

Abstract
DNA damage is a constant threat to genome integrity. DNA repair and damage signaling networks play a central role maintaining genome stability, suppress tumorigenesis, and determine tumor response to common cancer chemotherapeutic agents and radiotherapy. DNA double-strand breaks (DSBs) are critical lesions induced by ionizing radiation and when replication forks encounter damage. DSBs can result in mutations and large-scale genome rearrangements reflecting mis-repair by non-homologous end joining or homologous recombination. Ionizing radiation induces genetic change immediately, and it also triggers delayed events weeks or even years after exposure, long after the initial damage has been repaired or diluted through cell division. This review covers DNA damage signaling and repair pathways and cell fate following genotoxic insult, including immediate and delayed genome instability and cell survival/cell death pathways.
1. Introduction
DNA is subject to constant threat of damage from endogenous and exogenous genotoxic agents, such as reactive oxygen species (ROS) arising during normal cellular metabolism, and ionizing radiation (IR) which causes DNA damage directly, and indirectly through ROS production. IR creates a variety of DNA lesions including base damage, and single- and double-strand breaks (DSBs) with immediate effects such as point mutations, chromosomal aberrations, homologous recombination (HR) and cell death. IR also triggers delayed effects, apparent many cell generations or even years after the initial exposure (Fig. 1). In general, immediate effects are due to targeted DNA damage, whereas delayed effects are most likely non-targeted, akin to bystander effects (see article by T. Hei in this issue). DNA damage triggers signaling (checkpoint) pathways that are critical for genome stability and are frequently defective in cancer. Nearly all DNA lesions block DNA replication, and stalled replication forks can be processed to DSBs if not restarted in timely manner [1–3]. DNA lesions are processed by a variety of DNA repair mechanisms, some of which restore the chemical integrity of damaged DNA in a relatively error-free manner, which maintains genome integrity. Other repair mechanisms restore the chemical integrity of DNA, but not the genetic information, causing mutations of various types, and thus destabilize the genome. Mutations range from the smallest single-base changes to large alterations including chromosome translocation, deletion, insertion, inversion, and amplification events. Some large-scale genetic alterations arise from very complex mechanisms that involve interactions among multiple chromosomes [4–6].
Fig. 1.
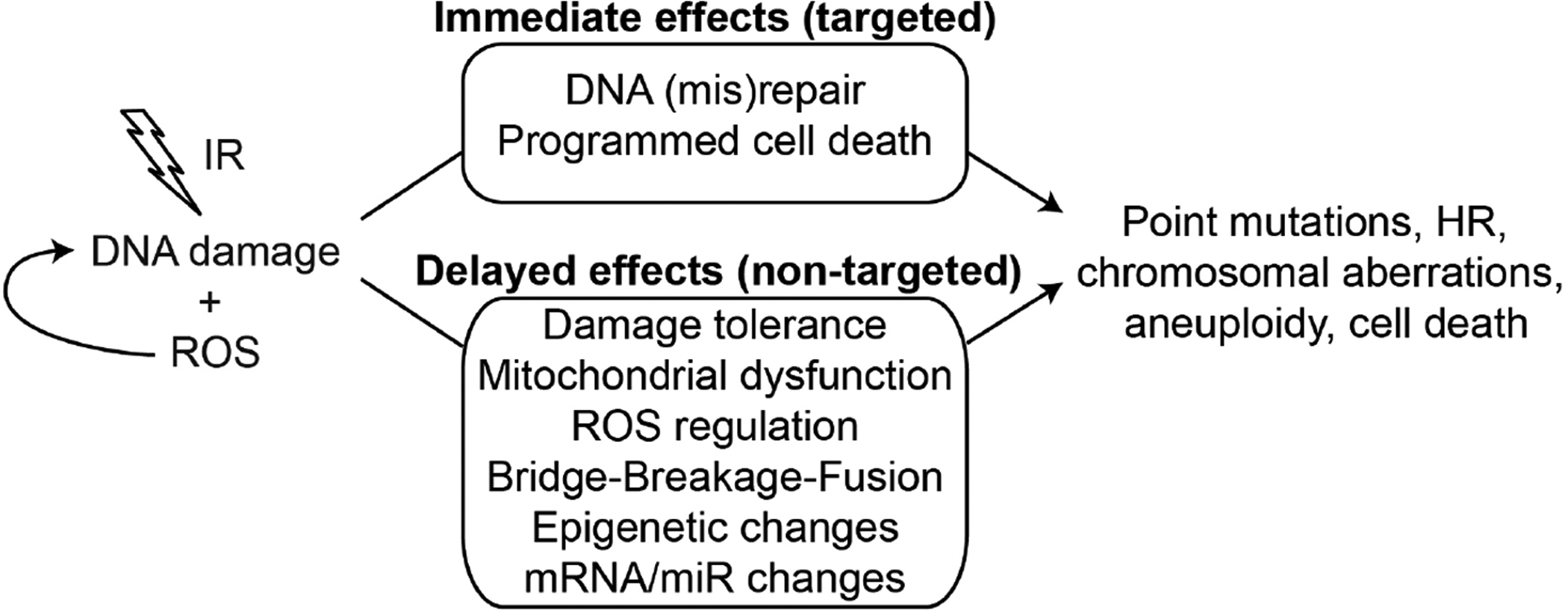
IR causes DNA damage directly and through ROS production, causing a wide variety of genetic changes. Some changes appear immediately after exposure and are most likely targeted effects. IR also induces delayed effects that can lead to the same types of genetic changes, but are likely non-targeted effects and may reflect changes in specific organelles or processes that have broad impact on genome stability.
This review focuses on DNA damage signaling and DNA repair processes that regulate genome stability, with an emphasis on homologous recombination repair. The DNA damage response (DDR) comprises interacting networks of DNA damage signaling and DNA repair pathways [7–12]. DNA damage signaling is initiated by proteins that detect and bind to DNA lesions (sensors), triggering signaling pathways mediated by post-translational modifications of downstream proteins (signal transducers), including phosphorylation by protein kinases, PARsylation by PARP, and other modifications [13, 14]. These pathways activate cell cycle checkpoint (effector) proteins that mediate cell cycle arrest and regulate DNA repair and programmed cell death and senescence pathways [15–22]. Here we explore both the spatial and temporal aspects of genetic changes observed in irradiated cells, and relate these topics to cancer etiology and therapy. Dr. William F. ‘Bill’ Morgan played an important pioneering role in the genome instability field, making critical contributions over more than two decades. I was very fortunate to meet Dr. Morgan when he visited Los Alamos National Laboratory while I was completing my postdoctoral training in the late 1980s. I developed a strong personal and professional relationship with him over nearly three decades, and we performed several collaborative projects that led to new insights in the delayed genome instability field. Dr. Morgan’s vast knowledge of radiobiology and cancer biology had a tremendous impact on my career, and the careers of many investigators world-wide. It is certain that Dr. Morgan’s impressive body of work will have a major impact in the radiation sciences far into the future.
Delayed radiation effects on chromosomes were first reported in the 1950s [23, 24], but the vast majority of radiobiological studies, beginning with the pioneering work of Muller nearly 90 years ago [25] focused on immediate effects of radiation on various endpoints including mutagenesis, chromosome aberrations, and cell death. Studies in the 1950s and 1960s indicated that cells exposed to a lethal dose of radiation could divide several times before cell division terminated [26, 27], and that lethal mutations could arise several generations after irradiation [28]. In the early 1990s the Little lab demonstrated that specific (HPRT) mutations can arise up to 7 cell generations after irradiation, and that non-specific, lethal mutations can arise up to 50 cell generations after irradiation, revealed as reduced cell cloning efficiency and termed “delayed reproductive death” [29, 30]. By this time, the Morgan laboratory had made significant contributions to the genome instability field, and in 1993 published the first report describing delayed chromosomal instability (DCI) revealed through advanced cytogenetic analysis of hamster cells carrying a single human chromosome [31]. This set the stage for more than 20 years of progress by Dr. Morgan’s group elucidating the molecular mechanisms of DCI. The present review focuses delayed genome instability arising via HR, and includes a discussion of DNA damage response (DDR) pathways that are well-known regulators of immediate cell responses to radiation, and are also likely to be involved in delayed responses to radiation. DDR pathways have gained significant attention in recent years as targets for cancer therapy, either in mono-therapy or as adjunct to traditional chemo- and radiotherapy strategies [9, 32]. Genome instability, in its many forms, is a potent factor in cancer etiology, and the idea that radiation and other genotoxic agents can induce genome instability many generations after the initial genotoxic insult, has significant implications with respect to risk assessment for populations exposed to radiation from environmental sources, in the workplace, and during medical procedures. A major concern is secondary tumor induction as a result of genotoxic cancer chemotherapy and radiotherapy, particularly in light of delayed or persistent induction of genome instability by radiation.
2. DNA damage signaling
Cells respond to DNA damage by activating checkpoint pathways mediated by protein kinases that arrest cell cycle progression, stimulate repair, promote survival and genome stability, and suppress cancer (Fig. 2). Two major checkpoint signaling pathways include one centered on ATM that responds to DSBs leading to Chk2 activation and p53 stabilization, and one centered on ATR that activates Chk1 in response to single-strand breaks and gaps (i.e., at replication forks). RPA bound to single-stranded DNA (ssDNA) recruits ATRIP-ATR, activating ATR. A third signaling pathway involves ATM, p38MAPK and MK2 kinases, and converges on similar cell cycle regulation targets as Chk1/2 [8, 33]. Checkpoint and repair pathways display substantial crosstalk, through protein-protein interactions and phosphorylation, methylation, acetylation, etc.). Many proteins originally defined for their roles in DNA repair, such as BLM, DNA-PK, MRE11, RAD51, and RAD52, or for their roles in DNA damage checkpoints, such as ATM and ATR play key roles in stabilizing the replisome when DNA lesions are encountered during DNA replication, and these proteins also promote restart of stalled or collapsed replication forks [34]. Through these functions, these DNA repair/checkpoint/fork restart proteins maintain genome stability in response to endogenous DNA damage arising during normal cellular metabolism (reflecting chemical lability of DNA or attack by ROS) or from exogenous genotoxins including the majority of cancer chemotherapeutics and radiation. DDR pathways operate in redundant fashion, and a major goal in the field is to identify synthetic (genetic) lethal interactions to exploit in cancer therapy [35].
Fig. 2.
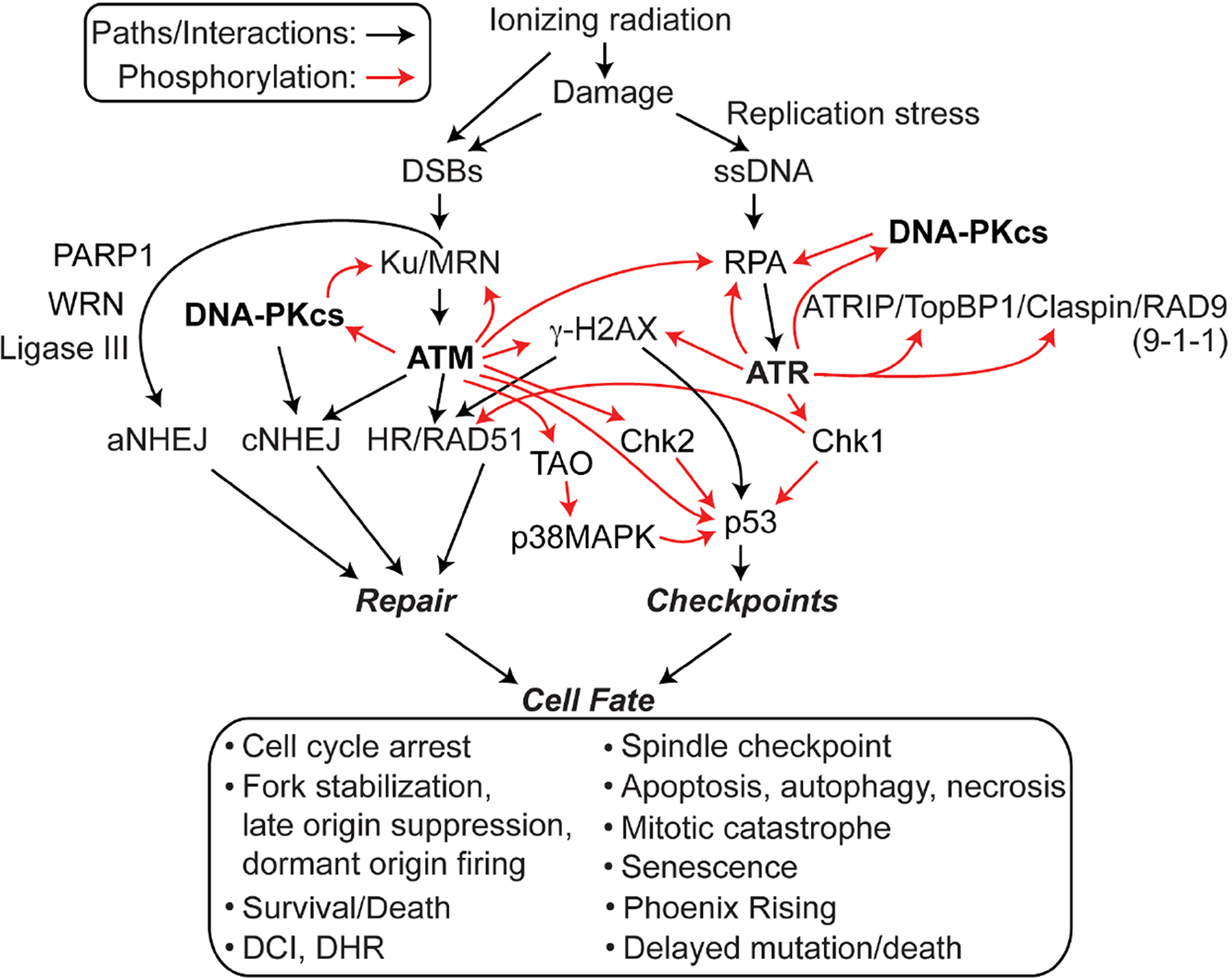
Core factors in DNA repair and DDR networks. Ionizing radiation causes DNA damage that activates PIKs (bold) which transmit signals to both downstream and upstream targets that regulate DNA repair by aNHEJ, cNHEJ, and HR, and activate checkpoint response pathways that arrest the cell cycle and trigger programmed cell death pathways, all of which regulate cell fate.
DDR pathways are not “on or off” but show graded responses depending on the level of damage. DDR thresholds are genetically regulated [36, 37], and thresholds may vary for each checkpoint [38]. With minimal damage, cells may activate repair but not arrest. At higher levels of damage, cells may arrest in G1 or S to prevent replication fork encounters with lesions [39, 40], in G2 to prevent mitotic catastrophe [41], and still more damage can cause cells to enter one of several programmed death pathways (see below).
An early step in DNA damage signaling is recruitment of the Ku70/Ku80 heterodimer and MRE11/RAD50/NBS1 (MRN) complex to DSBs; these proteins are early DNA damage sensors (Fig. 2). ATR, ATM, and DNA-PKcs are PI3-like kinases (PIKs) that play central roles in DDR signaling (Fig. 2, bold font). Although early studies suggested compartmentalized functions for ATM and ATR in response to frank DSBs and replication stress, respectively, this is an oversimplification as PIKs show considerable functional overlap and crosstalk in the DDR (Fig. 2). For example, DNA-PKcs (bound to DNA end-bound Ku) and ATM are both activated by DSBs, and they phosphorylate at least six shared targets including H2AX, RPA, and c-abl, and ATR/ATM regulate DNA-PKcs via phosphorylation [42–48]. ATR was originally thought to be primarily responsible for RPA phosphorylation during replication stress, but there is now clear evidence that DNA-PKcs phosphorylates RPA32 Ser4/Ser8 during replication stress [49–58]. These results account for the observation that DNA-PKcs defects sensitize cells to replication stress agents [48, 59, 60]. RPA has important roles in HR, and mutations in either DNA-PKcs or the Ser4/Ser8 phosphorylation targets in RPA32 confer similar phenotypes: in response to replication stress, both types of mutants show defects in replication checkpoint arrest, accelerated replication fork restart upon release from stress, defective suppression of late origin firing, hyper-recombination, and increased genome instability (mitotic catastrophe) [58].
DSBs can be induced by direct action of IR, but they also arise indirectly when other lesions such single-strand breaks and base damage block replication forks. DSBs also arise when stalled forks regress to “chicken foot” structures, or when they are cleaved by the structure-specific endonucleases MUS81, EEPD1, and Metnase [34, 61–68]. Broken DNA ends at DSBs are subject to 5’ to 3’ resection which creates 3’ single-stranded DNA (ssDNA) tails. Resection proceeds in stages, with limited resection initiated by CtIP and Mre11 nucleases, and more extensive resection catalyzed by Exo1 and Dna2 in collaboration with BLM and other accessory factors [63, 68–72]. The extent of end-resection is a key determinant of DSB repair pathway choice [63, 69, 73]. Resection is initially suppressed by 53BP1 and RIF1 bound to DNA ends; when CDK1 phosphorylates CtIP in S/G2 phase, phospho-CtIP collaborates with BRCA1 to dissociate 53BP1/RIF1 from ends allowing resection to proceed [69, 70, 74, 75].
The 3’ ssDNA tails at resected ends are rapidly coated with the abundant, heterotrimeric ssDNA binding protein RPA. ssDNA bound by RPA is a major signal for fork repair and replication checkpoint activation [39, 40, 61]. ssDNA coated with RPA recruits ATRIP and ATR, leading to ATR phosphorylation/activation by a mechanism that involves TopBP1, Claspin, RAD17-RFC, and the RAD9-RAD1-HUS1 (9-1-1) complex, via a RAD9-RPA interaction, and a host of other proteins [8, 76–78] (Fig. 2). Recently a RAD17-indepenent, NBS1-dependent ATR activation pathway was elucidated that depends on an NBS1-RPA interaction [79]. Once activated, ATM, ATR, and DNA-PKcs phosphorylate many targets including the RPA32 subunit of RPA, and Chk1 and Chk2 checkpoint effector kinases that in turn phosphorylate proteins that function in DNA repair (e.g., BLM, H2AX, RAD51, FANCD2, 53BP1, Ku70,RAD51 paralogs, and Metnase) [80–88]; regulate cell cycle progression, checkpoint arrest, and cell death pathways (e.g., p53, p21, RB, CDC25) [12, 33, 89–91]; stabilize or repair stalled/collapsed forks; and prevent late origin firing - presumably to prevent further encounters of forks with DNA damage [8, 92]. The PIK targets noted above represent a tiny fraction of the total: it has been reported that ATM and ATR phosphorylate at least 900 targets on 700 different proteins [7], and this is certainly an underestimate. Chk1 roles in checkpoint and repair responses make it an attractive target for enhancing chemotherapy and radiotherapy [93, 94].
In addition to enhancing DNA repair and regulating the cell cycle, PIK-Chk1-Chk2 signaling also plays a major role in death pathway activation (Section 3). By enhancing DNA repair and regulating cell cycle progression, DDR signaling through PIK pathways plays a major role in maintaining genome stability in the face of DNA damage and thus suppressing tumorigenesis [95].
As noted above, IR and other genotoxic agents induce lesions in DNA, nearly all of which block replicative polymerases. Replisomes are stabilized at stalled forks by many proteins with roles in DNA repair and DNA damage checkpoints, including RPA, ATR-ATRIP, ATM, BLM, and INO80 [40, 96–98]. In some cases, lesions can be bypassed translesion synthesis (TLS) polymerases, which are error-prone and not highly processive, so TLS creates mutations in nascent DNA strand opposite, and in the immediate vicinity of, DNA lesions [99–101]. Persistent stalling at blocking lesions can lead to fork collapse and one-ended DSBs (“double-strand ends” - DSEs), and replication fork encounters with single-strand breaks (induced by IR far more frequently than DSBs) may cause direct fork collapse to a DSE. As with frank DSBs, ATM and ATR phosphorylate histone H2AX (γ-H2AX) in the vicinity of DSEs [87], activating checkpoint and repair processes [88, 95]. While intra-S checkpoint arrest serves to minimize replication fork encounters with DNA lesions induced by IR, it is likely that once the checkpoint is released and replication resumes, residual damage may remain, triggering replication stress at later times (~8 h) after irradiation [102]. It is difficult to determine the relative contributions of immediate (mis)repair events vs. later replication stress to genome destabilization. As discussed in Section 5, genome stability is also threatened at much later times (weeks to years) in a fraction of cells that survive low to moderate doses of IR. We have a limited understanding of the mechanisms responsible for these delayed effects.
3. Regulation of cell fate after DNA damage
Cell fate following genotoxic insult can be divided into cell survival vs. death, and among survivors, cells may retain a stable genome or succumb to genome instability. Low to moderate levels of genome instability can be tolerated (yet trigger tumorigenesis and/or tumor progression to more aggressive stages) but extreme genome instability is incompatible with cell viability, because of gross gene expression imbalance due to numerical changes in chromosome content due to segregation defects, or from lethal mutations (induced or uncovered via loss of heterozygosity. Regulated cell death pathways guard against massive genome instability and cancer by eliminating highly damaged cells. Apoptosis is a set of well-characterized programmed (regulated) cell death pathways [21, 103]. There are two other programmed death pathways, autophagy and necrosis, as well as senescence, an anti-proliferation pathway in which cells remain metabolically active. From the standpoint of cancer therapy, inducing any of these death/senescence pathways serves to eradicate tumors or control tumor growth. Autophagy (“self-eating”) is a stress response pathway conserved from yeast to humans [17, 103] mediated by catabolic processes that degrade proteins and organelles, including mitochondria which helps maintain metabolic homeostasis. Autophagy is inhibited by oncogenic proteins that activate mTOR (e.g., Ras and AKT), and autophagy is stimulated by tumor suppressor proteins that inhibit mTOR (e.g., LKB1, PTEN, and AMPK), thus autophagy operates as a tumor suppressor pathway in normal cells [104, 105]. Consistent with this view, direct chemical or genetic inhibition of autophagy increases tumorigenesis which is associated with genome instability and increased reactive oxygen species. However, once a tumor is established, autophagy can actually protect tumor cells from endogenous stress common in tumors (nutrient deprivation, hypoxia, “oncogenic stress”), and exogenous stress associated with chemo- or radiotherapy. Once considered a passive death pathway, necrosis is instead a genetically regulated pathway, which has given rise to the term “necroptosis” [18, 19]. Similar to autophagy, necroptosis can influence both tumorigenesis and tumor responses to therapy [106]. Senescence was originally defined as growth arrest due to telomere shortening, which can be bypassed by telomerase in stem cells and cancer stem cells. However, an alternative senescence pathway of permanent checkpoint arrest blocks tumor re-growth after radiotherapy [20]. It is possible to define death pathway spectra for various stress conditions by using pathway-specific markers: caspase-3 cleavage, annexin-V, TUNEL and others for apoptosis [107], LC3-II for autophagy [108, 109], secreted HMGB1 for necrosis [110, 111], and checkpoint/SA-β-Gal expression (senescence) [112].
Some types of DNA damage and defects in chromosome decatenation or segregation cause cell death via mitotic catastrophe, which is revealed as giant cells, nuclear dysmorphism, multinucleate cells, micronuclei, and anaphase bridges [20]. Mitotic catastrophe is not programmed, per se, but it is regulated in the sense that it is suppressed by the DDR, and it may trigger cell death by other programmed death pathways including apoptosis and necrosis [41].
ATM phosphorylates/activates Chk2 kinase, which then phosphorylates and stabilizes p53, altering p53 transcriptional activity, and promoting apoptosis [113]. There are also p53-independent apoptotic pathways [114]. Chk1 also phosphorylates p53 on both shared and distinct residues targeted by Chk2. Chk1 appears to balance Chk2 by suppressing apoptosis, and Chk1 and Chk2 also regulate apoptosis through phosphorylation of the ubiquitin ligase Mdm4/X which promotes p53 degradation [113].
Apoptosis has long been considered a desirable outcome in radio- and chemotherapy, but this paradigm has recently shifted in light of studies demonstrating that a common upstream apoptotic event, caspase 3 activation, stimulates release of the paracrine growth factor prostaglandin E2 (PGE2) which enhances proliferation of nearby surviving cells. This pathway, termed “Phoenix Rising” is akin to wound healing [115]. Phoenix Rising is has clinical importance as patients with caspase 3-defective breast and head/neck cancers survive longer than those with functional caspase 3 [115]. Caspases act early in apoptosis [116] and Phoenix Rising depends on caspase-3 activation (cleavage) which activates the iPLA2-Cox1/2-PGE2 pathway [115]. Phoenix Rising can be blocked at many steps along the pathway from caspase-3 cleavage to PGE2 production/receptor binding [115, 117]. Although blocking apoptosis might enhance cell survival, sufficient DNA damage will likely induce death by another pathway. Inducing cell death without activating Phoenix Rising may be key to improved local tumor control. Although much is known about DDR proteins and their roles in cell survival and genome stability, and there are clear connections between the DDR and apoptosis, virtually nothing is known about how the DDR regulates other cell death pathways. Given the connection between apoptosis and Phoenix Rising [115, 118], it is critical to gain a better understanding of DDR regulation of all death pathways to simultaneously optimize tumor cell killing while preventing accelerated repopulation via Phoenix Rising and thus enhance local tumor control by radiotherapy. To summarize, cell fate depends on the level and complexity of damage, DDR signaling, DNA repair, programmed cell death pathway choices, and bystander effects like Phoenix Rising in which damaged or dying cells influence growth or genome stability of nearby cells. All of these factors contribute to normal tissue and tumor responses to IR.
4. DNA repair and genome instability
Genome instability takes many forms and arises from mis-repair of endogenous or exogenous DNA damage. Defects in DNA repair pathways can dramatically increase genome instability, with specific mutational spectra dependent on the types of damage and repair outcomes. All DNA repair pathways can restore the proper chemical structure of DNA, but they vary widely in their ability to restore the original genetic sequence, hence the term “mis-repair” denotes restoration of DNA chemical structure without restoration of the genetic sequence. Such errors span the spectrum from single-base changes and small insertion/deletion mutations, to gene-level changes (gene duplication, deletion, amplification, and rearrangement), to chromosome-level changes including translocations, loss of large regions of chromosome arms, and gain or loss of whole chromosomes. Defects in base excision repair and nucleotide excision repair increase point mutagenesis and are associated with many cancers including those of the gastrointestinal tract, breast, bladder, and skin [119]. Defects in mismatch repair greatly increase point mutagenesis including frameshift mutations arising from replication slippage in homo-polynucleotide runs, and slippage at triplet repeats, predisposing to colon cancer and other cancers [120].
Defects in DSB repair may reflect problems with NHEJ or HR (Fig. 3). Although cNHEJ is inherently error-prone, introducing short deletions or insertions at joints, there are greater risks associated with cNHEJ defects because the back-up aNHEJ pathway creates larger deletions and mediates translocations [121–128]. The Jasin lab showed that defects in core cNHEJ factors Ku, XRCC4, and Lig4 suppress translocations stimulated by simultaneous DSBs introduced into two chromosomes [125, 127]. Similarly, translocations were suppressed when cNHEJ was enhanced by overexpression of Metnase [129]. These results appear counterintuitive given that at least some translocations arise via end-joining, but can be explained by a model in which cNHEJ promotes rapid DSB repair, and when cNHEJ is defective DNA ends have a greater propensity to migrate through the nucleus and join with ends from other chromosomes to create translocations. This model gained additional support when it was determined that translocations frequently result from aNHEJ, which is mediated by MRE11 and CtIP (to effect limited resection), NBS1, PARP1, Lig3 and FEN1 [130, 131]. The enhanced genome instability with cNHEJ defects can also be understood in terms of repair accuracy, as aNHEJ requires microhomology and therefore typically creates larger deletions than cNHEJ.
Fig. 3.
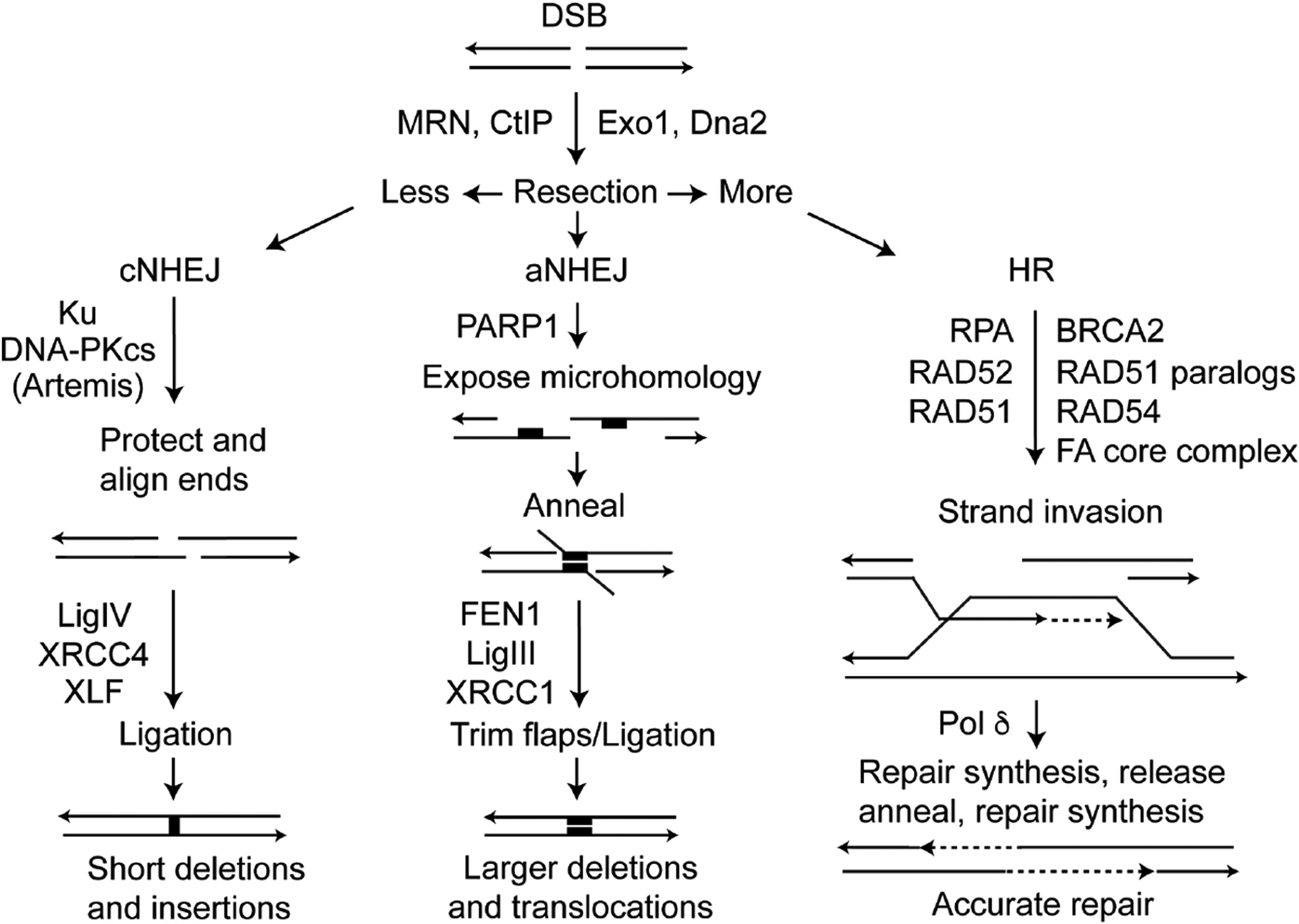
DSB repair pathways. DSB repair pathway choice is controlled by resection, mediated by several nucleases. cNHEJ involves little or no resection, aNHEJ limited resection to expose microhomologies near the DSB (black rectangles), and HR involves extensive resection creating long, 3’ single-stranded tails that invade homologous sequences (typically sister chromatids) that serve as accurate repair templates.
In contrast to error-prone cNHEJ/aNHEJ, HR is often described as an error-free DSB repair pathway. Extensively resected broken ends are first bound by RPA, as discussed above, and RPA is subsequently replaced by RAD51 with assistance from “mediator” proteins including BRCA2 and RAD51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3). The RAD51-ssDNA nucleoprotein filament seeks and invades homologous duplexes elsewhere in the genome which serve as (relatively) accurate repair templates. HR is largely suppressed in G1 phase and upregulated in S and G2, when sister chromatids can serve as highly accurate (and proximal) repair templates. However, homologous sequences anywhere in the genome may serve as a repair template, including homologous chromosomes and repetitive elements (Alu, MIRs, SINEs, LINEs, etc.) which are extremely common in higher eukaryotes, comprising >50% of human genomic DNA [132]. Because non-sister homologous sequences are often not 100% identical, HR repair from such templates can transfer divergent sequence information to the broken chromosome, a process termed gene conversion [133]. HR between non-sister sequences can also lead to significant structural changes in the genome when HR intermediates are resolved with a reciprocal exchange or crossover, including gene deletions, duplications, inversions, large-scale loss of heterozygosity, and translocations [63, 126, 133–136]. For this reason, mitotic crossovers are suppressed [137–140] and defects in proteins that suppress crossovers, like BLM, cause massive genome instability and cancer predisposition [141]. Thus, some HR events cause genome instability [126, 142], drive tumor evolution, and cause other diseases [136, 143, 144]. Although HR has the potential to destabilize the genome, instability is far more pronounced in HR defective cells, as seen with defects in BRCA1, BRCA2, and FANC proteins, which predispose to breast and other cancers [145–148]. Thus, HR is tightly regulated, and mutations that dysregulate HR, causing hyper-recombination or hypo-recombination phenotypes, generally have detrimental effects on genome stability.
HR plays a critical role in maintaining genome stability in the face of replication stress [3, 34, 40]. When replication forks encounter damage, they can assume a variety of branched structures including 5’ and 3’ flaps, single-strand gaps, and 4-way junctions akin to Holliday junctions in homologous recombination (HR) intermediates [64]. These branched structures are cleaved by structure-specific nucleases (Mus81-Eme2, EEPD1, Metnase) [66–68, 149–151], creating DSEs that are extensively resected to long 3’ ssDNA tails by the combined action of Mre11/CtiP, Exo1, and Dna2 (in association with BLM) to effect fork repair/restart via HR [64, 152]. Unlike DSBs, DSEs have no proximal end with which to join via NHEJ, thus if DSEs were to be subject to repair by NHEJ the outcome would be large-scale structural changes to the genome (deletions and translocations). This explains why HR is of utmost importance in replication fork restart: the alternatives are simply too risky. It also explains why severe HR defects, such as RAD51 null mutations, are cell lethal and cause embryonic lethality in mice [153, 154].
5. Radiation-induced delayed HR
As discussed in Section 1, IR induces both immediate and delayed effects, including mutations, chromosomal aberrations, homologous recombination, and cell death. Between 1996–2003 the Morgan laboratory led the field in defining mechanistic aspects of radiation-induced delayed chromosomal instability, offering insights into the types of DNA damage capable of inducing the effect; chromosomal structures involved (e.g., telomeres); its relationship to other delayed effects such as mutation and cell death; dose/dose rate, and radiation quality effects; the relevant target and target size; and early studies demonstrating that whole genome transcription profiles were similar in stable vs. chromosomally unstable cells [155–168]. This impressive body of work was a direct precursor to collaborative studies between the Morgan and Nickoloff laboratories, initiated in the early 2000s and focused on a different type of radiation-induced delayed genomic instability, namely delayed homologous recombination (DHR; also termed delayed hyper-recombination). In particular, we were interested in whether IR induced DHR and if so, whether DHR correlated with other delayed effects such as DCI and cell death [169]. To address these questions we constructed derivatives of RKO (human colon carcinoma) and GM10115 (hamster cells with a single human chromosome), each carrying a single integrated copy of a GFP direct repeat HR substrate. RKO cells were chosen because they show normal p53 and p21 induction by IR and have a near-diploid chromosome complement, and GM10115 cells were chosen because they are p53 mutant and had been extensively used in past DCI studies. In each case, parent cells are GFP− because neither of the two GFP genes were functional, but HR can convert these to GFP+ (Fig. 4A). We found that X-ray doses from 1–10 Gy induced immediate HR, evidenced as fully GFP+ colonies, as well as mixed GFP+/− colonies which by definition were evidence of DHR (Fig. 4B). Surprisingly, DHR was induced at very high frequencies: nearly 10% of surviving cells displayed this phenotype (Fig. 4C). DCI was monitored in the same exposed cell population, and consistent with prior studies, DCI was also induced at frequencies approaching 10% and was correlated with delayed death. Interestingly, cells that expressed DHR did not display DCI, and vice versa. Moreover, cells that expressed DHR showed no evidence of delayed death. Thus, although DCI and DHR and both induced by IR at similar, high frequencies, DCI and DHR are mechanistically distinct [169]. Dysregulation of HR is observed as hypo-HR, reflecting defects in HR proteins [170–177], or as hyper-HR, reflecting defects in HR regulatory systems such as in BLM-mutant cells [98, 178, 179]. Hyper-HR can also more generally reflect DNA repair defects or metabolic disorders that increase ROS, which increase HR associated with replication stress due to more frequent encounters of replication forks with DNA lesions [2, 3, 34]. Genome instability is associated with both hypo- and hyper-HR, thus, it is important for cells to maintain appropriate levels of HR in order to maintain genome stability. The relatively high efficiency by which radiation induces DHR suggests that DHR poses significant risks to genome stability at late times after low- to moderate-doses of IR. This risk is further exacerbated by the fact that the DHR phenotype correlates with high viability, as opposed to DCI, in which cells are often sick and prone to delayed death [169].
Fig. 4.
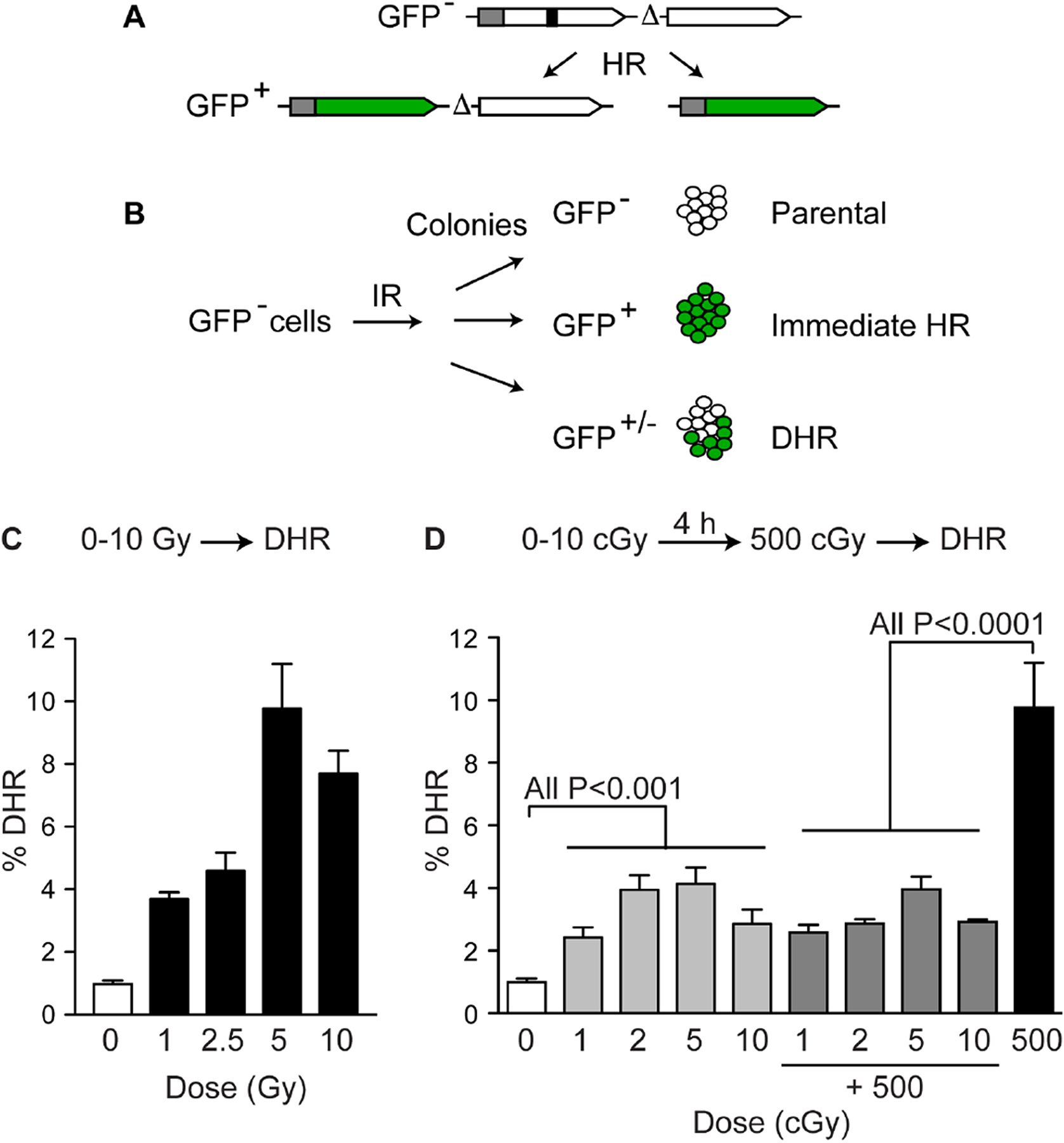
IR-induced DHR. (A) Cells carry a single copy of a direct repeat HR substrate with two inactive copies of GFP (GFP−). HR creates GFP+ products that retain both copies or delete one of the copies via crossover or single-strand annealing. (B) GFP− cells treated with IR produce colonies that are GFP− (parental, non-recombinant), fully GFP+ (immediate HR (C) Moderate to high doses of X-rays stimulate DHR at high frequencies. (D) Low (cGy) doses of X-rays stimulate DHR at high frequencies and induce an adaptive response to a later challenge dose of 500 cGy. Data compiled from refs. [169, 185].
The next question was whether DHR is induced by non-ionizing ultraviolet light (UV) radiation. The RKO-GFP cells were exposed to either UV-B, or the shorter wavelength and more damaging/more lethal UV-C and DHR was again scored as mixed GFP+/− colonies. As with IR, moderate doses of UV-C (5 J/m2, yielding ~20% survival) induced DHR in ~15% of cells; in contrast, no induction of DHR was observed with equitoxic doses of UV-B, or higher UV-B doses [180]. Thus, both IR and non-ionizing radiation induce DHR at very high frequencies, and in both cases, the DHR phenotype was not associated with delayed death [180].
As noted above, IR triggers several delayed effects including DCI, DHR, mutation, and cell death. RKO cells are derived from a male donor and carry a single, functional X-linked HPRT gene that allows facile detection of hprt mutants. As expected, hprt mutant frequencies were low in parental (non-exposed) cells. Mutant frequencies increased several-fold among UV-C survivors in which HR was induced immediately (full GFP+ colonies), but there was a dramatic, ~100-fold increase in hprt mutations among all DHR (GFP+/−) colonies tested (n=9). Note that all of the hprt mutations scored in this study arose after UV-C (or mock) exposure, hence the modest increase in mutagenesis in immediate HR cells, and the dramatic increase in DHR cells, all reflect delayed mutation. These results indicate that UV-C induced DHR and delayed mutation are strongly correlated and mostly likely stem from the same source. Sequencing of individual hprt mutant genes yielded yet another striking result. The most common types of spontaneous mutation in HPRT are individual point mutations comprising single-based changes or short frameshift mutations, with less frequent large-scale deletions [181, 182], although mutation spectra vary depending on mutator genotype among cancer cell lines [183]. Interestingly, the spectrum of delayed hprt mutations arising in immediate HR cells were typical, largely comprising individual point mutations or deletions, yet among 7 delayed hprt mutants arising in DHR cells, 5 had compound point mutations, with three displaying 5 or more mutations [180]. The specific types of delayed mutations arising in DHR cells were indicative of mutagenesis resulting oxidative DNA damage, providing a likely link between the UV-C induced DHR and delayed mutation phenotypes.
Having established that moderate doses of IR and non-ionizing radiation efficiently induce DHR, the next question was whether DHR was induced by low doses of IR, a topic relevant to risk assessment for low dose medical (e.g., diagnostic) exposures. Three key observations were made in this study. First, DHR was indeed stimulated by low dose IR, with significant increases seen with doses as low as 1 cGy (Fig. 4D). This is in the upper range for CT scans [184]. The fact that DHR is induced at high frequencies with extremely low IR doses indicates that the target is very large, either the nucleus or the entire cell, or perhaps that DHR may actually be a non-targeted (bystander) effect. Second, low dose exposures of several cGy showed roughly the same induction of DHR as doses 100-fold higher (compare Fig. 4C and 4D). Although the results did suggest that DHR increased with dose, the dose response was highly non-linear, raising the possibility of a threshold effect. Finally, low dose IR exposures of 1–5 cGy suppressed the greater induction of DHR by a 500 cGy dose delivered 4 h later, relative to a single 500 cGy dose, providing evidence that IR-induced DHR is subject to an adaptive response (Fig. 4D) [185]. Interestingly, these RKO-GFP cells did not show an adaptive response with respect to cell killing, that is, low dose pre-exposures did not reduce the cytotoxic effect of the subsequent challenge dose [185]. Clearly DHR and cell death are independently regulated, and it is also likely that these endpoints will differentially respond to modifications to the DDR, i.e., with PIK inhibitors commonly used as radiosensitizing agents.
Despite the fact that IR-induced DCI is associated with delayed cell death, DCI can persist for many years [166]. We recently undertook a project to determine whether DHR is similarly induced by low LET X-rays and high LET carbon ion radiation, and the length of time that DHR persisted in each case. As with DHR induced by X-rays, DHR was induced to high frequencies by carbon ions, and both showed atypical dose responses, although very low dose carbon ion exposures have not yet been tested. Importantly, DHR was found to persist for 2 weeks before resolving to background levels by the third week after irradiation, and this was true for both X-ray and carbon ion exposures (Allen, Hirakawa, Nakajima, Moore, Nie, Sugiura, Hoki, Araki, Abe, Okayasu, Fujimori, and Nickoloff, manuscript in preparation). These results indicate that if there is indeed a risk of genome instability and/or cancer associated with DHR, this risk appears more time-limited than that from DCI, and that there is no significant difference in risks associated with low vs. high LET exposures.
6. Future perspectives
Our knowledge of DNA repair and DDR networks has greatly expanded in recent years, yet as we tease out the details through traditional reductionist approaches, the “omics” revolution is providing new insights into the vast complexity of these critical cellular processes. DNA repair and DDR signaling systems are capable of sensing and processing a wide range of lesion types caused by different types of endogenous and exogenous genotoxic agents, and while it is clear that these response systems are fundamental to triggering the various cell fates of stressed cells, the astonishing genetic heterogeneity of tumors poses significant challenges to harnessing our knowledge to create better targeted and more effective, and safe, cancer therapies. Genome instability takes many forms, and often reflects defects in DNA repair and/or DDR signaling pathways. It has long been known that cancer cells harbor unstable genomes, but for many years it was unclear whether instability was a cause or an effect of the tumorigenic phenotype. It has been argued that genome instability is an “enabling characteristic” of cancer [186], and it is clear that instability can precede neoplastic transformation [187–190]. Delayed mutation, DCI, and DHR are still relatively poorly understood mechanistically, but the fact that moderate radiation doses, and in some cases very low doses, trigger these types of instability is certainly a concern.
Many questions remain in the delayed genome instability field, including the most basic question: what is the ultimate trigger? While it is clear that the target is as large as the nucleus and may be larger [166, 191, 192], the persistence of delayed effects may reflect persistent high levels of ROS [193, 194], at least in part reflecting mitochondrial dysfunction [195, 196]. This suggested paths to mitigate these effects through radical scavengers, as shown for IR-induced DCI [197], but parallel studies with DHR have not been performed. Moreover, there is little to no information about how modulating DNA repair or the DDR might influence, positively or negatively, various delayed genome instability phenotypes. Such information is critical as there is growing interest in targeting DNA repair and/or DDR proteins as mono-therapy to treat cancer, or as adjuncts to chemo- or radiotherapy [9, 10, 35, 91, 120, 124, 198–204].
Finally, a understanding the fundamentals that underlie IR-induced delayed genome instability, particularly those induced with high efficiency at low doses, is important with regard to radiation risk assessment. This is particularly true for low dose medical exposures during diagnostic imaging given the frequent use of such procedures. Among cancer patients there are also potential risks associated with genome instability triggered by moderate to high radiation doses delivered to normal and cancer tissues during radiotherapy, including progression of rare surviving tumor cells to a more aggressive state, and secondary cancer induction. Whether chronic low dose (e.g., environmental) exposures pose similar or different risks of delayed effects is poorly understood, but should be explored and factored into risk assessments [205–211]. Dr. Bill Morgan had much to say about these and many other topics in the radiobiology and radiation risk assessment spheres. We would do well to heed his messages as we drive these fields forward.
Acknowledgements
I thank Bill Morgan for his constant encouragement over several decades, and the many members of my laboratory for their countless contributions.
Funding
Research in the Nickoloff lab is supported by the National Institutes of Health [grant number R01 GM084020].
References
- [1].Costes A, Lambert SA, Homologous recombination as a replication fork escort: fork-protection and recovery, Biomolecules, 3 (2012) 39–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Carr AM, Lambert S, Replication stress-induced genome instability: the dark side of replication maintenance by homologous recombination, J. Mol. Biol, 425 (2013) 4733–4744. [DOI] [PubMed] [Google Scholar]
- [3].Zeman MK, Cimprich KA, Causes and consequences of replication stress, Nat. Cell Biol, 16 (2014) 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Loucas BD, Cornforth MN, Complex chromosome exchanges induced by gamma rays in human lymphocytes: an mFISH study, Radiat. Res, 155 (2001) 660–671. [DOI] [PubMed] [Google Scholar]
- [5].Albertson DG, Gene amplification in cancer, Trends Genet, 22 (2006) 447–455. [DOI] [PubMed] [Google Scholar]
- [6].Matsui A, Ihara T, Suda H, Mikami H, Semba K, Gene amplification: mechanisms and involvement in cancer, Biomol. Concepts, 4 (2013) 567–582. [DOI] [PubMed] [Google Scholar]
- [7].Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ, ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage, Science, 316 (2007) 1160–1166. [DOI] [PubMed] [Google Scholar]
- [8].Ciccia A, Elledge SJ, The DNA damage response: making it safe to play with knives, Mol. Cell, 40 (2010) 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].O’Connor MJ, Targeting the DNA damage response in cancer, Mol. Cell, 60 (2015) 547–560. [DOI] [PubMed] [Google Scholar]
- [10].Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FM, Therapeutic opportunities within the DNA damage response, Nat. Rev. Cancer, 15 (2015) 166–180. [DOI] [PubMed] [Google Scholar]
- [11].Jeggo PA, Pearl LH, Carr AM, DNA repair, genome stability and cancer: a historical perspective, Nat. Rev. Cancer, 16 (2016) 35–42. [DOI] [PubMed] [Google Scholar]
- [12].Mladenov E, Magin S, Soni A, Iliakis G, DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation, Semin. Cancer Biol, 37–38 (2016) 51–64. [DOI] [PubMed] [Google Scholar]
- [13].Pellegrino S, Altmeyer M, Interplay between Ubiquitin, SUMO, and Poly(ADP-Ribose) in the Cellular Response to Genotoxic Stress, Front. Genet, 7 (2016) 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wei H, Yu X, Functions of PARylation in DNA damage repair pathways, Genomics Proteomics Bioinformatics, 14 (2016) 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Finn K, Lowndes NF, Grenon M, Eukaryotic DNA damage checkpoint activation in response to double-strand breaks, Cell. Mol. Life Sci, 69 (2012) 1447–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Guenole A, Srivas R, Vreeken K, Wang ZZ, Wang S, Krogan NJ, Ideker T, van Attikum H, Dissection of DNA damage responses using multiconditional genetic interaction maps, Mol. Cell, 49 (2013) 346–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Stephan JS, Herman PK, The regulation of autophagy in eukaryotic cells: do all roads pass through Atg1?, Autophagy, 2 (2006) 146–148. [DOI] [PubMed] [Google Scholar]
- [18].Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G, Molecular mechanisms of necroptosis: an ordered cellular explosion, Nat. Rev. Mol. Cell Biol, 11 (2010) 700–714. [DOI] [PubMed] [Google Scholar]
- [19].Galluzzi L, Kroemer G, Necroptosis: a specialized pathway of programmed necrosis, Cell, 135 (2008) 1161–1163. [DOI] [PubMed] [Google Scholar]
- [20].Eriksson D, Stigbrand T, Radiation-induced cell death mechanisms, Tumour Biol, 31 (2010) 363–372. [DOI] [PubMed] [Google Scholar]
- [21].Roos WP, Kaina B, DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis, Cancer letters, 332 (2013) 237–248. [DOI] [PubMed] [Google Scholar]
- [22].Meyn RE, Milas L, Ang KK, The role of apoptosis in radiation oncology, Int. J. Radiat. Biol, 85 (2009) 107–115. [DOI] [PubMed] [Google Scholar]
- [23].Bora KC, Delayed effects in chromosome breakage by X rays, Br. J. Radiol, 27 (1954) 124–127. [DOI] [PubMed] [Google Scholar]
- [24].Rugh R, The immediate and delayed morphological effects of x-radiations on meiotic chromosomes, J. Cell. Physiol, 36 (1950) 185–203. [DOI] [PubMed] [Google Scholar]
- [25].Muller HJ, Artificial transmutation of the gene, Science, 66 (1927) 84–87. [DOI] [PubMed] [Google Scholar]
- [26].Puck TT, Marcus PI, Action of x-rays on mammalian cells, J. Exp. Med, 103 (1956) 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Elkind MM, Han A, Volz KW, Radiation response of mammalian cells grown in culture. IV. Dose dependence of division delay and postirradiation growth of surviving and nonsurviving Chinese hamster cells, J. Natl. Cancer Inst, 30 (1963) 705–721. [Google Scholar]
- [28].Thompson LH, Suit HD, Proliferation kinetics of x-irradiated mouse L cells studied with time-lapse photography, Int. J. Radiat. Biol, 15 (1969) 347–362. [DOI] [PubMed] [Google Scholar]
- [29].Chang WP, Little JB, Delayed reproductive death as a dominant phenotype in cell clones surviving X-irradiation, Carcinogenesis, 13 (1992) 923–928. [DOI] [PubMed] [Google Scholar]
- [30].Little JB, Gorgojo L, Vetrovs H, Delayed appearance of lethal and specific gene mutations in irradiated mammalian cells, Int. J. Radiat. Oncol. Biol. Phys, 19 (1990) 1425–1429. [DOI] [PubMed] [Google Scholar]
- [31].Marder BA, Morgan WF, Delayed chromosomal instability induced by DNA damage, Mol. Cell. Biol, 13 (1993) 6667–6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lord CJ, Ashworth A, The DNA damage response and cancer therapy, Nature, 481 (2012) 287–294. [DOI] [PubMed] [Google Scholar]
- [33].Reinhardt HC, Yaffe MB, Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2, Curr. Opin. Cell Biol, 21 (2009) 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Allen C, Ashley AK, Hromas R, Nickoloff JA, More forks on the road to replication stress recovery, J. Mol. Cell. Biol, 3 (2011) 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shaheen M, Allen C, Nickoloff JA, Hromas R, Synthetic lethality: exploiting the addiction of cancer to DNA repair, Blood, 117 (2011) 6074–6082. [DOI] [PubMed] [Google Scholar]
- [36].Putnam CD, Jaehnig EJ, Kolodner RD, Perspectives on the DNA damage and replication checkpoint responses in Saccharomyces cerevisiae, DNA Repair, 8 (2009) 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Peng A, Lewellyn AL, Schiemann WP, Maller JL, Repo-man controls a protein phosphatase 1-dependent threshold for DNA damage checkpoint activation, Curr. Biol, 20 (2010) 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fernet M, Megnin-Chanet F, Hall J, Favaudon V, Control of the G2/M checkpoints after exposure to low doses of ionising radiation: implications for hyper-radiosensitivity, DNA Repair, 9 (2010) 48–57. [DOI] [PubMed] [Google Scholar]
- [39].Branzei D, Foiani M, The checkpoint response to replication stress, DNA Repair, 8 (2009) 1038–1046. [DOI] [PubMed] [Google Scholar]
- [40].Budzowska M, Kanaar R, Mechanisms of dealing with DNA damage-induced replication problems, Cell Biochem. Biophys, 53 (2009) 17–31. [DOI] [PubMed] [Google Scholar]
- [41].Vakifahmetoglu H, Olsson M, Zhivotovsky B, Death through a tragedy: mitotic catastrophe, Cell Death Differ, 15 (2008) 1153–1162. [DOI] [PubMed] [Google Scholar]
- [42].Kastan MB, Lim DS, Kim ST, Yang D, ATM--a key determinant of multiple cellular responses to irradiation, Acta Oncol, 40 (2001) 686–688. [DOI] [PubMed] [Google Scholar]
- [43].Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB, Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway, Genes Dev, 18 (2004) 1423–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Baskaran R, Wood LD, Whitaker LL, Canman CE, Morgan SE, Xu Y, Barlow C, Baltimore D, Wynshaw-Boris A, Kastan MB, Wang JY, Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation, Nature, 387 (1997) 516–519. [DOI] [PubMed] [Google Scholar]
- [45].Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ, ATM phosphorylates histone H2AX in response to DNA double-strand breaks, J. Biol. Chem, 276 (2001) 42462–42467. [DOI] [PubMed] [Google Scholar]
- [46].Chen BP, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, Yajima H, Lobrich M, Shiloh Y, Chen DJ, Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break, J. Biol. Chem, 282 (2007) 6582–6587. [DOI] [PubMed] [Google Scholar]
- [47].Shrivastav M, Miller CA, De Haro LP, Durant ST, Chen BP, Chen DJ, Nickoloff JA, DNA-PKcs and ATM co-regulate DNA double-strand break repair, DNA Repair, 8 (2009) 920–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yajima H, Lee K-J, Chen BPC, ATR-dependent DNA-PKcs phosphorylation in response to UV-induced replication stress Mol. Cell. Biol, 26 (2006) 7520–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y, Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes, EMBO J, 18 (1999) 1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang H, Guan J, Perrault AR, Wang Y, Iliakis G, Replication protein A2 phosphorylation after DNA damage by the coordinated action of ataxia telangiectasia-mutated and DNA-dependent protein kinase, Cancer Res, 61 (2001) 8554–8563. [PubMed] [Google Scholar]
- [51].Anantha RW, Vassin VM, Borowiec JA, Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair, J. Biol. Chem, 282 (2007) 35910–35923. [DOI] [PubMed] [Google Scholar]
- [52].Block WD, Yu Y, Lees-Miller SP, Phosphatidyl inositol 3-kinase-like serine/threonine protein kinases (PIKKs) are required for DNA damage-induced phosphorylation of the 32 kDa subunit of replication protein A at threonine 21, Nucleic Acids Res, 32 (2004) 997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cruet-Hennequart S, Glynn MT, Murillo LS, Coyne S, Carty MP, Enhanced DNA-PK-mediated RPA2 hyperphosphorylation in DNA polymerase eta-deficient human cells treated with cisplatin and oxaliplatin, DNA Repair, 7 (2008) 582–596. [DOI] [PubMed] [Google Scholar]
- [54].Cruet-Hennequart S, Coyne S, Glynn MT, Oakley GG, Carty MP, UV-induced RPA phosphorylation is increased in the absence of DNA polymerase eta and requires DNA-PK, DNA Repair, 5 (2006) 491–504. [DOI] [PubMed] [Google Scholar]
- [55].Stephan H, Concannon C, Kremmer E, Carty MP, Nasheuer HP, Ionizing radiation-dependent and independent phosphorylation of the 32-kDa subunit of replication protein A during mitosis, Nucleic Acids Res, 37 (2009) 6028–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liaw H, Lee D, Myung K, DNA-PK-dependent RPA2 hyperphosphorylation facilitates DNA repair and suppresses sister chromatid exchange, PLoS ONE, 6 (2011) e21424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Liu S, Opiyo SO, Manthey K, Glanzer JG, Ashley AK, Troksa K, Shrivastav M, Nickoloff JA, Oakley GG, Distinct roles for DNA-PK, ATM, and ATR in RPA phosphorylation and checkpoint activation in response to replication stress, Nucleic Acids Res, 40 (2012) 10780–10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ashley AK, Shrivastav M, Nie J, Amerin C, Troksa K, Glanzer JG, Liu S, Opiyo SO, Dimitrova DD, Le P, Sishc B, Bailey SM, Oakley GG, Nickoloff JA, DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe, DNA Repair, 21 (2014) 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Arnaudeau C, Lundin C, Helleday T, DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells, J. Mol. Biol, 307 (2001) 1235–1245. [DOI] [PubMed] [Google Scholar]
- [60].Lundin C, Erixon K, Arnaudeau C, Schultz N, Jenssen D, Meuth M, Helleday T, Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells, Mol. Cell. Biol, 22 (2002) 5869–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Petermann E, Helleday T, Pathways of mammalian replication fork restart, Nat. Rev. Mol. Cell Biol, 11 (2010) 683–687. [DOI] [PubMed] [Google Scholar]
- [62].Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, Cejka P, Stewart S, Lopes M, Vindigni A, DNA2 drives processing and restart of reversed replication forks in human cells, J. Cell Biol, 208 (2015) 545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Symington LS, Gautier J, Double-strand break end resection and repair pathway choice, Annu. Rev. Genet, 45 (2011) 247–271. [DOI] [PubMed] [Google Scholar]
- [64].Rass U, Resolving branched DNA intermediates with structure-specific nucleases during replication in eukaryotes, Chromosoma, 122 (2013) 499–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Schwartz EK, Heyer WD, Processing of joint molecule intermediates by structure-selective endonucleases during homologous recombination in eukaryotes, Chromosoma, 120 (2011) 109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].De Haro LP, Wray J, Williamson EA, Durant ST, Corwin L, Gentry AC, Osheroff N, Lee SH, Hromas R, Nickoloff JA, Metnase promotes restart and repair of stalled and collapsed replication forks, Nucleic Acids Res, 38 (2010) 5681–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kim H-S, Chen Q, Kim S-K, Nickoloff JA, Hromas R, Georgiadis MM, Lee S-K, The DDN catalytic motif is required for Metnase functions in NHEJ repair and replication restart, J. Biol. Chem, 289 (2014) 10930–10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wu Y, Lee SH, Williamson EA, Reinert BL, Cho JH, Xia F, Jaiswal AS, Srinivasan G, Patel B, Brantley A, Zhou D, Shao L, Pathak R, Hauer-Jensen M, Singh S, Kong K, Wu X, Kim HS, Beissbarth T, Gaedcke J, Burma S, Nickoloff JA, Hromas RA, EEPD1 rescues stressed replication forks and maintains genome stability by promoting end resection and homologous recombination repair, PLoS Genet., 11 (2015) e1005675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chapman JR, Taylor MR, Boulton SJ, Playing the end game: DNA double-strand break repair pathway choice, Mol. Cell, 47 (2012) 497–510. [DOI] [PubMed] [Google Scholar]
- [70].Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, Razavian N, Berns MW, Wu X, Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells, Proc. Natl. Acad. Sci. USA, 110 (2013) 7720–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC, BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair, Genes Dev, 25 (2011) 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhu Z, Chung WH, Shim EY, Lee SE, Ira G, Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends, Cell, 134 (2008) 981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kakarougkas A, Jeggo PA, DNA DSB repair pathway choice: an orchestrated handover mechanism, Br. J. Radiol, 87 (2014) 20130685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Feng L, Fong KW, Wang J, Wang W, Chen J, RIF1 counteracts BRCA1-mediated end resection during DNA repair, J. Biol. Chem, 288 (2013) 11135–11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T, 53BP1 regulates DSB repair using Rif1 to control 5’ end resection, Science, 339 (2013) 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Nam EA, Cortez D, ATR signalling: more than meeting at the fork, Biochem. J, 436 (2011) 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Shiotani B, Zou L, ATR signaling at a glance, J. Cell Sci, 122 (2009) 301–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Awasthi P, Foiani M, Kumar A, ATM and ATR signaling at a glance, J. Cell Sci, 128 (2015) 4255–4262. [DOI] [PubMed] [Google Scholar]
- [79].Shiotani B, Nguyen HD, Hakansson P, Marechal A, Tse A, Tahara H, Zou L, Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1, Cell Rep, 3 (2013) 1651–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Sørensen CS, Hansen LT, Dziegielewski J, Syljuåsen RG, Lundin C, Bartek J, Helleday T, The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair, Nat. Cell Biol, 7 (2005) 195–201. [DOI] [PubMed] [Google Scholar]
- [81].Bashkirov VI, King JS, Bashkirova EV, Schmuckli-Maurer J, Heyer WD, DNA repair protein Rad55 is a terminal substrate of the DNA damage checkpoints, Mol. Cell. Biol, 20 (2000) 4393–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Herzberg K, Bashkirov VI, Rolfsmeier M, Haghnazari E, McDonald WH, Anderson S, Bashkirova EV, Yates JR 3rd, Heyer WD, Phosphorylation of Rad55 on serines 2, 8, and 14 is required for efficient homologous recombination in the recovery of stalled replication forks, Mol. Cell. Biol, 26 (2006) 8396–8409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Krejci L, Altmannova V, Spirek M, Zhao X, Homologous recombination and its regulation, Nucleic Acids Res, 40 (2012) 5795–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Goudelock DM, Jiang K, Pereira E, Russell B, Sanchez Y, Regulatory interactions between the checkpoint kinase Chk1 and the proteins of the DNA-dependent protein kinase complex, J. Biol. Chem, 278 (2003) 29940–29947. [DOI] [PubMed] [Google Scholar]
- [85].Sengupta S, Robles AI, Linke SP, Sinogeeva NI, Zhang R, Pedeux R, Ward IM, Celeste A, Nussenzweig A, Chen J, Halazonetis TD, Harris CC, Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest, J. Cell Biol, 166 (2004) 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Hromas R, Williamson E, Fnu S, Lee Y-J, Park S-J, Beck BD, You J-S, Laitao A, Nickoloff JA, Lee S-H, Chk1 phosphorylation of Metnase enhances DNA repair but inhibits replication fork restart, Oncogene, 31 (2012) 4245–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ward IM, Chen J, Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress, J. Biol. Chem, 276 (2001) 47759–47762. [DOI] [PubMed] [Google Scholar]
- [88].Downey M, Durocher D, gH2AX as a checkpoint maintenance signal, Cell Cycle, 5 (2006) 1376–1381. [DOI] [PubMed] [Google Scholar]
- [89].Tapia-Alveal C, Calonge TM, O’Connell MJ, Regulation of Chk1, Cell Div, 4 (2009) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Merry C, Fu K, Wang J, Yeh IJ, Zhang Y, Targeting the checkpoint kinase Chk1 in cancer therapy, Cell Cycle, 9 (2010) 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Smith J, Tho LM, Xu N, Gillespie DA, The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer, Adv. Cancer Res, 108 (2010) 73–112. [DOI] [PubMed] [Google Scholar]
- [92].Kemp MG, Akan Z, Yilmaz S, Grillo M, Smith-Roe SL, Kang TH, Cordeiro-Stone M, Kaufmann WK, Abraham RT, Sancar A, Unsal-Kacmaz K, Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress, J. Biol. Chem, 285 (2010) 16562–16571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Begg AC, Stewart FA, Vens C, Strategies to improve radiotherapy with targeted drugs, Nat. Rev. Cancer, 11 (2011) 239–253. [DOI] [PubMed] [Google Scholar]
- [94].Dai Y, Grant S, New insights into checkpoint kinase 1 in the DNA damage response signaling network, Clin. Cancer Res, 16 (2010) 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Chanoux RA, Yin B, Urtishak KA, Asare A, Bassing CH, Brown EJ, ATR and H2AX cooperate in maintaining genome stability under replication stress, J. Biol. Chem, 284 (2008) 5994–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zou Y, Liu Y, Wu X, Shell SM, Functions of human replication protein A (RPA): from DNA replication to DNA damage and stress responses, J. Cell. Physiol, 208 (2006) 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Shimada K, Oma Y, Schleker T, Kugou K, Ohta K, Harata M, Gasser SM, Ino80 chromatin remodeling complex promotes recovery of stalled replication forks, Curr. Biol, 18 (2008) 566–575. [DOI] [PubMed] [Google Scholar]
- [98].Davies SL, North PS, Hickson ID, Role for BLM in replication-fork restart and suppression of origin firing after replicative stress, Nat. Struct. Mol. Biol, 14 (2007) 677–679. [DOI] [PubMed] [Google Scholar]
- [99].Moldovan GL, Pfander B, Jentsch S, PCNA, the maestro of the replication fork, Cell, 129 (2007) 665–679. [DOI] [PubMed] [Google Scholar]
- [100].Niimi A, Brown S, Sabbioneda S, Kannouche PL, Scott A, Yasui A, Green CM, Lehmann AR, Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells, Proc. Natl. Acad. Sci. USA, 105 (2008) 16125–16130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Sale JE, Translesion DNA synthesis and mutagenesis in eukaryotes, Cold Spring Harb. Perspect. Biol, 5 (2013) a012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Wray J, Liu J, Nickoloff JA, Shen Z, Distinct RAD51 associations with RAD52 and BCCIP in response to DNA damage and replication stress, Cancer Res, 68 (2008) 2699–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Bitomsky N, Hofmann TG, Apoptosis and autophagy: Regulation of apoptosis by DNA damage signalling - roles of p53, p73 and HIPK2, FEBS J., 276 (2009) 6074–6083. [DOI] [PubMed] [Google Scholar]
- [104].Gozuacik D, Kimchi A, Autophagy as a cell death and tumor suppressor mechanism, Oncogene, 23 (2004) 2891–2906. [DOI] [PubMed] [Google Scholar]
- [105].Avalos Y, Canales J, Bravo-Sagua R, Criollo A, Lavandero S, Quest AF, Tumor suppression and promotion by autophagy, Biomed. Res. Int, 2014 (2014) 603980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Meng MB, Wang HH, Cui YL, Wu ZQ, Shi YY, Zaorsky NG, Deng L, Yuan ZY, Lu Y, Wang P, Necroptosis in tumorigenesis, activation of anti-tumor immunity, and cancer therapy, Oncotarget, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Martinez MM, Reif RD, Pappas D, Detection of apoptosis: A review of conventional and novel techniques, Anal. Methods, 2 (2010) 996–1004. [Google Scholar]
- [108].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T, LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing, EMBO J, 19 (2000) 5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T, ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation, Cell Death Differ, 14 (2007) 230–239. [DOI] [PubMed] [Google Scholar]
- [110].Steer SA, Scarim AL, Chambers KT, Corbett JA, Interleukin-1 stimulates beta-cell necrosis and release of the immunological adjuvant HMGB1, PLoS Med, 3 (2006) e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Thorburn J, Horita H, Redzic J, Hansen K, Frankel AE, Thorburn A, Autophagy regulates selective HMGB1 release in tumor cells that are destined to die, Cell Death Differ, 16 (2009) 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Gewirtz DA, Holt SE, Elmore LW, Accelerated senescence: an emerging role in tumor cell response to chemotherapy and radiation, Biochem. Pharmacol, 76 (2008) 947–957. [DOI] [PubMed] [Google Scholar]
- [113].Stracker TH, Usui T, Petrini JH, Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response, DNA Repair, 8 (2009) 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Mori E, Takahashi A, Yamakawa N, Kirita T, Ohnishi T, High LET heavy ion radiation induces p53-independent apoptosis, J. Radiat. Res, 50 (2009) 37–42. [DOI] [PubMed] [Google Scholar]
- [115].Huang Q, Li F, Liu X, Li W, Shi W, Liu F-F, O’Sullivan B, He Z, Peng Y, Tan A-C, Zhou L, Shen J, Han G, Wang X-J, Thorburn J, Thorburn A, Jimeno A, Raben D, Bedford JS, Li C-Y, Caspase-mediated paracrine signaling from dying cells potently stimulate tumor cell repopulation during cancer radiotherapy, Nat. Med, 17 (2011) 860–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Yi CH, Yuan J, The Jekyll and Hyde functions of caspases, Dev. Cell, 16 (2009) 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS, Li CY, Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration, Sci. Signal, 3 (2010) ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Allen CP, Tinganelli W, Sharma N, Nie J, Sicard C, Natale F, King M 3rd, Keysar SB, Jimeno A, Furusawa Y, Okayasu R, Fujimori A, Durante M, Nickoloff JA, DNA damage response proteins and oxygen modulate prostaglandin E2 growth factor release in response to low and high LET ionizing radiation, Front. Oncol, 5 (2015) 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Wallace SS, Murphy DL, Sweasy JB, Base excision repair and cancer, Cancer letters, 327 (2012) 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Li SK, Martin A, Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored, Trends Mol. Med, 22 (2016) 274–289. [DOI] [PubMed] [Google Scholar]
- [121].Corneo B, Wendland RL, Deriano L, Cui X, Klein IA, Wong SY, Arnal S, Holub AJ, Weller GR, Pancake BA, Shah S, Brandt VL, Meek K, Roth DB, Rag mutations reveal robust alternative end joining, Nature, 449 (2007) 483–486. [DOI] [PubMed] [Google Scholar]
- [122].Deriano L, Roth DB, Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage, Annu. Rev. Genet, 47 (2013) 433–455. [DOI] [PubMed] [Google Scholar]
- [123].Nussenzweig A, Nussenzweig MC, A backup DNA repair pathway moves to the forefront, Cell, 131 (2007) 223–225. [DOI] [PubMed] [Google Scholar]
- [124].Palmbos PL, Hussain MH, Targeting PARP in prostate cancer: novelty, pitfalls, and promise, Oncology, 30 (2016) 377–385. [PubMed] [Google Scholar]
- [125].Simsek D, Jasin M, Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation, Nat. Struct. Mol. Biol, 17 (2010) 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Weinstock DM, Richardson CA, Elliott B, Jasin M, Modeling oncogenic translocations: distinct roles for double-strand break repair pathways in translocation formation in mammalian cells, DNA Repair, 5 (2006) 1065–1074. [DOI] [PubMed] [Google Scholar]
- [127].Weinstock DM, Brunet E, Jasin M, Formation of NHEJ-derived reciprocal chromosomal translocations does not require Ku70, Nat. Cell Biol, 9 (2007) 978–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Bunting SF, Nussenzweig A, End-joining, translocations and cancer, Nat. Rev. Cancer, 13 (2013) 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Wray J, Williamson EA, Farrington J, Chester S, Kwan L, Weinstock D, Jasin M, Lee S-H, Nickoloff JA, Hromas R, The transposase domain protein Metnase/SETMAR suppresses chromosomal translocations, Cancer Genet. Cytogenet, 200 (2010) 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC, Homology and enzymatic requirements of microhomology-dependent alternative end joining, Cell Death Dis, 6 (2015) e1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Wray J, Williamson EA, Singh SB, Wu Y, Cogle CR, Weinstock DM, Zhang Y, Lee SH, Zhou D, Shao L, Hauer-Jensen M, Pathak R, Klimek V, Nickoloff JA, Hromas R, PARP1 is required for chromosomal translocations, Blood, 121 (2013) 4359–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD, Repetitive elements may comprise over two-thirds of the human genome, PLoS Genet., 7 (2011) e1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Nickoloff JA, Recombination: mechanisms and roles in tumorigenesis, in: Bertino JR (Ed.) Encyclopedia of Cancer, Second Edition, Elsevier Science (USA), San Diego, 2002, pp. 49–59. [Google Scholar]
- [134].Abeysinghe SS, Chuzhanova N, Krawczak M, Ball EV, Cooper DN, Translocation and gross deletion breakpoints in human inherited disease and cancer I: Nucleotide composition and recombination-associated motifs, Hum. Mutat, 22 (2003) 229–244. [DOI] [PubMed] [Google Scholar]
- [135].Elliott B, Richardson C, Jasin M, Chromosomal translocation mechanisms at intronic alu elements in mammalian cells, Mol. Cell, 17 (2005) 885–894. [DOI] [PubMed] [Google Scholar]
- [136].Kolomietz E, Meyn MS, Pandita A, Squire JA, The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors, Genes Chromosomes Cancer, 35 (2002) 97–112. [DOI] [PubMed] [Google Scholar]
- [137].Lo Y-C, Paffett KS, Amit O, Clikeman JA, Sterk R, Brenneman MA, Nickoloff JA, Sgs1 regulates gene conversion tract lengths and crossovers independently of its helicase activity, Mol. Cell. Biol, 26 (2006) 4086–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Bussen W, Raynard S, Busygina V, Singh AK, Sung P, Holliday junction processing activity of the BLM-Topo IIIalpha-BLAP75 complex, J. Biol. Chem, 282 (2007) 31484–31492. [DOI] [PubMed] [Google Scholar]
- [139].Raynard S, Bussen W, Sung P, A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIalpha, and BLAP75, J. Biol. Chem, 281 (2006) 13861–13864. [DOI] [PubMed] [Google Scholar]
- [140].Oh SD, Lao JP, Hwang PY, Taylor AF, Smith GR, Hunter N, BLM ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules, Cell, 130 (2007) 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Croteau DL, Popuri V, Opresko PL, Bohr VA, Human RecQ helicases in DNA repair, recombination, and replication, Annu. Rev. Biochem, 83 (2014) 519–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Shen Z, Nickoloff JA, Mammalian homologous recombination repair and cancer intervention, in: Wei Q, Li L, Chen DJ (Eds.) DNA Repair, Genetic Instability, and Cancer, World Scientific Publishing Co., Singapore, 2007, pp. 119–156. [Google Scholar]
- [143].Deininger PL, Batzer MA, Alu repeats and human disease, Mol. Genet. Metab, 67 (1999) 183–193. [DOI] [PubMed] [Google Scholar]
- [144].Strout MP, Marcucci G, Bloomfield CD, Caligiuri MA, The partial tandem duplication of ALL1 (MLL) is consistently generated by ALU-mediated homologous recombination in acute myeloid leukemia, Proc. Natl. Acad. Sci. USA, 95 (1998) 2390–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Roy R, Chun J, Powell SN, BRCA1 and BRCA2: different roles in a common pathway of genome protection, Nat. Rev. Cancer, 12 (2012) 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Deans AJ, West SC, DNA interstrand crosslink repair and cancer, Nat. Rev. Cancer, 11 (2011) 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Luebben SW, Kawabata T, Akre MK, Lee WL, Johnson CS, O’Sullivan MG, Shima N, Helq acts in parallel to Fancc to suppress replication-associated genome instability, Nucleic Acids Res, 41 (2013) 10283–10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, Wang ZQ, Jasin M, Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair, Proc. Natl. Acad. Sci. USA, 102 (2005) 1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Blanco MG, Matos J, Hold your horSSEs: controlling structure-selective endonucleases MUS81 and Yen1/GEN1, Front. Genet, 6 (2015) 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Doe CL, Ahn JS, Dixon J, Whitby MC, Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks, J. Biol. Chem, 277 (2002) 32753–32759. [DOI] [PubMed] [Google Scholar]
- [151].Pepe A, West SC, MUS81-EME2 promotes replication fork restart, Cell Rep, 7 (2014) 1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Symington LS, End resection at double-strand breaks: mechanism and regulation, Cold Spring Harb. Perspect. Biol, 6 (2014) a016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchiiwai Y, Takeda S, Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death, EMBO J, 17 (1998) 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Lim DS, Hasty P, A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53, Mol. Cell. Biol, 16 (1996) 7133–7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Marder BA, Morgan WF, Delayed chromosomal instability induced by DNA damage, Mol. Cell. Biol, 13 (1993) 6667–6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Morgan WF, Day JP, Kaplan MI, McGhee EM, Limoli CL, Genomic instability induced by ionizing radiation, Radiat. Res, 146 (1996) 247–258. [PubMed] [Google Scholar]
- [157].Bouffler SD, Morgan WF, Pandita TK, Slijepcevic P, The involvement of telomeric sequences in chromosomal aberrations, Mutat. Res, 366 (1996) 129–135. [DOI] [PubMed] [Google Scholar]
- [158].Limoli CL, Kaplan MI, Corcoran J, Meyers M, Boothman DA, Morgan WF, Chromosomal instability and its relationship to other end-points of genomic instability, Cancer Res, 57 (1997) 5557–5563. [PubMed] [Google Scholar]
- [159].Limoli CL, Kaplan MI, Phillips JW, Adair GM, Morgan WF, Differential induction of chromosomal instability by DNA strand-breaking agents, Cancer Res, 57 (1997) 4048–4056. [PubMed] [Google Scholar]
- [160].Limoli CL, Day JP, Ward JF, Morgan WF, Induction of chromosome aberrations and delayed genomic instability by photochemical processes, Photochem. Photobiol, 67 (1998) 233–238. [DOI] [PubMed] [Google Scholar]
- [161].Day JP, Limoli CL, Morgan WF, Recombination involving interstitial telomere repeat-like sequences promotes chromosomal instability in Chinese hamster cells, Carcinogenesis, 19 (1998) 259–266. [DOI] [PubMed] [Google Scholar]
- [162].Morgan WF, Corcoran J, Hartmann A, Kaplan MI, Limoli CL, Ponnaiya B, DNA double-strand breaks, chromosomal rearrangements, and genomic instability, Mutat. Res, 404 (1998) 125–128. [DOI] [PubMed] [Google Scholar]
- [163].Limoli CL, Corcoran JJ, Milligan JR, Ward JF, Morgan WF, Critical target and dose and dose-rate responses for the induction of chromosomal instability by ionizing radiation, Radiat. Res, 151 (1999) 677–685. [PubMed] [Google Scholar]
- [164].Limoli CL, Ponnaiya B, Corcoran JJ, Giedzinski E, Morgan WF, Chromosomal instability induced by heavy ion irradiation, Int. J. Radiat. Biol, 76 (2000) 1599–1606. [DOI] [PubMed] [Google Scholar]
- [165].Morgan WF, Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects, Radiat. Res, 159 (2003) 581–596. [DOI] [PubMed] [Google Scholar]
- [166].Morgan WF, Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiation-induced genomic instability and bystander effects in vitro, Radiat. Res, 159 (2003) 567–580. [DOI] [PubMed] [Google Scholar]
- [167].Smith LE, Nagar S, Kim GJ, Morgan WF, Radiation-induced genomic instability: radiation quality and dose response, Health Phys, 85 (2003) 23–29. [DOI] [PubMed] [Google Scholar]
- [168].Morgan WF, Is there a common mechanism underlying genomic instability, bystander effects and other nontargeted effects of exposure to ionizing radiation?, Oncogene, 22 (2003) 7094–7099. [DOI] [PubMed] [Google Scholar]
- [169].Huang L, Grimm S, Smith LE, Kim PM, Nickoloff JA, Goloubeva OG, Morgan WF, Ionizing radiation induces delayed hyperrecombination in mammalian cells, Mol. Cell. Biol, 24 (2004) 5060–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Brenneman MA, Weiss AE, Nickoloff JA, Chen DJ, XRCC3 is required for efficient repair of chromosome breaks by homologous recombination, Mutat. Res, 459 (2000) 89–97. [DOI] [PubMed] [Google Scholar]
- [171].Brenneman MA, Wagener BM, Miller CA, Allen C, Nickoloff JA, XRCC3 controls the fidelity of homologous recombination: roles for XRCC3 in late stages of recombination, Mol. Cell, 10 (2002) 387–395. [DOI] [PubMed] [Google Scholar]
- [172].Cui X, Brenneman MA, Meyne J, Oshimura M, Goodwin EH, Chen DJ, The XRCC2 and XRCC3 genes are required for chromosome stability in mammalian cells, Mutat. Res, 434 (1999) 75–88. [DOI] [PubMed] [Google Scholar]
- [173].Lio YC, Schild D, Brenneman MA, Redpath JL, Chen DJ, Human Rad51C deficiency destabilizes XRCC3, impairs recombination, and radiosensitizes S/G2-phase cells, J. Biol. Chem, 279 (2004) 42313–42320. [DOI] [PubMed] [Google Scholar]
- [174].Schoenmakers EFPM, Huysmans C, Van de Ven WJM, Allelic knockout of novel splice variants of human recombination repair gene RAD51B in t(12;14) uterine leiomyomas, Cancer Res, 59 (1999) 19–23. [PubMed] [Google Scholar]
- [175].Sigurdsson S, Van Komen S, Bussen W, Schild D, Albala JS, Sung P, Mediator function of the human Rad51B-Rad51C complex in Rad51/RPA-catalyzed DNA strand exchange, Genes Dev, 15 (2001) 3308–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Takata M, Sasaki MS, Sonoda E, Fukushima T, Morrison C, Abala JS, Swagemakers SMA, Kanaar R, Thompson LH, Takeda S, The Rad51 paralog Rad51B promotes homologous recombinational repair, Mol. Cell. Biol, 20 (2000) 6476–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Wiese C, Hinz JM, Tebbs RS, Nham PB, Urbin SS, Collins DW, Thompson LH, Schild D, Disparate requirements for the Walker A and B ATPase motifs of human RAD51D in homologous recombination, Nucleic Acids Res, 34 (2006) 2833–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Bohm S, Bernstein KA, The role of post-translational modifications in fine-tuning BLM helicase function during DNA repair, DNA Repair, 22 (2014) 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, Slovek LE, Capobianco AJ, German J, Boivin GP, Groden J, Enhanced tumor formation in mice heterozygous for Blm mutation, Science, 297 (2002) 2051–2053. [DOI] [PubMed] [Google Scholar]
- [180].Durant ST, Paffett KS, Shrivastav M, Timmins GS, Morgan WF, Nickoloff JA, UV radiation induces delayed hyperrecombination associated with hypermutation in human cells, Mol. Cell. Biol, 26 (2006) 6047–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Cariello NF, Skopek TR, Analysis of mutations occurring at the human hprt locus, J. Mol. Biol, 231 (1993) 41–57. [DOI] [PubMed] [Google Scholar]
- [182].Zhang LH, Vrieling H, van Zeeland AA, Jenssen D, Spectrum of spontaneously occurring mutations in the hprt gene of V79 Chinese hamster cells, J. Mol. Biol, 223 (1992) 627–635. [DOI] [PubMed] [Google Scholar]
- [183].Malkhosyan S, McCarty A, Sawai H, Perucho M, Differences in the spectrum of spontaneous mutations in the hprt gene between tumor cells of the microsatellite mutator phenotype, Mutat. Res, 316 (1996) 249–259. [DOI] [PubMed] [Google Scholar]
- [184].Hall EJ, Brenner DJ, Cancer risks from diagnostic radiology, Br. J. Radiol, 81 (2008) 362–378. [DOI] [PubMed] [Google Scholar]
- [185].Huang L, Kim PM, Nickoloff JA, Morgan WF, Targeted and non-targeted effects of low-dose ionizing radiation on delayed genomic instability in human cells, Cancer Res, 67 (2007) 1099–1104. [DOI] [PubMed] [Google Scholar]
- [186].Hanahan D, Weinberg RA, The hallmarks of cancer, Cell, 100 (2000) 57–70. [DOI] [PubMed] [Google Scholar]
- [187].Hanks S, Coleman K, Reid S, Plaja A, Firth H, FitzPatrick D, Kidd A, Méhes K, Nash R, Robin N, Shannon N, Tolmie J, Swansbury J, Irrthum A, Douglas J, Rahman N, Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B, Nat. Genet, 36 (2004) 1159–1161. [DOI] [PubMed] [Google Scholar]
- [188].Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW, Aneuploidy acts both oncogenically and as a tumor suppressor, Cancer cell, 11 (2007) 25–36. [DOI] [PubMed] [Google Scholar]
- [189].Shih IM, Zhou W, Goodman SN, Lengauer C, Kinzler KW, Vogelstein B, Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis, Cancer Res, 61 (2001) 818–822. [PubMed] [Google Scholar]
- [190].Lengauer C, Kinzler KW, Vogelstein B, Genetic instabilities in human cancers, Nature, 396 (1998) 643–649. [DOI] [PubMed] [Google Scholar]
- [191].Baverstock K, Radiation-induced genomic instability: a paradigm-breaking phenomenon and its relevance to environmentally induced cancer, Mutat. Res, 454 (2000) 89–109. [DOI] [PubMed] [Google Scholar]
- [192].Kaplan MI, Morgan WF, The nucleus is the target for radiation-induced chromosomal instability, Radiat. Res, 150 (1998) 382–390. [PubMed] [Google Scholar]
- [193].Clutton SM, Townsend KM, Walker C, Ansell JD, Wright EG, Radiation-induced genomic instability and persisting oxidative stress in primary bone marrow cultures, Carcinogenesis, 17 (1996) 1633–1639. [DOI] [PubMed] [Google Scholar]
- [194].Limoli CL, Hartmann A, Shephard L, Yang CR, Boothman DA, Bartholomew J, Morgan WF, Apoptosis, reproductive failure, and oxidative stress in Chinese hamster ovary cells with compromised genomic integrity, Cancer Res, 58 (1998) 3712–3718. [PubMed] [Google Scholar]
- [195].Kim GJ, Fiskum GM, Morgan WF, A role for mitochondrial dysfunction in perpetuating radiation-induced genomic instability, Cancer Res, 66 (2006) 10377–10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Kim GJ, Chandrasekaran K, Morgan WF, Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: a review, Mutagenesis, 21 (2006) 361–367. [DOI] [PubMed] [Google Scholar]
- [197].Limoli CL, Kaplan MI, Giedzinski E, Morgan WF, Attenuation of radiation-induced genomic instability by free radical scavengers and cellular proliferation, Free Radic. Biol. Med, 31 (2001) 10–19. [DOI] [PubMed] [Google Scholar]
- [198].Vens C, Begg AC, Targeting base excision repair as a sensitization strategy in radiotherapy, Semin. Radiat. Oncol, 20 (2010) 241–249. [DOI] [PubMed] [Google Scholar]
- [199].Kelley MR, Logsdon D, Fishel ML, Targeting DNA repair pathways for cancer treatment: what’s new?, Future Oncol., 10 (2014) 1215–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, Gil J, Leung AY, Ashworth A, So CW, Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors, Nat. Med, 21 (2015) 1481–1490. [DOI] [PubMed] [Google Scholar]
- [201].Jo U, Kim H, Exploiting the Fanconi anemia pathway for targeted anti-cancer therapy, Mol. Cells, 38 (2015) 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Killock D, Targeted therapies: DNA polymerase theta-a new target for synthetic lethality?, Nat. Rev. Clin. Oncol, 12 (2015) 125. [DOI] [PubMed] [Google Scholar]
- [203].Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, Turchi JJ, DNA repair targeted therapy: The past or future of cancer treatment?, Pharmacol. Ther, 160 (2016) 65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Liu FW, Tewari KS, New targeted agents in gynecologic cancers: synthetic lethality, homologous recombination deficiency, and PARP inhibitors, Curr. Treat. Options Oncol, 17 (2016) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Hill CK 3, Bull. Atomic Scientists, 68 (2012) 51–58. [Google Scholar]
- [206].Morgan WF, Bair WJ, Issues in low dose radiation biology: the controversy continues. A perspective, Radiat. Res, 179 (2013) 501–510. [DOI] [PubMed] [Google Scholar]
- [207].Morgan WF, Sowa MB, Non-targeted effects of ionizing radiation: implications for risk assessment and the radiation dose response profile, Health Phys, 97 (2009) 426–432. [DOI] [PubMed] [Google Scholar]
- [208].Feinendegen LE, Brooks AL, Morgan WF, Modeling of dose-risk functions, Health Phys, 100 (2011) 331. [DOI] [PubMed] [Google Scholar]
- [209].Morgan WF, International Commission on Radiological Protection Committee 1: current status and future directions, Ann. ICRP, 44 (2015) 8–14. [DOI] [PubMed] [Google Scholar]
- [210].Morgan WF, Compelling issues compounding the understanding of low dose radiation effects: but do they matter?, Health Phys, 110 (2016) 291–292. [DOI] [PubMed] [Google Scholar]
- [211].Morgan WF, Overview of ICRP Committee 1: radiation effects, Ann. ICRP, 45 (2016) 9–16. [DOI] [PubMed] [Google Scholar]