

MITOCHONDRIA AND CELL DEATH
The major mode of apoptosis in vertebrates is the mitochondrial pathway. This pathway of cell death is engaged by a vast array of cell stresses, including deprivation of growth factors, disruption of the cytoskeleton, DNA damage, accumulation of unfolded proteins, hypoxia, and many others. It is also activated by developmental signals, such as hormones, that instruct cells to die. In this review, we discuss how mitochondria are involved in the activation of caspases in this type of cell death.
A JUST-SO STORY
Before examining the remarkable way in which mitochondria effect apoptosis, it might be worth considering how this organelle might have become involved in cell death—after all, in nearly all eukaryotes, mitochondria are essential for life, providing not only energy via the tricarboxylic acid cycle and oxidative phosphorylation, but also many other essential services to the cell. These include lipid metabolism and the ability to live in the toxic world of oxygen.
Rudyard Kipling, in his Just So Stories, provided fanciful explanations of biological phenomena, such as how the elephant got its trunk, that were scientifically untestable. In evolutionary biology, too, we have similarly untestable just-so stories, such as the one that follows. But it may have value in helping to frame what is to come, and we will return to this fantasy at later stages in this and other reviews in this subject collection.
Approximately 2 billion years ago, an α-proteobacterium invaded an archeon cell. The subsequent endosymbiotic relationship produced what we now know as a eukaryotic cell, and the bacterium became the first mitochondrion.1 Although this idea of a grand symbiosis is strongly supported by data, the initial relationship might well have started off far from cooperatively. After all, the bacterium was infecting the archeon.
As we have noted, a very good strategy for host defense against intracellular pathogens is for the infected cell to die. This even seems to apply to single-cell organisms that die altruistically to avoid spreading infection to their identical clone mates.2 Of course, a pathogen that can prevent such proactive suicide gains the upper hand. The initial infection that led to the formation of the first mitochondrion might therefore have triggered a suicide response in the invaded cell (probably not apoptosis, but a precursor to this form of cell death). In turn, this was checked by the infecting bacterium (Fig. 1).
Figure 1.

A just-so story about mitochondria and cell death. According to this tale, an early archaeal cell defends itself by committing suicide when infected (upper pathway), but, when faced by a variant bacterium possessing by chance the wherewithal to interfere with the archaeal cell's defenses (lower pathway), the archaeal cell is subjugated by the invader. Over time, the invader repays the cell by developing into the cell's mitochondrial powerhouses that still retain the ability to act as lynchpins of cell death regulation.
As the bacteria became mitochondria, decisions regarding the life and death of the cell might have resided under the control of this evolving organelle. In time, the control moved from mitochondria to the eukaryotic cell as a whole. The mitochondria, however, remained the focus for the effects.
HOW MITOCHONDRIA ACTIVATE CASPASES
The mitochondrial pathway of apoptosis has been described primarily in vertebrates, although there is compelling evidence for its existence in echinoderms (such as sea urchins and sand dollars) and in the platyhelminthes (flatworms). As we know, apoptotic pathways involve mechanisms that activate an initiator caspase, which in turn activates the executioner caspases to orchestrate cell death. In the mitochondrial pathway, the initiator caspase is caspase-9.
In Green (2022a), we discussed how activation of initiator caspases by dimerization of inactive monomers requires an adapter protein to bind to their prodomains. In the case of caspase-9, the adapter that activates the caspase is APAF1 (apoptotic protease activating factor-1). APAF1 binds to caspase-9 by means of the APAF1 caspase-recruitment domain (CARD), which binds to the CARD domain in caspase-9 (Fig. 2).
Figure 2.

A CARD–CARD interaction. (PDB 3YGS [Qin et al. 1999].)
APAF1, like caspase-9, preexists in the cell as a cytosolic, inactive monomer that cannot bind to or cause dimerization of the caspase. Several events must occur to change this during the initiation of apoptosis. To understand these, it is useful to look at the different domains of APAF1 (Fig. 3).
Figure 3.
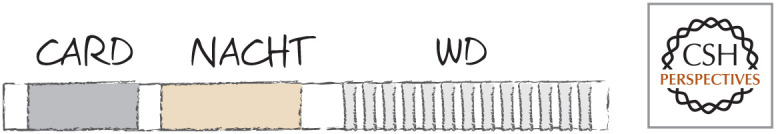
Domains of APAF1. A CARD and NACHT domain are followed by a series of WD repeats at the carboxy-terminal end.
The CARD region of APAF1, which binds to caspase-9 (via its own CARD), and the oligomerization domain are both buried in the inactive APAF1 monomer. The NACHT3 domain contains a nucleotide-binding site that is inaccessible, because access to it is blocked by the WD domain.4 If the WD domain is removed, the nucleotide deoxy-ATP (dATP) can bind to the nucleotide-binding site in the NACHT domain. This triggers a conformational change, exposing the oligomerization and CARD domains. Binding of cytochrome c (see below) to the WD domain has the same effect, changing the conformation of APAF1, allowing the binding of dATP. APAF1 molecules then assemble into the complex shown in Figure 4. The center of the APAF1 oligomeric complex contains the CARD domains that recruit caspase-9 molecules to activate the caspase. This APAF1–caspase-9 complex is called the “apoptosome.”
Figure 4.

Apoptosome formation in vertebrates. Interaction of cytochrome c with the WD region of APAF1 allows dATP binding to the NACHT, resulting in a conformational change. This drives oligomerization and exposure of the CARD that interacts with the CARD of caspase-9, resulting in the activation of the caspase.
Removal of the WD region results in formation of the apoptosome, but this is not what occurs in cells undergoing apoptosis. Instead, another protein binds to the WD region, producing a conformational change that exposes the nucleotide-binding site (presumably by moving the WD region away from it). The protein responsible is cytochrome c, a protein present in mitochondria, where it has a central role in electron transport and energy production.
Cytochrome c is a nuclear-encoded protein that is synthesized in the cytosol as apocytochrome c and then transported into mitochondria to the space between the inner and outer mitochondrial membranes (the intermembrane space). Here, the enzyme heme lyase attaches a heme group to create the mature protein (holocytochrome c). Apocytochrome c cannot activate APAF1, but holocytochrome c (which can do so) is normally sequestered away from the cytosol (and APAF1) by the mitochondrial outer membrane. For apoptosis to occur by the mitochondrial pathway, this barrier must be disrupted.
The ability of cytochrome c to engage APAF1 to induce apoptosome formation and caspase activation is independent of its function in electron transport. We can introduce mutations in a particular amino acid (lysine 72) that do not affect electron transport but impair apoptosome formation. Mammalian cells engineered to lack cytochrome c do not activate caspases when the mitochondrial pathway is engaged, nor do cells in which cytochrome c is mutated at this key residue.
Mice engineered to lack APAF1 or caspase-9 often die during development or just after birth, owing to extensive developmental abnormalities. These include large outgrowths of the brain occurring because of the presence of excess neurons. Cells from these mice do not activate caspases in response to signals that normally would engage the mitochondrial pathway of apoptosis. These defects in development are also seen in mice lacking caspases-3 and -7, the executioner caspases downstream of caspase-9. In addition, a mouse has been generated in which cytochrome c was mutated at lysine 72. This mutation produced an animal with the same defects as in those lacking APAF1, caspase-9, or the executioner caspases (Fig. 5). This serves as a formal demonstration that the mitochondrial pathway of apoptosis requires the function of cytochrome c.
Figure 5.

Mouse fetuses engineered to lack expression of APAF1, caspase-9, or caspases-3 and -7 or with a cytochrome K72A (lysine-to-alanine) mutation experience overgrowth of the forebrain (right) compared with a wild-type control (left). (Images courtesy of Dr. Christopher Dillon, St. Jude Children's Research Hospital, Memphis, Tennessee.)
DISRUPTING THE MITOCHONDRIAL OUTER MEMBRANE
If cytochrome c is to trigger apoptosis by engaging APAF1, it must move from the mitochondrial intermembrane space to the cytosol, where both APAF1 and caspase-9 reside. It is able to do this during apoptosis because upstream signals that induce cell death cause the outer membranes of all (or nearly all) mitochondria in the cell to become permeable by a process called “mitochondrial outer membrane permeabilization” (MOMP). This results in the release, by diffusion, of any soluble molecules residing in the intermembrane space, including cytochrome c. An example is shown in Figure 6.
Figure 6.

Mitochondrial outer membrane permeabilization (MOMP). Cells expressing a fusion between cytochrome c and green fluorescent protein (GFP) undergoing MOMP in response to an apoptosis-inducing stress (left to right). As MOMP occurs (cell on the right in each pair), the distribution of fluorescence changes from localization in the mitochondria to being diffuse throughout the cytoplasm. (The cell to the left underwent MOMP at a later time.) The time between images is 5 min, with the first image taken several hours after the initial stress. (Images provided by Dr. Stephen Tait, St. Jude Children's Research Hospital, Memphis, Tennessee.)
Through time-lapse imaging of cells expressing fluorescent fusion proteins, we know that, during apoptosis, MOMP is usually sudden, rapid, and irreversible. That is, when a cell is induced to undergo apoptosis, an indeterminate time passes, and then suddenly nearly all of the mitochondria undergo MOMP within a very short time period (∼5–10 min). Shortly after this, caspases become active, leading to apoptosis.
Because MOMP does not involve a loss of integrity of the inner mitochondrial membrane, mitochondrial function is not destroyed by this process, although electron transport is greatly reduced because cytochrome c becomes diluted by diffusion into the cell cytosol. However, as seen in Green (2022b), as executioner caspases become active, they gain access to proteins that are exposed on the inner membrane. Consequently, complex I of the electron-transport chain is destroyed and mitochondrial physiology is altered dramatically.
OTHER PROTEINS ARE RELEASED TOGETHER WITH CYTOCHROME c
When MOMP occurs during apoptosis upstream of caspase activation by the apoptosome, all soluble proteins of the intermembrane space are free to diffuse out of the mitochondria and into the cytosol. Of these hundreds of different proteins, some have roles in apoptosis and possibly other forms of cell death.
Among these is a protein called “second mitochondrial activator of caspases” (Smac, also called Diablo). This protein performs a function in caspase activation that is distinct from that of cytochrome c. Like the latter, it is produced by a nuclear gene, and the protein is imported into the mitochondrial intermembrane space. In the process, the amino-terminal region is proteolytically removed, revealing a short sequence at the new amino terminus with an important function.
Remember that vertebrate cells express XIAP, an endogenous inhibitor of caspase-9 and the executioner caspases. This can block caspase activation and apoptosis even if cytochrome c is released to trigger the formation of the APAF1 apoptosome. The function of Smac is to prevent XIAP from exerting this inhibitory effect. The amino terminus of mature Smac binds to the same region of XIAP that binds to caspase-9, preventing the inhibition of the caspase by XIAP and allowing caspase activation to proceed. This is illustrated in Figure 7.
Figure 7.

MOMP releases proteins that inhibit XIAP. Proteins that bind to XIAP, which include Smac and Omi, reside in the mitochondrial intermembrane space and are released upon MOMP. These block the ability of XIAP to inhibit caspase-3, caspase-7, and caspase-9.
Smac is a large protein dimer. Is the only function of this protein to present the small amino-terminal peptide to XIAP following MOMP? It seems unlikely, but no other mitochondrial functions of this protein have been identified. Peptides and drugs that mimic the amino terminus of Smac bind to and inhibit two other IAPs (in addition to XIAP)—cIAP1 and cIAP2—that have roles in other types of (nonapoptotic) signaling. However, at this point, we simply do not know whether Smac has a function beyond that in apoptosis.
Smac is not the only protein with this XIAP-neutralizing activity that is released following MOMP. Another is the serine protease Omi (also called HtrA2), which like Smac has an amino-terminal sequence that inhibits XIAP. Omi has other functions in the mitochondria that appear to be conserved not only in animals but in other eukaryotes as well.5 In some animals, Omi does not carry the XIAP-neutralizing sequence at all (one example is the Omi found in bovids). We return to Omi and Smac in Green (2022c) when we consider another pathway of vertebrate apoptosis.
MOMP CAN CAUSE CASPASE-INDEPENDENT CELL DEATH
Cells in which the mitochondrial pathway of apoptosis is engaged, but in which caspase activation is disrupted or blocked, can still die as a consequence of MOMP. This mode of cell death is often referred to as “caspase-independent cell death” (CICD). The name is a bit problematic because any cell death that is not apoptotic is generally “caspase independent.” But CICD has come to imply that MOMP is involved. In conditions under which MOMP is inhibited, CICD does not occur (the regulation of MOMP is discussed in detail in Green 2022d).
Mice lacking APAF1 have a number of developmental abnormalities, as we have seen (Fig. 5). However, in many tissues in which developmental apoptosis6 would normally occur, these mice also display cell death, but it has a different appearance. In particular, the cells die without the characteristic appearance of apoptosis. An example is cell death that is seen in the interdigital webs of the developing mice, an event required for the formation of digits (Fig. 8).
Figure 8.

Apoptosis and caspase-independent cell death (CICD) in interdigital webs of developing mice. During development, cells in the region between what will become digits die with the characteristics of apoptosis (note the chromatin condensation in the center panel). In mice engineered to lack APAF1, these cells still die, but without apoptotic morphology (right panel). (Reprinted from Chautan et al. 1999, ©1999 with permission from Elsevier.)
In fact, when we look more closely at developmental cell death in wild-type animals, some cell deaths more closely resemble CICD than apoptosis. It might be that, in some cells, APAF1 or the caspases are not efficiently expressed or engaged following MOMP , but, because the cell dies anyway, it does not matter.
Not all cells undergo CICD in response to MOMP if caspases are not active. In neurons, for example, the cells can survive MOMP if caspases are not activated, and the cells can eventually recover. In culture, most cells that undergo MOMP are generally doomed, but, under certain conditions, cells can recover if caspase activation is blocked or disrupted. CICD therefore appears to be less efficient than apoptosis, which might help to explain the developmental abnormalities in knockout mice that cannot engage caspase activation through the mitochondrial pathway.
MECHANISMS OF CICD
One likely explanation for CICD is “mitochondrial catastrophe.” Once MOMP occurs, the outer membranes of the mitochondria are compromised, and therefore all of the soluble proteins from the intermembrane space become severely diluted, affecting mitochondrial function. ATP production, lipid biogenesis, and other important functions of mitochondria no longer occur efficiently, and the cell reaches a point of no return and expires.
An alternative explanation for CICD is that some of the proteins released following MOMP can kill the cell regardless of whether caspases are activated. Two of these potential killers are endonuclease G and apoptosis-inducing factor (AIF).
Endonuclease G is a mitochondrial enzyme that can cleave DNA between nucleosomes, similarly to CAD/DFF40 (discussed in Green 2022b). Cells lacking CAD or iCAD, or cells in which caspases are inhibited, generally fail to fragment their DNA during cell death. It is not clear, therefore, that endonuclease G cleaves nuclear chromatin during either apoptosis or CICD, and the role of endonuclease G in CICD is not well established. Nevertheless, it remains possible that limited cutting of DNA by this enzyme contributes to CICD, but, at this point, we cannot say with certainty that it is involved.
The case for AIF is more intriguing. AIF is an essential protein that appears to be important for the proper transport and function of the mitochondrial electron-transport chain, and homologs of AIF are found throughout the eukaryotes. It resides in the intermembrane space and is tethered to the inner membrane. Following MOMP, proteases gain access to the intermembrane space as we described and then free AIF from its tether. Caspases can do this, as well as other proteases, such as calpain. It has been suggested that AIF then locates to the nucleus to effect CICD, possibly by causing DNA fragmentation (Fig. 9).
Figure 9.

Apoptosis-inducing factor (AIF) model of caspase-independent cell death (CICD). MOMP, mitochondrial outer membrane permeabilization.
Lack of AIF appears to be incompatible with tissue development, and most cells lacking AIF are severely compromised, which makes it difficult to rigorously test the importance of AIF in CICD. Although there are many studies concluding that the release of AIF from mitochondria is a cause of cell death in various systems, this remains controversial as other studies report no role for AIF in cell death of cells that do not require it for survival.
MITOCHONDRIAL PERMEABILITY TRANSITION
How does MOMP occur? If isolated mitochondria are exposed to high concentrations of calcium, this causes a channel—the permeability transition pore (PTP)—to open in the mitochondrial inner membrane. As a result, the transmembrane voltage potential across the inner membrane immediately dissipates, solutes enter the central matrix, and water swells the mitochondria until the inner membrane ruptures the outer membrane. This change in the inner membrane is referred to as the mitochondrial permeability transition (MPT).
Many signals in addition to calcium fluxes can cause the MPT, including reactive oxygen species (ROS), changes in cellular pH, and certain drugs. Because of this, it was widely believed that the MPT was the event that initiates apoptosis by breaking the outer membrane and releasing cytochrome c (and other proteins). This view persists in some quarters, and could well be correct in some settings. However, although the MPT is likely to have important roles in some forms of cell death, the available evidence is that it has no (or little) role in most forms of apoptosis, including the mitochondrial pathway of apoptosis, as we will see.
Unfortunately, we know very little about what comprises the PTP. Most schemes representing the PTP show several characterized molecules involved in forming a channel through both the inner and outer membranes, such as that depicted in Figure 10.
Figure 10.

Not the PTP. This is often the scheme used to explain the phenomenology of the PTP, but experimental evidence strongly suggests that VDAC and ANT are unlikely to represent key elements of the mechanism. How the PTP actually occurs remains controversial.
In this view, the PTP is mostly composed of the adenosine nucleotide transporter (ANT, which shuttles ADP and ATP across the inner membrane), with roles for the voltage-dependent anion channel (VDAC) in the outer membrane. The PTP forms when ANT opens a channel in the inner membrane. However, mitochondria from cells lacking different ANTs display normal permeability transitions (as well as apoptosis), as do cells lacking different VDACs. So currently, we do not know which membrane molecules comprise the PTP. Some evidence suggests that proteins comprising the ATP synthase in mitochondria are themselves responsible for the PTP. However, as this complex is generally essential for cell survival, it is difficult to prove this idea, or to test the importance of the PTP in different forms of cell death.
One component of the PTP for which there is some consensus is a matrix protein called cyclophilin D. Cyclophilin D is a peptidylprolyl isomerase, an enzyme that reconfigures proline residues in proteins.7 Mitochondria from mice lacking cyclophilin D display a defective MPT response to calcium (although, at much higher concentrations of calcium, an MPT can be detected). Significantly, however, no defects in apoptosis, either developmental or induced, can be detected in these mice, and they are developmentally normal. Interestingly, the mice are somewhat resistant to a nonapoptotic form of cell death arising from ischemic injury (discussed in more detail in Green 2022f).
Given these observations, the MPT is unlikely to be a major mechanism of MOMP in apoptosis. The calcium levels needed to trigger MPT are not achieved in routine cell signaling but might arise under pathological conditions. A more likely set of molecules that control MOMP in the mitochondrial pathway of apoptosis is the subject of Green (2022d).
APOPTOSOMES OF FLIES, WORMS, AND OTHER BEASTS
The adapter proteins responsible for the activation of initiator caspases in the fly (Dronc) and nematode (CED3) are homologs of APAF1 (Fig. 11). In Drosophila, this homolog is called APAF1-related killer (ARK), and, in Caenorhabditis elegans, it is CED4.
Figure 11.

Domain structures of apoptotic protease activating factors (APAFs) in humans (top), flies (center), and worms (lower).
In each case, a CARD in the adapter protein binds to the CARD in the prodomain of the caspase to activate the latter. However, unlike APAF1, neither of these appears to be activated by cytochrome c.
For CED4, the reason for this is pretty clear. CED4 does not possess a WD region (see above), and, as a consequence, it can spontaneously oligomerize to form an octameric apoptosome and activate CED3. In healthy cells, this is prevented by another protein, CED9, that holds CED4 as an inactive monomer. Curiously, this occurs on the surfaces of mitochondria for reasons that are not known (but we speculate on this in Green 2022e). During apoptosis, CED4 is released from CED9. CED4 then oligomerizes, recruits CED3, and activates the caspase to promote apoptosis (Fig. 12).
Figure 12.

Caspase activation in nematodes. Remember that this illustration is misleading: CED4 oligomerizes near its nucleotide-binding region into an octameric apoptosome; the CED4 CARD binds to the CARD of CED3, which is then activated by proximity.
In flies, the APAF1 homolog, ARK, appears to be constitutively active. That is, the protein might spontaneously oligomerize to bind and activate the initiator caspase Dronc. We say “might” because ARK, like APAF1, has a WD region, and therefore another molecule (a protein?) might have a role in the activation of ARK, but currently this is not known.8 However, if ARK is constitutively active, what holds apoptosis in check? The answer is the Drosophila inhibitor of apoptosis protein, DIAP1, which prevents Dronc activation. It is the disruption of the DIAP1–Dronc interaction that triggers apoptosis. For now, we can create the scheme shown in Figure 13 for apoptosis in the fly.
Figure 13.

Caspase activation in Drosophila. ARK is constitutively active, and forms the apoptosome, which activates the initiator caspase Dronc. Dronc is held inactive by the inhibitor of apoptosis protein DIAP1. Disruption of DIAP1 permits the active Dronc caspase to cleave executioner caspases, and apoptosis proceeds.
Furthermore, what do the apoptotic pathways in these animals say about our just-so story of an ancient role for mitochondria in controlling cell death? Is the nematode pathway ancestral, giving rise to the arthropod pathway in insects, and then the mitochondrial pathway in vertebrates? If so, then our story is simply a fantasy without value.
Flies did not evolve from nematodes, however, and humans did not evolve from flies. Thus, it is possible that the mitochondrial pathway, as it appears in vertebrates, is the ancestral mechanism. For some reason, nematodes and insects might have subsequently lost it.
Currently, to guide us we have only sequence data from different animal phyla, with limited functional information. APAF1 homologs are found throughout the animal kingdom, and, in those examined so far, only in nematodes do any of these lack the WD region that interacts with cytochrome c in the vertebrate pathway. At this point, we do not know whether most of these actually interact with cytochrome c. As noted at the beginning of this review, there is compelling evidence that MOMP occurs and cytochrome c activates APAF1 in echinoderms and platyhelminthes, and therefore it remains possible that the mitochondrial pathway of apoptosis, as we have described it, is widespread in the animals. However, what we can say is that the pathway in nematodes is unlikely to be ancestral.
IAP INHIBITION IN DROSOPHILA APOPTOSIS
As we have discussed, the APAF1 homolog in flies appears to be constitutively active, and apoptosis is controlled by the action of DIAP1 to block the initiator caspase Dronc. In flies, MOMP does not occur upstream of caspase activation, and therefore no IAP antagonist is released from mitochondria to promote cell death. Instead, proteins that use a strategy similar to that of Smac and Omi are transcriptionally expressed and neutralize DIAP1 to cause apoptosis. These proteins are encoded within a complex of functionally related genes that all have suitably morbid names, including Grim, Reaper, Sickle, and the less evocative Hid (because it was originally identified in a different setting). These proteins, like Smac (and other IAP antagonists), share the small amino-terminal region that allows them to bind to the IAP and take it out of action. The resulting Drosophila pathway for apoptosis is shown in Figure 14.
Figure 14.

Apoptotic signals in flies induce the expression of inhibitors of DIAP1, including Reaper, Hid, Grim, and Sickle.
Smac, Omi, and these Drosophila proteins all interact with IAP proteins in similar ways, through the binding of their amino-terminal sequences; Figure 15 shows these sequences for comparison. The same sequence is found in caspase-9, and the binding of this sequence to XIAP is necessary for inhibition of the caspase. The ability of proapoptotic proteins to neutralize IAP proteins therefore appears to be simple competition with the caspases for binding. If so, this small peptide region should be sufficient to produce the effect. This appears to be true in experimental systems.
Figure 15.

IAP-binding sequences (single-letter amino acid code) in humans and Drosophila. The amino-terminal sequences of Smac and Omi are produced following cleavage of the mitochondrial-localizing sequence in mitochondria. That of caspase-9 is produced by caspase cleavage. The similarities among these sequences is evidence for an evolutionarily conserved mechanism of IAP binding.
So far, we have considered only the part of the mitochondrial pathway of apoptosis that is downstream from MOMP, and MOMP is clearly an important event that determines life or death, at least in vertebrate cells. What causes MOMP and how it is regulated are considered next.
Footnotes
From the recent volume Cell Death: Apoptosis and Other Means to an End by Douglas R. Green
Additional Perspectives on Cell Death available at www.cshperspectives.org
This is the endosymbiont hypothesis of mitochondrial evolution.
There is a literature on active cell death (although not apoptosis) in bacteria and evidence that this is an effective response to infection by phage.
NACHT is an acronym based on several proteins that contain this domain: NAIP, CIITA, HET-E, and TP1. Although these proteins do not concern us here, APAF1 contains this domain. It is also referred to as a nucleotide-binding domain (NBD).
WD domains (also called WD40) contain multiple motifs of approximately 40 amino acids that often end with a tryptophan–aspartic acid (“WD” in single-letter amino acid code). Many proteins have WD domains that have functions in signaling and cell cycle control (and in the case of APAF1, apoptosis).
This is based on studies in organisms lacking the gene. In knockout mice, for example, the animals display cell death in the brain and immune systems ∼1 month after birth, but the reasons for this are not fully elucidated. It is clear, however, that this is not due to the role of Omi discussed here.
Discussed in more detail in Green (2022e).
Cyclophilin D interacts with ANT and other proteins, and it has been suggested that its reconfiguration of prolines in the target proteins is what opens the PTP.
There is some controversy about this. Genetic evidence supports a role for cytochrome c in the activation of caspases in spermatogenesis in the fly and perhaps apoptosis in some cells. However, experiments with Drosophila cells and cell extracts do not support a role for cytochrome c or its release from mitochondria in apoptosis.
ADDITIONAL READING
Mitochondria and Cell Death
Tait SWG, Green DR. 2010. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Cell Mol Biol 11: 621–632.
A detailed review of MOMP and its consequences.
Green DR, Kroemer G. 2004. The pathophysiology of mitochondrial cell death. Science 305: 626–629.
Wallace DC, Fan W. 2009. The pathophysiology of mitochondrial disease as modeled in the mouse. Genes Dev 23: 1714–1736.
This review discusses how defects in mitochondrial function affect cell survival and death.
Green DR, Fitzgerald P. 2016. Just so stories about the evolution of apoptosis. Curr Biol 26: R620–R627.
Cytochrome c and the Apoptosome
Green DR. 1998. Apoptotic pathways: the roads to ruin. Cell 94: 695–698.
An early review describing the mitochondrial pathway of apoptosis.
Green DR. 2005. Apoptotic pathways: ten minutes to dead. Cell 121: 671–674.
A later review that incorporates MOMP and additional players into the mitochondrial pathway of apoptosis.
Shakeri R, Kheirollahi A, Davoodi, J. 2017. Apaf-1: regulation and function in cell death. Biochimie 135: 111–125.
Liu X, Kim CN, Yang J, Jemmerson R, Wang X. 1996. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86: 147–157.
The original paper showing that cytochrome c induces caspase activation.
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. 1997. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c–dependent activation of caspase-3. Cell 90: 405–413.
The identification and characterization of APAF1.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479–489.
The interaction between APAF1 and caspase-9 leading to caspase activation was first described in this paper.
Cheng TC, Hong C, Akey IV, Yuan S, Akey CW. 2016. A near atomic structure of the active human apoptosome. Elife 5: 17755.
Li Y, Zhou M, Hu Q, Bai XC, Huang W, Scheres SH, Shi Y. 2017. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc Natl Acad Sci 114: 1542–1547.
The above two papers provide insights into the structure of the apoptosome and its activity.
MOMP
Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. 2000. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol 2: 156–162.
The original paper demonstrating MOMP in cells using fluorescent cytochrome c.
Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD, Green DR. 2001. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol 153: 319–328.
The immediate consequences of MOMP for mitochondria are explored.
Smac/DIABLO and Omi
Verhagen AM, Kratina TK, Hawkins CJ, Silke J, Ekert PG, Vaux DL. 2007. Identification of mammalian mitochondrial proteins that interact with IAPs via N-terminal IAP binding motifs. Cell Death Differ 14: 348–357.
Green DR. 2000. Apoptotic pathways: paper wraps stone blunts scissors. Cell 102: 1–4.
Du C, Fang M, Li Y, Li L, Wang X. 2000. Smac, a mitochondrial protein that promotes cytochrome c–dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42.
The original description of Smac, published with the original description of the mouse homolog DIABLO.
Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. 2000. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102: 43–53.
See above.
Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T, et al. 2002. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein–caspase interaction. J Biol Chem 277: 432–438.
One of several papers initially describing Omi and its role in apoptosis.
Caspase-Independent Cell Death
Chipuk JE, Green DR. 2005. Do inducers of apoptosis trigger caspase-independent cell death? Nat Rev Mol Cell Biol 6: 268–275.
Lartigue L, Kushnareva Y, Seong Y, Lin H, Faustin B, Newmeyer DD. 2009. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol Biol Cell 20: 4871–4884.
A study showing that MOMP-induced, caspase-independent cell death is a consequence of loss of mitochondrial function.
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, et al. 1999. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446.
The initial characterization of AIF.
Hangen E, Féraud O, Lachkar S, Mou H, Doti N, Fimia GM, Lam NV, Zhu C, Godin I, Muller K, et al. 2015. Interaction between AIF and CHCHD4 regulates respiratory chain biogenesis. Mol Cell 58: 1001–1014.
Characterization of the role of AIF in mitochondrial function.
Milasta S, et al. 2016. Apoptosis-inducing-factor-dependent mitochondrial function is required for T cell but not B cell function. Immunity 44: 88–102.
Further insights into AIF function, and evidence that it may not be directly involved in cell death.
Li LY, Luo X, Wang X. 2001. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412: 95–99.
Initial description of endonuclease G and its proposed role in caspase-independent cell death.
Mitochondrial Permeability Transition
Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. 2009. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 1787: 1395–1401.
An overview of the permeability transition in cell death, but one that suggests roles in apoptosis.
Bernardi P, Rasola A, Forte M, Lippe G. 2015. The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev. 95: 1111–1155.
He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE. 2017. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci 114: 3409–3414.
The above two papers highlight continued controversies in the nature of the MPTP.
Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, Galluzzi L. 2016. Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol. 26: 655–667.
Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. 2005. Cyclophilin D–dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652–658.
One of several papers showing that MPT has roles in necrosis but not apoptosis.
Apoptosomes in Worms, Flies, and Other Beasts
Bao Q, Shi Y. 2007. Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ 14: 56–65.
Qi S, Pang Y, Hu Q, Liu Q, Li H, Zhou Y, He T, Liang Q, Liu Y, Yuan X, et al. 2010. Crystal structure of the Caenorhabditis elegans apoptosome reveals an octameric assembly of CED-4. Cell 141: 446–457.
Cheng TC, Akey IV, Yuan S, Yu Z, Ludtke SJ, Akey CW. 2017. A near-atomic structure of the Dark apoptosome provides insight into assembly and activation. Structure 25: 40–52.
Rodriguez A, Chen P, Oliver H, Abrams JM. 2002. Unrestrained caspase-dependent cell death caused by loss of Diap1 function requires the Drosophila Apaf-1 homolog, Dark. EMBO J 21: 2189–2197.
The role of DIAP1 in restraining ARK (called “Dark”)-induced caspase activity in flies.
Rodriguez A, Oliver H, Zou H, Chen P, Wang X, Abrams JM. 1999. Dark is a Drosophila homologue of Apaf-1/CED-4 and functions in an evolutionarily conserved death pathway. Nat Cell Biol 1: 272–279.
The identification of ARK (called “Dark”).
Yuan J, Horvitz HR. 1992. The Caenorhabditis elegans cell death gene ced-4 encodes a novel protein and is expressed during the period of extensive programmed cell death. Development 116: 309–320.
The original characterization of CED4.
Chinnaiyan AM, Chaudhary D, O'Rourke K, Koonin EV, Dixit VM. 1997. Role of CED-4 in the activation of CED-3. Nature 388: 728–729.
One of the original papers showing that CED4 biochemically activates CED3.
Bender CE, Fitzgerald P, Tait SW, Llambi F, McStay GP, Tupper DO, Pellettieri J, Sanchez Alvarado A, Salvesen GS, Green, DR. 2012. Mitochondrial pathway of apoptosis is ancestral in metazoans. Proc Natl Acad Sci 109: 4904–4909.
Evidence for the mitochondrial pathway of apoptosis, including cytochrome c activation of apoptosome function, in platyhelminths (flatworms) and echinoderms.
REFERENCES
*Reference is also in this collection.
- *.Green DR. 2022a. Caspase activation and inhibition. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022b. Caspases and their substrates. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022c. The death receptor pathway of apoptosis. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a04105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022d. The mitochondrial pathway of apoptosis, Part II: the BCL-2 protein family. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2022e. Cell death in development. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Green DR. 2021f. Nonapoptotic cell death pathways. Cold Spring Harb Perspect Biol 10.1101/cshperspect.a041079 [DOI] [PMC free article] [PubMed] [Google Scholar]
FIGURE CREDITS
- Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P. 1999. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr Biol 9: 967–970. 10.1016/s0960-9822(99)80425-4 [DOI] [PubMed] [Google Scholar]
- Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y. 1999. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399: 549–557. 10.1038/21124 [DOI] [PubMed] [Google Scholar]