

Abstract
Background
Next generation sequencing has progressed rapidly, characterizing microbial communities beyond culture-based or biochemical techniques. 16S ribosomal RNA gene sequencing (16S) produces reliable taxonomic classifications and relative abundances, while shotgun metagenome sequencing (WMS) allows higher taxonomic and functional resolution at greater cost. The purpose of this study was to determine if 16S and WMS provide congruent information for our patient population from paired fecal microbiome samples.
Results
Comparative indices were highly congruent between 16S and WMS. The most abundant genera for 16S and WMS data did not overlap. Overlap was observed at the Phylum level, as expected. However, relative abundances correlated poorly between the two methodologies (all P-value>0.05). Hierarchical clustering of both sequencing analyses identified overlapping enterotypes. Both approaches were in agreement with regard to demographic variables.
Conclusion
Diversity, evenness and richness are comparable when using 16S and WMS techniques, however relative abundances of individual genera are not. Clinical associations with diversity and evenness metrics were similarly identified with WMS or 16S.
Keywords: 16S rRNA gene sequencing, whole genome shotgun sequencing, gut microbiome, cervical cancer
1. Introduction
The gut microbiome is increasingly recognized as a critical determinant of health and disease (Lynch and Pedersen, 2016). The vast majority of microbiome analyses have utilized 16S rRNA gene sequencing (16S) which uses variable regions of the 16S ribosomal RNA gene to assign taxonomic classification and read abundance to calculate the relative frequency of the organisms within a sample (Vogtmann et al., 2016). 16S sequencing via any amplicon sequencing-based method offers advantages over WMS in terms of precision (specific gene targeting). Additionally, 16S has historically been less costly due to the simplicity of library preparation and it does not require the same level read coverage as WMS. 16S is a reliable method for identifying the relative frequency of organisms but does not provide reliable functional information about the genes encoded by these organisms (Laudadio et al., 2018). As a consequence, whole-metagenome sequencing (WMS) data has been increasingly utilized with the goal of providing functional information about the organisms present. WMS analyzes large swaths of genomic information, which confers several advantages over 16S. Most notably, WMS allows for an increased depth and specificity of sequenced species as well as insights into gene abundance and metabolic capacity (Salipante et al., 2015). Since WMS yields genomic information beyond the 16S rRNA gene, it also confers a better assessment of the true diversity of the sample. Thus, WMS can be used to provide species level resolution, as well as differences in presence of microbial genes, articulated pathways and metabolic functions (Salipante et al., 2015). Yet, a limitation of shotgun sequence data is the large number of sequence reads which must be mapped to databases, which requires significant expertise to balance classification accuracy with discarded reads. Now, it is possible to analyze 16S and WMS microbiome data side-by-side to investigate bacterial communities as well as the abundance of associated genes and metabolic pathways (Franzosa et al., 2019, 2018; Pasolli et al., 2019; Vatanen et al., 2019; Vila et al., 2018; Zolfo et al., 2018). Still, the extent to which these two sequencing methods correlate with one another is a critical assumption, which should be explored thoroughly.
Few studies have had the opportunity to compare previously observed 16S gene associations with data from WMS on the same cohort of patients (Vogtmann et al., 2016). By subjecting the same sample to both sequencing methods, we aim to investigate the reliability, validity and reproducibility of these different approaches. To do so we utilized baseline gut microbiome analysis from patients receiving standard chemoradiation therapy for cervical cancer in order to examine and compare 16S microbiome associations with WMS data on a variety of clinical variables. We deployed commonly used alpha diversity metrics (Inverse Simpson Diversity, Shannon Diversity, Camargo Evenness, Pielou Evenness, Observed Operational Taxonomic Units, and the Low Abundance Rarity Index) as well as abundance measures, to draw comparisons between the two datasets. Additionally, we submitted the datasets to unsupervised hierarchical clustering in order to assess if the microbiome profiles associated together in a similar manner, as would be expected from two datasets derived from a single sample source.
2. Materials and Methods
2.1. Study design and participants
We collected rectal swab samples from a cohort of 41 patients with newly diagnosed, locally advanced cervical cancer undergoing treatment at The University of Texas MD Anderson Cancer Center and Harris Health System Lyndon B. Johnson clinic. We collected a swab sample from each patient before they received chemoradiation treatment. Patients with previous pelvic radiation or treatment for cervical cancer were excluded. This study was part of an IRB approved protocol (MDACC 2014–0543).
2.2. Patient population and treatment characteristics
Patients were enrolled in an IRB-approved (2014–0543) multi-institutional prospective clinical trial at The University of Texas MD Anderson Cancer Center and the Harris Health System, Lyndon B. Johnson General Hospital Oncology Clinic. Inclusion criteria were newly diagnosed cervical cancer per the Federation of Gynecology and Obstetrics (FIGO) 2009 staging system, clinical stage IB1-IVA cancers, visible, exophytic tumor on speculum examination with planned definitive treatment of intact cervical cancer with external beam radiation therapy, cisplatin and brachytherapy. Patients with any previous pelvic radiation therapy were excluded.
Patients underwent standard-of-care pretreatment evaluation for disease staging, including tumor biopsy to confirm diagnosis; pelvic magnetic resonance imaging (MRI) and positron emission tomography/computed tomography (PET/CT); and standard laboratory evaluations, including a complete blood cell count, measurement of electrolytes, and evaluation of renal and liver function. Patients received pelvic radiation therapy to a total dose of 40‒45 Gy delivered in daily fractions of 1.8 to 2 Gy over 4 to 5 weeks. Thereafter, patients received intracavitary brachytherapy with pulsed-dose-rate or high-dose-rate treatments. Patients received cisplatin (40 mg/m2 weekly) during external beam radiation therapy according to standard institutional protocol. Patients underwent repeat MRI at the completion of external beam radiation therapy or at the time of brachytherapy, as indicated by the extent of disease. Patients with no residual tumor on repeat MRI were considered to be exceptional responders while those with residual MRI tumor volumes ≤20% and >20% of initial volumes after 4 to 5 weeks after initiation of RT were considered to be standard and poor responders, respectively.
2.3. Sample collection and sequencing
Rectal swabs were collected in clinic at the time of rectal examination prior to treatment using quick release matrix designed Isohelix swabs (Isohelix cat. SK-2S, Cell Projects LTD, Kent, United Kingdom). We placed the swabs in 400 μL of Isohelix Lysis buffer and stored them at −80°C within 1 hour of sample collection. One portion of each sample was sequenced using 16SV4 rRNA sequencing targeting the V4 region with primers 515F-806R (Thompson et al., 2017), while another portion was sequenced using WMS. 16S rRNA gene sequencing was performed through the Alkek Center for Metagenomics and Microbiome Research (CMMR) at Baylor College of Medicine. 16S rRNA gene sequencing methods were adapted from the methods developed for the Earth Microbiome Project (Thompson et al., 2017). Briefly, bacterial genomic DNA was extracted using MO BIO PowerSoil DNA Isolation Kit (MO BIO Laboratories, QIAGEN, San Diego, CA, USA). The 16S rDNA V4 region was amplified by PCR and sequenced on the MiSeq platform (Illumina) using the 2×250 bp paired-end protocol yielding pair-end reads that overlap almost completely. The primers used for amplification contain adapters for MiSeq sequencing and single-end barcodes allowing pooling and direct sequencing of PCR products. Then gene sequences were clustered into OTUs at a similarity cutoff value of 97% using the UPARSE algorithm (Edgar, 2013). To generate taxonomies, OTUs were mapped to an optimized version of the SILVA rRNA database containing the 16S V4 region and then rarefied at 6989 reads. A custom script was used to construct an OTU table from the output files generated as described above for downstream analyses. Here, OTUs were selected as a basis for further analysis because this method is currently the most common approach to 16S analysis in the clinical research setting.
For WMS data, genomic bacterial DNA (gDNA) extraction methods optimized to maximize the yield of bacterial DNA from specimens while keeping background amplification to a minimum were employed (Huttenhower et al., 2012; The Human Microbiome Project Consortium, 2012). Metagenomic shotgun sequencing was performed on extracted total gDNA on Illumina sequencers using chemistries that yielded paired-end reads. Sequencing reads were derived from raw BCL files which were retrieved from the sequencer and called into fastqs by Casava v1.8.3 (Illumina). Then, paired-end reads (fastq format) were filtered to remove Illumina PhiX sequences and trimmed for the Illumina adapters by using bbduk in BBTools (version 38.34) (Bushnell et al., 2017). To remove host DNA contamination, the trimmed reads were then mapped to a human reference sequence database (hg38) by using Bowtie2 (version 2.3.5) (Langmead and Salzberg, 2012). Taxonomic classification was performed through MetaPhlAn2 (McDonald et al., 2012). Also based on Bowtie2, we mapped the cleaned (unmapped to host genome) reads to a marker gene database (mpa_v295_CHOCOPhlAn_201901, updated 11/11/2019) to get an individual relative abundance table for each sample. Relative abundance tables for all samples were merged and converted to a BIOM format (version 1.0) (Truong et al., 2015), which was then imported into ATIMA (Agile Toolkit for Inclusive Microbial Analysis) (The Human Microbiome Project Consortium, 2012) for statistical and diversity analysis. Additionally, we obtained the functional annotation of the microbial community by using HUMAnN2 (Franzosa et al., 2018). The pipeline used for sequencing the 16S rRNA data has been previously described (Sims et al., 2021).
2.4. Alpha Diversity Indices
We then analyzed data from both WMS and 16S using several alpha diversity metrics provided in the Microbiome R package (Lahti et al., n.d.) (R version 3.6.2), in order to assess the richness, divergence and evenness of the microbial communities within each patient sample. We calculated several index measures from observed operational taxonomic unit (OTU) counts for 16S data and WMS data collected from MetaPhlAn2 analysis independently. The Shannon Diversity (SD) (Mouillot and Leprêtre, 1999) accounts for both the abundance and the evenness of the taxa within a given sample and Inverse Simpson Diversity (ISD)[21] index provides a measure of the relative abundance of the different species within a sample making up the sample richness. The Camargo (Camargo, 1995) and Pielou (Pielou, 1966) Evenness indices are designed to calculate the proportionality of individual species within a sample population. A high degree of evenness would imply that the abundances of all individuals are roughly the same, or in equal proportions. Finally, the richness of the datasets was calculated using observed OTU counts and the Low Abundance Rarity (LAR) Index measures. The observed OTUs index provides a count based on the presence of at least one read for a given species within a sample. The LAR index (Lahti et al., n.d.) instead characterizes the concentration of species within a sample which have low abundance, defined as those falling below the detection level of 0.2%.
2.5. Comparative Statistical Analysis of 16S and WMS
We then paired each patient value from one dataset with its corresponding value for the same patient in the other dataset. The amount of agreement between the two datasets, in terms of alpha diversity measures, was then quantified using Spearman’s rank correlation coefficient (R or rho) with value of 1 indicating a perfect agreement, between two sets of variables.
To assess the consistency in reporting microbial abundance between 16S and WMS, we identified the most abundant taxa at the genus level for each sequencing method independently. Then, we compiled a list of organisms on either of the lists. Thus, the next set of comparisons were drawn using the total number of possible taxa identified by taxonomic name at all phylogenetic levels (with the exception of species).
We also analyzed the datasets individually while considering patient demographic and clinical characteristics. We analyzed six clinical variables to assess differences in diversity, evenness, and richness between groups. Binary classifications were analyzed using the independent t-test (Age, Smoking History, Histology) while multivariable classifications were analyzed using One-Way ANOVA (Ethnicity, Node Level, FIGO Stage). We also studied age and BMI as continuous variables in relation to Inverse Simpson Diversity and Pielou evenness for both 16S and WMS datasets. Consensus between the two datasets was defined as a P-value<=0.1. The cut-off P-value of 0.1 was chosen since we aimed to test for a consensus between the two sequencing methods, and not only for significance. All analyses were conducted using R version 3.6.2 and Microsoft Excel (2016).
2.6. Hierarchical Clustering
To further explore the consistency of the two datasets, specifically the sample grouping according to the putative taxa abundance profiles, we used unsupervised hierarchical clustering of each OTU table by the cluster software with default settings (Bronstein and Deutsch, 1991). We performed the clustering using correlation as the similarity metric and the centroid linkage as the clustering method. The data used for clustering was limited by only using OTUs found in more than 14 samples. The obtained heatmaps were visualized by the Java TreeView software (Saldanha, 2004).
3. Results
3.1. Taxonomic composition and abundance using 16S and WMS
The number of putative taxa compiled in OTU tables was dramatically different between the technologies and included 984 OTUs in the 16S OTU table yet only 451 in the WMS table. The WMS OTU table was not as sparse as the 16S table and had a different abundance distribution frequency display, which was close to normal (Figure 1, A). The sparse 16S OTU table had significantly more rare low-abundance taxa; this feature is evident from the frequency distribution (Figure 1, B) and is well-characterized for this type of dataset.
Figure 1. Different distribution of putative taxa species abundance in WMS and in 16S OTU tables.
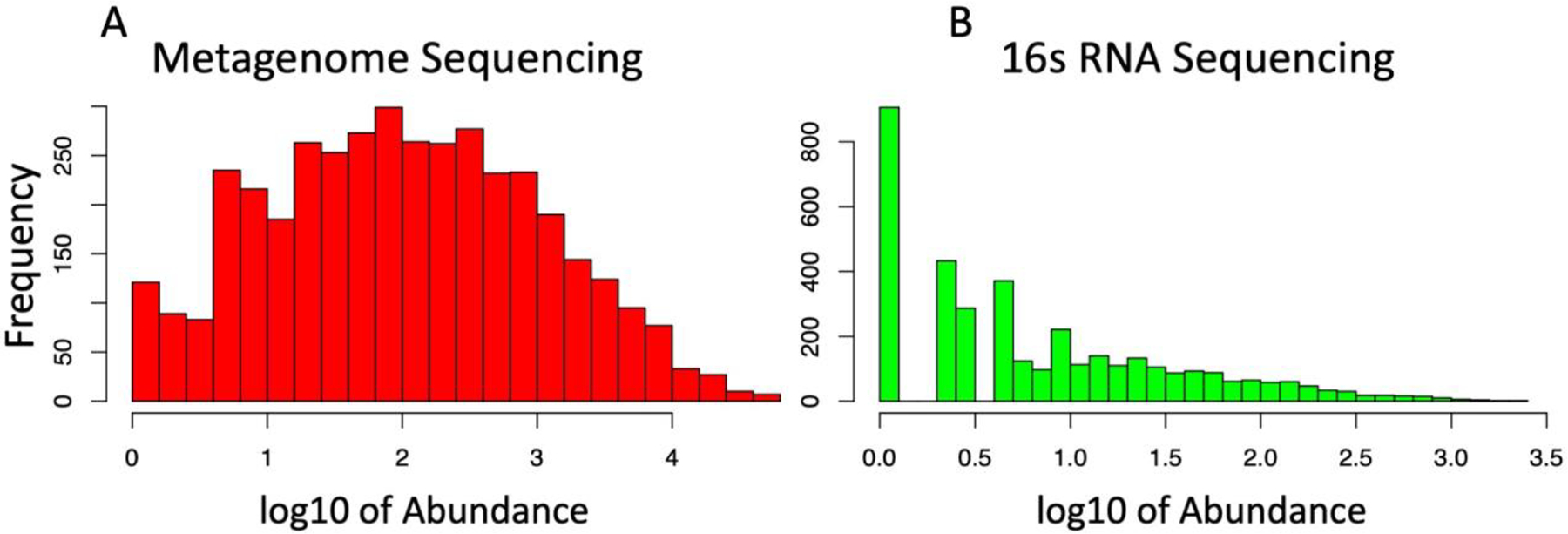
To display differences in abundances in terms of OTU frequencies, OTU counts were log transformed and presented here as histograms. WMS (A) OTU identifiers displayed reduced overall frequency compared to 16S (B), however the distribution of species abundance showed a more normal distribution.
The top 10 most abundant phyla and genera found in 16S and WMS are shown in Figure 2. There was better consensus between 16S and WMS datasets on the phyla level than on the genus level (Figure 2). The most abundant phyla identified in both 16S and WMS were Bacteroides, Firmicutes, Proteobacteria, Actinobacteria and Fusobacteria. Interestingly, Verrucomicrobia were found to be highly abundant only by WMS. Tenericutes were ranked third most abundant by 16S but had low abundance according to WMS. There was a significant mid-level association (rho=0.69, P-value=0.03) between the phyla abundances (Figure 2, A–B). No significant associations between the abundances were identified at other taxonomic levels.
Figure 2. Comparison of top 10 taxa at highest and lowest taxonomic levels for WMS and 16sRNA.
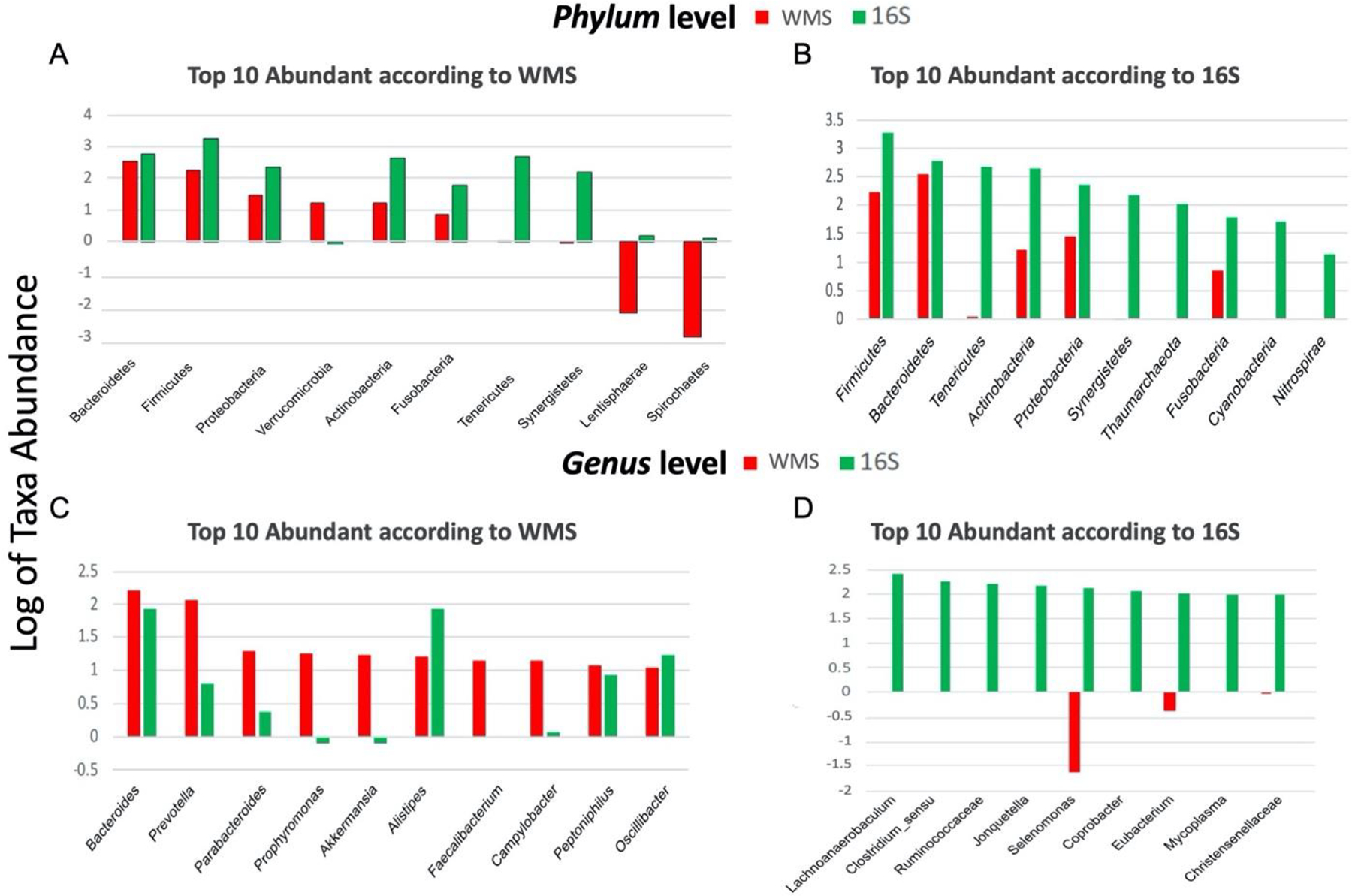
The bar plots presented show the top ten most abundant taxa present in the WMS (red), 16sRNA (green) as identified at the Phylum (A,B) and Genus (C,D) levels of taxa. The two datasets have a greater level of consensus in terms of microbial abundance at higher taxonomic levels (eg. Phylum) than lower levels (eg. Genus).
None of the top abundant genera according to 16S were identified as the most abundant genera according to WMS (Figure 2, C–D). There was no overlap between the top 10 most abundant genera in 16S and WMS. The top 10 genera in 16S or WMS are listed in Table 1 with ranked relative abundances in each data set. Twelve genera were present in either the top 10 of 16S or WMS and present at any rank level in the other data set, and thus no abundance comparisons were made. Most genus level abundances correlated poorly between 16S and WMS (rho<0.15). The only genus, with relative abundance correlated well between the data sets, was Peptoniphilus (rho=0.68, P-value<0.01).
Table 1.
Comparisons of Top Ranked WMS and 16S Genera.
| Genus Name | WMS Rank | 16S Rank | Spearman R | p Value |
|---|---|---|---|---|
| Bacteroides | 1* | 12 | 0.29 | 0.0639 |
| Prevotella | 2* | 70 | 0.21 | ns |
| Parabacteroides | 3* | 88 | 0.09 | ns |
| Porphyromonas | 4* | 126 | −0.1 | ns |
| Akkermansia | 5* | 129 | 0.18 | ns |
| Alistipes | 6* | 13 | −0.27 | 0.0828 |
| Faecalibacterium | 7* | - | - | - |
| Campylobacter | 8* | 115 | 0.05 | ns |
| Peptoniphilus | 9* | 56 | 0.68 | 1.14E-06 |
| Oscillibacter | 10* | 34 | 0.12 | ns |
| Mycoplasma | 58 | 9* | 0.24 | ns |
| Coprobacter | 73 | 7* | 0.22 | ns |
| Jonquetella | 119 | 5* | −0.18 | ns |
| Nocardioides | - | 1* | - | - |
| Lachnoanaerobaculum | - | 2* | - | - |
| Clostridium sensu stricto 1 | - | 3* | - | - |
| Ruminococcaceae UCG_014 | - | 4* | - | - |
| Selenomonas_4 | - | 6* | - | - |
| Eubacterium coprostanoligenes group | - | 8* | - | - |
| Christensenellaceae R_7_group | - | 10* | - | - |
The top ten most abundant Genera identified in WMS or 16S (identified with *) are shown next to their counterpart and the associated rank. Genera present in both lists were correlated in terms of abundance using Spearman R. Resulting p values are shown as not significant (ns), less than 0.1, or less than 0.05 (bold). Genera present in one dataset, but not the other ( - ) could not be correlated.
Consistent with the difference in frequency distribution of species abundances, the 16S dataset included more putative species annotated at different taxonomic levels. Furthermore, a high percentage of the taxa identified via 16S (58–67%) were not identified by WMS, potentially as a result of using marker-gene classifiers. Conversely, most taxa found in the WMS table were also identified by 16S (Figure 3, A–E). This percentage decreased at low taxonomic levels.
Figure 3. Comparison of number of overlapping taxa at each phylogenetic level for WMS and 16S.
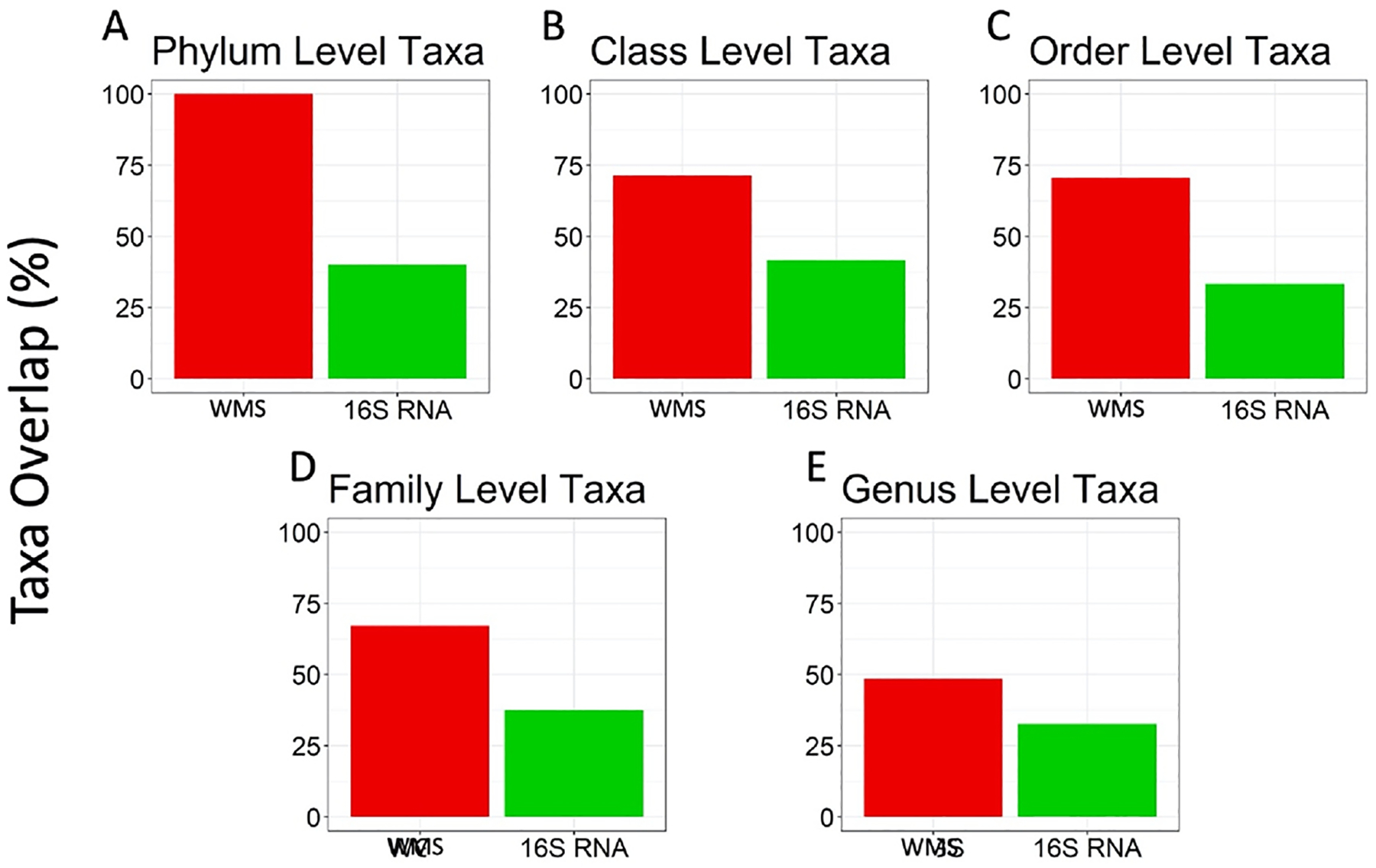
The bar plots show the percentage of (A) Phylum, (B) Class, (C) Order, (D)Family, and (E) Genus level taxa present in WMS (red), and 16S (green) which overlap in both lists. Across all levels, many of the WMS taxa identified were also identified in the list of 16S taxa.
3.2. Diversity, evenness, and richness by 16S and WMS
To further investigate the varied microbial compositions and abundances of taxa at most phylogenetic levels, we next explored the effects of different general characteristics of species diversity within the gut microbiomes. Surprisingly, we found that most indices of diversity, evenness, and richness showed significant correlation between 16S and WMS datasets (Figure 4). All of the diversity and richness measures were tightly correlated between 16S and WMS (ISD rho=0.89, P-value<0.01; SD rho=0.90, P-value=<0.01; observed OTUs rho=0.76, P-value<0.01; LAR rho=0.72, P-value<0.001) (Figure 4, A–F). Evenness indices had a weaker correlation between 16S and WMS (Camargo rho=0.41, P-value<0.01; Pielou rho=0.84, P-value<0.01; Figure 4, C–D), which is not surprising considering there was greater similarity in taxa abundances in the WMS dataset then in the 16S dataset (Figure 1). Despite significant differences in the rare OTUs, low abundance rarity indexes also significantly correlated between the datasets. The slope of the regression line of the association was also consistent with a significantly greater number of rare low abundance species in the 16S dataset than in WMS (Figure 4, E–F).
Figure 4. Correlation between WMS and 16S in terms of diversity, evenness, and richness.
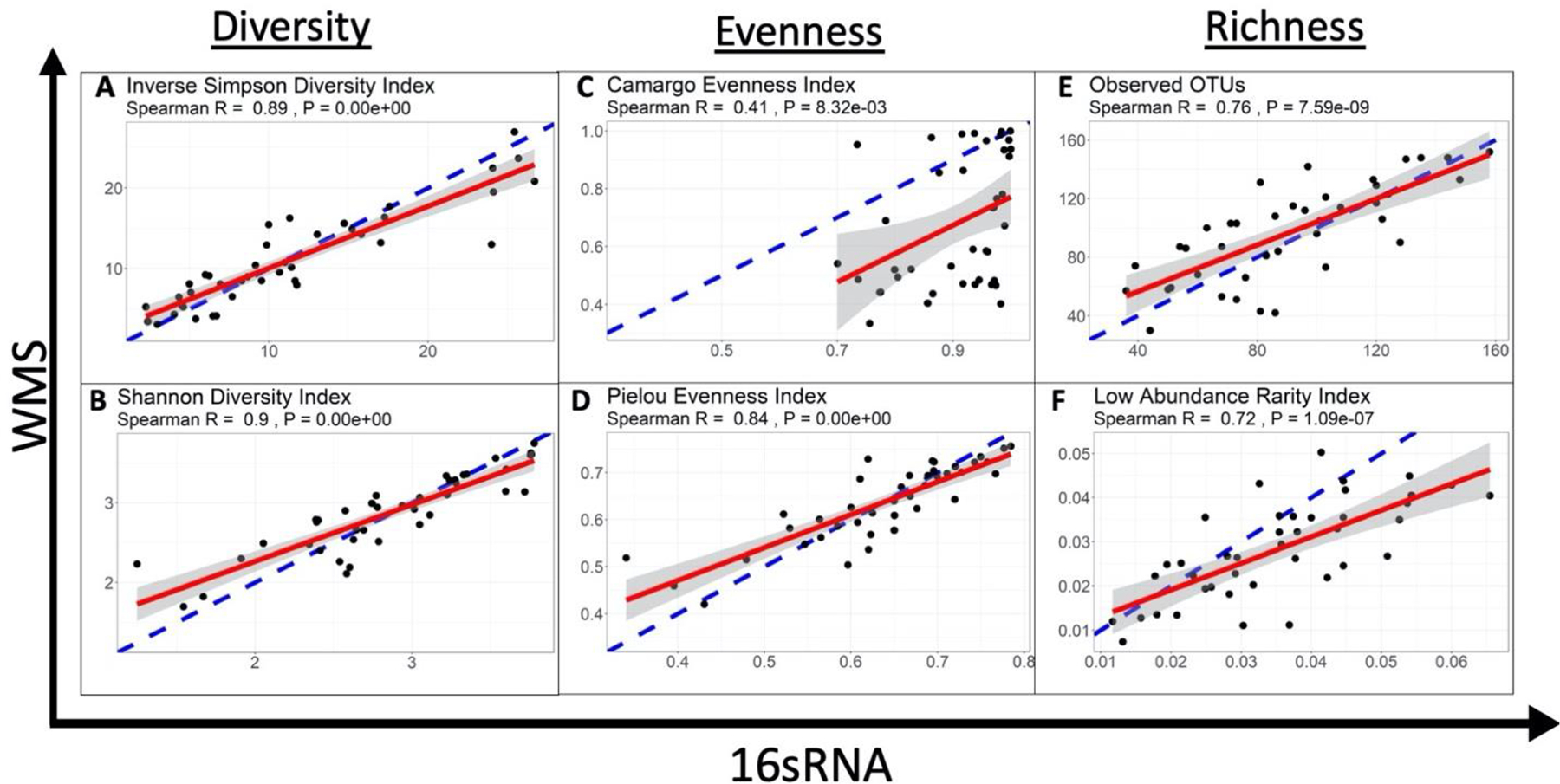
In this figure, each data point represents a single patient. Consensus between both sequencing methods in terms of alpha diversity is calculated by a Spearman Correlation (R). The slope of the correlation is represented by a red line, while the blue dotted line represents the ideal correlation (R=1) and the 95% confidence interval is represented by a grey shaded area. The data derived from 16S rRNA gene sequencing correlates well with the diversity assessment values derived from WMS for diversity and richness. The evenness measures suggest that the sequencing methods differ in terms of the proportionality of individual bacterial taxa.
3.3. Association of demographic characteristics with diversity of microbiomes and specific taxa
In our next step, we investigated whether the differences and similarities considered above affected biological conclusions drawn from each dataset. Namely, we explored demographic variables (Supplementary Table 1) in association with gut microbiome diversity and specific taxa using either the 16S or WMS dataset. When diversity, evenness and richness indices were compared to baseline characteristics using both 16S and WMS (Table 2, [see Additional file 1]), only age was associated with ISD in both WMS and 16S (P-value<0.1). Age was associated with SD (P-value=0.04), and Pielou evenness (P-value=0.01) using 16S, but not WMS. Camargo evenness was associated with age only using WMS (P-value=0.008). LAR richness was associated with BMI using WMS (P-value=0.05) but not 16S. Other baseline demographic variables were not associated with diversity, evenness or richness using any metric. Overall, there was consensus between methods (both P-value<=0.1 or >0.1) across all demographics for ISD only. A positive correlation between age and gut diversity, and between age and evenness was identified in both 16S (ISD; rho=0.37, P-value=0.02. Pielou; rho=0.39, P-value=0.01) and WMS (ISD; rho=0.29, P-value=0.06. Pielou; rho=0.28, P-value=0.08) data (Figure 5). Both datasets failed to find a difference between patient populations regarding ethnicity, smoking status, tumor histology, nodal involvement, or FIGO stage.
Table 2.
Correlation between WMS and 16S in terms of Diversity, Evenness, and Richness.
| Clinical Variable | Diversity | Evenness | Richness | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inverse Simpson | Shannon | Camargo | Pielou | Observed OTUs | LAR | |||||||
| WMS | 16sRNA | WMS | 16sRNA | WMS | 16sRNA | WMS | 16s RNA | WMS | 16SRNA | WMS | 16sRNA | |
| Age | 0.0631 | 0.0166 | ns | 0.0396 | 0.0077 | ns | ns | 0.0111 | ns | ns | ns | ns |
| Ethnicity (W,B,H,O) | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns |
| Smoking History (Y/N) | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns |
| Histology (Adeno/Squam) | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns |
| Node level | ns | ns | ns | ns | ns | ns | ns | ns | 0.0748 | ns | ns | ns |
| FIGO Stage | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns |
| BMI | ns | ns | ns | ns | ns | ns | ns | ns | ns | ns | 0.0468 | ns |
In this table, patient demographics and clinical assessments were collected and used as classification criteria to investigate differences between these characteristics in terms of alpha diversity measurements discussed earlier. Both datasets were analyzed using either a parametric t-test [Smoking History (Yes/No), Histology (Adenocarcinoma/Squamous Cell Carcinoma)], linear regression [Age and BMI], or One-Way ANOVA [Ethnicity (White, Black, Hispanic, Other), Node Level (Common Iliac/External Iliac/Internal Iliac/None/Para-Aortic), FIGO Stage (IA1, IB1, IB2, IBI, IIA, IIB, IIIB, IVA)]. The resulting p value measures are indicated on the table as being either non-significant (ns), less than 0.1 or less than 0.05 (bold). Consensus between both methods, whole-metagenome sequencing and 16sRNA sequencing, indicates the validity of using either method for exploring that clinical variable.
Figure 5. Correlation between age and Inverse Simpson Diversity and Pielou Evenness for 16S vs WMS.
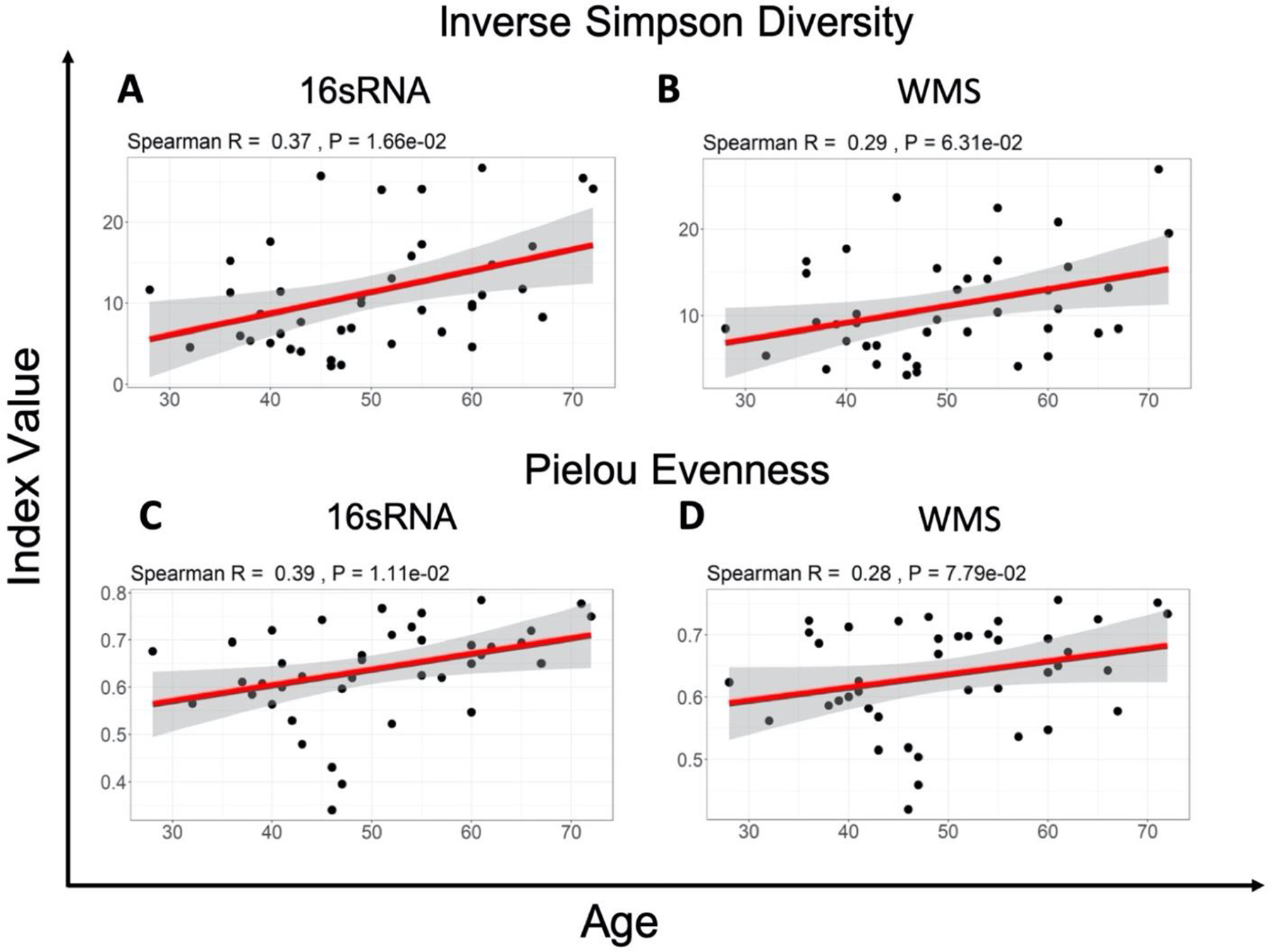
The slope of the correlation is shown in red while the 95% confidence interval is indicted by the grey shaded region. Spearman Correlation shows a weak association between age and the Inverse Simpson Diversity Index value (A,B) as well as the Pielou evenness Index value (C,D), for both 16S and WMS data.
We further explored specific taxa associated with the age of cervical cancer patients using Linear Discriminant Analysis (LDA) Effect Size (LEfSe). The clinical variable of age, was classified in three different ways: over vs under 50 years of age, over vs under the median age (49 years), and finally the patients were split into three sections where the 14 youngest and 14 oldest patients were compared against each other, with middle age group omitted (SFig. 1). We applied the one-against-all strategy with a threshold of 3 on the logarithmic LDA score for discriminative features and α of 0.05 for factorial Kruskal-Wallis test among classes. Regardless of the classification method used, the taxa identified as significantly enriched in older or younger patients were not consistent between 16S and WMS datasets.
3.4. Grouping of cervical cancer patients in terms of putative species abundances
Unsupervised hierarchical clustering of samples based on the species abundances in WMS and 16S (Figure 6, A–B) and OTU tables revealed 2 broad groups of patients in each hierarchy with significant overlap among patients comprising each group (Fisher’s Exact Test P-value is 0.004). Despite the significant differences in the number of genera identified by 16S and WMS, the hierarchical clustering of OTUs was consistent between datasets and revealed a set of OTUs enriched with Prevotella, Peptoniphilus, and Porphyromonas. These genera were more abundant in both 16S and WMS Cluster 1, but less abundant in 16S and WMS Cluster 2. Most notably, the grouping of patients in Cluster 1 and 2 was associated with the BMI index of the patients (Fisher’s Exact Test P-value is 0.002 for WMS and 0.06 for 16S). There were significantly more patients with BMI<median (28.63) in Cluster 1 (WMS and 16S) than in Cluster 2 (both datasets). There were 20 patients in the 16S Cluster 1 and 24 patients in the WMS Cluster 1. Thirteen of which were in common between those clusters, and they were grouped close to one another. Indicating a greater degree of similarity in terms of their OTU abundance profiles.
Figure 6. Unsupervised hierarchical clustering of samples in terms of putative species abundances.
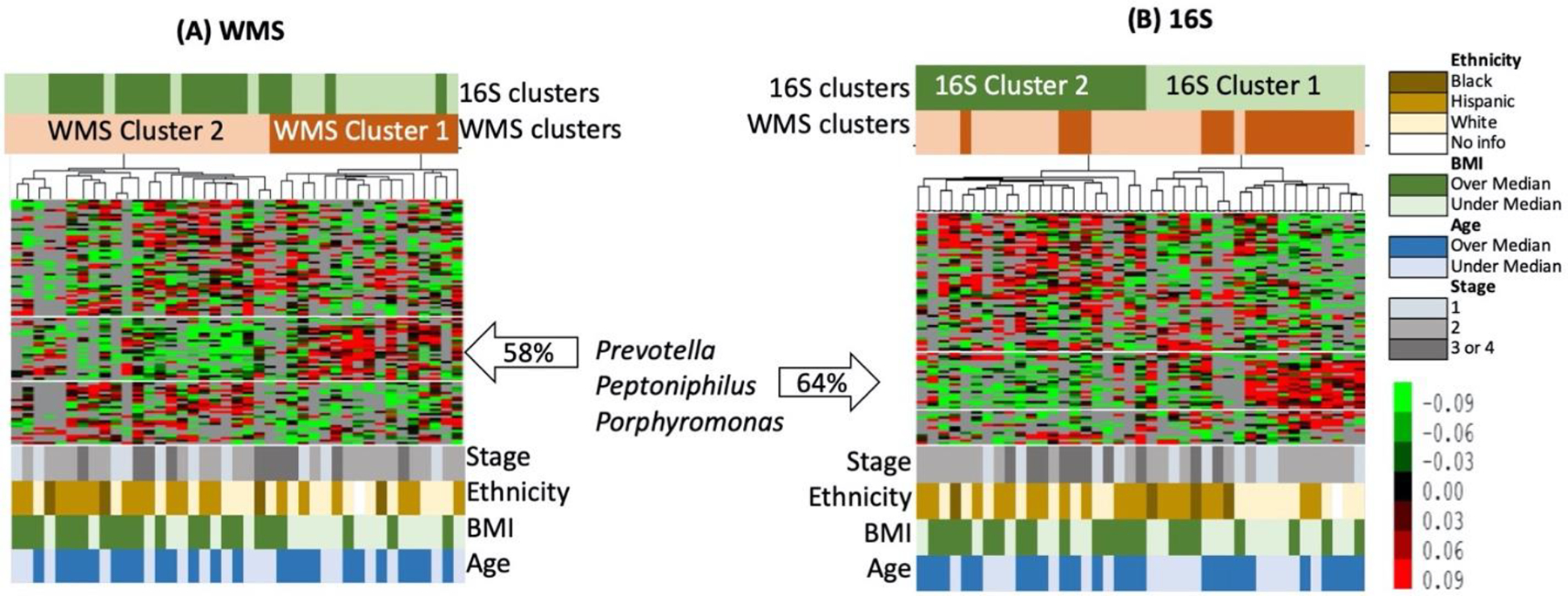
To generate these heat maps, only OTUs found in more than 14 samples were considered; 91 OTUs in 16S OTU table (A) and 103 in the WMS OTU table (B). Overlay between 16S and WMS sample Clusters are shown at the bars above the heat map, while demographic data is presented at the bottom.
4. Discussion
This study is limited by our analytic pipelines and available samples, but the results suggest that 16S with OTU clustering provides a similar description of sample diversity and composition for gut microbiomes using paired specimens from one rectal swab from cervical cancer patients versus WMS. This finding is important as it allows researchers to analyze a larger number of samples using 16S at a fraction of the cost of WMS. Our developed 16S pipeline is robust and compares well to WMS as an alternative to QIIME2, and is a valid approach for assessing taxonomic distribution. Camargo evenness and skewness were the least correlated indices between the two methodologies, which suggests that the sequencing methods differ in terms of the proportionality of in dividual bacterial taxa. This might be improved using a 16S analysis pipeline that uses amplicon sequence variants, such as QIIME2, to retain more reads. The Camargo index has low sensitivity for variation in species diversity for sample sizes <3000, while the Pielou index is a sensitive assessment index for smaller sample sizes (<1000). Thus, the Pielou evenness index is more appropriate in terms of this sample size, and correlates well between the two datasets (Mouillot and Leprêtre, 1999). With regards to rare taxa (LAR), WMS provides more noise in a dataset by identifying individual genes, which may be linked to unidentified bacterial species. 16S combined with OTU clustering can at best provide information at the genus level with a high degree of confidence and relies on 97% similarity clustering at the OTU level. This difference is to be expected, and could be exploited in specific analyses, such as searching for previously identified species or particular gene functions. It is reassuring that there was significant consensus between the methodologies on the higher order levels. Much of the focus in next generation sequencing analysis is placed on the smallest taxonomic level available (i.e. the genus or species level), but higher order taxa also provide valuable information.
Previous work has also posited a sizable amount of agreement between 16S and WMS sequencing techniques at higher orders of taxa (Vogtmann et al., 2016), consistent with these results. 16S and WMS have a significant degree of correlation; however, most of those studies utilize data derived from samples collected in similar but not identical contexts. This project provides a unique opportunity in that both 16S and WMS sequencing datasets were derived from a single sample collected from each patient and then bacterial DNA was extracted for both methods. Using this high-quality information, we investigated the correlation of these two datasets in terms of microbial composition abundance and alpha diversity, to precisely determine how well these sequencing methods corroborated. Since the two datasets are derived from the same samples, association with the clinical variables should also result in the same conclusion regardless of the sequencing method used, which was again confirmed. Age is perhaps the variable most strongly associated with microbiome diversity, which was confirmed in both datasets in our study. It is also important to note, hierarchical clustering analysis showed 9 (69%) out of 13 patients in Cluster1 were white, while only 8 (31%) of the 28 patients in the rest of the cohort were white. In addition, 12 (92%) out of the 13 patients in Cluster1 had a disease stage of 1 or 2, compared to 19 (68%) out of the 28 patients in the rest of the cohort.
An important limitation of the study is that we focus solely on taxonomic characteristics of the gut community. The major advantage of WMS is that it provides an opportunity to assay functional diversity of the microbiome, a capability severely lacking in 16S data. Tools such as PICRUSt (Langille et al., 2013) can infer metabolic profiles from 16S data, but they cannot truly assemble functional pathways. Yet, the most fundamental drawback of this study is due to the limitations of analytic pipelines used in each approach and the databases available for both 16S and WMS data. Tools for analyzing 16S have been developed and successfully deployed far longer than WMS analysis software, while the WMS analysis pipelines and databases are continually being developed and shared. The differences in alignment techniques and databases would account for a lot of the variation in taxa names herein. For example, by calculating OTUs we recapitulated a popular method of alignment used in this field, but in doing so the data has been collapsed at the cost of potential diversity information. Additionally, tools used for metagenomic analysis vary based on techniques used such as distance metrics and clustering approaches (Bushnell et al., 2017). Here, we used OTU clustering at 97% similarity using previously described methodology from the Human Microbiome Project (The Human Microbiome Project Consortium, 2012), but this data could be re-analyzed using QIIME2 and amplicon sequence variant (ASV) calling (Bolyen et al., 2019) and result in variations in ASV vs. OTU assignment that could affect the analysis. Amplicon sequence variant calling with DADA2 denoising (Callahan et al., 2016) may be a preferable system for WMS comparisons as the pipeline is more similar to how WMS reads are treated. Another important consideration is that the MetaPhlAn2 tool inherent in the Humann2 pipeline uses a relatively small fraction of the data generated, whereas another non-marker gene based identifier such as QIIME2, Kraken 2 (Wood and Salzberg, 2014) or the mothur software (Schloss et al., 2009) will generate a larger and more varied, spread of results. Still, MetaPhlAn2 outperformed IGGsearch (Nayfach et al., 2019) which was also deployed on our WMS dataset, and it remains the most popular marker-gene based tool in the metagenome field.
Another limitation to address, for this work and many others, is establishing a confident rarefication cut off for analysis. Usually this cut off value would be validated by utilizing a mock microbial community dataset to be analyzed alongside experimental data. Here, we were unable to acquire complete mock communities as such information is privileged and difficult to attain. However, the cut off value we used was selected because it was consistently stringent across both 16S and WMS datasets while retaining as much information as possible.
All this is to say, variations in approaches to metagenome assembly pipelines similarly could affect taxonomic assignment in 16S and WMS data. It is possible that a particular sequence relevant to both datasets would be classified differently during preprocessing, highlighting the necessity of universal reference databases and sequencing alignment tools and protocol consensus.
Given this variability in sequencing and data processing pipelines, the use of multiple techniques across different types of sequencing data is an excellent way to confirm consistency in conclusions. However, limited resources (e.g. material from clinical samples, bioinformatics support, time and finances) hamper the ability for this expansive and in-depth microbiome profiling for all studies. Although WMS has been demonstrated to confer significant advantages over 16S, this work suggests there is very little additional taxonomic information identified from WMS that was not identified in 16S data (Laudadio et al., 2018; Salipante et al., 2015). This can vary depending on the context of analysis, for example method of sample collection (i.e., whole stool vs swabs) and determining the functional components of the microbiome in question (Langille et al., 2013; Ranjan et al., 2016) It is even possible that extracting DNA from the same sample at two different times, instead of splitting a single extraction as was done here, may yield slightly different results.
There have been multiple studies conducted that have reported on the differences between 16S rRNA and WGS (Chan et al., 2015; Escobar-Zepeda et al., 2018; Poretsky et al., 2014; Ranjan et al., 2016; Shah et al., 2011). A study conducted by Escobar-Zepeda et al. used multiple datasets to evaluate public databases and showed that the overall performance of almost all methods they evaluated using WMS was better than 16S rRNA but with a trade-off between sensitivity and specificity (Escobar-Zepeda et al., 2018). Another study comparing the databases of 16S and WMG showed that the two methods differ significantly in terms of community structure for most of the bacterial communities sampled (Shah et al., 2011). In one study, stool samples were collected from one participant and the two methods were compared (Ranjan et al., 2016) WMS had enhanced detection of bacterial species, diversity, and prediction of genes as compared to 16S rRNA sequencing. Our study presents novel findings as it is the first to compare the two sequencing methods in patients with cervical cancer using rectal swabs as opposed to stool samples.
In our work, alpha diversity assessments such as overall diversity, evenness and richness can provide meaningful, and more important, comparable information (Figure 1) when obtained with either 16S or WMS. Furthermore, the two datasets provided a high degree of consensus when these indices were subjected to statistical analysis. This suggests that for studies where overall microbiome diversity, richness and evenness are the goals of an analysis, 16S is more than sufficient to provide this information. For basic taxonomic descriptions, there was a meaningful agreement on the phyla and higher taxa levels, suggesting that 16S is also sufficient in this setting for hypothesis-generating data. Nonetheless, these two datasets did provide some differences in taxonomic assignment, particularly on the genus level, and relative abundances of individual taxonomies. This suggests that for studies where a broader repertoire of potential species are needed, both techniques may be necessary.
A good agreement in richness and diversity between 16S and WMS, in spite of the difference in the specific taxonomies, is not surprising. It confirms that both technologies are actually good in picking differences among samples within a particular ecosystem. If sample 1 is less diverse than sample 2 according to 16S, it will also be less diverse according to WMS, even if specific genera identified by the technologies are different. The technologies are inherently different, and each relies on technology-specific computational algorithms and databases. In the case of WMS we sample all DNA and may even assemble a complete genome if it is dominated in the environment. In the case of 16S, we sample only one gene and identify only putative species. The fact that an organism is identified by 16S but not WMS, does not mean that it is an ‘incorrect’ annotation, and it should be discarded.
The results of our study show that if we use 16S and WMS as currently employed, we can expect broad, high level conclusions, such as grouping samples from a particular ecosystem according to their diversity or according to the taxa abundance profiles, will be likely consistent between technologies. Significant differences may emerge when we go in more detail, such as low-level taxonomic classification. Both technologies are fundamentally different and have technology-specific advantages and disadvantages that should be taken into consideration when designing a study
5. Conclusions
In all, this evidence suggests that using 16S alone may be sufficient in the clinical cancer research setting, where available patient material, time and money can be scarce. Based on these findings, we suggest 16S for the gut microbiome of cancer patients for initial diversity, richness and evenness metrics along with higher level taxonomic classification. WMS can provide a large swath of detailed microbial information, albeit with less sensitivity than 16S, and may be ideal when additional information on genus and species level identification is needed or to confirm conclusions drawn from 16S data.
Supplementary Material
Highlights.
WMS and 16S were more similar in high- then low-level taxonomic characteristics
Diversity metrics were tightly correlated between the technologies
Consistency was also found in biological inferences
Significant difference was observed in individual taxa and their abundances
16S may satisfy the initial diversity characterization of the gut microbiome
Funding
This research was supported in part by the Radiological Society of North America Resident/Fellow Award (to L.E.C.), the National Institutes of Health (NIH) through MD Anderson’s Cancer Center Support Grant P30CA016672, the Emerson Collective and the National Institutes of Health T32 grant #5T32 CA101642-14 (T.T.S). This study was partially funded by The University of Texas MD Anderson Cancer Center HPV-related Cancers Moonshot (L.E.C and A.K.).
List of abbreviations
- 16S
16S rRNA gene sequencing
- WMS
shotgun metagenome sequencing
- MRI
magnetic resonance imaging
- PET/CT
positron emission tomography/computed tomography
- MDACC
MD Anderson Cancer Center
- CMMR
Alkek Center for Metagenomics and Microbiome Research
- gDNA
genomic bacterial DNA
- ATIMA
Agile Toolkit for Inclusive Microbial Analysis
- ISD
Inverse Simpson Diversity
- SD
Shannon Diversity
- OTU
Observed operational Taxonomic Unit
- LAR
Low Abundance Rarity Index
- LEfSe
Linear Discriminant Analysis Effect Size
- ASV
amplicon sequence variant
- FIGO
Federation of Gynecology and Obstetrics
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethics approval and consent to participate and for publication
This study was part of an IRB approved protocol (MDACC 2014–0543) and Patients were enrolled in an IRB approved (2014–0543) multi-institutional prospective clinical trial at The University of Texas MD Anderson Cancer Center and the Harris Health System, Lyndon B. Johnson General Hospital Oncology Clinic.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Availability of data and material
Both 16S and WMS datasets will be available upon study completion and publication via the database of Genotypes and Phenotypes (dbGaP). Similarly, proprietary code will be available upon study completion and publication through GitHub.
References
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu Y-X, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG, 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology 37, 852–857. 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronstein M, Deutsch M, 1991. Early diagnosis of proximal femoral deficiency. Gynecologic and Obstetric Investigation 34, 246–248. 10.1159/000292772 [DOI] [PubMed] [Google Scholar]
- Bushnell B, Rood J, Singer E, 2017. BBMerge – Accurate paired shotgun read merging via overlap. PLOS ONE 12, e0185056. 10.1371/journal.pone.0185056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP, 2016. DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods 13, 581–583. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo JA, 1995. On Measuring Species Evenness and Other Associated Parameters of Community Structure. Oikos 74, 538–542. 10.2307/3546000 [DOI] [Google Scholar]
- Chan CS, Chan K-G, Tay Y-L, Chua Y-H, Goh KM, 2015. Diversity of thermophiles in a Malaysian hot spring determined using 16S rRNA and shotgun metagenome sequencing. Front. Microbiol 6. 10.3389/fmicb.2015.00177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods 10, 996–998. 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- Escobar-Zepeda A, Godoy-Lozano EE, Raggi L, Segovia L, Merino E, Gutiérrez-Rios RM, Juarez K, Licea-Navarro AF, Pardo-Lopez L, Sanchez-Flores A, 2018. Analysis of sequencing strategies and tools for taxonomic annotation: Defining standards for progressive metagenomics. Scientific Reports 8, 12034. 10.1038/s41598-018-30515-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, Huttenhower C, 2018. Species-level functional profiling of metagenomes and metatranscriptomes. Nature Methods 15, 962–968. 10.1038/s41592-018-0176-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, Vatanen T, Hall AB, Mallick H, McIver LJ, Sauk JS, Wilson RG, Stevens BW, Scott JM, Pierce K, Deik AA, Bullock K, Imhann F, Porter JA, Zhernakova A, Fu J, Weersma RK, Wijmenga C, Clish CB, Vlamakis H, Huttenhower C, Xavier RJ, 2019. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nature Microbiology 4, 293–305. 10.1038/s41564-018-0306-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, Creasy HH, Earl AM, FitzGerald MG, Fulton RS, Giglio MG, Hallsworth-Pepin K, Lobos EA, Madupu R, Magrini V, Martin JC, Mitreva M, Muzny DM, Sodergren EJ, Versalovic J, Wollam AM, Worley KC, Wortman JR, Young SK, Zeng Q, Aagaard KM, Abolude OO, Allen-Vercoe E, Alm EJ, Alvarado L, Andersen GL, Anderson S, Appelbaum E, Arachchi HM, Armitage G, Arze CA, Ayvaz T, Baker CC, Begg L, Belachew T, Bhonagiri V, Bihan M, Blaser MJ, Bloom T, Bonazzi V, Paul Brooks J, Buck GA, Buhay CJ, Busam DA, Campbell JL, Canon SR, Cantarel BL, Chain PSG, Chen I-MA, Chen L, Chhibba S, Chu K, Ciulla DM, Clemente JC, Clifton SW, Conlan S, Crabtree J, Cutting MA, Davidovics NJ, Davis CC, DeSantis TZ, Deal C, Delehaunty KD, Dewhirst FE, Deych E, Ding Y, Dooling DJ, Dugan SP, Michael Dunne W, Scott Durkin A, Edgar RC, Erlich RL, Farmer CN, Farrell RM, Faust K, Feldgarden M, Felix VM, Fisher S, Fodor AA, Forney LJ, Foster L, Di Francesco V, Friedman J, Friedrich DC, Fronick CC, Fulton LL, Gao H, Garcia N, Giannoukos G, Giblin C, Giovanni MY, Goldberg JM, Goll J, Gonzalez A, Griggs A, Gujja S, Kinder Haake S, Haas BJ, Hamilton HA, Harris EL, Hepburn TA, Herter B, Hoffmann DE, Holder ME, Howarth C, Huang KH, Huse SM, Izard J, Jansson JK, Jiang H, Jordan C, Joshi V, Katancik JA, Keitel WA, Kelley ST, Kells C, King NB, Knights D, Kong HH, Koren O, Koren S, Kota KC, Kovar CL, Kyrpides NC, La Rosa PS, Lee SL, Lemon KP, Lennon N, Lewis CM, Lewis L, Ley RE, Li K, Liolios K, Liu B, Liu Y, Lo C-C, Lozupone CA, Dwayne Lunsford R, Madden T, Mahurkar AA, Mannon PJ, Mardis ER, Markowitz VM, Mavromatis K, McCorrison JM, McDonald D, McEwen J, McGuire AL, McInnes P, Mehta T, Mihindukulasuriya KA, Miller JR, Minx PJ, Newsham I, Nusbaum C, O’Laughlin M, Orvis J, Pagani I, Palaniappan K, Patel SM, Pearson M, Peterson J, Podar M, Pohl C, Pollard KS, Pop M, Priest ME, Proctor LM, Qin X, Raes J, Ravel J, Reid JG, Rho M, Rhodes R, Riehle KP, Rivera MC, Rodriguez-Mueller B, Rogers Y-H, Ross MC, Russ C, Sanka RK, Sankar P, Fah Sathirapongsasuti J, Schloss JA, Schloss PD, Schmidt TM, Scholz M, Schriml L, Schubert AM, Segata N, Segre JA, Shannon WD, Sharp RR, Sharpton TJ, Shenoy N, Sheth NU, Simone GA, Singh I, Smillie CS, Sobel JD, Sommer DD, Spicer P, Sutton GG, Sykes SM, Tabbaa DG, Thiagarajan M, Tomlinson CM, Torralba M, Treangen TJ, Truty RM, Vishnivetskaya TA, Walker J, Wang L, Wang Z, Ward DV, Warren W, Watson MA, Wellington C, Wetterstrand KA, White JR, Wilczek-Boney K, Wu Y, Wylie KM, Wylie T, Yandava C, Ye L, Ye Y, Yooseph S, Youmans BP, Zhang L, Zhou Y, Zhu Y, Zoloth L, Zucker JD, Birren BW, Gibbs RA, Highlander SK, Methé BA, Nelson KE, Petrosino JF, Weinstock GM, Wilson RK, White O, The Human Microbiome Project Consortium, 2012. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahti L, Shetty S, Obenchain V, Turaga N, n.d. Tools for microbiome analysis in R. Version 1.9.19 [WWW Document]. URL http://microbiome.github.com/microbiome
- Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C, 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology 31, 814–821. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL, 2012. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudadio I, Fulci V, Palone F, Stronati L, Cucchiara S, Carissimi C, 2018. Quantitative Assessment of Shotgun Metagenomics and 16S rDNA Amplicon Sequencing in the Study of Human Gut Microbiome. OMICS 22, 248–254. 10.1089/omi.2018.0013 [DOI] [PubMed] [Google Scholar]
- Lynch SV, Pedersen O, 2016. The Human Intestinal Microbiome in Health and Disease [WWW Document]. https://doi.org/10.1056/NEJMra1600266. 10.1056/NEJMra1600266 [DOI] [PubMed]
- McDonald D, Clemente JC, Kuczynski J, Rideout JR, Stombaugh J, Wendel D, Wilke A, Huse S, Hufnagle J, Meyer F, Knight R, Caporaso JG, 2012. The Biological Observation Matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. Gigascience 1. 10.1186/2047-217X-1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouillot D, Leprêtre A, 1999. A comparison of species diversity estimators. Population Ecology 41, 203–215. 10.1007/s101440050024 [DOI] [Google Scholar]
- Nayfach S, Shi ZJ, Seshadri R, Pollard KS, Kyrpides NC, 2019. New insights from uncultivated genomes of the global human gut microbiome. Nature 568, 505–510. 10.1038/s41586-019-1058-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, Collado MC, Rice BL, DuLong C, Morgan XC, Golden CD, Quince C, Huttenhower C, Segata N, 2019. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 176, 649–662.e20. 10.1016/j.cell.2019.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielou EC, 1966. The measurement of diversity in different types of biological collections. Journal of Theoretical Biology 13, 131–144. 10.1016/0022-5193(66)90013-0 [DOI] [Google Scholar]
- Poretsky R, Rodriguez-R LM, Luo C, Tsementzi D, Konstantinidis KT, 2014. Strengths and Limitations of 16S rRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLOS ONE 9, e93827. 10.1371/journal.pone.0093827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan R, Rani A, Metwally A, McGee HS, Perkins DL, 2016. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochemical and Biophysical Research Communications 469, 967–977. 10.1016/j.bbrc.2015.12.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ, 2004. Java Treeview—extensible visualization of microarray data. Bioinformatics 20, 3246–3248. 10.1093/bioinformatics/bth349 [DOI] [PubMed] [Google Scholar]
- Salipante SJ, SenGupta DJ, Cummings LA, Land TA, Hoogestraat DR, Cookson BT, 2015. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J Clin Microbiol 53, 1072–1079. 10.1128/JCM.03385-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Horn DJV, Weber CF, 2009. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol 75, 7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Tang H, Doak TG, Ye Y, 2011. Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics, in: Pacific Symposium on Biocomputing 2011, PSB 2011. pp. 165–176. 10.1142/9789814335058_0018 [DOI] [PubMed] [Google Scholar]
- Sims TT, El Alam MB, Karpinets TV, Dorta-Estremera S, Hegde VL, Nookala S, Yoshida-Court K, Wu X, Biegert GWG, Delgado Medrano AY, Solley T, Ahmed-Kaddar M, Chapman BV, Sastry KJ, Mezzari MP, Petrosino JF, Lin LL, Ramondetta L, Jhingran A, Schmeler KM, Ajami NJ, Wargo J, Colbert LE, Klopp AH, 2021. Gut microbiome diversity is an independent predictor of survival in cervical cancer patients receiving chemoradiation. Communications Biology 4, 1–10. 10.1038/s42003-021-01741-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium, 2012. A framework for human microbiome research. Nature 486, 215–221. 10.1038/nature11209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, Navas-Molina JA, Janssen S, Kopylova E, Vázquez-Baeza Y, González A, Morton JT, Mirarab S, Zech Xu Z, Jiang L, Haroon MF, Kanbar J, Zhu Q, Jin Song S, Kosciolek T, Bokulich NA, Lefler J, Brislawn CJ, Humphrey G, Owens SM, Hampton-Marcell J, Berg-Lyons D, McKenzie V, Fierer N, Fuhrman JA, Clauset A, Stevens RL, Shade A, Pollard KS, Goodwin KD, Jansson JK, Gilbert JA, Knight R, 2017. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463. 10.1038/nature24621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N, 2015. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nature Methods 12, 902–903. 10.1038/nmeth.3589 [DOI] [PubMed] [Google Scholar]
- Vatanen T, Plichta DR, Somani J, Münch PC, Arthur TD, Hall AB, Rudolf S, Oakeley EJ, Ke X, Young RA, Haiser HJ, Kolde R, Yassour M, Luopajärvi K, Siljander H, Virtanen SM, Ilonen J, Uibo R, Tillmann V, Mokurov S, Dorshakova N, Porter JA, McHardy AC, Lähdesmäki H, Vlamakis H, Huttenhower C, Knip M, Xavier RJ, 2019. Genomic variation and strain-specific functional adaptation in the human gut microbiome during early life. Nature Microbiology 4, 470–479. 10.1038/s41564-018-0321-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila AV, Imhann F, Collij V, Jankipersadsing SA, Gurry T, Mujagic Z, Kurilshikov A, Bonder MJ, Jiang X, Tigchelaar EF, Dekens J, Peters V, Voskuil MD, Visschedijk MC, Dullemen H.M. van, Keszthelyi D, Swertz MA, Franke L, Alberts R, Festen EAM, Dijkstra G, Masclee AAM, Hofker MH, Xavier RJ, Alm EJ, Fu, Wijmenga C, Jonkers DMAE, Zhernakova A, Weersma RK, 2018. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Science Translational Medicine 10. 10.1126/scitranslmed.aap8914 [DOI] [PubMed] [Google Scholar]
- Vogtmann E, Hua X, Zeller G, Sunagawa S, Voigt AY, Hercog R, Goedert JJ, Shi J, Bork P, Sinha R, 2016. Colorectal cancer and the human gut microbiome: Reproducibility with whole-genome shotgun sequencing. PLoS ONE 11. 10.1371/journal.pone.0155362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DE, Salzberg SL, 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biology 15, R46. 10.1186/gb-2014-15-3-r46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolfo M, Asnicar F, Manghi P, Pasolli E, Tett A, Segata N, 2018. Profiling microbial strains in urban environments using metagenomic sequencing data. Biology Direct 13, 9. 10.1186/s13062-018-0211-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Both 16S and WMS datasets will be available upon study completion and publication via the database of Genotypes and Phenotypes (dbGaP). Similarly, proprietary code will be available upon study completion and publication through GitHub.