

Summary
With a growing number of available de novo sequenced genomes, protocols for their applications to population genetics will benefit our understanding of the human genome. Here we detail analytic steps to apply an example de novo reference genome to map and detect variants of short-read sequences from corresponding populations and to discover variants of disease-relevant genes. Using this protocol, we can improve variant discovery, better investigate population-specific genome properties, and evaluate the potential of sequenced genomes in medical studies.
For complete details on the use and execution of this protocol, please refer to Lou et al. (2022).
Subject areas: Bioinformatics, Health Sciences, Genetics, Genomics, Systems biology
Graphical abstract
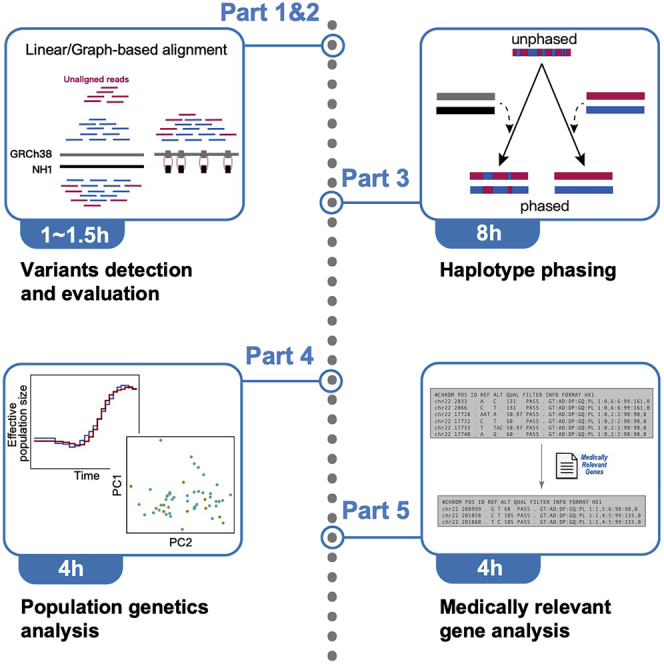
Highlights
-
•
Protocol for mapping and variants detection of short-read sequences
-
•
Advantages of using a population-specific reference genome in population genomic studies
-
•
Analytic steps to discover potential variants of disease-relevant genes
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
With a growing number of available de novo sequenced genomes, protocols for their applications to population genetics will benefit our understanding of the human genome. Here, we detail analytic steps to apply an example de novo reference genome to map and detect variants of short-read sequences from corresponding populations and to discover variants of disease-relevant genes. Using this protocol, we can improve variants' discovery and better investigate population-specific genome properties and evaluate the potential of sequenced genomes in medical studies.
Before you begin
Download the test dataset
Timing: 1 h
-
1.
In this protocol, we used the publicly available genome data of Han Chinese as the example data, including the draft genome in scaffold level of a northern Han Chinese (NH1) (Du et al., 2019), and the polished primary contigs assembled from the long-read sequences of a southern Han Chinese (HX1) (Shi et al., 2016). We also used the human reference genome GRCh38 in this protocol. The Human Genome Diversity Project (HGDP) data were incorporated into the population genetic analysis (Bergström et al., 2020). We provide links to download these data in Table 1.
-
2.
For a rapid test of this protocol, we extracted and analyzed chromosome 22 from the downloaded genome data (Table 1), and calculated the time of execution for each step. Make sure that the input data are consistent in the chromosome identifier, e.g., “chr22”, “Chr22”, or “22”. The test dataset is freely available at https://www.picb.ac.cn/PGG/resource_download.php?id=44&file=PGG_Web_Data/protocol_test_data_chr22.tar.gz and at GitHub: https://github.com/Shuhua-Group/TJ1_STARProtocols.
Table 1.
Steps to generate the test dataset
| Input data | Source | Description | Code | Output data |
|---|---|---|---|---|
| hx1f4.3rdfixedv2.fa.gz | http://www.openbioinformatics.org/hx1/data/hx1f4.3rdfixedv2.fa.gz | HX1 genome assembly | >gunzip hx1f4.3rdfixedv2.fa.gz >samtools faidx hx1f4.3rdfixedv2.fa 000604F 000707F 000300F 000247F 000361F 000443F 000220F > HX1.chr22.fa >samtools faidx HX1.chr22.fa |
HX1.chr22.fa; HX1.chr22.fai |
| GWHAAAS00000000.genome.fasta.gz | GWH (https://ngdc.cncb.ac.cn/gwh/): GWHAAAS00000000 | NH1 genome assembly | >gunzip GWHAAAS00000000.genome.fasta.gz >samtools faidx GWHAAAS00000000.genome.fasta GWHAAAS00000500 > NH1.chr22.fa >samtools faidx NH1.chr22.fa |
NH1.chr22.fa; NH1.chr22.fai |
| Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz | http://ftp.ensembl.org/pub/release-105/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz | Human reference genome GRCh38 | >gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz >samtools faidx Homo_sapiens.GRCh38.dna.primary_assembly.fa 22 > GRCh38.chr22.fa >sed -i 's/ˆ>/>chr/g' GRCh38.chr22.fa >samtools faidx GRCh38.primary_assembly.chr22.fa |
GRCh38.chr22.fa; GRCh38.chr22.fai |
| hgdp_wgs.20190516.full.chr22.vcf.gz; hgdp_wgs.20190516.full.chr22.vcf.gz.tbi | ftp://ngs.sanger.ac.uk/production/hgdp | Genotype data from HGDP | >bcftools view --force-samples -S <samples.txt> --threads 20 -f PASS -m 2 -M 2 -v snps hgdp_wgs.20190516.full.chr22.vcf.gz |bgzip -@20 -c > <out.vcf.gz> >tabix <out.vcf.gz> |
<4Han/4Tujia/9Tujia_43Han>.HGDP.snp.chr22.b38.vcf.gz; <4Han/4Tujia/9Tujia_43Han>.HGDP.snp.chr22.b38.vcf.gz.tbi |
| <sample>_1.fastq.gz; <sample>_2.fastq.gz |
ENA (https://www.ebi.ac.uk/ena/browser/home): PRJEB6463 | Raw sequences of 4 Han Chinese and 4 Tujia samples from HGDP | The raw sequencing reads were aligned to the human reference genome assembly GRCh38 with BWA and output BAM records. The BAM files were then sorted with SAMtools. Duplicated reads were marked with MarkDuplicates (Picard) in GATK (see key resources table). The code for short-reads mapping and duplicate removal is given in part 1: variants detection from the short-read sequences using linear alignment. | <HGDP00776/HGDP00784/HGDP00812/HGDP00819/HGDP01096/HGDP01100/HGDP01102/HGDP01104>.dedup.chr22.sorted.bam; <HGDP00776/HGDP00784/HGDP00812/HGDP00819/HGDP01096/HGDP01100/HGDP01102/HGDP01104>.dedup.chr22.sorted.bam.bai |
The input data are the original data downloaded from the public resources; the output data are those included in the test dataset.
Download the software and scripts
-
3.
Most of the analyses in this protocol are performed using existing software, which is listed in the key resources table and can be downloaded via the links provided.
-
4.
We provide scripts to run some of the programs sequentially and to analyze or evaluate the output data. Some analytic script files used in reads filtering, genome alignment, variants detection and annotation, and result plots are available on GitHub and Zenodo (see key resources table).
Compile a list of medically relevant genes
-
5.
Wagner et al. (Wagner et al., 2021) provide a list of medically relevant genes in GRCh38 coordinates, including 4,701 autosomal genes. It is available at GitHub: https://github.com/usnistgov/cmrg-benchmarkset-manuscript/blob/master/data/gene_coords/unsorted/GRCh38_mrg_full_gene.bed. You can also curate a list of known disease and phenotype-associated genes related to your trait of interest. There is no strict threshold for the number of genes in the list.
-
6.
Save all the genes in a BED file, with four columns containing the chromosome, the start position (bp), the end position (bp), and the gene identifier (Figure 1).
Figure 1.

Screenshot of a medically relevant gene list
Key resources table
Materials and equipment
-
•
Human reference genome assemblies (FASTA files for the human reference genome GRCh38 and the population-specific reference genome) and the short-read sequences (BAM files) and genotypes (VCF files) of samples from corresponding populations (see download the test dataset in before you begin).
-
•
Software and scripts are used in this protocol (see software and algorithms section of key resources table).
-
•
Basic knowledge about Python, R, and bash scripting is required to understand and apply this protocol. Getting the basic operations of SAMtools and BCFtools may also be helpful as they are essential to the sequencing data process. VCFtools (Danecek et al., 2011) is an alternative to BCFtools in analyzing the VCFs.
-
•
All tests are run on 64-core Intel Xeon CPU E7-4850 v4 2.10 GHz Linux servers. We recommend using computing clusters to perform the data analyses. Assuming 10 CPUs, at least 100 GB of RAM is required for analyzing the whole-genome data.
Step-by-step method details
Part 1: Variants detection from the short-read sequences using linear alignment
A population-specific reference genome is of importance in detecting variants. We first apply a linear approach for the short read mapping and variant calling, using NH1 as the reference genome. To evaluate the variant call rate and genotyping accuracy, we use the simulated short reads from the HX1 sequences in this step. The path of the scripts and files should be properly indicated when running the code provided in this protocol.
-
1.Simulate the short-read sequences (SRS) using EAGLE.
-
a.Filter out the HX1 contigs < 1 Kb in length and create a sequence dictionary.>python Filter_HX1_fasta_contig_length.py HX1.chr22.fa HX1.chr22.fa.faiHX1.chr22.filtered.fa>gatk CreateSequenceDictionary -R HX1.chr22.filtered.fa -O HX1.chr22.filtered.dict
 CRITICAL: Although not required by EAGLE, contig filtration is necessary for this step, as we find EAGLE is always interrupted when dealing with those short contigs (troubleshooting 1).
CRITICAL: Although not required by EAGLE, contig filtration is necessary for this step, as we find EAGLE is always interrupted when dealing with those short contigs (troubleshooting 1). -
b.Input the filtered HX1 sequences to EAGLE to simulate the raw reads of short-read sequencing, with a read length of 101 bp and sequencing depth of 30×.>path/to/configureEAGLE.pl--run-info=path/to/RunInfo_PairedReads1x1Tiles.xml--reference-genome=HX1.chr22.filtered.fa--coverage-depth=30--motif-quality-drop-table=path/to/MotifQualityDropTables/DefaultMotifQualityDropTable.tsv--quality-table=path/to/QualityTables/DefaultQualityTable.read1.length101.qval--quality-table=path/to/QualityTables/DefaultQualityTable.read2.length101.qval>cd EAGLE>make fastq -j 15>cd 111206_EAS987_0567_FC1234TST>bgzip EAGLE_S1_L001_R1_001.fastq>bgzip EAGLE_S1_L001_R2_001.fastq
-
a.
-
2.Map the simulated reads to the reference genome, and detect variants.
-
a.Map the simulated HX1 short reads to the NH1 genome using the BWA package.>bwa index NH1.chr22.fa>bwa mem -M -t 10 -R "@RG\tID:HX\tSM:HX1\tLB:HX1\tPU:HX1\tPL:ILLUMINA" NH1.chr22.faEAGLE_S1_L001_R1_001.fastq.gz EAGLE_S1_L001_R2_001.fastq.gz | samtools view -bS - > HX1.pe.bam>samtools sort -@ 5 -m 4G HX1.pe.bam -T HX1 -o HX1.bam>samtools index HX1.bam
-
b.Remove duplicated reads.>gatk --java-options "-Xmx4g -Djava.io.tmpdir=HX1/" MarkDuplicates -I HX1.bam -OHX1.dedup.bam --VALIDATION_STRINGENCY SILENT --REMOVE_DUPLICATES true -M metrics_HX1.txt-AS true --CREATE_INDEX true
-
c.Detect variants based on the short-read mapping using GATK.Note: Despite that GATK consumes a large amount of RAM, we do not suggest any alternative software as it is the most widely used toolkit for sequencing reads processing and variants calling with good performance. The users can refer to the Best Practices Workflows (https://gatk.broadinstitute.org/hc/en-us/sections/360007226651-Best-Practices-Workflows) for more instructions to apply GATK.#Variants calling for each chromosome>ref_chr_list=`cat NH1.chr22.fa.fai | awk '{print $1}'`>gatk CreateSequenceDictionary -R NH1.chr22.fa -O NH1.chr22.dict>gatk --java-options "-Xmx3G -XX:ParallelGCThreads=2 -Dsamjdk.compression_level=5"HaplotypeCaller -R NH1.chr22.fa -ploidy 1 -L GWHAAAS00000500 -I HX1.dedup.bam -OHX1.GWHAAAS00000500.g.vcf.gz -ERC GVCF -GStandardAnnotation -G AS_StandardAnnotation -G StandardHCAnnotation --seconds-between-progress-updates 30>sh Combine_list.sh>sh 170.JointCalling.sh>cd 170.GenotypeGVCFs.joint.calling>for chr in $ref_chr_listdosh 170.${chr}.shdone#Combine the VCFs of all chromosomes>sh 170.combine.sh>tabix -p vcf HX1.genomewide.hc.vcf.gz#An optional step to rename the VCF files as we only analyze chromosome 22 here>mv HX1.genomewide.hc.vcf.gz HX1.chr22.vcf.gz>mv HX1.genomewide.hc.vcf.gz.tbi HX1.chr22.vcf.gz.tbi
-
d.Perform GATK hard-filtering to filter out probable artifacts from the call set (De Summa et al., 2017).Note: Here we do not recommend using the GATK VQSR module to do variant filtering, as it relies on known and highly validated variant resources. This step does not guarantee that all the variants in the filtered calls are reliable, and some variants of particular interest need careful check.>gatk SelectVariants -select-type SNP -V HX1.chr22.vcf.gz -O HX1.chr22.snp.vcf.gz>gatk VariantFiltration -V HX1.chr22.snp.vcf.gz--filter-expression "QD < 2.0 || MQ < 40.0 || FS > 60.0 || SOR > 3.0"--filter-name "Filter" -O HX1.chr22.snp.filter.vcf.gz>gatk SelectVariants -select-type INDEL -V HX1.chr22.vcf.gz -O HX1.chr22.indel.vcf.gz>gatk VariantFiltration -V HX1.chr22.indel.vcf.gz--filter-expression "QD < 2.0 || FS > 200.0 || SOR > 10.0"--filter-name "Filter" -O HX1.chr22.indel.filter.vcf.gz>gatk MergeVcfs -I HX1.chr22.snp.filter.vcf.gz -I HX1.chr22.indel.filter.vcf.gz -OHX1.chr22.filter.vcf.gz>bcftools view -f PASS HX1.chr22.filter.vcf.gz | bgzip -c > HX1.chr22.filtered.vcf.gz>tabix -p vcf HX1.chr22.filtered.vcf.gz
-
a.
-
3.Evaluate the accuracy of variants detection by comparing the results of simulated data to that obtained in real data.
-
a.Align the NH1 and HX1 genomes using Minimap2, which has been integrated into the script below.Note: BWA does not support the alignment of two genomes, and thus should not be used in this step as an alternative software package to Minimap2.>paftools_wgs_call.sh NH1.chr22.fa HX1.chr22.fa>bgzip NH1.chr22.HX1.chr22.vcf>tabix -p vcf NH1.chr22.HX1.chr22.vcf.gz
-
b.Run RTGtools to compare the two VCF files – one callset of real data generated in this step, and another obtained from simulated data in the above step, and output summary metrics on the screen (Figure 2).>rtg format -o NH1.sdf NH1.chr22.fa>rtg vcfeval -b NH1.chr22.HX1.chr22.vcf.gz -c HX1.chr22.filtered.vcf.gz -o output -tNH1.sdf
-
a.
Figure 2.

Screenshot of the output summary metrics of RTGtools
“Threshold” represents the genotyping quality (GQ) threshold, and you can refer to the row where the threshold is “None”. “baseline” represents the true data, and “call” represents the GATK callset. Therefore, “True-pos-baseline” means baseline variants that match between the baseline and calls; “True-pos-call” means called variants that match between the baseline and calls; “False-pos” means called variants not matched in the baseline; “False-neg” means baseline variants not matched in the call set. “Precision” means the precision rate of called variants; “Sensitivity” means the recall rate of baseline variants; “F-measure” means the weighted harmonic mean of its precision and sensitivity.
Part 2: Variants detection from the short-read sequences using the graph-based approach
The graph-based method is an alternative to the linear method for variants detection in the SRS data, and it has a much better performance, especially on the genotyping of insertions. The performance of the linear method only relies on the input sequencing data, while that of the graph-based method is additionally related to the quality of graph construction. Here we apply the vg toolkit and use NH1, which provides a comprehensive and reliable variant list representing Han Chinese, to construct a genome graph.
Note: The vg toolkit is updated frequently, and the results vary largely across different updates. Make sure to use an identical version of vg (≥1.23 is recommended) to complete the entire process, and that your commands conform to the syntax requirements of that version.
-
4.
Construct a graph genome using vg with a reference genome in a FASTA file (e.g., the current human reference genome assembly, GRCh38) and a set of variants in a VCF file (e.g., the NH1 variant call set named “NH1.GRCh38.chr22.vcf.gz” in the test dataset) (troubleshooting 2 and 3).
Note: The information lines of the VCF file will affect the output as the merged information generated by different software may conflict in vg processing, so we recommend keeping only necessary information, such as “##fileformat” and “##contig”. The option “-S” should be specified in “vg construct” if structural variants are needed in constructing the genome graph.
>workdir=`pwd`
>vg construct -C -S -a -R chr22 -r GRCh38.chr22.fa -v NH1.GRCh38.chr22.vcf.gz -t 1 -m 32
--flat-alts 1>chr22.vg 2>chr22.vg.log
-
5.
Build an xg index and a gcsa index for the output graph in the .vg file.
Note: The xg index is a compressed version of the graph that allows fast node, edge and path lookups; the gcsa index is a pruned substring index used only for read mapping.
#Build xg index
>vg ids -j chr22.vg
>vg index -b ${workdir} -x wg.xg chr22.vg
#Prune the graph and build gcsa index
>vg prune -r chr22.vg > chr22.pruned.vg
>vg index -b ${workdir} -g wg.gcsa chr22.pruned.vg
-
6.
Map the paired SRS to the graph for variants detection and genotyping. The augmentation step is for detecting rare variants.
Note: The earlier versions of vg only read input sequencing data in FASTQ files, while recent versions also support BAM files, which will potentially save the overall analysis time.
#Map the short-read sequences
>vg map -x wg.xg -g wg.gcsa -f EAGLE_S1_L001_R1_001.fastq.gz -f
EAGLE_S1_L001_R2_001.fastq.gz > 001.mapped.gam
>vg convert wg.xg -p > wg.xg.pg
#Augmentation
>vg augment wg.xg.pg 001.mapped.gam -m 4 -q 5 -Q 5 -A wg-001.aug.gam > wg-001.aug.pg
>vg snarls wg-001.aug.pg > wg-001.aug.snarls
>vg pack -x wg-001.aug.pg -g wg-001.aug.gam -o wg-001.aug.pack
>vg call wg-001.aug.pg -r wg-001.aug.snarls -k wg-001.aug.pack -s vg-001 > wg-001.aug.vcf
Part 3: Haplotype phasing using the population-specific genome as a reference
Haplotype phasing benefits from population-specific contexts like appropriate reference panels. Here we phase the HGDP Han Chinese genomes using the variant callset of the Han Chinese genome assembly as reference. We then provide a script to estimate the switch error rate, which can be further compared across the haplotypes inferred by using different reference panels.
-
7.
Perform variant calling of the NH1 and HX1 genomes based on the human reference genome GRCh38.
>run-dipcall HX1NH1_refb38 GRCh38.chr22.fa NH1.chr22.fa HX1.chr22.fa > HX1NH1_refb38.mak
>make -j2 -f HX1NH1_refb38.mak
>python prepare_HX1NH1_bcf.py HX1NH1_refb38.dip.vcf.gz
-
8.
Run SHAPEIT4 to phase the genotypes of 4 randomly selected Han Chinese samples from the HGDP dataset (Table 1). A genetic map can be optionally specified with “--map”, and SHAPEIT4 provides the HapMap genetic map in GRCh38 coordinates (https://github.com/odelaneau/shapeit4/raw/master/maps/genetic_maps.b38.tar.gz) (The International HapMap Consortium, 2007).
>shapeit4 --input 4Han.HGDP.snp.chr22.b38.vcf.gz --region chr22
--reference HX1NH1_refb38.dip.filtered.bcf --thread 5
--output 4Han.HGDP.snp.chr22.b38.phased.vcf.gz --sequencing
-
9.
Estimate the switch error rate for each sample (provided in a “.switch_error.txt” file, Figure 3).
Note: This estimation is based on the allelic configuration for the adjacent heterozygotes (Lou et al., 2022), and requires the input of .bam files. It takes more than 7 h.
>python run_switch_script.py
Figure 3.

Screenshot of the output file of the switch error estimation for phased genotypes
Part 4: Resolve fine-scale population structure
The population-specific reference genome may also facilitate population genetic analyses, and in particular, resolve the population differentiation and genetic structure at a fine scale. This part illustrates the analyses of population genetic relationship and demographic history using whole-genome variants called from SRS with the population-specific assembly as reference. Here we use 9 Tujia and 43 Han Chinese genome sequences from HGDP as the test data (see key resources table).
-
10.Principal component analysis (PCA).
-
a.Select biallelic SNPs with missing rate < 0.01 and minor allele frequency > 0.05 for further analyses.>bcftools view -i 'F_MISSING<0.01 && MAF>0.05' 9Tujia_43Han.HGDP.snp.chr22.b38.vcf.gz |bgzip > 9Tujia_43Han.HGDP.snp.chr22.b38.miss001.maf005.vcf.gz
-
b.Carry out SNP downsampling according to the physical distance of 50 Kb to roughly exclude possible linkage between loci.>plink -vcf 9Tujia_43Han.HGDP.snp.chr22.b38.miss001.maf005.vcf.gz --make-bed --double--bp-space 50000 --thin 0.99 --out 9Tujia_43Han.HGDP.snp.chr22.b38.miss001.maf005.thin50
-
c.Run FlashPCA2, and plot the first two PCs using a script provided (Figure 4). The script requires an additional file assigning the color of each individual on the plot – two columns denoting the name of individuals and the colors, respectively. An R package named “hash” needs to be installed in R before applying this script. This analysis only takes a few minutes.>flashpca --bfile 9Tujia_43Han.HGDP.snp.chr22.b38.miss001.maf005.thin50k-f .9Tujia_43Han.pca>Rscript pc_plot.r pcs.9Tujia_43Han.pca 9Tujia_43Han.color pcs.9Tujia_43Han.pca.pdf
-
a.
-
11.Infer population demographic history. We apply a multiple sequentially Markovian coalescent approach (MSMC2) to estimate the effective population size (Ne) of the Tujia and Han Chinese populations over time. We select 4 samples (8 haplotypes) from each population (Table 1).Note: Both MSMC and MSMC2 work for this analysis, while the memory usage and time consumption are less for the latter. In addition, MSMC loses power in ancient times with increasing numbers of input genomes (Schiffels and Wang, 2020).
-
a.Genotype phasing of the Tujia and Han Chinese samples using population-specific reference genomes, following part 3: haplotype phasing using the population-specific genome as a reference. Here we use the Han Chinese genomes (HX1 and NH1) as a reference to infer the Tujia haplotypes, while we suggest using the Tujia reference genome (Lou et al., 2022) instead to achieve better performance.
-
b.Perform GenMap to generate a list of low-mappability genomic regions for custom reference genomes (NH1).Note: In principle, read mapping tools like BWA can be used to compute the genome mappability. However, they rely on the read alignment to the reference genome and thus are not applicable to analyzing the NH1 genome assembly. The GEM mappability program is also widely used (Derrien et al., 2012). Here we recommend GenMap as it is a magnitude faster than GEM.>genmap index -F NH1.chr22.fa -I index>genmap map -K 35 -E 1 -I index/ -O ./ -r -t -w -bg 2> err.log>awk -F '\t' '$4==1' NH1.chr22.genmap.bedgraph |cut -f1-3 > NH1.chr22.mappability.bed
-
c.Generate a VCF file for each individual, and run BamCaller.py implemented in MSMC2 for quality control.>for i in {1..4}dobcftools view -s sample${i} -r chr22 <4Han/4Tujia>.HGDP.snp.chr22.b38.phased.vcf.gz |bgzip > sample${i}.chr22.vcf.gzsamtools mpileup -q 20 -Q 20 -C 50 -u -r <chr> -f <ref.fa> <bam> | bcftools call -c -Vindels | python bamCaller.py <mean_cov> <out_mask.bed.gz> | gzip -c > <out.vcf.gz>done
-
d.Mask the low mappability regions in the individual files and make an input file for MSMC2.>generate_multihetsep.py --mask=NH1.chr22.mappability.bed--mask=<sample1_mask.bed.gz> --mask=<sample2_mask.bed.gz>--mask=<sample3_mask.bed.gz> --mask=<sample4_mask.bed.gz>sample1.chr22.vcf.gz sample2.chr22.vcf.gz sample3.chr22.vcf.gz sample4.chr22.vcf.gz > msmc_input
-
e.Run MSMC2 to estimate Ne and plot the Ne dynamics. The Tujia and Han Chinese populations should be analyzed independently.Note: The final output (.final.txt) contains the scaled begin and end time of the interval and scaled inverse Ne of the interval. The ne_plot.r script allows for converting scaled times and Ne to real numbers (saved in .converted.txt file, Figure 5) with given parameters including a mutation rate of the human genome (1.25 × 10−8 per bp per generation by default) and a generation time (25 years per generation by default), and to plot the dynamic changes of Ne (saved in a .final.converted.pdf file, Figure 6). This script limits the plot of Ne dynamics during the period of 1,000–1,000,000 years ago.>msmc2 --fixedRecombination -o msmc_output msmc_input>Rscript ne_plot.r msmc_output <mutation_rate> <generation_time> <color>
-
a.
Figure 4.

PCA plot of the Tujia and Han Chinese populations, represented by orange and green dots, respectively
Figure 5.

Screenshot of the converted results of Ne estimation by MSMC2
Figure 6.

Plot of the Ne dynamics in Han Chinese (red) and Tujia (blue) populations
Part 5: Discover variants in medically relevant genes
In this part, we focus on the recall of genome variants in the previously compiled medically relevant genes (see compile a list of medically relevant genes in before you begin). We examine the HX1 variants detected by mapping the simulated SRS to the NH1 genome, and generate a list of medically relevant variants genotyped. These variants may provide special insights into the genetic basis of some phenotypes or diseases in the corresponding population, and deserve further investigations.
-
12.
Analysis of the medically relevant genes requires a liftover of the gene coordinates to match the NH1 genome coordinates. We apply Liftoff to convert the GFF formatted gene annotation (e.g., GENCODE human release 34, see key resources table) file to NH1, using GRCh38 as the reference genome and the NH1 assembly as the target genome.
>liftoff -g gencode.v34.annotation.gff3 -a 0.9 -s 0.9 -exclude_partial -p 10 -o
NH1.gencode.v34.gff -u NH1_unmapped.txt NH1.chr22.fa GRCh38.chr22.fa
>python get_NH1_medically_genes.py chr22 GRCh38_mrg_full_gene.bed NH1.gencode.v34.gff
>mv NH1.chr22.fa /path/to/snpeff_test/sequences.fa
-
13.
Extract the variants located in the medically relevant genes in the HX1 callset generated in part 1: variants detection from the short-read sequences using linear alignment, and then perform variants annotation using SnpEff.
>bcftools view -R NH1.medically_gene.bed HX1.chr22.filtered.vcf.gz | bgzip >
HX1.chr22.medically_gene.filtered.vcf.gz
>python snpeff_config.py /path/to/snpEff/ /path/to/snpeff_test/
>java -Xmx4g -jar snpEff.jar -v NH1 HX1.chr22.medically_gene.filtered.vcf.gz | bgzip 1>
HX1.chr22.medically_gene.filtered.snpeff_ann.vcf.gz
Expected outcomes
SRS variant call sets (part 1 and 2)
The first two parts of our protocol generate two VCF files, containing two variant call sets of SRS by linear and graph-based methods, respectively, with a population-specific assembly as reference. In addition, Part 1 provides an evaluation outcome of the accuracy of variants detection by simulation (Figure 2). Figure 2 shows that the precision rate of variants detected in the simulated SRS data is 91.7%, with a sensitivity of 86.0%. It is to be expected that the sequence similarity between the target and the reference genome may contribute to the improvement of variant discovery in SRS.
Inferred haplotypes of the SRS data (part 3)
Phasing of the SRS data using the population-specific genome as reference yields the haplotype information, which can be stored in VCF or BCF files according to SHAPEIT4. Each file contains the haplotype of one chromosome. To evaluate the performance of phasing, we further calculated the switch error rate. The outcome is a “.switch_error.txt” file (Figure 3) providing the estimated switch error rate for each sample, which can be compared across independent phasing analyses on an identical genome dataset but using different reference panels. According to our previous study (Lou et al., 2022), using a population-specific genome as a reference may greatly improve the genome phasing accuracy of that population.
Plots of the estimated principal components and population demography (part 4)
Pipelines of PCA and population demographic analyses are included in this part, and accordingly, the output files generated by running the software (e.g., FlashPCA2 and MSMC2) are provided. It is possible to visualize the results using our scripts. Figure 4 depicts a dot plot of Han Chinese and Tujia samples according to the top 2 PCs; Figure 6 shows the dynamic changes in effective population size of Han Chinese and Tujia populations, based on the converted results in the “.final.converted.txt” file (Figure 5). When using the SRS variant call sets generated using population-specific assemblies as a reference, we are expected to obtain a high resolution of population stratification between the two closely related populations. However, Figures 4 and 6 do not show significant genetic differentiation between the Han Chinese and Tujia populations as those in (Lou et al., 2022), possibly because we use the original HGDP callset as test data, which were generated by the alignment to the human reference genome GRCh38, and chromosome 22 provides limited information compared with the whole genome.
A list of medically relevant variants (part 5)
The outcome of Part 5 is a VCF file listing the medically relevant variants that could be potentially detected using the population-specific reference genome. Rare or population-specific variants are expected to be detected with high accuracy using population-specific assembly, and these variants deserve further analyses as they may have large impacts on biological function and abnormality.
Intermediate files
This protocol compiles multiple analytic steps, and therefore generates a lot of intermediate files. Here we list some important intermediate files used as input to reach the outcomes in Table 2.
Table 2.
A list of intermediate files is generated in this workflow
| Files | Description | File generation |
File usage |
||
|---|---|---|---|---|---|
| Parts (Steps) | Software/Scripts | Parts (Steps) | Software/Scripts | ||
| EAGLE_S1_L001_R1_001.fastq.gz; EAGLE_S1_L001_R2_001.fastq.gz | Simulated short reads of HX1 | 1(1b) | EAGLE | 1(2a) | BWA |
| 2(6) | vg map | ||||
| HX1.dedup.bam | Mapping and alignment output of the simulated reads of HX1. | 1(2b) | gatk MarkDuplicates | 1(2c) | gatk HaplotypeCaller |
| HX1.chr22.vcf.gz | Genome variants called from the simulated reads of HX1 | 1(2c) | gatk HaplotypeCaller | 1(2d) | gatk VariantFiltration |
| HX1.chr22.filtered.vcf.gz | Genome variants called from the simulated reads of HX1 with hard-filtering | 1(2d) | gatk VariantFiltration | 1(3b) | RTGtools |
| 5(13) | BCFtools | ||||
| wg.xg; wg.gcsa | Index files of a graph genome reference of GRCh38 constructed based on the NH1 assembly | 2(5) | vg construct∗; vg index | 2(6) | vg map |
| <4Han/4Tujia>.HGDP.snp.chr22.b38.phased.vcf.gz | Phased haplotypes of the HGDP samples | 3(8) | SHAPEIT4 | 3(9) | run_switch_script.py |
| 4(11c) | BCFtools; MSMC2∗ | ||||
| NH1.chr22.mappability.bed | A list of low-mappability regions of NH1 that can be removed to keep high confidence variants | 4(11b) | GenMap | 4(11d) | generate_multihetsep.py |
Intermediate files that are not used in subsequent steps are not shown. We indicate the software or scripts that do not directly process these files but are key analyses in corresponding steps with asterisks (∗).
Limitations
The practical use of a population-specific reference genome relies heavily on the quality of the genome assembly. It can be expected that an increasing number of population-specific reference genomes will facilitate human genome studies, but at the current stage, there is no available data for most the human populations. This protocol provides a comprehensive strategy to explore the benefits of a population-specific reference genome, which will help bridge the gap between constructing a de novo genome assembly and applying it to gain more insights into the human genome. However, we are not able to include all the applicable methods and algorithms in this protocol, and we only show the procedures to run some of the commonly used software and analyses. Using published software packages may lead to limitations on managing the process and improving the results. We commented on some critical steps and limitations of the software packages, and the users need to reach the original sources for more information (see key resources table). The users should make their own decisions on some parameters and standards of quality control based on the data properties. For instance, we need to balance between limiting false discovery and enabling true discovery when applying GATK hard-filtering to the variant callset. According to the GATK recommendation, any variant with a mapping quality (MQ) value less than 40 should be removed. Increasing the MQ cutoff to 50 to reduce the false positive rate is also acceptable. Similar considerations need to be taken to remove low-quality sites when performing vg augment (e.g., “-m”, “-q”, and “-Q”), samtools mpileup (e.g., “-q”, “-Q”, and “-C”) or Liftoff (e.g., “-a” and “-s”). Although most of the analytical steps can be done using software and scripts provided, this protocol requires good knowledge of programming. New users may need to learn some skills underlying the protocol to execute it fully, including the basic knowledge of Python and bash scripting, at least.
Troubleshooting
Problem 1
EAGLE commands fail (Figure 7) (step 1a in part 1: variants detection from the short-read sequences using linear alignment).
Figure 7.

Screenshot of the error message of EAGLE
Potential solution
Filter out contigs < 1 kb in the input data.
Problem 2
Vg commands fail with an error message: “ERROR: Signal 6 occurred. VG has crashed” (step 4 in part 2: variants detection from the short-read sequences using the graph-based approach).
Potential solution
Set --flat-alts in vg construct.
Problem 3
Vg commands fail with an error message: “error:[vg::Constructor] non-ATGCN characters found in chr22” (step 4 in part 2: variants detection from the short-read sequences using the graph-based approach).
Potential solution
Filter out the alleles that do not match with A, T, G, C, or N in the human reference genome GRCh38.
Problem 4
SnpEff commands fail (Figure 8) (step 13 in part 5: discover variants in medically relevant genes).
Figure 8.

Screenshot of the error message of SnpEff
Potential solution
Remove the genes listed in the error message from the GFF file.
Problem 5
Incorrect annotations at some loci in the output of SnpEff without any error message when executing this software. (step 13 in part 5: discover variants in medically relevant genes).
Potential solution
Input the GFF file in an uncompressed form.
Resource availability
Lead contact
Further information and requests for resources and code should be directed to and will be fulfilled by the lead contact, Shuhua Xu (xushua@fudan.edu.cn).
Materials availability
This study did not generate new materials.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC) grant (32030020, 32041008, 31900418, and 31961130380), the Strategic Priority Research Program (XDPB17, XDB38000000) of the Chinese Academy of Sciences (CAS), the Science and Technology Commission of Shanghai Municipality (19YF1455200), the UK Royal Society-Newton Advanced Fellowship (NAF\R1\191094), and the Shanghai Municipal Science and Technology Major Project (2017SHZDZX01). We thank Dr. Haiyi Lou for his advice on constructing this protocol. We are grateful to Yang Gao for his help in conducting the population genetic analyses.
Author contributions
S.X. conceived and initiated this study. B.X., Y.W., and L.D. prepared the pipeline. B.X. and X.Z. prepared the test dataset. L.D., B.X., and Y.W. wrote the first draft. L.D. and S.X. revised the paper.
Declaration of interests
The authors declare no competing interests.
Data and code availability
The test dataset is freely available at https://www.picb.ac.cn/PGG/resource_download.php?id=44&file=PGG_Web_Data/protocol_test_data_chr22.tar.gz and at GitHub: https://github.com/Shuhua-Group/TJ1_STARProtocols. The code generated during this study is available at GitHub: https://github.com/Shuhua-Group/TJ1_STARProtocols and at Zenodo: https://zenodo.org/record/6520447#.YnO6gC-KHUp.
References
- Abraham G., Qiu Y., Inouye M. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics. 2017;33:2776–2778. doi: 10.1093/bioinformatics/btx299. [DOI] [PubMed] [Google Scholar]
- Bergström A., McCarthy S.A., Hui R., Almarri M.A., Ayub Q., Danecek P., Chen Y., Felkel S., Hallast P., Kamm J., et al. Insights into human genetic variation and population history from 929 diverse genomes. Science. 2020;367:eaay5012. doi: 10.1530/ey.17.14.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute . GitHub Repository. 2019. Picard toolkit.https://broadinstitute.github.io/picard/ [Google Scholar]
- Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary J.G., Braithwaite R., Gaastra K., Hilbush B.S., Inglis S., Irvine S.A., Jackson A., Littin R., Rathod M., Ware D., et al. Comparing variant call files for performance benchmarking of next-generation sequencing variant calling pipelines. bioRxiv. 2015 doi: 10.1101/023754. Preprint at. [DOI] [Google Scholar]
- Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Summa S., Malerba G., Pinto R., Mori A., Mijatovic V., Tommasi S. GATK hard filtering: tunable parameters to improve variant calling for next generation sequencing targeted gene panel data. BMC Bioinformatics. 2017;18:119. doi: 10.1186/s12859-017-1537-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O., Zagury J.F., Robinson M.R., Marchini J.L., Dermitzakis E.T. Accurate, scalable and integrative haplotype estimation. Nat. Commun. 2019;10:5436. doi: 10.1038/s41467-019-13225-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T., Estelle J., Marco Sola S., Knowles D.G., Raineri E., Guigo R., Ribeca P. Fast computation and applications of genome mappability. PLoS One. 2012;7:e30377. doi: 10.1371/journal.pone.0030377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z., Ma L., Qu H., Chen W., Zhang B., Lu X., Zhai W., Sheng X., Sun Y., Li W., et al. Whole genome analyses of Chinese population and de novo assembly of A northern Han genome. Dev. Reprod. Biol. 2019;17:229–247. doi: 10.1016/j.gpb.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng J. Improving the interactivity and functionality of Web-based radiology teaching files with the Java programming language. Radiographics. 1997;17:1567–1574. doi: 10.1148/radiographics.17.6.9397464. [DOI] [PubMed] [Google Scholar]
- Garrison E., Siren J., Novak A.M., Hickey G., Eizenga J.M., Dawson E.T., Jones W., Garg S., Markello C., Lin M.F., et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 2018;36:875–879. doi: 10.1038/nbt.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J., Denoeud F., Frankish A., Reymond A., Chen C.K., Chrast J., Lagarde J., Gilbert J.G.R., Storey R., Swarbreck D., et al. GENCODE: producing a reference annotation for ENCODE. Genome Biol. 2006;7:1–9. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin L. Github Repository. 2014. Eagle - enhanced artificial genome engine.https://github.com/sequencing/EAGLE [Google Scholar]
- Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Bloom J.M., Farjoun Y., Fleharty M., Gauthier L., Neale B., MacArthur D. A synthetic-diploid benchmark for accurate variant-calling evaluation. Nat. Methods. 2018;15:595–597. doi: 10.1038/s41592-018-0054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H., Gao Y., Xie B., Wang Y., Zhang H., Shi M., Ma S., Zhang X., Liu C., Xu S. Haplotype-resolved de novo assembly of a Tujia genome suggests the necessity for high-quality population-specific genome references. Cell Syst. 2022;13:321–333.e6. doi: 10.1016/j.cels.2022.01.006. [DOI] [PubMed] [Google Scholar]
- Malaspinas A.S., Westaway M.C., Muller C., Sousa V.C., Lao O., Alves I., Bergström A., Athanasiadis G., Cheng J.Y., Crawford J.E., et al. A genomic history of aboriginal Australia. Nature. 2016;538:207–214. doi: 10.1038/nature18299. [DOI] [PubMed] [Google Scholar]
- Pockrandt C., Alzamel M., Iliopoulos C.S., Reinert K. GenMap: ultra-fast computation of genome mappability. Bioinformatics. 2020;36:3687–3692. doi: 10.1093/bioinformatics/btaa222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R Foundation for Statistical Computing; 2020. R: A Language and Environment for Statistical Computing. [Google Scholar]
- Schiffels S., Wang K. In: Statistical Population Genomics. Dutheil J.Y., editor. Springer US); 2020. MSMC and MSMC2: the multiple sequentially Markovian coalescent; pp. 147–166. [DOI] [PubMed] [Google Scholar]
- Shi L., Guo Y., Dong C., Huddleston J., Yang H., Han X., Fu A., Li Q., Li N., Gong S., et al. Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun. 2016;7:12065. doi: 10.1038/ncomms12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumate A., Salzberg S.L. Liftoff: Accurate Mapping of Gene Annotations. Bioinformatics. 2021;37(12):1639–1643. doi: 10.1093/bioinformatics/btaa1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;43:11.10.11–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rossum G., Drake F.L. CreateSpace; 2009. Python 3 Reference Manual. [Google Scholar]
- Wagner J., Olson N.D., Harris L., McDaniel J., Cheng H., Fungtammasan A., Hwang Y.-C., Gupta R., Wenger A.M., Rowell W.J., et al. Towards a comprehensive variation benchmark for challenging medically-relevant autosomal genes. bioRxiv. 2021 doi: 10.1101/2021.06.07.444885. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The test dataset is freely available at https://www.picb.ac.cn/PGG/resource_download.php?id=44&file=PGG_Web_Data/protocol_test_data_chr22.tar.gz and at GitHub: https://github.com/Shuhua-Group/TJ1_STARProtocols. The code generated during this study is available at GitHub: https://github.com/Shuhua-Group/TJ1_STARProtocols and at Zenodo: https://zenodo.org/record/6520447#.YnO6gC-KHUp.