

Abstract
Introduction and importance
Primary hepatic neuroendocrine tumors (PHNETs) are extremely rare, and the clinical symptoms, test results, and imaging characteristics are nonspecific in most patients; thus, it is difficult to differentiate from other liver masses before surgery. Histopathology and immunohistochemistry are the main basis for the diagnosis. PHNETs and colon tumors co-occur in a patient and are non-homologous, as reported in the English-language literature for the first time.
Case presentation
We present a case of a 60-year-old woman with right hepatic lobe mass accidentally discovered on abdominal ultrasonography during a routine examination. Preoperative liver contrast-enhanced computed tomography suggested hepatocellular carcinoma; then, surgery were performed. Pathological results revealed a Grade 2 neuroendocrine tumor of the liver. In search of the primary tumor, upper and lower endoscopy of the GI tract was performed and revealed a mass in the ascending colon. Ascending colon cancer was considered; then, laparoscopic right hemicolectomy was performed. Pathological results suggested tubular villous adenoma of the ascending colon. The final diagnosis was not colon cancer with liver metastases but was PHNETs with colon adenoma.
Clinical discussion
PHNETs are rare cancers that are difficult to diagnose, requiring not only differentiation from other liver masses but also exclusion of metastases from extrahepatic sources. The pathological results play an important in making an accurate diagnosis.
Conclusion
Pathology, postoperative follow-up, and comprehensive imaging examinations are powerful tools in the diagnosis of PHNETs. Currently, surgery is the best treatment to achieve a potential cure and prolong the patient's survival.
Keywords: PHNETs, NETs, Diagnosis, Colon adenoma, Case report, Pathology
Highlights
-
•
PHNETs and a colon tumor co-occur in a patient and are non-homologous, reported in the English-language literature for the first time.
-
•
PHNETs invaded the diaphragm and underwent radical surgical resection, which are rarely reported.
1. Introduction
Neuroendocrine tumors (NETs) are a heterogeneous group of tumors originating from enterochromaffin cells throughout the body, which most commonly develop in the gastrointestinal tract, lungs, pancreas, gallbladder, thymus, and ovaries [1]. The liver is the most common metastatic site of NETs but a rare site of tumor origin. Primary hepatic NETs (PHNETs) are extremely rare and account for approximately 0.3% of all NETs, which was first reported by Edison in 1958, and less than 200 cases have been reported in the literature, yet the incidence rate has shown an upward trend recently [2]. Due to rarity and the lack of specific clinical symptoms and imaging findings, PHNETs are difficult to differentiate from hepatocellular carcinoma and cholangiocarcinoma preoperatively. Pathological results of PHNET lesions do not differ from those of secondary metastatic NETs; thus, further examination to exclude metastatic lesions from extrahepatic origins and long-term follow-up are needed to confirm the diagnosis. This report presents a case of successfully resected PHNET. Further colonoscopy revealed a colonic adenoma, and pathological analysis showed that colonic surgical specimens were non-homologous to the liver tumor. This case has been reported in line with the SCARE criteria [3].
2. Case presentation
A 60-year-old woman was admitted to our hospital on December 8, 2021 for a suspicious mass in the right liver lobe that had been found by B-ultrasonography more than half a month before admission. She had a past medical history of right oophorectomy 40 years ago. She denied any relevant family history, including genetic information, drug history, and psychosocial history. She had no symptoms of fever, nausea, vomiting, diarrhea, or jaundice before admission. Physical examination did not show obvious abnormality. Laboratory examinations showed that the patient's routine blood test, coagulation function, and liver and renal function were normal, hepatitis B antigen and anti-hepatitis C virus antibodies were negative, and tumor markers, such as AFP, CEA, and CA199, were within the normal range. Abdominal ultrasonography showed hyperechoic foci in the right hepatic lobe (Fig. 1). Contrast computed tomography (CT) of the abdomen revealed a mass of approximately 6.0 cm in the right hepatic lobe involving segments 6 and 7, which was enhanced partially in the arterial phase and washout in the delayed phase (Fig. 1). Both CT and ultrasonography suggested signs of liver cirrhosis. Chest CT showed no space-occupying lesions. Based on the abovementioned test results, our diagnosis is presumed to be hepatocellular carcinoma.
Fig. 1.

Ultrasonography and contrast-enhanced computed tomography showing hepatic solid mass. A. Ultrasonography showed a hyperechoic mass. B. The lesion had low density in the plain scan phase. C. The lesion showed partial enhancement in the arterial phase. D. The lesion was washout in the delayed phase.
On December 13, 2021, we performed surgical treatment and found that the liver cancer invaded the diaphragm accidentally during the surgery. Therefore, partial liver lobectomy, partial resection of the diaphragm, and diaphragm repair were performed. The surgery lasted 105 min, and intraoperative blood loss was approximately 300 mL. The patient recovered well postoperatively, and she was discharged from the hospital on the 7th postoperative day. The macroscopic evaluation of resected specimen showed a 9.0 × 6.0 × 2.5 cm tumor. Hematoxylin-eosin staining showed that the tumor had a nest-like and trabecular-like structure and granular cytoplasm, and the tumor cells were uniform in size and rich in blood sinuses (Fig. 2). Postoperative histopathology and immunohistochemistry (IHC) revealed that the specimen was an intermediate-grade neuroendocrine tumor classified as G2. The mitotic count was 10 per 10 high-power fields. IHC staining showed that the tumor was positive for chromogranin A (CgA), synaptophysin (Syn) (Fig. 2), CK, CD31, CD34, hepatocyte, and Arg-1, and the Ki67 index was approximately 20% (Fig. 2). However, IHC staining for P53 was negative.
Fig. 2.

pathological results. A: Microscopically, the tumoral lesion had a nest-like and Trabecular-like structure and granular cytoplasm, and the tumor cells were uniform in size and rich in blood sinuses (HE×100); B: Strong immunoreactivity for CgA in tumor cells. Original magnification: ×100; C: The Ki67 proliferation index is about 20% in tumor cells. Original magnification: ×100; D: Strong immunoreactivity for Syn in tumor cells. Original magnification: ×100.
This patient was diagnosed with a hepatic neuroendocrine tumor based on the IHC findings. To exclude metastatic lesions from extrahepatic primary origins, chest CT, gastroscopy, colonoscopy, and enhanced MRI of the upper abdomen were performed during the postoperative follow-up period. The chest CT finding was negative for any tumor, but colonoscopy and abdominal contrast-enhanced MRI revealed a mass in the ascending colon (Fig. 3). Endoscopic biopsy specimen of the ascending colon showed glandular epithelial tubular and papillary hyperplasia with mild to moderate dysplasia. Then, the patient was admitted to our hospital again on February 26, 2022. She denied abdominal pain, melena, weight loss, and other symptoms. No obvious positive signs were found in physical examination, and no obvious abnormality was found in the laboratory tests, including tumor markers. The presumed diagnosis was ascending colon cancer according to the abovementioned examination results. Subsequently, laparoscopic right hemicolectomy was performed. On March 3, 2022, pathological and IHC analysis on the surgical resection specimens unexpectedly revealed ascending colon tubular villous adenoma measuring 3.0 × 2.0 × 1.5 cm. The patient was finally diagnosed with ascending colon adenoma with primary liver neuroendocrine tumor. The patient recovered well and was discharged on postoperative day 12. The patient was grateful for the timely discovery of colon adenomas. She did not receive chemoradiotherapy and has been undergoing regular follow-up in our institution until now.
Fig. 3.
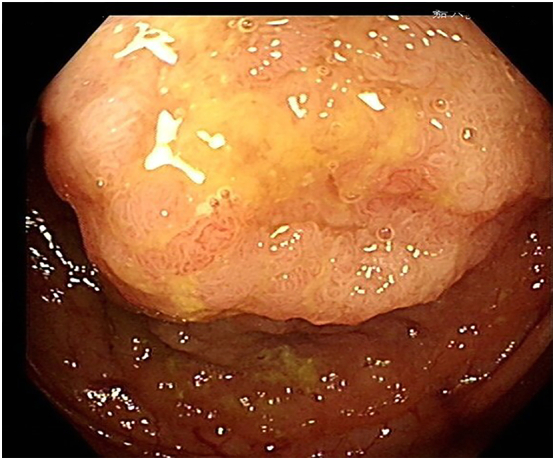
Colonoscopy results. A mass was found in the ascending colon.
3. Discussion
NETs account for only 1–2% of all gastrointestinal tumors, and most hepatic NETs metastasize from the gastrointestinal tract and bronchopulmonary tract; however, PHNETs account for 0.32% of all NETs. Presently, the origin of PHNETs is controversial. There are three hypotheses about the origin of PHNET [4]: (1) It is transformed from neuroendocrine cells of intrahepatic bile duct epithelium. (2) It originates from multifunctional stem cells of the liver. (3) It originates from the ectopic adrenal and pancreatic tissues in the liver. Therefore, PHNETs are extremely rare; thus, the clinical symptoms, treatment methods, and prognosis are not fully understood. Patients are usually asymptomatic at the early stages and often discovered incidentally during physical examination. At the middle and late stages, patients may present symptoms such as abdominal pain, bloating, loss of appetite, weight loss, and obstructive jaundice as the tumor grows, and very few patients show signs of carcinoid syndrome, such as flushing, diarrhea, asthma, fever, and palpitations [5], [6]. Patients with PHNETs do not show obvious carcinoid syndrome-related symptoms, while patients with liver metastatic NETs usually have typical carcinoid syndrome-related symptoms. NETs can be divided into two groups according to the presence of hormone secretion function and clinical symptoms caused by hormones: functional NETs and non-functional NETs. PHNETs are usually nonfunctional and fail to produce a biological effect. Preoperative tumor marker detection showed no obvious abnormality. Certainly, some studies pointed out that tumor markers are not helpful in the diagnosis. Serum 5-hydroxytryptamine or 24-h urine of 5-hydroxyindoleacetic acid may be useful in the diagnosis, but we do not routinely test for it due to the rarity of PHNETs. Most patients have a single lesion (76.6%) located commonly in the right liver (48.4%) [7], but there can also be multiple lesions. According to previous reports, the incidence of PHNETs did not significantly differ between men and women with a mean age of occurrence of 51.9 years [8]. PHNETs have a slow growth and are often diagnosed in the middle and late stages due to the lack of clinical symptoms in the early stage. Similar to those in previous reports, PHNETs in the right hepatic lobe in our patient presented no specific clinical features, and diagnosis was delayed on the routine health examination, which lead to late treatment.
Currently, there is no classification system for PHNETs, but NET classification in the digestive system established by the WHO in 2019 can be used to evaluate the malignancy of tumors and prognosis based on the number of mitotic cells found in 10 high-power fields and the Ki67 index. NETs can be divided into three grades: low-grade malignancy (G1) intermediate grade malignancy (G2), and high-grade malignancy (G3). As the grade increases, it indicates poor differentiation and worse prognosis [9]. As shown in our case, a Ki67 index of approximately 20% and 10 mitotic counts per 10 high-power fields are graded as G2 according to the 2019 WHO classification, but the tumor had invaded the diaphragm, in which a worse diagnosis is expected.
The diagnosis of PHNETs is a continuous process. Pathological confirmation should also include comprehensive preoperative examination, careful intraoperative exploration, and long-term postoperative follow-up. PHNETs exhibit miscellaneous radiological features and are difficult to distinguish from hepatocellular carcinoma and cholangiocarcinoma. PHNETs have an abundant blood supply. Kim reported an analysis of dynamic contrast-enhanced CT findings of 38 PHNETs and revealed that hepatocellular carcinoma-like patterns account for almost half of tumors and cholangiocarcinoma-like or combined patterns account for the other half of tumors [10]. In our case, contrast-enhanced CT of the abdomen showed that the tumor exhibited a fast-in and fast-out hepatocellular carcinoma-like pattern: low-density foci in the plain scan, partial enhancement in the early phase, and washout in the delayed phase. Therefore, it is extremely difficult to diagnose PHNETs only by imaging tests, and the patient was easily misdiagnosed with liver cancer before surgery.
Needle biopsy for diagnosis has been reported in several studies [7], [11], [12], but the diagnostic accuracy is not very high. In a study of 124 patients undergoing needle biopsy, only 14 cases were consistent with the final diagnosis postoperatively [8]. Therefore, post-surgical histological and IHC evaluation is the best method to confirm PHNETs. Microscopically, tumors show a nest- and trabecular-like structure and granular cytoplasm. Neuron-specific enolase, CgA, and Syn are generally considered highly sensitive IHC markers for the diagnosis of NETs. The best option in the diagnosis of PHNETs is CgA due to its much higher sensitivity and specificity than other IHC markers. A previous report revealed that CgA was detected by IHC in 94.7% of cases [8]. In our case, the tumor cells were positive for CgA and Syn with a Ki67 index of approximately 20%. Even with histopathological and IHC findings, PHNETs are often indistinguishable from metastatic hepatic NETs. Therefore, long-term follow-up is required to exclude extrahepatic primary lesions. In our patient, she underwent chest CT, contrast-enhanced MRI of the upper abdomen, and upper and lower endoscopy of the GI tract postoperatively. Pathological findings suggest that the rectal mass is nonhomologous to liver NETs, so it has more reasons to believe that we diagnose this patient with PHNETs. Currently, there is no uniform treatment guideline for PHNETs, but surgical resection is the best treatment option for resectable lesions. Li et al. reported that the 5-year survival rates were 71.9% and 15.6%, respectively, in the analysis of 291 patients, with or without undergoing surgery [13]. The study by Givi et al. showed that the median survival time for patients who underwent surgery was approximately 159 months, compared to only 47 months for those who did not [14]. These results show that surgical patients have better prognosis than non-surgical patients. However, high postoperative tumor recurrence has been reported in the literature. For example, Knox and Quarter et al. reported postoperative recurrence rates of 18% and 19–20%, respectively [8], [15]. Therefore, regular follow-up after surgery is necessary to timely detect tumor recurrence. There is still no consensus on whether to use chemotherapy drugs postoperatively. For unresectable lesions, trans-arterial chemoembolization (TACE), radiofrequency ablation (RAF), percutaneous ethanol injection treatment, systemic chemotherapy, somatostatin hormone therapy or its analogs, and liver transplantation are also alternative treatment options [16], [17], [18], but the limited number of cases makes it difficult to evaluate the effectiveness of the interventions. According to Yao et al., hepatic chemoembolization for metastatic gastrointestinal NETs can effectively improve clinic symptoms and may achieve tumor control [19]. According to Park et al., the median survival of four patients who received chemotherapy was 11.3 months (range, 3.0–26.4 months) [20]. The current outcomes are questionable, but with the increase in the number of reported cases, we believe that, in the future, we will have a better understanding of the diagnosis, treatment methods, and prognosis of PHNETs.
4. Conclusion
PHNETs are extremely rare and present with nonspecific clinical symptoms and various imaging manifestations; thus, preoperative diagnosis is extremely difficult. Histopathology and IHC play an important role in the diagnosis of hepatic NETs, extrahepatic metastases still need to be excluded. Surgical resection is the mainstay treatment. For unresectable lesions or metastatic lesions, TACE, RAF, liver transplantation, etc., are also treatment options.
Funding
The work was supported by the following financial support: Science and Technology Bureau of Jiaxing City (grant no. 2019AY32029); Science and Technology Bureau of Jiaxing City (grant no. 2019AD32208); and Medical Science and Technology Project of Zhejiang Province (grant no. 2021KY1101).
Ethical approval
This paper is a case report; therefore, it does not require ethics approval.
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Registration of research studies
Not applicable.
Provenance and peer review
Not commissioned, externally peer-reviewed.
Guarantor
Jie Zhang
CRediT authorship contribution statement
Liu Xu and Xiaohuan Li: data collection and analysis.
Yifan Lu: concept and design of the manuscript and revision of the manuscript.
Jie Zhang: main surgical provider for the patient, operator, and lead of this project; provided final approval of the version to be published.
Declaration of competing interest
The authors declare no potential conflict of interest related to this manuscript.
References
- 1.Foster D.S., Jensen R., Norton J.A. Management of liver neuroendocrine tumors in 2018. JAMA Oncol. 2018;4(11):1605–1606. doi: 10.1001/jamaoncol.2018.3035. [DOI] [PubMed] [Google Scholar]
- 2.Yao J.C., Hassan M., Phan A., et al. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008;26(18):3063–3072. doi: 10.1200/jco.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 3.Agha R.A., Franchi T., Sohrabi C., Mathew G., Kerwan A. The SCARE 2020 guideline: updating consensus Surgical CAse REport (SCARE) guidelines. International journal of surgery (London, England). 2020;84:226–230. doi: 10.1016/j.ijsu.2020.10.034. [DOI] [PubMed] [Google Scholar]
- 4.Yu W.M., Li R., Sun B.L., Du J.K., Tuo H.F. Primary hepatic neuroendocrine tumour with multiple liver metastases: a case report with literature review. Int. J. Surg. Case Rep. 2021;89 doi: 10.1016/j.ijscr.2021.106590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang Y.Q., Xu F., Yang J.M., Huang B. Primary hepatic neuroendocrine carcinoma: clinical analysis of 11 cases. Hepatobiliary Pancreat. Dis. Int. 2010;9(1):44–48. [PubMed] [Google Scholar]
- 6.Shetty P.K., Baliga S.V., Balaiah K., Gnana P.S. Primary hepatic neuroendocrine tumor: an unusual cystic presentation. Indian J. Pathol. Microbiol. 2010;53(4):760–762. doi: 10.4103/0377-4929.72078. [DOI] [PubMed] [Google Scholar]
- 7.Lin C.W., Lai C.H., Hsu C.C., et al. Primary hepatic carcinoid tumor: a case report and review of the literature. Cases J. 2009;2(1):90. doi: 10.1186/1757-1626-2-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quartey B. Primary hepatic neuroendocrine tumor: what do we know now? World J. Oncol. 2011;2(5):209–216. doi: 10.4021/wjon341w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klimstra D.S., Modlin I.R., Coppola D., Lloyd R.V., Suster S. The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems. Pancreas. 2010;39(6):707–712. doi: 10.1097/MPA.0b013e3181ec124e. [DOI] [PubMed] [Google Scholar]
- 10.Kim J.E., Lee W.J., Kim S.H., Rhim H., Song H.J., Park C.K. Three-phase helical computed tomographic findings of hepatic neuroendocrine tumors: pathologic correlation with revised WHO classification. J. Comput. Assist. Tomogr. 2011;35(6):697–702. doi: 10.1097/RCT.0b013e318231c6d8. [DOI] [PubMed] [Google Scholar]
- 11.Hsueh C., Tan X.D., Gonzalez-Crussi F. Primary hepatic neuroendocrine carcinoma in a child. Morphologic, immunocytochemical, and molecular biologic studies. Cancer. 1993;71(8):2660–2665. doi: 10.1002/1097-0142(19930415)71:8<2660::aid-cncr2820710835>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 12.Dala R., Shoosmith J., Lilenbaum R., Cabello-Inchausti B. Primary hepatic neuroendocrine carcinoma: an underdiagnosed entity. Ann. Diagn. Pathol. 2006;10(1):28–31. doi: 10.1016/j.anndiagpath.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 13.Li Y.F., Zhang Q.Q., Wang W.L. Clinicopathological characteristics and survival outcomes of primary hepatic neuroendocrine tumor: a surveillance, epidemiology, and end results (SEER) population-based study. Med. Sci. Monit. 2020;26 doi: 10.12659/msm.923375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Givi B., Pommier S.J., Thompson A.K., Diggs B.S., Pommier R.F. Operative resection of primary carcinoid neoplasms in patients with liver metastases yields significantly better survival. Surgery. 2006;140(6):891–897. doi: 10.1016/j.surg.2006.07.033. discussion 7–8. [DOI] [PubMed] [Google Scholar]
- 15.Knox C.D., Anderson C.D., Lamps L.W., Adkins R.B., Pinson C.W. Long-term survival after resection for primary hepatic carcinoid tumor. Ann. Surg. Oncol. 2003;10(10):1171–1175. doi: 10.1245/aso.2003.04.533. [DOI] [PubMed] [Google Scholar]
- 16.Wängberg B., Nilsson O., Johanson V.V., et al. Somatostatin receptors in the diagnosis and therapy of neuroendocrine tumor. Oncologist. 1997;2(1):50–58. [PubMed] [Google Scholar]
- 17.Komatsuda T., Ishida H., Furukawa K., Miyauchi T., Heianna J. Primary carcinoid tumor of the liver: report of a case with an emphasis on contrast-enhanced ultrasonographic findings. J. Clin. Ultrasound. 2005;33(6):302–304. doi: 10.1002/jcu.20132. [DOI] [PubMed] [Google Scholar]
- 18.Shah D., Mandot A., Cerejo C., Amarapurkar D., Pal A. The outcome of primary hepatic neuroendocrine tumors: a single-center experience. J. Clin. Exp. Hepatol. 2019;9(6):710–715. doi: 10.1016/j.jceh.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao K.A., Talamonti M.S., Nemcek A., et al. Indications and results of liver resection and hepatic chemoembolization for metastatic gastrointestinal neuroendocrine tumors. Surgery. 2001;130(4):677–682. doi: 10.1067/msy.2001.117377. discussion 82–5. [DOI] [PubMed] [Google Scholar]
- 20.Park C.H., Chung J.W., Jang S.J., et al. Clinical features and outcomes of primary hepatic neuroendocrine carcinomas. J. Gastroenterol. Hepatol. 2012;27(8):1306–1311. doi: 10.1111/j.1440-1746.2012.07117.x. [DOI] [PubMed] [Google Scholar]