

Summary
Argonautes are nucleic acid-guided proteins that perform numerous cellular functions across all domains of life. Little is known about how distinct evolutionary pressures have shaped each Argonaute’s biophysical properties. We applied high-throughput biochemistry to characterize how Thermus thermophilus Argonaute (TtAgo), a DNA-guided, DNA endonuclease, finds, binds, and cleaves its targets. We find that TtAgo uses similar biophysical adaptations to eukaryotic Argonautes for rapid association, but requires more extensive complementarity to achieve high-affinity target binding. Using these data, we constructed models for TtAgo association rates and equilibrium binding affinities that estimate the nucleic acid- and protein-mediated components of the target interaction energies. Finally, we show that TtAgo cleavage rates vary widely based on the DNA guide, suggesting that only a subset of guides cleave targets on physiologically relevant time-scales.
eTOC Blurb
High-throughput measurements of binding and cleavage of >3,000 unique targets for each of five TtAgo guides reveals guide-specific functional adaptations of a bacterial Argonaute. Quantitative models of association kinetics and binding affinity decompose protein- and nucleic-acid- mediated components of TtAgo binding.
Graphical Abstract
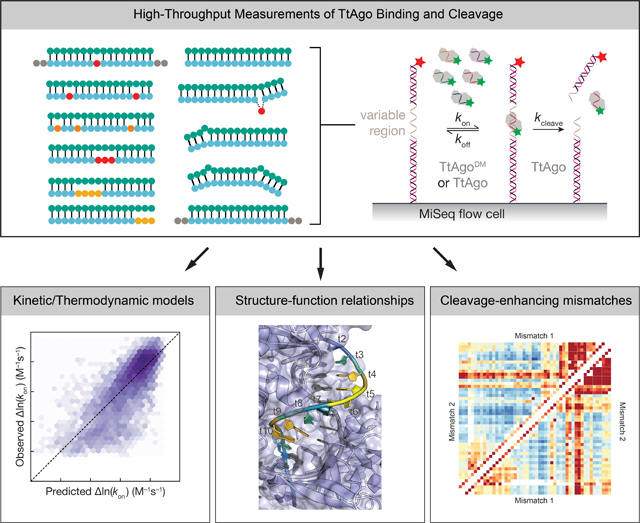
Introduction
Argonaute proteins constitute a diverse family of nucleic acid-guided proteins present in eukaryotes, archaea, and bacteria(Swarts et al., 2014a). In eukaryotes, small RNA guides direct Argonaute (Ago) proteins to bind complementary RNA targets and repress their expression (Bartel, 2018; Ghildiyal and Zamore, 2009; Ozata et al., 2019). In contrast, some prokaryotic Argonaute (pAgo) proteins, such as Thermus thermophilus Ago (TtAgo), use DNA guides to bind and cleave DNA or RNA targets(Hegge et al., 2019; Kuzmenko et al., 2020; Sheng et al., 2014; Swarts et al., 2014b, 2015; Wang et al., 2008a; Zander et al., 2017). TtAgo has been proposed to defend the bacterium against plasmids and to assist in disentangling the catenated circular chromosomes produced during DNA replication(Jolly et al., 2020; Swarts et al., 2014b).
All Ago proteins investigated to date alter the biochemical properties of their nucleic acid guides. Consequently, the speed and affinity with which they find and bind target sequences cannot be predicted from the principles of nucleic acid hybridization. Both prokaryotic and eukaryotic Ago proteins pre-organize guide bases within the seed sequence to accelerate target finding(Elkayam et al., 2012; Parker et al., 2005, 2009; Salomon et al., 2016; Schirle et al., 2014; Wang et al., 2008b; Yuan et al., 2005). After binding, Ago proteins require defined guide:target interactions to enable target cleavage. The effects on cleavage of mismatches at specific positions is best understood for the miRNA-guided mammalian protein AGO2 and the siRNA-directed, arthropod-specific protein Ago2(Becker et al., 2019; Wee et al., 2012). The effects of select mismatches on TtAgo cleavage have been reported previously(Sheng et al., 2017; Wang et al., 2008a), and a recent high-throughput study examined cleavage efficiency for ~2,000 TtAgo guides against predominantly perfectly complementary targets(Hunt et al., 2021). However, a systematic exploration of the effect of mismatches on TtAgo cleavage behavior has not yet been reported.
Here, we deploy multiple, high-throughput biochemical approaches to characterize how TtAgo uses its DNA guide to bind and cleave target DNA. We measure relative association rates, binding affinities, and cleavage rates of >3,000 targets for five DNA guides in complex with TtAgo. Target finding by TtAgo depends on stable seed binding, which itself depends on the sequence composition of the seed region and accurate pairing at specific positions within the guide:target duplex. For multiple guides, we observe that unlike eukaryotic Ago proteins, TtAgo requires more than just seed complementarity to achieve high-affinity target binding(Smith et al., 2019). Using our high-throughput biophysical data, we constructed separate, quantitative models to predict kinetic and thermodynamic binding parameters for TtAgo loaded with any DNA guide sequence. We used these models to estimate the nucleic acid- and protein-mediated components of the target interaction energies, revealing that TtAgo binding affinity relies significantly on nearest-neighbor predicted nucleic acid binding energy for target interactions outside the proximal seed region. As has been previously reported(Hunt et al., 2021), rates of TtAgo-catalyzed target cleavage vary widely depending on guide sequence: the fastest guide cleaves a fully complementary target >25 times faster than the slowest. Like plant and animal Argonaute proteins(Anzelon et al., 2021; Schirle et al., 2014; Sheng et al., 2017; Sheu-Gruttadauria et al., 2019; Tomari and Zamore, 2005; Wang et al., 2008a), TtAgo cleaves many mismatched targets faster than the corresponding fully complementary targets, suggesting that these mismatches increase the proportion of time the protein spends in its catalytically active conformation(Schirle et al., 2014; Sheng et al., 2017; Sheu- Gruttadauria et al., 2019; Tomari and Zamore, 2005; Wang et al., 2008a).
Results
High-throughput, multiplexed measurements of TtAgo binding and cleavage
We chose five unique single-stranded DNA (ssDNA) guide sequences to profile TtAgo binding and cleavage (Fig. 1): three endogenous guides with >60% GC content, previously identified as being naturally loaded in TtAgo in vivo (guides 1, 2 and 3)(Jolly et al., 2020), a guide corresponding to the first 16 nucleotides of the let-7a miRNA (guide 4); and a guide with an AT-rich seed sequence (guide 5). For each, we designed target libraries of >3,000 unique targets (16,169 total targets). These libraries contained all singly and doubly mismatched targets, a subset of triply mismatched targets, insertions of up to five nucleotides, all single and double nucleotide deletions, and targets with stretches of mismatches starting at every target position and ending at every other position(Supplementary Fig. 1a). The five target libraries were sequenced on an Illumina MiSeq, and the resulting sequenced flow cell was used to measure (1) target binding of a 3′ Cy3-labeled, naked DNA guide, and (2) target binding and (3) cleavage by the same guide loaded into TtAgo (Fig. 1a)(Becker et al., 2019; Buenrostro et al., 2014; She et al., 2017). To minimize differences in target site accessibility due to secondary structure, DNA oligonucleotides with TM >78°C were annealed to the invariant regions of the target libraries. A 5′ Alexa647-labeled oligonucleotide (TM >90°C) was annealed to the distal end of each target sequence to monitor target cleavage. Each guide-loaded TtAgo bound only its own targets and did not show measurable binding to targets of the other four guides (Supplementary Fig. 1b), allowing us to pool the five guide-loaded TtAgo complexes and measured their kinetic properties simultaneously. We used catalytically inactive TtAgoD478A,D546A (Double point-mutant, TtAgoDM) to measure binding kinetics and thermodynamics, and wild-type TtAgo to determine cleavage rates. To measure relative association rates (krel; Fig. 1b), multiple concentrations of either the unloaded DNA guide or loaded TtAgoDM was flowed continuously through the MiSeq flow cell at 37°C. To measure binding affinity, we used multiple concentrations of the unloaded guides or loaded TtAgoDM and measured the dissociation constant (KD) for each guide:target pair at equilibrium (Fig. 1c). To estimate the KD of low affinity targets, we used an empirical distribution of maximum fluorescence values to constrain our binding curve fits(Becker et al., 2019; Denny et al., 2018)(Methods). These constrained fits enabled estimation of KD values between 85 pM–8.6 μM for unloaded ssDNA guides and 10 pM–90 nM for loaded TtAgoDM. Guide affinities were measured at 37°C, and TtAgoDM affinities were measured at both 37°C and at 55°C (higher temperatures resulted in poorer quality data in our equilibrium binding experiments); T. thermophilus naturally grows at 47–85°C (Oshima and Imahori, 1974). Single-turnover cleavage rates at 55°C or 65°C were measured using 25 nM DNA guide-loaded TtAgo (Fig. 1a,d). Loss of red fluorescent signal, signifying release of the 3′ cleavage product correlated well with loss of green fluorescent signal, indicating departure of TtAgo-loaded guide from the cleaved target (Fig. 1e).
Figure 1 |.

High-throughput Characterization of TtAgo binding and cleavage. (a) Schematic of TtAgo loaded with a fluorescent guide binding and cleaving ssDNA targets in a sequenced MiSeq flow cell. Target cleavage was scored by loss of red fluorescent signal, signifying release of 3′ cleavage product, or loss of green fluorescent signal, indicating TtAgo departure from cleaved targets. (b) A representative set of association data for a single target. Error bars correspond to the 95% confidence interval on the median fluorescence. The plot to the right shows the relationship between concentration of unloaded guide or guide-loaded TtAgo and observed rate, from which the association rate was determined. (c) Representative binding isotherms for unloaded guide at 37ºC and guide-loaded TtAgo measured at 37ºC and 55ºC. Error bars correspond to the 95% confidence interval on the normalized fit maximum fluorescence from each association experiment. (d) Representative cleavage curves for three different targets (shown in corresponding color in schematic) containing different degrees of complementarity to the guide (in gray). Cleavage was measured at 55ºC. Error bars correspond to the 95% confidence interval on the normalized fit maximum fluorescence from each cleavage experiment. (e) Correlation of fitted cleavage rates using the departure of TtAgo (green), or release of the cleaved product (red) as the signal for cleavage. See also Figure S1.
Acceleration of ssDNA binding by TtAgo requires stable binding in the seed region
Like eukaryotic Ago proteins, TtAgo pre-organizes the seed of its ssDNA guide (guide nucleotides g2–g7) to accelerate target finding(Chandradoss et al., 2015; Salomon et al., 2016; Wang et al., 2008a, 2008b, 2009; Wee et al., 2012) (Supplementary Fig. 2a–c). Target mismatches within the seed disproportionately slowed the relative rate of TtAgoDM association with target (Fig. 2a). While seed mismatches resulted in significantly slower relative association rates for all guides, the position-specific effect of seed mismatches varied with guide sequence. For example, mismatches involving target strand bases 2 and 3 (t2 and t3) significantly slowed association for guides 1 and 3, whereas mismatches at t3 and t4 had the largest effect for guide 2 and mismatches at t4 and t5 had the largest effect for guide 4.
Figure 2 |.

Sequence determinants of TtAgo association kinetics. (a) Change in association rates for tandem double mismatches of TtAgo targets relative to a fully complementary target (dashed line). Numbers near the bottom of each plot indicate the number of targets (out of a possible 9) in each group that were within the limits of detection. The region corresponding to the eukaryotic Ago seed region (bases t2–t8) is shaded in gray. (b) Comparison of model predicted TtAgoDM relative association rates to observed relative association rates. Color bar indicates the number of targets in each bin. Guide 4 was excluded from all association model training and testing data due to its outlier association behavior (applies to b-e). (c) Target position penalty weights. Error bars indicate SEM of penalty weights across models fit with each guide held out in turn (n = 4). (d) t1 base identity penalty weights. Error bars as in (c). (e) Guide to target mismatch penalty weights. See also Figure S2.
For guides 1, 2, 3, and 5, target mismatches outside of the seed region had smaller effects on the association rate, generally slowing association rate less than twofold. Guide 4 did not follow this trend: both mismatches within and outside the seed region of this guide reduced the TtAgoDM relative association rate (Fig. 2a). These effects could not be explained by potential secondary structure in the target (Supplementary Fig. 2d). One possible explanation is that guide 4 may be loaded into TtAgoDM in a distinct conformation compared with other guides, contributing to this discordant association behavior, however further experimental and structural studies would be required to substantiate this possibility.
A general model for target finding by TtAgo
To define the features determining TtAgo association rates, we modeled target association using a set of biochemically interpretable parameters. (Because guide 4 was an outlier, we excluded it from our modeling data set). This model contained 33 biochemical parameters that can be interpreted as increasing or decreasing an energy barrier for association (Methods). To capture both the effects of different types of mismatches between the guide and target, as well as differences in sensitivity to mismatches along the target:guide duplex, we employed a scaling parameter for each position within the guide:target duplex. Structural studies show that the bases at guide position 1 (g1) and target position 1 (t1) do not base pair, but are instead bound in binding pockets in the TtAgo MID and PIWI domains, respectively(Sheng et al., 2014; Swarts et al., 2017a). Three additional parameters were used to account for the identity of target position 1, fixing t1G to have a penalty of 0. We also observed that the predicted nearest neighbor stability of the seed region contributed to the observed association rate, but only when the predicted binding energy was ≳−8 kBT (Supplementary Fig. 2e), consistent with a seed energy threshold for transitioning from an initial collision to stable target binding. To incorporate this observation into our model, we used NUPACK(Zadeh et al., 2011) to predict the nearest neighbor energy of the k-mer spanning the seed sequence for each target, and transformed this energy term into an association penalty using a logistic function. Because target secondary structure can sequester binding sites(Becker et al., 2019; Kedde et al., 2010), we also included the NUPACK-predicted target secondary structure. When jointly fit to all data, model predictions had Pearson correlation of 0.79 to measured values (Fig. 2b). The majority of model parameters were stable when fit to training datasets using leave-one-out cross validation (i.e., fit to data from three guides then tested on data generated from a guide not used in training), although some parameters, such as the importance of target position 4 (t4), tended to be more guide-sequence dependent (Fig. 2c and Supplementary Fig. 2f). The median Pearson correlation of models tested on held out guides was 0.71 (Supplementary Fig. 2g).
Our generalized TtAgo association model revealed several features influencing TtAgo association rates. A guanosine at target position 1 (t1G) increases the stability of TtAgo binding(Jolly et al., 2020; Smith et al., 2019; Swarts et al., 2017a). One therefore might expect that any non-t1G base could slow binding, but in fact only a t1A decreased the association rate (Fig. 2d), suggesting that a t1A decreases the probability of a collision transitioning to productive binding, possibly by reducing the stability of interactions between the t1 nucleotide and its binding pocket in the PIWI domain(Swarts et al., 2017a). After accounting for the overall stability of the seed region, the target positions most important for rapid association were t2–t4 (Fig. 2c), an observation consistent with available structures of the binary TtAgo complex showing guide bases g2–g6 stacked and exposed to solvent(Wang et al., 2008b), and with biochemical studies of TtAgo and eukaryotic Ago association rates demonstrating that these three seed bases are critical for the initial target search(Becker et al., 2019; Salomon et al., 2016; Schirle et al., 2014). Notably, in the case of mismatches in these three crucial guide:target base pairs, the identity of the mismatches significantly influenced association rates. Target mismatches opposite thymine bases in the guide (i.e. gT:tG, gT:tC, gT:tT vs gG:tG, gG:tA, gG:tT) had smaller effects on the relative association rate, while mispairings to guide guanine bases had larger effects (Fig. 2e). Additionally, purine-purine mismatches tended to be more disruptive than pyrimidine-pyrimidine mismatches.
A limitation of this association model was the necessity of removing the outlier guide 4 from our training and testing data. We anticipate that characterization of many more TtAgo guides may be necessary to determine whether this guide was a rare outlier, or if there exists a broader class of TtAgo guides that exhibit similarly discordant association behavior.
Proximal seed pairing is required for protein-mediated stabilization of TtAgo-bound targets
For the majority of mismatched targets, TtAgoDM enhanced the binding affinity of its ssDNA guide. We measured the affinity of each DNA guide alone and in complex with TtAgoDM for our entire 16,169 member target library. These paired measurements were performed at 37°C because the unloaded DNA guides did not bind most mismatched targets with high enough affinity to be measurable at higher temperatures. TtAgoDM increased the affinity of DNA guides for mismatched targets by a median of −5 kBT for guides 1, 2, and 5; −3 kBT for guide 4; and −6 kBT for guide 2, although the −ΔΔG values spanned a ~10 kBT range for all guides (Fig. 3a). A −5 kBT energy increase corresponds to a ~150-fold increase in binding affinity, and the −10 kBT of additional binding energy achieved for some TtAgoDM targets represents a >20,000-fold increase in affinity.
Figure 3 |.

Differential effects of mismatches on binding affinity of unloaded guide and guide-loaded TtAgo. (a) Difference in measured binding energies between TtAgoDM:guide and unloaded guide. Shown are targets for which binding affinities of both TtAgoDM:guide and unloaded guide were within limits of detection. The black dotted line indicates the median binding energy difference, while the gray dotted lines indicate the bottom and top 10% of binding energy differences. (b) Enrichment of mismatch positions in the 10% of least TtAgo-stabilized targets, relative to all measured targets. (c) Binding energies for unloaded guide 1 (upper left) and TtAgoDM:guide 1 (lower right) targets containing stretches of complementary nucleotide mismatches (e.g., A to T). E.g., for the 2–4 mismatches, the corresponding targets in the heatmap are located at the intersection of 2 on the “beginning complement mismatch” axis and 4 on the “ending complement mismatch” axis. White asterisks on the upper left heatmap indicate the minimum measurable binding affinity for the unloaded guide. (d–e) Binding energies for guide 1 (d) and guide 2 (e) targets containing progressively more complementarity to the DNA guide. Target mismatches progress from the 5′ end (left panel) or the 3′ end (right panel) of the target. Error bars indicate the 95% confidence interval on the binding energy. The dotted line indicates the minimum measurable binding affinity for unloaded guide. See also Figure S3.
TtAgo magnifies the energetic penalty for target mispairing to the seed sequence, thereby enhancing the binding specificity of this region and minimizing occupancy at off-target sequences. As for other Ago proteins, target mismatches with the seed sequence were more disruptive to binding than mismatches with the 3′ portion of the TtAgoDM-bound DNA guide. To define the positions that contribute the most to the TtAgo-mediated enhancement in binding affinity, we examined all targets containing only point mismatches (i.e. those that did not contain indels) and identified positions at which mismatches substantially reduced the difference in binding affinity between each DNA guide alone and loaded into TtAgoDM. For all five guides, mismatches at target position 2 (t2), and to a lesser extent, positions t3–t4 were enriched in the top 10% of mismatched targets that substantially reduced TtAgoDM-mediated binding enhancement (Fig. 3b). By contrast, mismatches in the distal region of the target were depleted. The least disruptive target sequences—i.e., those that maintained large differences between binding energies of DNA guide alone vs. those of loaded TtAgoDM—generally had mismatches at t5, t6, t8, t10, t11, and t12, although the most enriched positions were unique to each guide (Supplementary Fig. 3a). To further explore the positional dependency of TtAgo-mediated enhancement of target affinity, we examined targets that contained stretches of mismatches at all starting and ending positions within the target sequence (Fig. 3c). As expected, mismatches starting from the 3′ end of the target, which pairs to the seed region of the guide, dramatically decreased TtAgoDM binding affinity. For guides 1, 2 and 5, mismatches starting at t1 and extending to t5 caused TtAgoDM to bind with an affinity lower than its respective unloaded DNA guide (Fig. 3c–e and Supplementary Fig. 3b).
Previous single-molecule experiments examined one specific guide sequence for which TtAgo bound a t2–t10 target with low-nanomolar affinity at 55°C (Jolly et al., 2020). We extended this single-molecule analysis to all five guides used here: TtAgoDM similarly bound t2–t10 seed-matched targets with low-nanomolar affinity at 37°C for all guides (Supplementary Fig. 3b). While base-pairing to nucleotides g2–g12 of the guide 4 is required for very stable binding (koff < 0.050 s–1, Supplementary Fig. 3c), some guides, particularly the naturally occurring guides with >60% GC-content in their seed, allowed surprisingly stable TtAgoDM binding to targets with as few as 8 contiguous matched seed bases. Indeed, TtAgoDM loaded with guide 1 bound to an 8-mer seed-matched target with a <2 nM affinity at 37°C, while 12 bases of contiguous complementarity were required to achieve a similar affinity for the unloaded guide (Fig. 3d).
Modeling nucleic acid and protein contributions to TtAgo binding
Inspection of all possible doubly mismatched targets for each guide revealed guide sequence-dependent positional mismatch sensitivities for both the unloaded DNA guide and for TtAgoDM (Fig. 4a,b). In many cases, the target mismatches most disruptive to TtAgoDM binding also had the largest effect on the unloaded DNA guide. Indeed, binding affinities for TtAgoDM correlated with the corresponding nearest neighbor ensemble DNA energies of the guide:target duplex (Pearson’s r = 0.58) (Supplementary Fig. 4a). Thus nucleic acid binding energy is partially responsible for TtAgoDM binding affinity, consistent with TtAgo structures showing guide and target strands engaging in base pairing from positions g2:t2 to g16:t16 (Sheng et al., 2014; Swarts et al., 2017a).
Figure 4 |.

Predictive model for TtAgo binding. (a) Measured binding energies for unloaded guide 4 (upper left) and TtAgoDM:guide 4 (lower right) binding to single and double mismatched targets at 37°C. Axes are labeled with the 3′ end of the target (5′ end of the guide) starting at position 1. White boxes represent missing data. White asterisks on the upper left heatmap indicate the minimum measurable binding affinity for the unloaded guide. (b) Same as in (a), but for guide 5. (c) Comparison of binding energies predicted by the mismatch-only model to the observed binding energies when trained on all data. (d) Mean parameters obtained when fitting the mismatch-only model with each guide held out in turn (n = 5). Error bars indicate SEM of fitted values across the five model fits. See also Figure S4.
We next developed a model that could predict the binding affinity of any target to any guide loaded into TtAgo. To estimate the overall TtAgoDM nucleoprotein affinity for any guide, we divided the binding energy into contributions from the protein and contributions from the nucleic acid. For the nucleic acid contributions, we used NUPACK to predict the energy for the guide:target ensemble. While these NUPACK predicted energies reflect energy changes resulting from mismatches between the guide and target, the protein contribution is also dependent on whether the guide and target are matched at a given position. To account for this, we included model parameters capturing energy defects to binding at positions 2–16 when any type of mismatch occurred. We also included a model parameter that reflects the protein contribution to binding to a fully complementary target with a G at target position t1, as well as model parameters reflecting the energy associated with each possible alternative nucleotide at target position t1. Finally, because target secondary structures can sequester the target sequence and reduce the observed binding affinity, we included a parameter to incorporate the predicted energy of the ensemble of target secondary structures incompetent for binding. This 20 parameter model was fit to data from the five guide sequences using leave-one-out cross-validation (i.e., fit to four guides and tested on the guide not used in training). The model was accurate (median Pearson correlation of 0.7) when predicting affinities for data from a held-out guide. When the model was fit to all of the data simultaneously, we observed a Pearson correlation of 0.72 between model predictions and experimental results (Fig. 4c). The fit parameters were stable among the different guides used to train the model and can be interpreted as the protein contributions to binding affinity (Fig. 4d).
When mismatches occur between guide and target, binding energy is lost from both base-pairing, and from protein:nucleic acid interactions. In our model decomposition, the positional mismatch parameters represent the additional binding energy lost due to protein:nucleic acid interactions. Consistent with previous findings, the presence of a guanine at position 1 increased the affinity of TtAgoDM for its target sequence(Smith et al., 2019; Swarts et al., 2017a). In many cases, the mismatch penalties of the model were slightly negative, suggesting that the protein partially offsets the energy lost with a mismatch. The protein-induced energetic penalty for mismatches was greatest for positions 2–4, and near zero for most of the remaining positions, again highlighting that most of the additional energy provided by the protein requires pairing at positions 2–4. Thus unlike many eukaryotic Ago proteins, which create a 3′ supplemental binding region from t13–t16 (Becker et al., 2019; Brennecke et al., 2005; Grimson et al., 2007; Lewis et al., 2005; Salomon et al., 2016; Sheu- Gruttadauria et al., 2019; Wee et al., 2012), the non-seed bases that most impact binding affinity for TtAgo corresponded to the bases most critical for the binding of an unloaded guide.
While this minimal binding model was able to capture many features of TtAgoDM nucleoprotein binding, we also tested models that considered the type of mismatch at each position. Accounting for transition and transversion mismatches separately (15 additional parameters; 37 total) did not improve model performance on held-out guides (median Pearson correlation = 0.69) or when fit to all guides (Pearson correlation = 0.72; Supplementary Fig. 4b).
TtAgo functionally divides its guide into two distinct helices
Structural and functional studies of TtAgo and other pAgos have reported various degrees of disruption by indels in the seed region of the guide:target duplex(Liu et al., 2018; Sheng et al., 2017). Furthermore, mammalian AGO2 tolerates large target insertions between the seed and supplemental region(Becker et al., 2019; Sheu-Gruttadauria et al., 2019). We measured the binding affinity for each of our five DNA guides alone or in complex with TtAgoDM for targets containing 1–5-nt insertions at each position (Fig. 5a). As expected for the DNA guides alone, central insertions in the target generally reduced hybridization affinity more than those near the ends of the target sequence, and larger insertions were more disruptive than smaller insertions. These effects were dramatically different for the guide loaded into TtAgoDM. For both guides 4 and 5, TtAgoDM displayed a bimodal sensitivity to target insertions at both 37°C and 55°C: insertions in the distal seed (t5–t7) or near the cleavage site (t9–t12) caused substantial reductions in binding affinity, while insertions between t7 and t9 caused more moderate losses in affinity.
Figure 5 |.

Effect of insertions and deletions of target nucleotides on binding energy. (a) Binding energies for unloaded guide at 37ºC (top), TtAgoDM:guide at 37ºC (middle) and TtAgoDM:guide at 55ºC (bottom) to targets with 1- to 5-nt insertions. Axes are labeled with the 3′ end of the target (5′ end of the guide) starting at position 1. Horizontal black lines indicate the limits of detection, and points below the bottom black line bound with higher affinity than the detection limit. The horizontal grey dotted lines in the unloaded guide plots indicate the TtAgoDM upper limit of detection and the horizontal grey lines in the TtAgoDM plots indicate the lower limit of detection for the unloaded guide. (b) The median binding energies of double insertions at each position mapped onto the target strand of a TtAgo crystal structure (PDB ID: 4NCB). Affinity of target insertions between t1/t2 are displayed on t2 of the target strand. (c) Binding energies for the three endogenous guides to targets with 1- to 5-nt insertions at 55°C. Color legend as in (a). (d) Binding energies of unloaded guide (upper left) and TtAgoDM:guide (lower right) to DNA targets with single and double deletions at 37°C. Color bar as in (b). White asterisks on the upper left heatmap indicate the minimum measurable binding affinity for the unloaded guide. See also Figure S5.
Mapping the median binding energy for targets with a 2-nt insertion at each target position for guide 5 onto the crystal structure of TtAgo(Sheng et al., 2014) (Fig. 5b) provides an explanation for these effects. The distal seed (t5–t7) is where the target strand comes into closer proximity with the L2 linker and PAZ domain, and marks the point at which the target strand begins to turn and enter the nucleic-acid binding channel of the protein. Similarly, the second sensitive region, t9–t12, represents the location at which the target backbone directly abuts the PIWI domain. We postulate that like mammalian AGO2(Bartel, 2018; Chandradoss et al., 2015; Salomon et al., 2016; Sheu-Gruttadauria et al., 2019; Wang et al., 2008a), TtAgo first nucleates binding with the proximal seed region, then zippers through the distal seed, next winds the target through the central cleft of the protein, and finally anchors binding by base pairing with the 3′ region of the guide. This divides the guide into two separate helices that make close contacts with the protein. Insertions up to 3-nt between t7 and t9 can be looped out of the central channel with a relatively small energetic penalty, but insertions at t5–t7 and t9–t12 are more difficult to accommodate without disrupting the structure of the protein itself.
For guides 1–3, binding was most disrupted by insertions positioned between bases t5 and t7, suggesting that sensitivity to target insertions in the distal seed is a general feature of TtAgo binding. Unlike guides 4 and 5, insertions in t9–t12 had little effect on binding by guides 1–3 (Fig. 5c). We speculated that guides 1–3 do not require sequence-specific stabilizing interactions outside of the seed region for high affinity binding. Guides 1–3 all bound with sub-nanomolar affinity to targets complementarity only to target bases t1–t10 (Fig. 3c–e and Supplementary Fig. 3c).
Target deletions result in unpaired guide nucleotides, which TtAgo structural studies have shown cannot be ‘looped-out’ of the guide:target duplex in regions where the guide strand makes extensive contacts with the protein(Sheng et al., 2017). While single-nucleotide deletions in the target sequence generally did not measurably decrease TtAgoDM binding affinity, many double deletions decreased TtAgoDM affinity to unmeasurably low levels (>90 nM), even at 37°C (Fig. 5d). A deletion in the proximal seed (t2, t3) in combination with a deletion in the central or distal region of the guide (t7–t12) reduced even the highest affinity guides (guides 1 and 3) to undetectable levels of binding. For guide 4, deletion of bases t2 or t3 in combination with t9 or t10 resulted in binding affinities lower than the unloaded ssDNA guide. Together, these findings highlight the positional sensitivities to helical disruptions in the guide-target duplex imposed by steric constraints of the protein itself(Sheng et al., 2014, 2017).
TtAgo cleavage rates vary widely between guides and are influenced by flanking sequence context
Cleavage rates spanned a 25-fold range for our different guide sequences—a magnitude of variation not observed for mammalian AGO2 (<2-fold range between two previously tested guides)(Becker et al., 2019). We used catalytically active TtAgo to measure the rate of target cleavage directed by each DNA guide at 55°C and 65°C. Although TtAgo is most efficient in the presence of Mn2+, cleavage experiments were performed using Mg2+, because Mn2+ caused excessive photobleaching of our fluorescent labels. At 55°C, little cleavage was detected even after 24 h using fully complementary targets for guides 1–3 (Fig. 6a). The fully complementary target sequence for guide 5 cleaved slowly at 55°C (half-life 11.4 h, kc = 0.001 min–1), and the perfectly matched target sequence for guide 4 cleaved significantly faster (half-life 1.5 h, kc = 0.0077 min–1). At 65°C target cleavage was readily detectable but slow for guides 2 and 3; both had estimated cleavage half-lives >11 h, (kc < 0.001 min–1). For Guide 4, the fastest cleaving guide at 55°C, the target half-life was 12 min (kc = 0.051 min–1) at 65°C. Although guides 1–3 are loaded into TtAgo in vivo(Jolly et al., 2020), they were considerably less active than the artificial sequence of guide 4. These data are consistent with the observation that a low GC content, and a dT at position g1, associated with faster cleavage rates(Hunt et al., 2021). Notably, our guide 4 was the fastest cleaving guide and the only guide that contained a dT at position g1.
Figure 6 |.

Influence of guide sequence and target mismatches on single turnover cleavage kinetics. (a) Cleavage rates of fully complementary targets for each of the five guides at 55°C and 65°C. Error bars indicate the 95% confidence interval on the fit cleavage rate. Light points indicate the cleavage rates of the fully complementary target in alternative flanking sequence contexts. The solid gray lines indicate the limits of detection, and the targets below the solid lines fell beyond those limits. (b) Cleavage rates measured for fully complementary targets of the guide 4 with different 5-nt flanking sequences. (c) Cleavage rates of mismatched targets for each of the five guides. The white dot indicates the cleavage rate of the fully complementary target. Only targets predicted to have saturated binding are shown. (d–e) Single turnover cleavage rates of targets complementary to the guide 2 (d) or guide 3 (e) containing single and double mismatches. Cleavage was measured at 55ºC and was scored by loss of red fluorescence (upper left), i.e. release of the cleaved product, or by loss of the green fluorescence (lower right), i.e. departure of TtAgo. Axes are labeled with the 3′ end of the target (5′ end of the guide) starting at position 1. Color bar as in (b). See also Figure S6.
Although sequence context had small effects on association rates or binding affinity for this guide (Supplementary Fig. 6a,b), flanking sequence context had a sizable influence on the rate of cleavage of targets fully complementary to guide 4, with an 11-fold (at 55°C) or 4.2-fold (at 65°C) change in rates when comparing favorable to unfavorable contexts (Fig. 6a,b). Flanking C and T nucleotides at the 5′ end of the target (furthest from the seed region) most significantly reduced the rate of target cleavage. Most 3′ sequence contexts had minor effects, except for the context with alternating A and G nucleotides. Together, these findings suggest that TtAgo—like AGO2(Ameres et al., 2007)—makes contacts with the flanking sequence outside the region complementary to the guide and some of these contacts influence cleavage activity. While a similar effect of flanking sequence context on cleavage rate was observed for guide 5, its slower cleavage rates made these measurements less precise (Supplementary Fig. 6c).
Many mismatched targets cleave faster than fully matched targets
For all five guides, hundreds of imperfectly complementary targets had TtAgo-catalyzed cleavage rates faster than that of the corresponding fully matched target (Fig. 6c). We confirmed the enhanced cleavage activity of several of these mismatched targets using an ensemble cleavage assay(Supplementary Fig. 6d–f). The effect of mismatches is unlikely to reflect the use of Mg2+: we observe the same rate enhancement of mismatches in the presence of 0.5 mM Mn2+ for each of two guides tested (Supplementary Fig. 6g). Similarly, RNA cleavage by RNA-guided, eukaryotic Ago proteins often also tolerates mismatches between the guide and target, with specific mismatches increasing the speed of single-turnover cleavage above that observed for perfectly matched substrates(Becker et al., 2019; Chen et al., 2017).
Surprisingly, we often observed increased cleavage rates when mismatches were present in the central region (t6–t10) of the target. To approximate single-turnover conditions for assaying cleavage, we used the 55°C TtAgoDM binding data to exclude targets not predicted to be fully bound by TtAgo under the conditions of the cleavage experiment (KD at 55°C >1.4 nM). Strikingly, for target sequences saturated with TtAgo-bound guide, perfectly matched target for guides 2 and 3 did not detectably cleave at 55°C, yet 83% (574 targets, guide 2) to 91% (935 targets, guide 3) of double mismatched targets had measurable cleavage rates (Fig. 6d,e). For these guides, mismatches in the central region of the target (t6–t10) most increased the single-turnover cleavage rate. These results show that mismatches in the central region of the guide can increase cleavage rates for a number of guide sequences, an observation reminiscent of cleavage rate-enhancements seen for mouse AGO2 with mismatches at t8 and t12 (Becker et al). Surprisingly, many targets with mismatches at positions t10 and t11 were also cleaved by TtAgo; mismatches at these positions disrupt target cleavage by mouse AGO2 or fly Ago2 (Becker et al., 2019; Elbashir et al., 2001; Hutvagner, 2002; Wee et al., 2012), highlighting both the similarities and differences between in the sequence preferences of TtAgo and eukaryotic Argonaute proteins.
Mismatches in the distal region of the target (t12–t16) did not enhance the cleavage rate, and mismatches at t13 were particularly poorly tolerated, even when paired with mismatches that enhanced cleavage (e.g., at positions t6–t10). Target position t13 is similarly critical for efficient cleavage by mouse AGO2(Becker et al., 2019). Therefore, base pairing in this region appears to be a universal requirement for cleavage by Ago proteins, suggesting this position may be necessary to permit cleavage-capable Ago proteins to adopt a cleavage-competent configuration.
Discussion
Nucleic acid hybridization under biological conditions is plagued by a lack of specificity and slow, highly variable on-rates(Cisse et al., 2012; Ross and Sturtevant, 1960; Wetmur and Davidson, 1968; Zhang et al., 2012, 2018). These characteristics present a challenge for biological processes that use nucleic acid guides to direct target binding. Indeed, Cas proteins also have a seed-like region that speeds target finding(Boyle et al.; Gorski et al., 2017; Liu et al., 2017; Swarts et al., 2017b). TtAgo, like other previously studied pAgos and eAgos, relies on a pre-organized seed region to accelerate the rate of nucleic acid hybridization (Fig. 2). Not all bases of the seed are equivalent however: like mouse AGO2, TtAgo on-rate enhancement depends on base pairing to guide bases g2–g4 in the initial target search (Fig. 2c). However, nucleotide composition and nucleic-acid derived binding energy of the larger seed region, g2–g7, also plays a role in effective, rapid target binding (Fig. 2a). The ability to accelerate and decrease the on-rate variability of nucleic acid association rates appears to be a universally conserved feature of Ago proteins.
Many eukaryotic Argonaute proteins require only short regions of complementarity to the seed region of the loaded RNA guide to bind targets with sub-nanomolar affinity(Salomon et al., 2016; Wee et al., 2012). This trait is well suited to Agos that load miRNAs to bind and regulate diverse mRNA targets(Baek et al., 2008; Selbach et al., 2008). In contrast, TtAgo serves two distinct biological functions: 1) it functions in host defense by directing cleavage of invading DNA elements using guides acquired from the same invaders, and 2) it aids in replication by resolving catenated chromosomes, potentially by recruiting recombination factors to the replication terminus using guides derived from that same region (Jolly et al., 2020; Swarts et al., 2014b). The greatest degree of TtAgo-mediated target affinity enhancement requires binding to the proximal seed region, and conversely, mispairing here can result in weaker binding than even the unloaded guide (Fig. 3b,d,e,4d). These effects likely minimize the time that TtAgo spends at off-target sequences lacking a proximal seed match, improving the specificity of target interactions. TtAgo binding is also sensitive to mismatches outside the seed, particularly for bases that provide the most energy to nearest neighbor base pairing of the constituent nucleic acids (Fig. 4a,b). Thus, like other Ago proteins, pairing to the seed region is generally necessary for high affinity target binding by TtAgo, but rarely suffices for sub-nanomolar affinity interactions.
Among those prokaryotic “long” Ago proteins whose domain architecture is similar to that of eAgos, less than one-quarter are predicted to retain catalytic activity(Ryazansky et al., 2018; Swarts et al., 2014a). While TtAgo is a catalytically active long pAgo, its cleavage activity varies dramatically among different guide sequences. Even at 65°C, only one of the five guides tested showed robust cleavage activity, and all three of the endogenous TtAgo guides cleaved slowly relative to AT-rich guides, except when targeting sequences containing central (t6–t10) mismatches (Fig. 6a,d,e). The relatively slow cleavage activity of endogenous guides is likely in part explained by their high overall GC content and predominance of guanosine at target position 1 (t1G) in naturally occurring complementary targets (Hunt et al., 2021). The degree to which mismatches enhanced cleavage by TtAgo was unexpected, given that TtAgo is thought to be loaded with guides fully complementary to its intended targets(Jolly et al., 2020; Swarts et al., 2014b). It is unclear how enhanced cleavage of mismatched targets would benefit TtAgo’s function in resolving catenated chromosomes, but it is possible that enhanced cleavage of incompletely matched targets helps suppress the evolution of resistance in invading DNA elements via single base changes. Because TtAgo reaches optimal cleavage activity at temperatures ≥ 65°C(Jolly et al., 2020; Swarts et al., 2014b), we speculate that mismatched targets of GC-rich endogenous guides may mimic the higher conformational flexibility near the center of the guide:target duplex of AT-rich guides at elevated temperatures. Mismatches near the cleavage site were surprisingly well-tolerated by TtAgo, suggesting that positioning of the target strand backbone into the cleavage site of TtAgo is not critically dependent on guide strand pairing at the cleavage site (Fig. 6d–e). However, pairing at position 13 is critical for cleavage by TtAgo, a property shared with mammalian AGO2 (Becker et al., 2019). For TtAgo specifically, pairing at position 13 may stabilize the catalytically competent “plugged-in” conformation of the protein: the backbone of guide base g13 makes contact with the backbone of the Glu residue (E512) that is repositioned into the catalytic pocket of the protein (Sheng et al., 2014). We note, however, that pairing at position 13 is observed in both the catalytically incompetent and catalytically competent structures of the protein, suggesting that pairing at position 13 is at most a necessary, but not sufficient condition to promote the required conformational transition (Supplementary Fig. 7). Furthermore, mammalian AGO2 does not appear to adopt the “unplugged” conformation of TtAgo in its catalytically inactive state(Schirle et al., 2014), indicating that pairing at position 13 for mammalian AGO2 likely promotes cleavage by some other mechanism.
With the caveat that in vitro experimental conditions that cannot perfectly recapitulate the T. thermophilus in vivo environment, the slow (relative to AT-rich guides) catalytic activity of TtAgo directed by the three naturally occurring guides derived from the T. thermophilus genome supports the possibility that direct cleavage is not the sole mechanism through which TtAgo resolves catenated replicated chromosomes. This conclusion is supported by the observation that a T. thermophilus strain with catalytically inactive TtAgo had a less severe filamentation phenotype than the strain with a full TtAgo knockout, and that TtAgo is capable of recruiting additional recombination factors to bound targets(Jolly et al., 2020; Swarts et al., 2014b). One possibility, supported by previous in vivo TtAgo studies(Jolly et al., 2020), is that less GC-rich guides acquired from invading viruses enable TtAgo to directly cleave invader DNA, while GC-rich guides derived from the TtAgo genome bind tightly to genomic targets to recruit recombination factors but are less capable of promoting physiologically-relevant target cleavage. Additionally, if these GC-rich guides do mediate target cleavage in vivo, the relatively slower cleavage rate enhances target specificity, reducing the risk of off-target cleavage of the T. thermophilus genome. In this manner, T. thermophilus might utilize TtAgo for multiple distinct biological functions by linking efficient cleavage activity to the source of the guide sequence itself. These findings highlight the potential value of studying multiple guides and targets for other nucleic-acid guided proteins, as different guide sequence(s) may reveal a surprising range of biochemical activities and lead to testable hypotheses for biological function.
Limitations of the study
For association experiments, our data quality filtering procedure is more likely to exclude low-affinity targets which had lower binding saturation and consequently lower confidence fit curves at the concentrations used for the association experiments (Methods). This filtering procedure could in theory introduce bias into our estimates of the effect of mismatches on the association rate if: 1) many targets with mismatches at a certain position were excluded due to being low affinity, and 2) the association rate was strongly confounded with the affinity of those targets.
Our TtAgo association experiments revealed that guide 4 did not follow the same trend in association behavior as the other four guides—mismatches outside of the seed region significantly slowed the association rate relative to the fully complementary target (Figure 2A). For this reason, we had to exclude this guide from the training and testing of our general TtAgo association model and thus the inferences obtained from this model are inherently limited to this majority class of TtAgo guides. With only five total guides profiled in this study, we were unable to determine whether the association behavior of this guide was a unique outlier, or if there exists a broader class of TtAgo guides that display similarly distinct association kinetics. Answering this question would require testing many more TtAgo guides, or development of higher-throughput experimental approaches to measure association rates.
In vitro experiments such as those performed here cannot perfectly recapitulate the T. thermophilus intracellular environment. For example, much of our TtAgo binding data was obtained at 37°C, a temperature that may be more useful for understanding how TtAgo alters the biophysical properties of its loaded guide, rather than most accurately recapitulating the binding behavior of TtAgo in vivo. This limitation is particularly relevant to hypotheses based on the absolute rates of TtAgo cleavage for various guides. While GC-rich endogenous TtAgo guides cleaved slowly compared to AT-rich or optimally mismatched guides in all conditions tested in this study, suggesting that cleavage may not be their primary biological role, in vivo T. thermophilus experiments will be required to further refine our understanding of how TtAgo uses both endogenous and exogenously derived guides in its native biological context.
STAR Methods
Resource Availability
Lead Contact and Materials Availability
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, William Greenleaf (wjg@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The kinetic and thermodynamic measurements generated in this paper are available for download (Supplementary Table 1) and on Mendeley Data (http://doi.org/10.17632/vrb6n8gm3x.1).
Custom software for determination of association rates, cleavage rates, and dissociation constants is available on GitHub (https://github.com/GreenleafLab/TtAgo) and on Mendeley Data (http://doi.org/10.17632/vrb6n8gm3x.1).
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
Method Details
Guide Selection and Library Design
Of the five TtAgo guides used, guide 1 (5′-CCATGGGCACGCAGAA), guide 2 (5′-CTCCGCCTCTTCCAGA), and guide 3 (5′-CGCTCCAGGAGGGAAA), are endogenous guides isolated from Thermus thermophilus(Jolly et al., 2020). Guide 4 is the first 16 DNA bases corresponding to the let-7a miRNA sequence (5′-TGAGGTAGTAGGTTGT), and guide 5 is a randomly generated sequence with an AT-rich seed region (5′-CAATTACCTGGCATCA). Guides were selected to have a high pairwise Levenshtein, or edit distance, with the closest pair of guides (guides 1 and 5) having an edit distance of 8, and the median edit distance of all pairs being 10. Guides were ordered from IDT with 5′ Phosphates and 3′ Cy3 fluorophores and underwent HPLC purification.
Target libraries were designed for each guide. Each target library included all single mismatches, all double mismatches, a subset of triple mismatches, all single insertions and deletions, homopolymer base insertions up to 5 bases, and a selection of higher-order mismatched targets. The target library for guide 5 was originally designed for the guide sequence 5′-AAATTACCTGGCATCA, and therefore all variants naturally have a thymine at target position 1 (t1). This is indicated with an asterisk in any figures with this guide, and the sequences provided in the data table correspond to the actual sequences used in all experiments. All targets were placed in the same flanking sequence context, which in most cases consisted of five flanking adenines on the 5′ and 3′ ends of the target. The total sizes of individual target libraries ranged between 3,224 and 3,256 unique targets, and the total number of unique targets was 16,169. A graphical overview of the library design for a single guide is shown in Supplementary Fig. 1a, and Supplementary Table 1 contains the full list of sequences used.
Assembly and sequencing of library
Target libraries were ordered from Custom Array (Bothell, WA). Each target variant was flanked by common 5′ and 3′ priming sequences. The synthesized libraries were assembled into full constructs compatible with Illumina paired end sequencing. Assembly reactions were carried out in a 20 μL volume of 1× NEBNext Master Mix (NEB, M0541) with ~10 nM synthesized library, 10 nM T7A1_NexteraR1 oligo, 50 nM of C_i7_bc_T7A1, 50 nM of D_designed_lib_R2, and 250 nM of both C and D primers (all oligo sequences available in Supplementary Table 2). SYBR green was added at a final concentration of 0.6× to assembly reactions so that assembly progress could be monitored. Assembly reactions underwent cycles of 98°C for 10 s, 63°C for 30 s, and 72°C for 30 s until the SYBR green signal of a reaction began to plateau (17 cycles), after which the reaction was stopped and assembled products were purified using a QIAquick PCR purification kit. A small amount of the purified product was visualized on an agarose gel to confirm assembly of the desired product. The assembled target library was quantified for sequencing by performing a qPCR against a PhiX standard curve, using 250 nM each of C and D primers. Fiducial mark constructs were assembled separately in the same manner described above, except that the fiducial sequences were ordered as single oligos from IDT.
The target library was sequenced on an Illumina MiSeq instrument using a custom read 2 primer complementary to the 3′ priming sequence of the library (the 5′ priming sequence was the same as the Nextera R1 sequence and therefore did not require a custom primer). Target libraries generally made up 20–25% of the total sequencing chip, with the remaining sequence space being high-complexity genomic libraries. Libraries were sequenced using paired end sequencing with 76-bp reads.
Processing sequencing data
Following sequencing, tile and x, y coordinates of each cluster were extracted from fastq files. Clusters that were members of the target library were identified by aligning to a portion of the common 3′ priming sequence (5′-CGGACGCGGGAAGACAGAAT). The fiducial marks were identified by aligning to the fiducial sequence (5′-TAGCCAGCCTGATAAGTAACACCACCACTG). Fiducial marks and library members identified in this manner were used for registering the sequence data to experimental images. Only clusters that exactly matched a known library sequence in both reads were fit in downstream data analysis.
TtAgo expression and purification
Wild-type or mutant TtAgo (TtAgoD478A,D546A or TtAgoDM) cloned and expressed from pET-SUMO (Invitrogen K30001) in E. coli BL21-DE3 was purified as described (Wang et al., 2008a) except: (1) After cleavage and removal of the 6×His-SUMO tag, wild-type or mutant TtAgo was additionally purified by HiTrap SP HP (GE Healthcare) chromatography, dialyzed against 3 × 2 L 20 mM HEPES-KOH pH 7.4, 250 mM potassium acetate, 3 mM magnesium acetate, 0.1 mM 2,2′,2′′,2′′′-(ethane-1,2-diyldinitrilo)tetraacetic acid, 10% glycerol (w/v), 5 mM DTT (TtAgo) or 30 mM HEPES-KOH, pH 7.4, 250 mM potassium acetate, 1 mM DTT, 0.01% Igepal CA-630, 20% (v/v) glycerol (TtAgoDM). Purified protein was aliquoted into tubes, flash frozen in liquid nitrogen, and stored at −80°C. Protein was quantified by Bradford Assay using BSA as standard. Purification of TtAgo with magnesium buffers yields guide-free TtAgo and purification with manganese containing buffers retains endogenous guides(Swarts et al., 2014b). TtAgo, purified as above with magnesium containing buffers, has an A260/A280 ratio of 0.56, indicating it to be free of nucleic acids. SYBR Gold (Invitrogen) staining did not detect nucleic acids.
Imaging station setup
High-throughput, quantitative biochemical measurements were made using the sequenced MiSeq flow cell and a custom imaging platform as described in (She et al., 2017) and (Becker et al., 2019). Briefly, the lasers, Z-stage, XY-stage, syringe pump, camera and objective lens were salvaged from Illumina GAIIx instruments and were combined with a fluidics adaptor designed to interface with Illumina MiSeq chips. The instrument was additionally outfitted with a temperature control system and laser control electronics. Imaging was performed using either a 400 ms exposure time at 150 mW fiber input power of a 660 nm laser and a 664 nm long pass emission filter (Semrock) or with a 600 ms exposure time at 150 mW input power of a 532 nm laser and a 590 nm center wavelength and 104 nm guaranteed minimum 93% bandwidth bandpass emission filter (Semrock).
Preparation of flow cell for binding experiments
Following sequencing, MiSeq flow cells containing the sequenced target library were loaded onto the custom imaging station. All pump, temperature change, stage movements and imaging steps were performed using custom xml scripts and MATLAB control software. Unless stated otherwise, pump volumes were 100 μL and were flowed at 100 μL/min.
Post-sequencing process of flow cell
For the first experiment following sequencing, DNA not covalently attached to the flow cell surface was removed by heating the flow cell to 55°C and washing with 100% (v/v) formamide. Residual fluorescence from sequencing reversible terminators was removed by further heating the flow cell to 60°C and incubating in 80 mM Tris-HCl, pH 8.0, 80 mM NaCl, 0.05% (v/v) Tween 20, 100 mM tris(2-carboxyethyl)phosphine (TCEP) for 10 min.
Hybridizing blocking and fiducial mark oligos
In preparation for experiments, the non-variable regions of the single stranded DNA clusters from the target library were blocked using short oligos complementary (Supplementary Table S2) to common regions of the library constructs. Fluorescently labeled fiducial mark oligos (Supplementary Table S2) were hybridized to the chip surface at the same time. Hybridization of both the blocking oligos and fiducial marks was carried out in multiple phases. First, the flow cell was heated to 60°C and washed with Hybridization buffer (5× SSC buffer (ThermoFisher 15557036), 5 mM EDTA, 0.05% (v/v) Tween 20). All blocking oligos and fluorescently labeled fiducial marks were diluted in Hybridization buffer to a final concentration of 500 nM each, and the flow cell was incubated in this blocking mixture for 12 min at 60°C. The temperature of the flow cell was then dropped to 40°C for an additional 12 min. The flow cell was then washed with Annealing buffer (1× SSC, 5 mM EDTA, 0.05% (v/v) Tween 20). The temperature was dropped to 37°C, after which the flow cell was incubated in Annealing buffer containing 500 nM of each oligo for 10 min. After hybridization of blocking and fiducial oligos was complete, the flow cell was washed with 300 μL of Annealing buffer.
Measurement of association rates and equilibrium dissociation constants
Loading TtAgo
All selected TtAgo guides had large pairwise edit distances, and therefore each TtAgo guide only appreciably bound targets within its own library (Supplementary Fig. 1b). Because of this, TtAgo and guide alone experiments were carried out with each of the five guides pooled together. Despite this, each of the five TtAgo guides was loaded in a separate loading reaction. Single-use aliquots of TtAgoDM were thawed on wet ice. Loading reactions were prepared in TtAgo sample buffer (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 3 mM MgCl2, 5 mM DTT, 0.05% (v/v) Tween 20) with 500 nM Cy3-labeled guide and 1 μM unloaded TtAgoDM in a final volume of 25 μL. Loading reactions were heated at 75°C for 30 min. Following loading, each TtAgoDM:guide complex was combined into a single volume with the final concentration of each guide at 100 nM. All subsequent TtAgo concentrations reference the loaded concentration, which corresponds to the amount of guide in the loading reaction.
TtAgo binding experiments on imaging station
TtAgoDM loaded with Cy3-labeled guides was flowed into the prepared MiSeq flow cell at various concentrations to measure association kinetics. TtAgoDM association was measured at 37°C at 55 pM, 131 pM, 314 pM, and 754 pM. During association, tiles within the flow cell were imaged continuously during the first 20 min, spaced approximately every 90 sec. Association experiments lasting longer than 20 min had additional images taken at log-spaced intervals until the end of the experiment. Between association experiments, the chip was washed with 500 μL TtAgo sample buffer and then all protein and DNA not covalently attached to the flow cell was removed by heating the chip to 55°C and flushing with 100% formamide. Blocking and fiducial oligos were re-annealed prior to beginning the next experiment.
TtAgoDM equilibrium binding experiments were performed at both 37°C and 55°C. For both temperatures, increasing concentrations of loaded TtAgoDM were introduced into the flow cell, allowed to equilibrate, and then imaged. The concentrations of loaded TtAgoDM used were 55 pM, 131 pM, 314 pM, 754 pM, 1.81 nM, 4.34 nM, and 10.42 nM. After the final concentration, the flow cell was regenerated as described above.
Unloaded guide binding experiments on imaging station
Binding experiments using unloaded guides were performed on the imaging station similarly to the TtAgoDM experiments. Cy3-labeled guides were diluted in TtAgo sample buffer to the required concentrations for association and equilibrium binding experiments. Both the association and equilibrium binding experiments were performed at 37°C. Unloaded guide association experiments were performed using 480 pM, 1.25 nM, 3.24 nM, 8.42 nM, and 21.9 nM of each guide. Unloaded guide equilibrium binding experiments were performed using 480 pM, 1.25 nM, 3.24 nM, 8.42 nM, 21.9 nM, 56.9 nM, 148 nM, 385 nM, and 1 μM of each guide.
Measurement of cleavage rates on imaging station
To measure cleavage rates of TtAgo for target libraries, the flow cell was prepared as described above. The distal end of each cluster was labeled using an oligo with a red fluorophore (Supplementary Table S2). Instead of the catalytically inactive TtAgoDM, wild-type TtAgo was loaded with guides as described above. The flow cell was heated to 55°C, and 25 nM guide-loaded TtAgo was applied to the flow cell. Tiles were imaged in log-spaced intervals over 24 h to measure cleavage.
Measurement of individual target cleavage rates
To validate cleavage rates measured on the imaging station, in-solution cleavage experiments were performed using a single TtAgo:guide and target per reaction. Targets were ordered from IDT and contained a common fluorescent probe binding site and a variable target region (Supplementary Table S2). Cleavage experiments were performed in TtAgo sample buffer (50 mM Tris-HCl, pH 8.0, 200 mM NaCl, 3 mM MgCl2, 5 mM DTT, 0.05% (v/v) Tween 20) supplemented with either 3 mM MgCl2 or 0.5 mM MnCl2. Before cleavage reactions were prepared, catalytically active TtAgo was loaded with the appropriate guide as described above. Cleavage reactions were prepared on wet ice in TtAgo sample buffer with 25 nM (f.c.) guide-loaded TtAgo 2.5 nM (f.c.) target ssDNA. Cleavage reactions were aliquoted into 10 μL volumes and then incubated at either 55°C or 65°C in a prewarmed thermocycler. After the indicated amount of time had elapsed, individual aliquots of cleavage reactions were quenched on dry-ice and stored at –80°C until all reactions had completed. For each experiment, one aliquot was quenched and stored immediately after mixing to serve as a zero time point. All reactions were thawed on wet ice and excess of Alexa-647-labeled ssDNA probe was added to a final concentration of 5 nM. Next, 1 μL Proteinase K (New England Biolabs, P8107S, 800 units/μL) was added to each reaction, incubated at 25°C for 15 min, and then denatured at 95°C for 10 min. Following denaturation, reactions were cooled slowly down to 25°C (10 min) to anneal the fluorescent probe. Each reaction was then mixed with loading dye and resolved by 10% polyacrylamide gel electrophoresis. Gels were visualized using a fluorescent gel imager and bands were quantified using GelAnalyzer 19.1 [(www.gelanalyzer.com) by Istvan Lazar Jr., PhD and Istvan Lazar Sr., PhD, CSc]. Cleavage rates were fit to a single exponential decay where indicates the uncleaved proportion of target at each time point:
with being the uncleaved proportion of target at each time point, uncleaved fraction at infinite time, and being the observed cleavage rate.
Co-Localization Single-Molecule Spectroscopy
TtAgoDM was assembled with guides bearing a 3′ Alexa Fluor 555. Concentration of active protein was measured using double filter binding assay as described(Wee et al., 2012). Single-stranded DNA targets were generated as previously described(Salomon et al., 2016). In a typical labeling procedure, 100 pmol DNA target (Supplementary Table S2) was mixed with a 1.5-fold molar excess of Klenow template oligonucleotide (Supplementary Table S2) in 7.5 μl of 10 mM HEPES-KOH, pH 7.4, 20 mM sodium chloride and 0.1 mM EDTA. Samples were incubated at 90°C for 5 min in a heat block. Then, the heat block was switched off and allowed to cool to room temperature. Afterwards, the annealed strands (30% of final reaction volume) were added without further purification to a 3′ extension reaction, comprising 1× NEB buffer 2 (New England Biolabs, Ipswich, MA), 1 mM dATP, 1 mM dCTP, 0.12 mM Alexa Fluor 647aminohexylacrylamido‐ dUTP (Life Technologies), and 0.2 U/μl Klenow fragment (3′-to5′ exo‐ minus, New England Biolabs) and incubated at 37°C for 1 h. The reaction was quenched with 500 mM (f.c.) ammonium acetate and 20 mM (f.c.) EDTA. A 15- fold molar excess of ‘trap’ oligonucleotide (Supplementary Table S2) was added to the Klenow template oligonucleotide. The entire reaction was precipitated overnight at ‒ 20°C with three volumes of ethanol. The labeled target was recovered by centrifugation, dried, dissolved in loading buffer (7 M Urea, 25 mM EDTA), and incubated at 95°C for 5 min. The samples were resolved on 6% polyacrylamide gel and isolated by electroelution.
Single-molecule experiments were performed and analyzed as described(Smith et al., 2019). Fresh cover glasses were prepared for each day of imaging. Cover glasses (Gold Seal 24 Å~ 60 mm, No. 1.5, Cat. #3423), and glass coverslips (Gold Seal 25 Å~ 25 mm, No. 1, Cat. #3307) were cleaned by sonicating for 30 min in NanoStrip (KMG Chemicals, Houston, TX), were washed with 10 changes of deionized water and were dried with a stream of nitrogen. Two ~1 mm diameter lines of high vacuum grease (Dow Corning, Midland, MI) were applied to the cover glass to create a flow cell. Three layers of adhesive tape were applied outside of the flow cell. The coverslip was placed on top of the cover glass, with a ~0.3 mm gap between the cover glass and coverslip. To minimize non-specific binding of protein and DNA molecules to the glass surface, microfluidic chambers were incubated with 2 mg/mL poly-L-lysine-graft-PEG-biotin in 10 mM HEPES- KOH, pH 7.4 at room temperature for 30 min and washed extensively with imaging buffer (30 mM HEPES-KOH, pH 7.9, 120 mM potassium acetate, 3.5 mM magnesium acetate, 20% (w/v) glycerol) immediately before use. To allow immobilization of biotinylated DNA targets, streptavidin (0.01 mg/mL, NEB) was incubated for 5 min in each microfluidic chamber. Unbound streptavidin was washed away with imaging buffer.
Immediately before each experiment, a flow cell was incubated with imaging buffer supplemented with 75 μg/mL heparin (Sigma H4784), oxygen scavenging system (2.5 mM protocatechuic acid (Aldrich 37580) and 0.5 U/mL Pseudomonas sp. protocatechuate 3,4- Dioxygenase (Sigma P8279)) and triplet quenchers (1 mM trolox (Aldrich 238813), 1 mM propyl gallate (Sigma P3130), and 1 mM 4 nitrobenzyl alcohol (Aldrich N12821)) for 2 min. Then, it was filled with ~100 pM target in imaging buffer supplemented with 75 μg/mL heparin, oxygen scavenging system and triplet quenchers. Target deposition was monitored by taking a series of images; once the desired density was achieved, the flow cell was washed three times with imaging buffer supplemented with oxygen scavenging system and triplet quenchers. A syringe pump (KD Scientific, Holliston, MA) running in withdrawal mode at 0.15 mL/min was applied to the flow cell outlet to introduce TtAgo:guide complex (pre-heated to 37°C) supplemented with an oxygen scavenging system and triplet quenchers. Typically, 1,500 frames were collected at 5 frames per s. A digitally controlled heater (TP-LH, Tokai Hit) maintained objective temperature at 42°C to achieve sample temperature of 37°C. Temperature on the surface of the cover glass was independently monitored with a Type E, 0.25 mm O.D. thermocouple (Omega Engineering Inc., Sutton, MA) inserted between the top and the bottom cover glasses.
Imaging was performed on an IX81- ZDC2 zero- drift inverted microscope equipped with a cell^TIRF motorized multicolor TIRF illuminator with 561 and 640 nm 100 mW lasers and a 100×, oil immersion, 1.49 numerical aperture UAPO N TIRF objective with FN = 22 (Olympus, Tokyo, Japan). Fluorescence signals were split with a main dichroic mirror (Olympus OSF-LFQUAD) and triple emission filter (Olympus U-CZ491561639M). The primary image was relayed to two ImagEM X2 EM-CCD cameras (C9100–23B, Hamamatsu Photonics, Hamamatsu, Japan) using a Cairn three-way splitter equipped with a longpass dichroic mirror (T635lpxr-UF2, Chroma). Illumination and acquisition parameters were controlled with cell^TIRF and MetaMorph software (Molecular Devices, Sunnyvale, CA), respectively.
Images were recorded as uncompressed TIFF files and merged into stacked TIFF files. Images were processed using the pipeline(Smith et al., 2019) as described in the manual. Co-localization events required that (1) the intensity of TtAgoDM:guide complex > 150 photons, (2) ratio intensity of the TtAgoDM:guide complex to the local background > 1, (3) the distance between the target and guide was < 1.2 pixel, and (4) sigma < 4.6. To exclude short, non-specific events, the minimal event duration was set to 5 frames. To overcome short temporary loss of TtAgoDM fluorescent signal due to blinking of the fluorescent dye, the gap parameter was set to 2 frames. Only the first binding event at each target location was used for estimation of arrival time and dwell time, in order to minimize errors caused by occupation of sites by photobleached molecules. The same analysis was automatically performed on ‘dark’ locations, i.e., regions that contained no target molecules; these served as a control for non-specific binding of TtAgoDM:guide complex to the surface of the cover glass. The individual experiments were saved, combined, and error evaluated by 1,000-cycle bootstrapping of 90% of the data.
Quantification and statistical analysis
Data processing and image fitting
For each experiment, sequence data was mapped to images collected on the imaging station. Individual sequenced clusters were mapped to each image through an iterative cross-correlation procedure that made use of fiducial marks and known target library members (Denny et al., 2018; She et al., 2017). After coordinate mapping, each cluster was fit to a 2D Gaussian to quantify fluorescence.
Association curve fitting
Following quantification of cluster intensity at each time point, association rates were fit for each variant. To account for variability between illumination and focus in each imaging cycle, the fluorescence intensity at each timepoint was normalized by dividing by the median fluorescence intensity of a fiducial mark (a fluorescent DNA oligonucleotide hybridized directly to single stranded DNA) that otherwise should have constant fluorescence intensity during the experiment. Observed rate constants were determined by fitting the median fluorescence of all clusters corresponding to a particular target sequence to the following single exponential:
with being the observed fluorescence intensity, the fluorescence intensity at infinite time, fmin is the fluorescence intensity at time 0, and being the observed rate. Least-squares fitting here and for the equilibrium and cleavage fitting below was carried out using the python package lmfit.
Error in the measurement of the observed rates was estimated by bootstrapping the clusters representing each molecular variant. All clusters representing a single variant were sampled with replacement and the median fluorescence of the resampled clusters was fit to the above equation. This was repeated 1,000 times to generate 95% confidence intervals on the observed rate constant fits.
Observed rates obtained from multiple association experiments were used to estimate association rates using the following equation:
with kobs being the observed rate, the association constant, the dissociation constant, and [guide] the concentration of guide-loaded TtAgoDM or the concentration of guide alone, depending on the experiment. To obtain estimates of error on association rates, values from the previous bootstrapping procedure were randomly sampled and used to refit the above equation. This was repeated 1,000 times to generate 95% confidence intervals for association rate fits. For downstream analyses, we filtered association data for fit quality by requiring each target to be defined by at least two binding rates from separate association experiments, and a fit with R2 ≥ 0.8. This filtering procedure resulted in reported measurements for 86.3% of possible TtAgo targets. Lower affinity targets were more likely to be filtered out of the final dataset, as these were more likely to have insufficient binding saturation for multiple association experiments.
We observed some differences in measured absolute association rates between single molecule experiments and binding experiments performed on the MiSeq imaging station (Supplementary Fig. 2A,B). It is possible that the more dense surface of the MiSeq flow cell produces more significant surface effects, and consequently relatively slower association rates, than observed in single-molecule experiments. For this reason, we chose to only report and focus our analyses on relative association rates for TtAgoDM measured on the MiSeq imaging station.
Equilibrium binding curve fitting
To better estimate dissociation constants for target sequences that were not fully bound at the highest guide or TtAgoDM concentration, binding curves for low affinity targets were fit using an estimated distribution of values. To obtain this distribution, the median fluorescence values for each variant were fit to:
with being the fluorescence intensity, the fluorescence intensity when the target is fully bound, the fluorescence intensity of the unbound target, [guide] the concentration of guide-loaded TtAgoDM or of guide alone, and Kd the dissociation constant. Because fmin did not vary appreciably between targets, it was constrained to be the median fluorescence intensity of all clusters in the absence of any labeled guide. The distribution of fmax was estimated by selecting all variants with KD values corresponding to >97.5% binding saturation at the highest experimental concentration.
For final KD and error estimation, the distribution was enforced for targets that (1) were not used in constructing the distribution, and (2) did not achieve a maximum median fluorescence value above the lower limit of the 95% confidence interval of the distribution. For targets that did not meet these criteria, the parameter was allowed to float during fitting. To get an estimate of the error for values, all clusters of a given target were sampled with replacement and the median fluorescence intensities of the sampled clusters was refit. This was repeated 1,000 times for each target, and used to obtain a 95% confidence interval on KD fit values. The median value for KD from bootstrapped fits was used as the final reported fit value. For downstream analyses, we quality filtered targets that did not have either a fit with R2 ≥ 0.9 or a fit RMSE < 0.1. We also thresholded data that fell above or below our limits of detection. Targets that were not at least 10% bound at the highest concentration (KD of 90 nM for TtAgo, 8.6 μM for unloaded guides) were labeled as exceeding the upper LOD and targets that were still greater than 85% bound at the lowest concentration (KD of 10 pM for TtAgo, 85 pM for unloaded guides) were labeled as being below the lower LOD. We converted KD to ΔG values in units of kBT by taking the natural log.
Cleavage rate fitting
To measure cleavage rates of catalytically active TtAgo, the distal ends of clusters were fluorescently labeled with a red fluorophore (Alexa-647N). Loss of fluorescence intensity in the red channel therefore reported on cleaved clusters being released from the flow cell surface. Raw fluorescence intensity was normalized at each timepoint by dividing by the median fluorescence of 105 highly degenerate target sequences with long stretches of central and seed mismatches to all guides. These sequences were confirmed to have no detectable TtAgo binding even at the highest concentration used for equilibrium binding experiments. Following normalization, cleavage rates and 95% confidence intervals were determined using a similar bootstrapping procedure as described for binding curve fitting, except that the median fluorescence of sampled clusters was fit to the following single exponential decay:
with fintensity being the observed fluorescence intensity, the fluorescence intensity at time 0, is the fluorescence intensity at infinite time, and being the observed cleavage rate. For downstream analyses, we quality filtered targets that did not have either a fit with R2 ≥ 0.9 or a fit RMSE < 0.1, as well as requiring that the fit fmax be at least 0.3 (corresponding to ~30% of maximum signal).
Association model fitting
The TtAgo association binding model was fit using only guides 1–3 and 5, since guide 4 was an outlier in our association data set. Using the remaining four guides, we first filtered association data for fit quality by requiring each target to be defined by at least two kobs binding rates from separate association experiments (see above), and a fit with R2 ≥ 0.8. We further restricted the targets examined to include only substitution variants, i.e. targets for which the guide and target were the same length. We calculated rates for each target from its guide-matched fully complementary target. We removed outlier targets defined as those that had rates above the 99th percentile or below the 1st percentile. This resulted in 7,879 total targets used for model fitting (2,450 for guide 1, 1,823 for guide 2, 2,417 for guide 3, and 1,189 for guide 5).
We observed that the predicted nearest neighbor stability of the seed region contributed to the observed association rate, but only when that energy dropped below a certain level (Supplementary Fig. 2e). This may reflect a seed energy threshold requirement for transitioning from an initial collision into stable target binding. We used NUPACK(Zadeh et al., 2011) to predict the nearest neighbor energy of the 5-mer spanning the seed sequence (g2–6) and transformed this energy term into an association penalty using a logistic function (scaled seed ΔG, 3 free parameters). In both the association and binding models, we used selected NUPACK to estimate thermodynamic parameters for nucleic acid interactions for several reasons: 1) it is able to estimate the full partition function and MFE structure for inter- and intra-molecular DNA strands at user-specified salt concentrations and temperatures, 2) it allows us to predict thermodynamics for subsequences of our actual targets, (e.g. for the seed region alone), 3) it has user-friendly command-line tools for estimating the parameters of thousands of sequences at a time, and 4) the software does not require a license for non-commercial uses.
This scaled k-mer energy alone poorly estimates the change in association rate for most targets, because TtAgo modifies the relative importance of base pairing at certain positions in the guide:target duplex. To account for this, we defined 15 guide-target duplex position weight parameters from g2:t2 through g16:t16 (w2–w16), 12 mismatch identity parameters (e.g. gA:tG, gC:tA, gT:tC, etc.), and 3 t1 base parameters (t1A, t1C, t1T). t1G and correct base-pairings (e.g. gA:tT, gG:tC) were defined to have no penalty, mismatch identity parameters were constrained during fitting to be negative, and w2 was set to 1. Thus, for each position, the position weight parameter (wp) is multiplied by the mismatch identity parameter (Mp) to give the overall association penalty at that position. Finally, we included a scaling parameter on the NUPACK-predicted target secondary structure energy to account for the effect of binding sites being sequestered in inaccessible folded conformations (internal structure ΔG). In total, this resulted in 33 parameters being fit in the association model. This model can be interpreted as a summation of modifications to an energy barrier for association.
Model performance and variability of model parameters were estimated using leave-one-out cross validation (i.e., model was trained on data from three guides and tested on the guide not used in training). The median Pearson correlation of models tested on held-out guides was 0.71, and the Pearson correlation of the model trained on all data was 0.79. All models were fit using Ridge regression to stabilize parameter estimates, and fitting was performed using the lmfit module in Python 3.6.
Binding model fitting
A general binding model was fit using all five guide sequences. To fit the parameters of the binding affinity model fitting, we only included target sequences that did not contain designed target insertions or deletions. We also filtered out target sequences with fit affinities near the boundaries (ΔG (kBT) ≥ −17 and ΔG (kBT) ≤ −24) or 95% confidence intervals on the fit affinity of greater than or equal to 4 kBT. This resulted in 7,647 total targets used for model fitting (1,827 for guide 1, 1,621 for guide 2, 1,043 for guide 3, 1,164 for guide 4, and 1,992 for guide 5). To predict the binding affinity of different TtAgo:guide complexes for target sequences we partitioned the affinity into energetic contributions from DNA:DNA interactions and protein specific contributions. To account for the nucleic acid contributions, we used NUPACK to predict the energy for the guide:target ensemble. To account for protein specific contributions, the mismatch only model included one parameter for binding of loaded TtAgo to a fully complementary target with a G at target position 1 (t1G), 15 parameters for whether there were DNA mismatches at positions 2–16, 3 parameters for the other three possible nucleotide identities at target position 1 (t1A, t1C, t1T), and 1 parameter that scaled the NUPACK predicted energy of the ensemble of target secondary structures incompetent for binding. This 20-parameter binding model was first fit with leave-one-out cross-validation (i.e., fit to four guides and tested on the guide not used in training) using scikit_learn version 0.19.0, and the mean and standard error of the fit parameters for these cross-validated fits were computed. A final version of the model was fit to data from all of the guides simultaneously. Pearson correlations between observed and predicted affinities were computed with scipy.stats.pearsonr for all fits. A more complex binding model accounting for transition and transversion mismatches at positions 2–16 separately (15 additional parameters; 37 total) was also fit following the procedure described above.
Supplementary Material
Table S1 | TtAgo Kinetic and Thermodynamic Data, Related to all Figures
Table S2 | Oligonucleotides used in this study, Related to STAR Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli BL21-DE3 | NEB | C2527H |
| Chemicals, peptides, and recombinant proteins | ||
| NEBNext High-Fidelity 2x PCR Master Mix | NEB | M0541 |
| SYBR Green | Invitrogen | S7563 |
| SYBR Gold | Invitrogen | S11494 |
| PhiX Control V3 | Illumina | 110-3001 |
| HiTrap SP HP | GE Healthcare | 17115201 |
| Proteinase K | NEB | P8107S |
| Alexa Fluor 647-aminohexylacrylamido-dUTP | Life Technologies | A32763 |
| Alexa Fluor 555 NHS Ester | Life Technologies | A20009 |
| Klenow fragment (3′-to-5′ exo‐minus) | NEB | M0212 |
| Cover glasses (24 Å~ 60 mm) | Gold Seal | Cat. #3423 |
| glass coverslips (25 Å~ 25 mm) | Gold Seal | Cat. #3307 |
| NanoStrip | KMG Chemicals | 210034 |
| Streptavidin | NEB | N7021S |
| Heparin | Sigma | H4784 |
| Protocatechuic acid | Aldrich | 37580 |
| Pseudomonas sp. protocatechuate 3,4-Dioxygenase | Sigma | P8279 |
| trolox | Aldrich | 238813 |
| propyl gallate | Sigma | P3130 |
| 4-nitrobenzyl alcohol | Aldrich | N12821 |
| Critical commercial assays | ||
| QIAquick PCR Purification Kit | QIAGEN | 28106 |
| MiSeq Reagent Kit v3 (150 cycle) | Illumina | MS-102-3001 |
| Deposited data | ||
| Mendeley Data: Fit parameters for TtAgo binding and cleavage | This Paper | http://doi.org/10.17632/vrb6n8gm3x.1 |
| Oligonucleotides | ||
| Table S2 | This Paper | |
| Recombinant DNA | ||
| pET-SUMO | Invitrogen | K30001 |
| Software and algorithms | ||
| NUPACK | J. N. Zadeh, C. D. Steenberg, J. S. Bois, B. R. Wolfe, M. B. Pierce, A. R. Khan, R. M. Dirks, N. A. Pierce. NUPACK: analysis and design of nucleic acid systems. J Comput Chem, 32:170-173, 2011 | http://www.nupack.org/ |
| GelAnalyzer 19.1 | GelAnalyzer 19.1 by Istvan Lazar Jr., PhD and Istvan Lazar Sr., PhD, CSc | www.gelanalyzer.com |
| CoSMoS pipeline | C. S. Smith, K. Jouravleva, M. Huisman, S.M. Jolly, P. D. Zamore, D. Grunwald. An automated Bayesian pipeline for rapid analysis of single-molecule binding data. Nat Commun. 2019 Jan 17;10(1):272. | https://github.com/quantitativenanoscopy/cosmos_pipeline |
| TtAgo curve fitting scripts | This Paper | https://github.com/GreenleafLab/TtAgo and Mendeley Data: http://doi.org/10.17632/vrb6n8gm3x.1 |
| Other | ||
Highlights.
Binding energies, association and cleavage rates measured for >16,000 TtAgo targets
Quantitative modeling of TtAgo association rates and binding energies
Target interactions outside the seed are required for high-affinity TtAgo binding
Specific guide:target mismatches enhance the single-turnover TtAgo cleavage rate
Acknowledgments
This work was supported in part by NIH grants GM65236 and P01HD078253 to P.D.Z. and R01GM111990, P50HG007735, R01HG009909, P01GM066275, UM1HG009436, and R01GM121487 to W.J.G. W.J.G. acknowledges support as a Chan-Zuckerberg Investigator. B.O.R. and W.R.B. were supported in part by the Stanford MSTP training grant (T32GM007365). K.J. was supported in part by a Charles A. King Trust Postdoctoral Fellowship. W.R.B. was supported in part by the SIGF affiliated with ChEM-H.
Footnotes
Declaration of Interests
P.D.Z. is a member of the scientific advisory boards of Alnylam Pharmaceuticals, Voyager Therapeutics, and ProQR. He is also a consultant for The RNA Medicines Company. W.J.G is a scientific co-founder of Protillion and a consultant for Guardant Health and 10x Genomics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ameres SL, Martinez J, and Schroeder R. (2007). Molecular basis for target RNA recognition and cleavage by human RISC. Cell 130, 101–112. [DOI] [PubMed] [Google Scholar]
- Anzelon TA, Chowdhury S, Hughes SM, Xiao Y, Lander GC, and MacRae IJ (2021). Structural basis for piRNA targeting. Nature 597, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villén J, Shin C, Camargo FD, Gygi SP, and Bartel DP (2008). The impact of microRNAs on protein output. Nature 455, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2018). Metazoan MicroRNAs. Cell 173, 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker WR, Ober-Reynolds B, Jouravleva K, Jolly SM, Zamore PD, and Greenleaf WJ (2019). High-Throughput Analysis Reveals Rules for Target RNA Binding and Cleavage by AGO2. Mol. Cell 75, 741–755.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EA, Andreasson JOL, Chircus LM, Sternberg SH, Wu MJ, Guegler CK, Doudna JA, and Greenleaf WJ High-throughput biochemical profiling reveals Cas9 off-target binding and unbinding heterogeneity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, and Cohen SM (2005). Principles of MicroRNA–Target Recognition. PLoS Biology 3, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD., Araya CL, Chircus LM, Layton CJ, Chang HY, Snyder MP, and Greenleaf WJ (2014). Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nat. Biotechnol. 32, 562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandradoss SD, Schirle NT, Szczepaniak M, MacRae IJ, and Joo C. (2015). A Dynamic Search Process Underlies MicroRNA Targeting. Cell 162, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GR, Sive H, and Bartel DP (2017). A Seed Mismatch Enhances Argonaute2Catalyzed Cleavage and Partially Rescues Severely Impaired Cleavage Found in Fish. Mol. Cell 68, 1095–1107.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse II, Kim H, and Ha T. (2012). A rule of seven in Watson-Crick base-pairing of mismatched sequences. Nat. Struct. Mol. Biol. 19, 623–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny SK, Bisaria N, Yesselman JD, Das R, Herschlag D, and Greenleaf WJ (2018). High-Throughput Investigation of Diverse Junction Elements in RNA Tertiary Folding. Cell 174, 377–390.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Martinez J, Patkaniowska A, Lendeckel W, and Tuschl T. (2001). Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 20, 6877–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkayam E, Kuhn C-D, Tocilj A, Haase AD, Greene EM, Hannon GJ, and Joshua-Tor L. (2012). The structure of human argonaute-2 in complex with miR-20a. Cell 150, 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M, and Zamore PD (2009). Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 10, 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski SA, Vogel J, and Doudna JA (2017). RNA-based recognition and targeting: sowing the seeds of specificity. Nat. Rev. Mol. Cell Biol. 18, 215–228. [DOI] [PubMed] [Google Scholar]
- Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, and Bartel DP (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegge JW, Swarts DC, Chandradoss SD, Cui TJ, Kneppers J, Jinek M, Joo C, and van der Oost J. (2019). DNA-guided DNA cleavage at moderate temperatures by Clostridium butyricum Argonaute. Nucleic Acids Res. 47, 5809–5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt EA, Tamanaha E, Bonanno K, Cantor EJ, and Tanner NA (2021). Profiling Thermus thermophilus Argonaute Guide DNA Sequence Preferences by Functional Screening. Frontiers in Molecular Biosciences 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvagner G. (2002). A microRNA in a Multiple-Turnover RNAi Enzyme Complex. Science 297, 2056–2060. [DOI] [PubMed] [Google Scholar]
- Jolly SM., Gainetdinov I, Jouravleva K, Zhang H, Strittmatter L, Bailey SM, Hendricks GM, Dhabaria A, Ueberheide B, and Zamore PD (2020). Thermus thermophilus Argonaute Functions in the Completion of DNA Replication. Cell 182, 1545–1559.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedde M, van Kouwenhove M, Zwart W, Oude JA, Elkon R, and Agami R. (2010). A Pumilio-induced RNA structure switch in p27–3′ UTR controls miR-221 and miR-222 accessibility. Nature Cell Biology 12, 1014–1020. [DOI] [PubMed] [Google Scholar]
- Kuzmenko A, Oguienko A, Esyunina D, Yudin D, Petrova M, Kudinova A, Maslova O, Ninova M, Ryazansky S, Leach D, et al. (2020). DNA targeting and interference by a bacterial Argonaute nuclease. Nature. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, and Bartel DP (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. [DOI] [PubMed] [Google Scholar]
- Liu L, Li X, Ma J, Li Z, You L, Wang J, Wang M, Zhang X, and Wang Y. (2017). The Molecular Architecture for RNA-Guided RNA Cleavage by Cas13a. Cell 170, 714–726.e10. [DOI] [PubMed] [Google Scholar]
- Liu Y, Esyunina D, Olovnikov I, Teplova M, Kulbachinskiy A, Aravin AA, and Patel DJ (2018). Accommodation of Helical Imperfections in Rhodobacter sphaeroides Argonaute Ternary Complexes with Guide RNA and Target DNA. Cell Rep. 24, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima T, and Imahori K. (1974). Description of Thermus thermophilus (Yoshida and Oshima) comb. nov., a Nonsporulating Thermophilic Bacterium from a Japanese Thermal Spa. International Journal of Systematic Bacteriology 24, 102–112. [Google Scholar]
- Ozata DM, Gainetdinov I, Zoch A, O’Carroll D, and Zamore PD (2019). PIWIinteracting RNAs: small RNAs with big functions. Nat. Rev. Genet. 20, 89–108. [DOI] [PubMed] [Google Scholar]
- Parker JS, Roe SM, and Barford D. (2005). Structural insights into mRNA recognition from a PIWI domain-siRNA guide complex. Nature 434, 663–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Parizotto EA, Wang M, Roe SM, and Barford D. (2009). Enhancement of the seed-target recognition step in RNA silencing by a PIWI/MID domain protein. Mol. Cell 33, 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross PD, and Sturtevant JM (1960). THE KINETICS OF DOUBLE HELIX FORMATION FROM POLYRIBOADENYLIC ACID AND POLYRIBOURIDYLIC ACID. Proc. Natl. Acad. Sci. U. S. A. 46, 1360–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryazansky S, Kulbachinskiy A, and Aravin AA (2018). The Expanded Universe of Prokaryotic Argonaute Proteins. MBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon WE., Jolly SM, Moore MJ, Zamore PD, and Serebrov V. (2016). Single-Molecule Imaging Reveals that Argonaute Reshapes the Binding Properties of Its Nucleic Acid Guides. Cell 166, 517–520. [DOI] [PubMed] [Google Scholar]
- Schirle NT, Sheu-Gruttadauria J, and MacRae IJ (2014). Structural basis for microRNA targeting. Science 346, 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, and Rajewsky N. (2008). Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63. [DOI] [PubMed] [Google Scholar]
- She R, Chakravarty AK, Layton CJ, Chircus LM, Andreasson JOL, Damaraju N, McMahon PL, Buenrostro JD, Jarosz DF, and Greenleaf WJ (2017). Comprehensive and quantitative mapping of RNA-protein interactions across a transcribed eukaryotic genome. Proc. Natl. Acad. Sci. U. S. A. 114, 3619–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng G, Zhao H, Wang J, Rao Y, Tian W, Swarts DC, van der Oost J, Patel DJ, and Wang Y. (2014). Structure-based cleavage mechanism of Thermus thermophilus Argonaute DNA guide strand-mediated DNA target cleavage. Proc. Natl. Acad. Sci. U. S. A. 111, 652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng G, Gogakos T, Wang J, Zhao H, Serganov A, Juranek S, Tuschl T, Patel DJ, and Wang Y. (2017). Structure/cleavage-based insights into helical perturbations at bulge sites within T. thermophilus Argonaute silencing complexes. Nucleic Acids Research 45, 9149–9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu- Gruttadauria J, Xiao Y, Gebert LFR, and MacRae IJ (2019). Beyond the seed: structural basis for supplementary micro RNA targeting by human Argonaute2. The EMBO Journal 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CS, Jouravleva K, Huisman M, Jolly SM, Zamore PD, and Grunwald D. (2019). An automated Bayesian pipeline for rapid analysis of single-molecule binding data. Nat. Commun. 10, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts DC, Makarova K, Wang Y, Nakanishi K, Ketting RF, Koonin EV, Patel DJ, and van der Oost J. (2014a). The evolutionary journey of Argonaute proteins. Nat. Struct. Mol. Biol. 21, 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts DC, Jore MM, Westra ER, Zhu Y, Janssen JH, Snijders AP, Wang Y, Patel DJ, Berenguer J, Brouns SJJ, et al. (2014b). DNA-guided DNA interference by a prokaryotic Argonaute. Nature 507, 258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts DC, Hegge JW, Hinojo I, Shiimori M, Ellis MA, Dumrongkulraksa J, Terns RM, Terns MP, and van der Oost J. (2015). Argonaute of the archaeon Pyrococcus furiosus is a DNA-guided nuclease that targets cognate DNA. Nucleic Acids Res. 43, 5120–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts DC, Szczepaniak M, Sheng G, Chandradoss SD, Zhu Y, Timmers EM, Zhang Y, Zhao H, Lou J, Wang Y, et al. (2017a). Autonomous Generation and Loading of DNA Guides by Bacterial Argonaute. Mol. Cell 65, 985–998.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts DC., van der Oost J, and Jinek M. (2017b). Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Mol. Cell 66, 221–233.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomari Y, and Zamore PD (2005). Perspective: machines for RNAi. Genes Dev. 19, 517–529. [DOI] [PubMed] [Google Scholar]
- Wang Y, Juranek S, Li H, Sheng G, Tuschl T, and Patel DJ (2008a). Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature 456, 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sheng G, Juranek S, Tuschl T, and Patel DJ (2008b). Structure of the guide-strand-containing argonaute silencing complex. Nature 456, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Juranek S, Li H, Sheng G, Wardle GS, Tuschl T, and Patel DJ (2009). Nucleation, propagation and cleavage of target RNAs in Ago silencing complexes. Nature 461, 754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee LM, Flores-Jasso CF, Salomon WE, and Zamore PD (2012). Argonaute divides its RNA guide into domains with distinct functions and RNA-binding properties. Cell 151, 1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmur JG, and Davidson N. (1968). Kinetics of renaturation of DNA. Journal of Molecular Biology 31, 349–370. [DOI] [PubMed] [Google Scholar]
- Yuan Y-R, Pei Y, Ma J-B, Kuryavyi V, Zhadina M, Meister G, Chen H-Y, Dauter Z, Tuschl T, and Patel DJ (2005). Crystal structure of A. aeolicus argonaute, a site-specific DNA-guided endoribonuclease, provides insights into RISC-mediated mRNA cleavage. Mol. Cell 19, 405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, Pierce MB, Khan AR, Dirks RM, and Pierce NA (2011). NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 32, 170–173. [DOI] [PubMed] [Google Scholar]
- Zander A., Willkomm S, Ofer S, van Wolferen M, Egert L, Buchmeier S, Stöckl S, Tinnefeld P, Schneider S, Klingl A, et al. (2017). Guide-independent DNA cleavage by archaeal Argonaute from Methanocaldococcus jannaschii. Nat Microbiol 2, 17034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DY, Chen SX, and Yin P. (2012). Optimizing the specificity of nucleic acid hybridization. Nature Chemistry 4, 208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JX, Fang JZ, Duan W, Wu LR, Zhang AW, Dalchau N, Yordanov B, Petersen R, Phillips A, and Zhang DY (2018). Predicting DNA hybridization kinetics from sequence. Nat. Chem. 10, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | TtAgo Kinetic and Thermodynamic Data, Related to all Figures
Table S2 | Oligonucleotides used in this study, Related to STAR Methods
Data Availability Statement
The kinetic and thermodynamic measurements generated in this paper are available for download (Supplementary Table 1) and on Mendeley Data (http://doi.org/10.17632/vrb6n8gm3x.1).
Custom software for determination of association rates, cleavage rates, and dissociation constants is available on GitHub (https://github.com/GreenleafLab/TtAgo) and on Mendeley Data (http://doi.org/10.17632/vrb6n8gm3x.1).
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.