidelines
The New Integrated Guidelines comprises 10 chapters and 24 parts, as shown in Table 1. We will go over the three practical changes introduced in the new guidelines that have relatively significantly impacted researchers: (1) centralized review, (2) electromagnetic consent and (3) research cooperating organization.
Table 1.
Chapter and sections of the New Integrated Guidelines
| Chapter 1 General Provisions Part 1 Purpose and Basic Principles Part 2 Glossary Part 3 Scope of Application |
| Chapter 2 Obligations of Investigators, etc. Part 4 Basic Obligations of Investigators, etc. Part 5 Obligations of the Chief Executive of Research Implementing Entity |
| Chapter 3 Proper Implementation of Research, etc. Part 6 Procedures Related to Research Protocol Part 7 Contents of Research Protocol |
| Chapter 4 Informed Consent, etc. Part 8 Procedures for Obtaining Informed Consent, etc. Part 9 Procedures, etc. for Obtaining Informed Consent from Legally Acceptable Representatives, etc. |
| Chapter 5 Handling of Results Obtained Through Research, etc. Part 10 Handling of Results Obtained Through Research, etc. |
| Chapter 6 Ensuring Reliability of Research Part 11 Proper Procedures and Reporting of Research Part 12 Managing Conflicts of Interest Part 13 Storage of Specimens and Information, etc. for Research Part 14 Monitoring and Audit |
| Chapter 7 Response to Serious Adverse Events Part 15 Response to Serious Adverse Events |
| Chapter 8 Ethical Review Committee Part 16 Organizing, etc. of Ethical Review Committee Part 17 Roles, Responsibilities, etc. of Ethical Review Committee |
| Chapter 9 Personal Information and Anonymously Processed Information, etc. Part 18 Basic Obligations about Personal Information, etc. Part 19 Security Control Measures Part 20 Disclosure, etc. of Retained Personal Information Part 21 Handling of Anonymously Processed Information |
| Chapter 10 Supplementary Provisions Part 22 Effective Date Part 23 Transitional Measures Part 24 Amendment |
Centralized review
The most significant impact of the New Integrated Guidelines on researchers is the centralized review. Under the previous ethical guidelines, the inefficiency and inconsistent quality of multiple reviews by ethical review committees at each institution in multi-institutional studies have been long-standing issues. In fact, the survey conducted by the Clinical Research Initiative for Global Health (CRIGH) from 2017 to 2018 to investigate the differences in ethics review systems among countries revealed that the number of ethics review committees in Japan was 1700, which was by far the highest among the countries surveyed (7). Although a central review had been acceptable under the previous ethical guidelines, each institution usually has its own institutional review board (IRB) that reviewed most cases. Centralized review was introduced under the CTA in 2018, and a certification system for the review board was introduced only for the CTA by the MHLW, with 97 boards certified as of January 2022. The specified clinical trials are required to undergo review by the certified review board. Hence, the New Integrated Guidelines mandate centralized review in principle.
One of the practical issues in implementing centralized review is the lack of a certification system for the review board under the New Integrated Guidelines. Thus, researchers can choose freely which ethics review committee to request for centralized review because no criteria for selecting an ethics review committee have been specified. Researchers can decide which ethics review committee to choose on the basis of whether the review fee is acceptable and whether the effort required for submitting the required documents is reasonable since the documents required for application vary depending on each IRB. To avoid so-called IRB shopping, some institutions actually limit the ethics review committees that researchers at their institution can request for centralized review so that they can ensure a certain level of quality in the review.
Notably, this centralized review requirement is only a ‘principle’. The New Integrated Guidelines state that centralized review does not preclude undergoing the review by an individual ethics review committee in multi-institutional studies. This means that multiple reviews by ethical review committees at each institution are allowed, as was done under the previous guidelines. Additionally, the guidelines state that a mix of centralized and institution-based reviews is acceptable in a multi-institutional study; hence, some institutions can undergo a single central review and others can undergo review by individual ethics review committees in one study. Therefore, various operations are possible depending on the circumstances of each institution and each study. This raises the concern that this will lead to complexity and inefficiency. Thus, the selection should at least be expected to be standardized, which is either a centralized review for all institutions or an institution-specific review for all institutions.
Electromagnetic consent
In the New Integrated Guidelines, electromagnetic consent (so-called eConsent), has been allowed. The process of informed consent comprises three phases, namely, explanation, obtaining consent and maintaining records. With the development of digital devices, the explanation and maintaining records phases have already been introduced. eConsent was not allowed in obtaining consent phase in the previous guidelines because of difficulty in identifying the research subjects but has been recently added as one of the options.
Examples of the implementation of eConsent envisioned in the process of developing the New Integrated Guidelines include explaining the research using a video and obtaining consent by electromagnetic signature on a tablet or sending the subject a link to a website that explains the research online and obtaining consent by clicking the consent button on the screen. The new ethical guidelines clearly state that special consideration must be given to the following three points: (i) research subjects shall be appropriately identified; (ii) research subjects shall be given the opportunity to ask questions regarding the content of the explanations, and such questions shall be fully answered; and (iii) the consent items shall be readily accessible, and documents shall be provided to research subjects as requested even after informed consent has been obtained. The New Integrated Guidelines also provide guidance on how to appropriately identify the research subject by, for example, self-reporting or presenting an ID card in situations that do not require personal appearance (8). Although eConsent has yet to be integrated into research practice because of the hurdles to overcome in implementing the above considerations, eConsent is expected to be more often adopted as a tool to facilitate research, as supported by the recent research digitalization movements and represented by the ‘decentralized clinical trial’ or ‘virtual clinical trial.’
Research cooperating organization
In the New Integrated Guidelines, research cooperating organizations, which exclusively provide research-implementing entities with ‘newly acquired’ specimens and information without or with only minor invasiveness, have been introduced. Minor invasiveness is defined as a procedure causing minor injury and/or distress on the research subjects’ body and/or mind. For example, blood sampling and irradiation that are equivalent to blood sampling and simple chest radiography in general health examinations and magnetic resonance imaging (MRI) imaging without contrast media are regarded as procedures with minor invasiveness (8). Research cooperating organizations are exempted from the responsibility of undergoing the ethical review and researcher education. Under the previous guidelines, the exemption is applied only to ‘existing’ specimens or information. Thus, the specifications newly introduced in the New Integrated Guidelines could be regarded as the expansion of the previous definition for existing specimens or information to newly acquired specimens or information.
Note worthily, individuals in a research cooperating organization are not considered researchers and therefore cannot obtain informed consent from research subjects. In other words, researchers at one of the research participating institutions typically must obtain informed consent remotely from research subjects at the research cooperating organizations by electromagnetic procedures. Since eConsent is not yet in widespread use, its maturation must be anticipated to fully utilize the system of research cooperating organizations.
One possible practical use of research cooperating organizations is to have researchers at the research participating institution obtain informed consent from the research subjects and then ask a research cooperating organization to perform tests with minor invasiveness such as MRI or follow-up observations with periodic blood sampling. Of note, the research cooperating organization system cannot be used to request medical procedures with major invasiveness such as computed tomography scans and positron emission tomography scans.
Future prospects
Some practical issues that must be addressed include encouraging the use of central review by simplifying the process of preparing and collecting required documents, determining the types of research and the manner eConsent should be allowed and promoting the use of research cooperating organizations by sharing and accumulating specific examples.
Regulatory requirements are expected to evolve with the diversification of clinical research due to evolving technology and changing social needs. In fact, in response to the 2020 and 2021 amendments of the Act on the Protection of Personal Information, the New Integrated Guidelines are scheduled to be revised and enforced in April 2022. To promote the conduct of scientifically and ethically valuable clinical research in a rapidly changing society and environment, easily comprehensible regulatory information must be provided and shared with researchers, and researchers must conduct clinical research with an understanding of the latest regulatory information.
Funding
This work was supported by the National Cancer Center Research and Development Fund (2020-A-13) and the Science Research Grant funded by the Ministry of Health, Labour and Welfare, Japan (21CA2010, 21KC2005).
Conflict of interest statement
All authors have no conflicts of interest to disclose.
Acknowledgement
We thank all the members of JCOG Data Center, JCOG Operations Office and Clinical Research Support Office in the National Cancer Center Hospital for their useful suggestions.
Contributor Information
Junko Eba, JCOG Operations Office, Clinical Research Support Office, National Cancer Center Hospital, Tokyo, Japan.
Kenichi Nakamura, JCOG Operations Office, Clinical Research Support Office, National Cancer Center Hospital, Tokyo, Japan.
References
- 1. Ministry of Education, Culture, Sports, Science and Technology, Ministry of Health, Labour and Welfare, and Ministry of Economy, Trade and Industry . Ethical guidelines for medical and biological research involving human subjects. https://www.mhlw.go.jp/content/000757566.pdf (4 January 2022, date last accessed) (in Japanese).
- 2. Nakamura K, Shibata T. Regulatory changes after the enforcement of the new Clinical Trials Act in Japan. Jpn J Clin Oncol 2020;50:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ministry of Health, Labour and Welfare . Explanatory documents for overview of the clinical trials Act. https://www.mhlw.go.jp/content/10800000/000647734.pdf (4 January 2022, date last accessed) (in Japanese).
- 4. Ministry of Education, Culture, Sports, Science and Technology, Ministry of Health, Labour and Welfare, and Ministry of Economy, Trade and Industry . Explanatory documents for ethical guidelines for medical and biological research involving human subjects. https://www.mhlw.go.jp/content/000769921.pdf (4 January 2022, date last accessed) (in Japanese).
- 5. Tanimoto T, Kami M, Shibuya K. Research misconduct and scientific integrity: a call for a global forum. Lancet 2013;382:940. [DOI] [PubMed] [Google Scholar]
- 6. McCurry J. Former Novartis employee arrested over valsartan data. Lancet 2014;383:2111. [DOI] [PubMed] [Google Scholar]
- 7. Nakada H, Hasthorpe S, IJsselmuiden C, et al. Recommendations for promoting international multi-site clinical trials—from a viewpoint of ethics review. Dev World Bioeth 2019;19:192–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ministry of Education, Culture, Sports, Science and Technology, Ministry of Health, Labour and Welfare, and Ministry of Economy, Trade and Industry . The guidance of ethical guidelines for medical and biological research involving human subjects. https://www.mhlw.go.jp/content/000769923.pdf (4 January 2022, date last accessed) (in Japanese).