

Abstract
Purpose of review
In Alport syndrome, over 1,700 genetic variants in the COL4A3, COL4A4, and COL4A5 genes cause the absence or malfunctioning of the collagen IVα345 scaffold – an essential component of the glomerular basement membrane (GBM). Therapies are limited to treatment with ACE inhibitors to slow progression of the disease. Here, we review recent progress in therapy development to replace the scaffold or restore its function.
Recent findings
Multiple approaches emerged recently for development of therapies that target different stages of production and assembly of the collagen IVα345 scaffold in the GBM. These approaches are based on 1) recent advances in technologies allowing to decipher pathogenic mechanisms that underlie scaffold assembly and dysfunction, 2) development of DNA editing tools for gene therapy, 3) RNA splicing interference, and 4) control of mRNA translation.
Summary
There is a growing confidence that these approaches will ultimately provide cure for Alport patients. Development of therapy will be accelerated by studies that provide a deeper understanding of mechanisms that underlie folding, assembly, and function of the collagen IVα345 scaffold.
Keywords: gene editing, exon skipping, stop codon readthrough, chemical and pharmacological chaperones, protein replacement
INTRODUCTION
In Alport syndrome, nearly two-thousand variants in the COL4A3, COL4A4 and COL4A5 genes cause a broad range of glomerulopathies affecting the function of the glomerular basement membrane (GBM) in millions of people worldwide (Fig. 1) [1••, 2••, 3, 4••]. The genes encode the assembly of the collagen IVα345 scaffold, the major GBM constituent, composed of α345 triple-helical protomers made of α3, α4 and α5 chains [4••, 5•] (Fig. 1B). The variants lead to a broad array of clinical manifestations, ranging from microscopic hematuria to end stage renal disease. Current therapy is limited to treatment with ACE inhibitors to slow progression [6]. Collagen IVα345 therapies aiming to repair the glomerular filter [7, 8] are in urgent need.
Figure 1. Alport pathology in kidney.
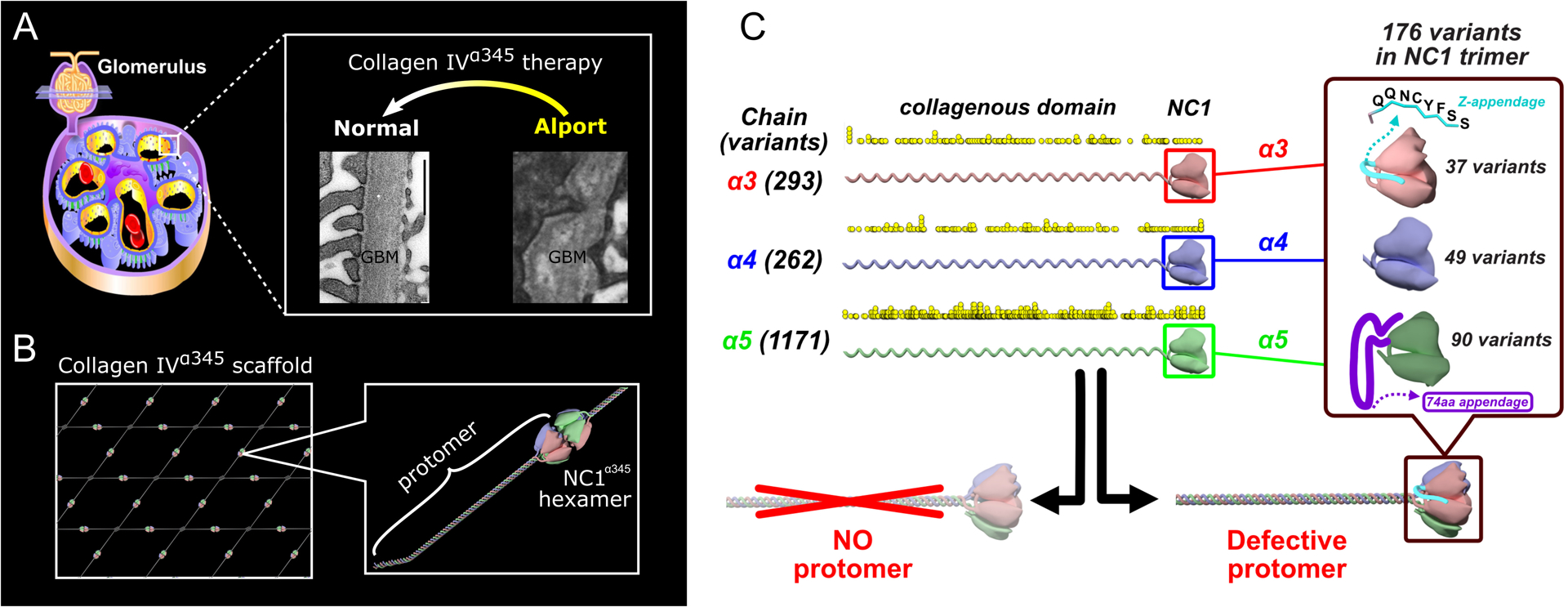
A. Alport syndrome is a glomerulopathy characterized by irregular thickening and splitting of glomerular basement membrane (GBM). Electron microscopy images of human GBM are from [7] for Alport and from [8] for normal. Collagen IVα345 therapies aim to restore the morphology and function of GBM. B. Schematic representation of the collagen IVα345 scaffold assembled from the protomers where NC1 domain hexamerization plays a crucial role. C. Alport variants (previously published in [4••]). The number and location of over 1700 genetic Alport-associated variants (indicated by yellow dots) in the α3, α4, and α5 chains of collagen IV. Pathogenic variants cause either loss of the α345 protomers in the GBM or assembly of defective α345 protomers that can incorporate into the GBM, causing a broad spectrum of GBM phenotypes. Significant number of Alport-associated variants occur in the NC1 domain of α3, α4, and α5 chains (right insert).
Development of an Alport cure hinges on answering the question of “How the plethora of pathogenic variants, encoding defective α3, α4, and α5 chains, causes GBM dysfunction?” Whereas certain variants result in absence of the collagen IVα345 in the GBM, others produce and deposit a scaffold with a defective structure (Fig. 1B). The answer requires basic knowledge of the pathobiology of collagen IVα345 in relation to its structure, assembly, stability, function, and dysfunction [9]. Here, we present recent breakthroughs that serve as groundwork for therapy development and summarize prospective therapies.
BREAKTHROUGHS IN THE PATHOBIOLOGY OF THE COLLAGEN IVA345 SCAFFOLD
A knock-in mouse model and the α345 hexamer as a focal point in GBM function
Several mouse models of Alport syndrome have been studied over the last 25 years. These include knockout mice for Col4a3, Col4a4 and Col4a5 [10–14], harboring variants that eliminate the α345 scaffold from the GBM function, as well as two models that incorporate defective scaffolds [15, 16]. However, the phenotypes in the latter mouse models were attributed to reduced amounts or proteolytic cleavage of α345. Collectively, these studies have verified the essentiality of the α345 scaffold for GBM function, but how the 1700 variants cause GBM dysfunction remains an enigma.
Our recent studies have focused on how the 176 known variants in NC1 domains cause GBM dysfunction (Fig. 1B). These domains have been extensively studied and found to function as key recognition modules in the assembly of the α345 scaffold [17, 18]. Two variants are distinct and encode an 8-residue appendage, called Zurich (Z-) variant, attached to the C-terminus of the α3NC1 domain, and a 74-residue appendage, attached to the α5NC1 domain (Fig. 1B). We developed a knock-in mouse model harboring the Z-appendage [4••]. This variant incorporated into the α345 scaffold, cause GBM ultrastructural abnormalities and proteinuria (Fig. 2A, middle panel), phenocopying a wide spectrum of glomerular phenotypes in human AS. The Z-appendage pathogenicity, in both homozygous and heterozygous mice, pinpointed the α345 hexamer as a critical structure of the collagen IV scaffold that governs morphology and ultrastructure of the GBM, features that enabled permselective ultrafiltration.
Figure 2. Pathobiology of the collagen IVα345 scaffold.
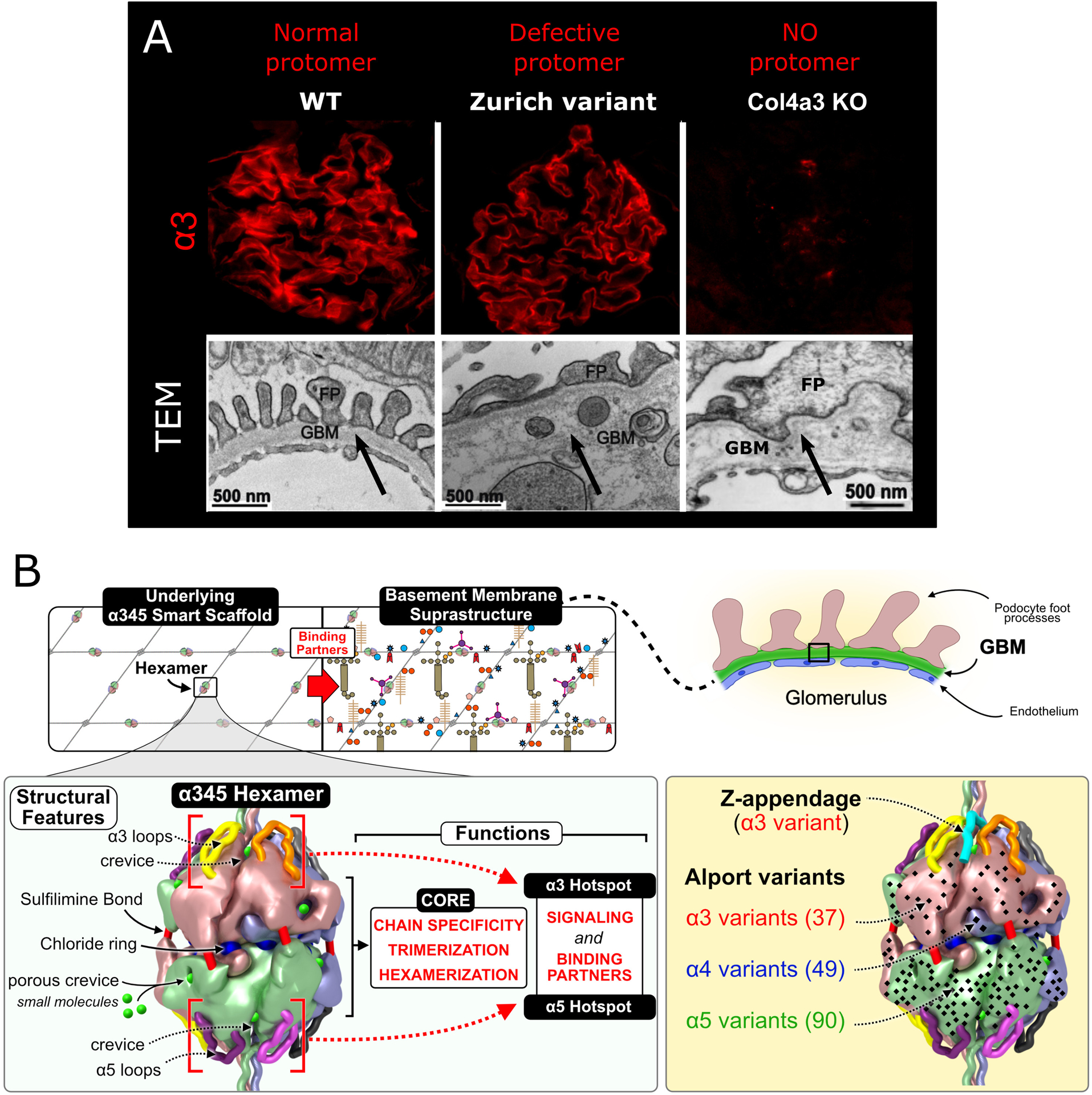
A. Mouse models for Alport syndrome. Two types of Alport animal models based on presence or absence of the collagen IVα345 scaffold in the GBM represented. Top panel demonstrates immunofluorescent staining for alpha 3 chain of collagen IV (previously published in [4••]). In the glomeruli of Col4a3 KO mouse the collagen IVα345 scaffold is absent while in Zurich variant mouse defective collagen IVα345 scaffold is incorporated in the GBM and staining intensity is similar to that of the wild type control. Lower panel shows defects in the GBM of both models observed by electron microscopy. EM image of Col4a3 KO GBM (lower right) is previously published in [19]. B. The α345 hexamer is a focal point of GBM function and is altered in Alport syndrome (modified from [4••]). The collagen IVα345 scaffold is populated with several classes of macromolecules - laminins, nidogens and proteoglycans, forming the glomerular basement membrane suprastructure. The α345 hexamer harbors a number of features involved in pathogenesis of Alport syndrome. Multiple Alport-associated variants occur within α3, α4, and α5 NC1 domains (black dots) including the Zurich variant of α3 NC1. The overall concept is presented in three companion papers in J Biol Chem. [4••, 21••, 23••].
Z-mouse model provides a unique strategy to acquire seminal information about the pathobiology of the collagen IVα345 scaffold at the molecular, cellular, and tissue levels. In particular, the model provides a trackable strategy to discover functions of the α345 hexamer at specific sites and pathogenic mechanisms that are relevant to other hypomorph variants in the NC1 domains. Moreover, this model provides a strategy for testing potential pharmacological chaperones and gene editing therapies.
Single-chain NC1 trimer technology and crystal structure of α345 hexamer
We developed a groundbreaking technology for the synthesis of recombinant single-chain NC1 trimers to explore how the “Zurich” and other variants causes GBM dysfunction [20••, 21••]. Importantly, this technology provided the pivotal advance to solve the crystal structure of the α345 hexamer [21••], a key assembly and connection module within the collagen IVα345 scaffold. The hexamer structure (Fig. 2B) revealed a ring of twelve chloride ions at the trimer-trimer interface, analogous to the collagen IVα121 scaffold [20••, 22••]. The surface of the α345 hexamer is marked by multiple pores and crevices that are potentially accessible to small molecules. Over 170 variants occur within the α3, α4 and α5 NC1 domains, including the Zurich variant of α3NC1, a C-terminal 8-amino acid appendage (Fig. 1B). Most variants are truncating and missense, both of which can cause total loss of hexamer in the GBM and renal dysfunction. Whereas, the Z-appendage, inserted at the apex of the hexamer, allowed the incorporation of the variant into the GBM causing renal dysfunction. The pathogenicity of the Z-appendage (vide supra) revealed that the α345 hexamer is a critical domain for GBM structure and function.
Furthermore, our single-chain trimer technology, coupled with the α345 hexamer structure, provides a platform to decipher how the multiple variants in the hexamer cause GBM dysfunction. The Z-appendage and various missense variants (hypomorphs) can alter the hexamer conformation. These sites, along with the pores and crevices at the hexamer surface are prospective targets for pharmacological chaperone therapy to restore native function.
Miniprotomer recombinant technology and assembly of the α345 hexamer
We explored the α345 hexamer as a step to decipher mechanisms that underlie the assembly of the collagen IVα345 scaffold. The α3, α4 and α5 NC1 monomers are encoded with structural determinants that govern both chain selection in protomer assembly and in protomer oligomerization into scaffold (Fig. 3A). Chloride ions signal protomer assembly and stabilize the α345 hexamer - an essential step in the oligomerization of protomers [23••]. Bromide ions function as cofactors in the formation of sulfilimine crosslinks, which reinforce the stability of the α345 hexamer [24••] (Figs. 2B, 3A). Furthermore, the loop-crevice-loop (LCL) bioactive sites on the hexamer surface exhibit conformational plasticity, supporting hexamer functions that include cell signaling and organizing macromolecular complexes in the assembly of the GBM, as summarized in Fig. 2B. We validated these in vitro results by expressing a mini version of the full-length α345 protomer in cell culture [23••], which undergoes the same assembly mechanism triggered by chloride ions (Fig. 3B). The miniprotomers technology is also a step towards production of recombinant full-length protomers suitable for protein replacement therapy [9].
Figure 3. Collagen IVα345 synthesis, folding, and assembly.
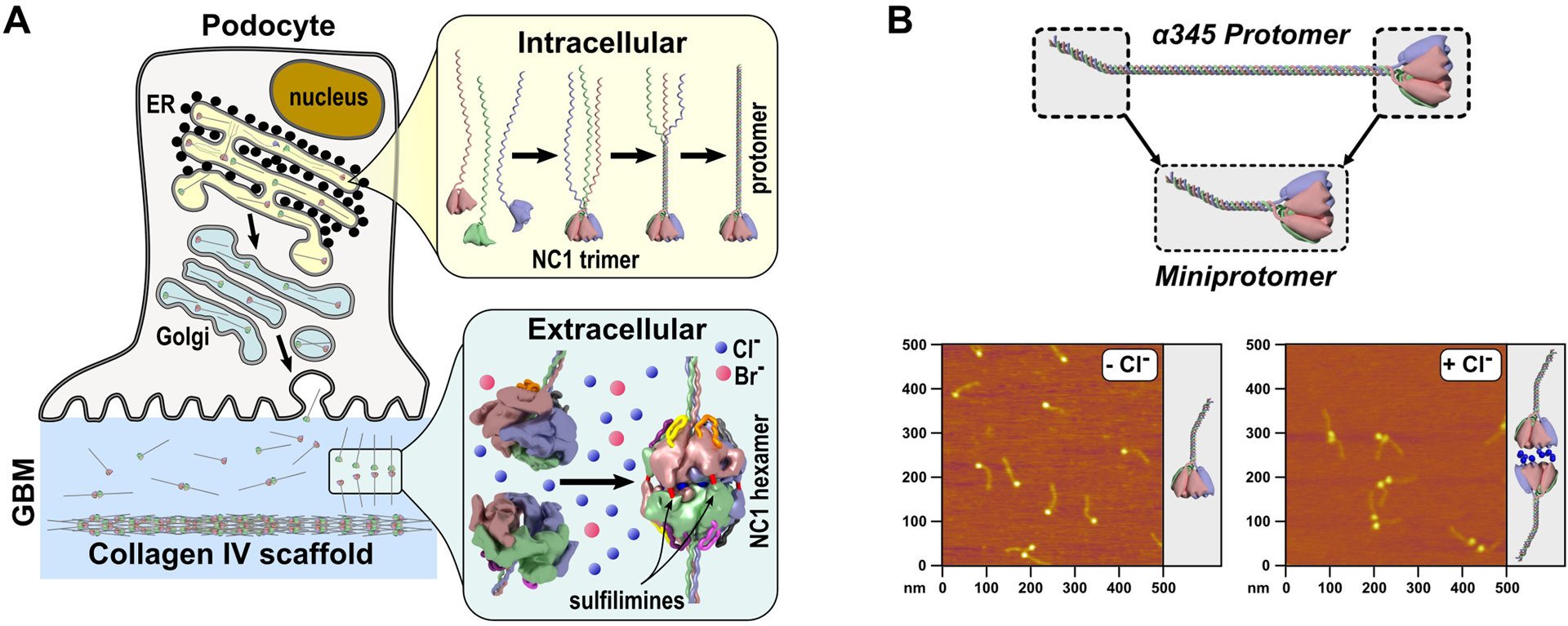
A. Schematic representation of collagen IV synthesis followed by intracellular and extracellular assembly steps. The NC1 domain guides individual chains to assemble into the trimeric protomer inside the cell and further initiate the assembly of the scaffold via hexamerization outside the cell. Chloride ions trigger and stabilize scaffold assembly and bromide ions act as co-factors in formation of sulfilimine bonds which reinforce hexamer stability. B. Schematic representation of miniprotomer and atomic force microscopy (AFM) images of miniprotomers (previously published in [23••]), which display the role of Cl− ions in hexamer formation (protomer-to-protomer interactions forming a scaffold).
PROSPECTIVE THERAPEUTIC APPROACHES
Premature termination codon readthrough therapy
Truncated α3, α4, and α5 chains lacking an intact NC1 domain due to a premature termination codon (PTC) cannot assemble into heterotrimers and lead to the absence of the α345 scaffold in the GBM. Achieving full-length protein expression via readthrough truncating nonsense mutations is a potential therapy for AS. Small molecule-based PTC readthrough (PTC-RT) therapy has been well studied in other genetic diseases.
Recently, 49 individual nonsense COL4A5 mutations found in AS patients were screened for PTC-RT in cell culture experiments [25••]. 11 mutations were found susceptible to PTC-RT induced by G418, which is known to have high readthrough activity in a class of aminoglycoside derivatives. These results suggest that PTC-RT therapy is a feasible approach for some patients with AS.
Exon skipping therapy
The COL4A5 gene consists of 51 exons with 44 of these exons belonging to the collagenous domain (exons 3–46). Among these 44 exons, 35 exons have nucleotide numbers that are a multiple of 3, which is a requirement not to have a frame shift upon exon skipping. When patients have truncating mutations in one of these exons, exon skipping can shift the truncation to a non-truncating mutation, that is, in-frame deletion mutations that can delay the development of ESRD in AS.
Recently, truncating variants in exon 21 of the COL4A5 gene were targeted by exon skipping, which enabled trimer formation, leading to clinical and pathological improvements including deposition of the α5 chain into the glomerular and tubular basement membrane [26••]. In addition, the survival period was clearly prolonged in the antisense oligonucleotide treated mice group. This data suggests that exon skipping may represent a promising therapeutic approach for treating severe AS cases.
Gene therapy
Alport syndrome caused by a pathogenic variant in one or more of three genes: COL4A3, COL4A4, or COL4A5. Progress in development of gene therapy for Alport syndrome is currently limited to early testing. Nevertheless, several key achievements are foundational and demonstrate potential for this approach.
An artificial chromosome transgenic line of mice carrying the human COL4A3-COL4A4 locus was generated [27•]. In the kidney, when expressed onto a Col4a3(−/−) background, the human α3 chain restored the expression of and co-assembled with the mouse α4 and α5 chains. The co-assembly of the human and mouse chains into a hybrid network in the GBM restored a functional GBM and rescued the Alport phenotype.
Lin et al. [28] demonstrated effective restoration of the missing α345 collagen IV network in a mouse model of Alport syndrome using an inducible transgene system. This proof-of-principle study demonstrated the plasticity of the mature GBM and validated the pursuit of therapeutic approaches aimed at normalizing the GBM to prolong kidney function.
Another proof-of-concept study used podocyte-lineage cells from two AS patients to test whether gene therapy can correct mutations in the COL4A3 and COL4A5 genes. Researchers used the CRISPR/Cas9 system to target the faulty genes in kidney cells isolated from urine [29••]. They achieved reversion of variants greater than 40% with undesired insertions/deletions lower than 15%.
Although still in its infancy, gene therapy may one day be part of clinical trials assessing its potential in Alport syndrome patients.
Pharmacological chaperones
The assembly of the α345 scaffold requires hexamer formation of the α3, α4, and α5 NC1 domains, suggesting that mutations within NC1 in Alport syndrome may disrupt this assembly [18]. The atomic structure of the α345 hexamer revealed multiple pores, crevices, and inner cavities [21••] (Fig. 2B), which may not be functionally important but are potential targets for small molecules (SMOLs) that can affect folding, assembly and stability of the quaternary structure without detrimental effect on the function. Trimerization of α345 collagen IV protomer is a culprit for multiple variants causing AS. Omachi et al. [30•] developed a system to identify α345 collagen IV protomer trimerization in cells. They demonstrated that chemical chaperones rescue the trimer formation ability of clinically reported α5 variants.
Pharmacological chaperones (pharmacoperones) initially defined as molecules correcting misfolding caused by pathogenic variants have recently been extended with molecules stabilizing final structure of a protein as they are also able to facilitate folding of aberrant variants and in addition slow down degradation (turnover rate) of the folded proteins [31•, 32]. The best class, also referred to as “second-generation” pharmacoperones, targets binding pockets that do not affect the function of the protein [31•]. This new class of molecules can potentially correct and stabilize the α345 hexamer, without affecting its function, and can be developed into a new therapy for Alport patients. Our single-chain trimer technology provides a strategy for high-throughput screening to identify potential drugs for Alport syndrome.
Protein replacement therapy
Protein replacement therapy remains an attractive and unexplored opportunity for Alport patients. Delivery of full-length recombinant laminin molecules to the GBM [33•] set up a possibility that a full-length or mini-α345 protomer can be delivered therapeutically to the glomerulus, where it can oligomerize forming the α345 scaffold in the GBM. Recent advancements in producing the α345 NC1 single-chain trimer [21••] and a miniature version of α345 collagen IV protomer [23••], named miniprotomer (Fig. 3B), provide tools to begin development of protein replacement therapy. Both trimers and miniprotomers may harbor sufficient activities to have a therapeutic effect. Further steps will include development and testing of full-length α345 protomers.
CONCLUSION
The discoveries of the α3, α4 and α5 chains of collagen IV set the foundation for unraveling the mystery of Alport syndrome and development of therapy [34•, 35•, 36•, 37•, 38•, 39•, 40•]. Over the preceding 30 years, sequences of cognate genes COL4A3, COL4A4 and COL4A5 were determined and shown to harbor nearly two thousand pathogenetic variants that cause GBM dysfunction [1••, 2••, 3, 4••]. In parallel, advances were made in understanding the structure and assembly of chains into a complex collagen IVα345 scaffold, as summarized in [5•, 39•, 41•]. Collectively, this knowledge has provided fundamental underpinnings for prospective therapeutic approaches that are summarized in Fig. 4.
Figure 4.
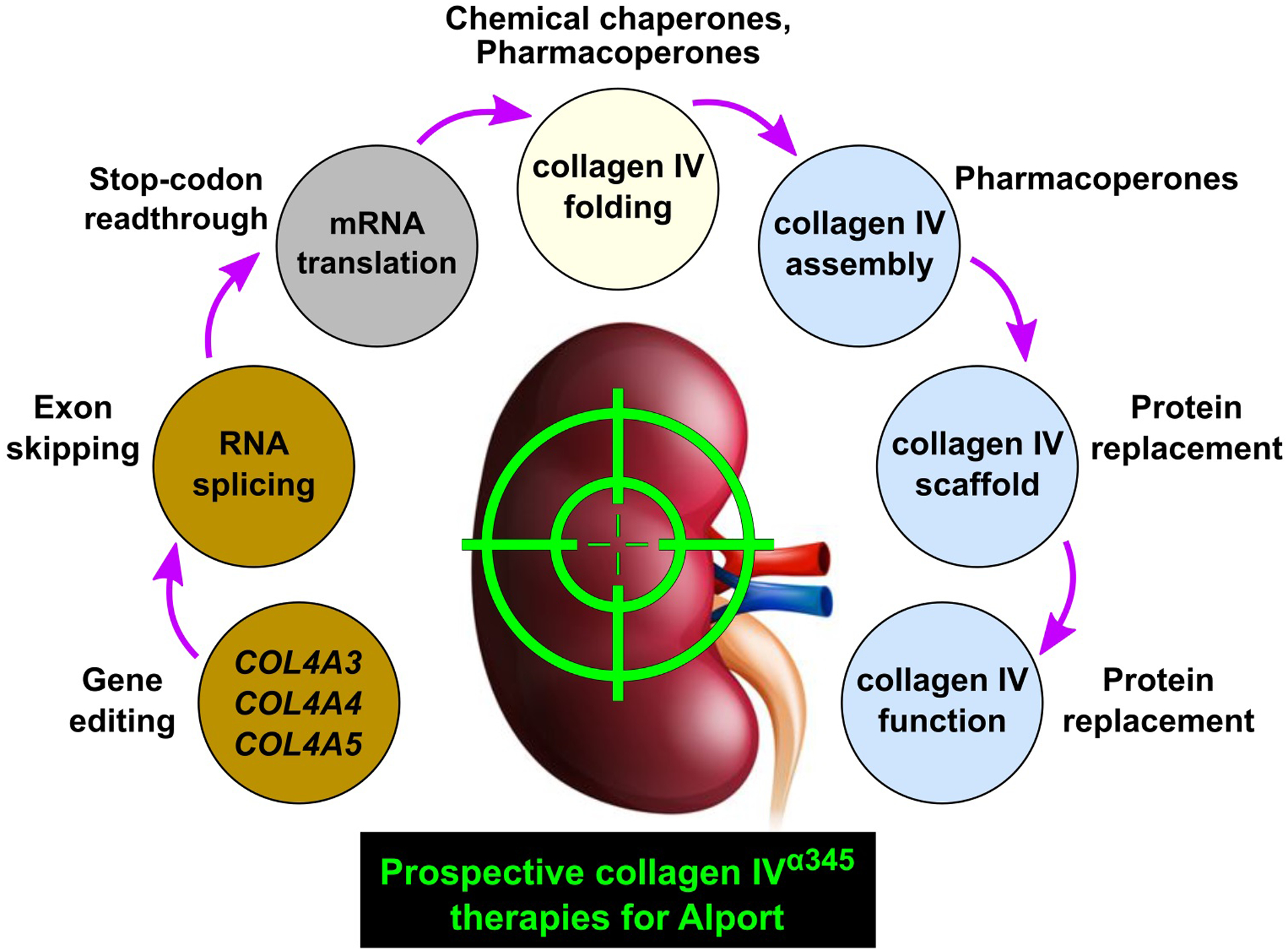
Prospective collagen IVα345 therapies for Alport syndrome.
Attractive approaches include protein replacement and pharmacological chaperones because the GBM is directly accessible to protein delivery via the bloodstream. Recent advances in protein design set up a possibility that a full-length or mini-α345 protomer can be delivered therapeutically to the glomerulus, where it can oligomerize forming the collagen IVα345 scaffold in the GBM. Moreover, because a significant number of hypomorph variants occurs within the α345 hexamer, the multiple pores, crevices, and cavities of the hexamer surface can be potential targets for pharmacological chaperones that correct misfolding or stabilize the protein from proteolytic degradation.
Yet only a paucity of information is known about how the chains assemble into a collagen IVα345 scaffold in the context of the complex supramolecular structure of the GBM. Moreover, how the scaffold functions at the molecular and cellular levels, and how pathogenetic variants cause GBM dysfunction remain obscure. This lack of knowledge impedes the development of precision therapies aimed at restoring/repairing function of the scaffold.
KEY POINTS.
Cure for Alport patients is to restore production and function of the collagen IVα345 scaffold in the GBM (collagen IV therapy)
Collagen IV therapies are possible at various stages of production and assembly of scaffold
A new mouse model of Alport syndrome with an aberrant scaffold incorporated into the GBM is valuable for studying pathobiology and developing therapies
Development of protein-based therapies hinge on a deep understanding of the pathobiology of the scaffold in GBM
Financial support and sponsorship
The authors are supported by the research grants from the NIH/NIDDK: R01-DK018381 (S.P.B, B.G.H), R56-DK131101 (S.P.B, B.G.H), and R01-DK DK065138 (B.G.H.).
Footnotes
Conflicts of interest
None.
References and recommended reading
Papers of particular interest have been highlighted as:
• of special interest
•• of outstanding interest
- 1.••.Kashtan CE, Ding J, Garosi G, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV alpha345: a position paper of the Alport Syndrome Classification Working Group. Kidney Int. 2018;93(5):1045–1051. [DOI] [PubMed] [Google Scholar]; A new classification of genetic disorders of the collagen IVα345 molecule was proposed with the goal of improving renal outcomes through regular monitoring and early treatment.
- 2.••.Groopman EE, Marasa M, Cameron-Christie S, et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N Engl J Med. 2019;380(2):142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]; Exome sequencing in a combined cohort of more than 3,000 patients with chronic kidney disease yielded a genetic diagnosis in just under 10% of cases, highlighting a prominence of collagen IVα345 variants.
- 3.Savige J, Lipska-Zietkiewicz BS, Watson E, et al. Guidelines for Genetic Testing and Management of Alport Syndrome. Clin J Am Soc Nephrol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.••.Pokidysheva EN, Seeger H, Pedchenko V, et al. Collagen IV(alpha345) dysfunction in glomerular basement membrane diseases. I. Discovery of a COL4A3 variant in familial Goodpasture’s and Alport diseases. J Biol Chem. 2021;296:100590. [DOI] [PMC free article] [PubMed] [Google Scholar]; A recent advance in deciphering pathogenic mechanims underlying Alport syndrome and functionality of collagen IV in glomerular ultrafiltration. Also first mouse model of Alport syndrome with full-length and normal quantity of collagen IVα345 scaffold highlighting functional role of the NC1α345 hexamer in GBM.
- 5.•.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N Engl J Med. 2003;348(25):2543–2556. [DOI] [PubMed] [Google Scholar]; A detailed review of the cannonical structural and assembly features of collagen IV and their relationship with pathogenesis of Alport and Goodpasture diseases.
- 6.Kashtan CE, Gross O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol. 2021;36(3):711–719. [DOI] [PubMed] [Google Scholar]
- 7.Bu L, Chen J, Nelson AC, et al. Somatic Mosaicism in a Male Patient With X-linked Alport Syndrome. Kidney Int Rep. 2019;4(7):1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tryggvason K, Patrakka J. Thin basement membrane nephropathy. J Am Soc Nephrol. 2006;17(3):813–822. [DOI] [PubMed] [Google Scholar]
- 9.Tryggvason K Complex genetics of Alport and Goodpasture syndromes. Nat Rev Nephrol. 2021;17(10):635–636. [DOI] [PubMed] [Google Scholar]
- 10.Rheault MN, Kren SM, Thielen BK, et al. Mouse model of X-linked Alport syndrome. J Am Soc Nephrol. 2004;15(6):1466–1474. [DOI] [PubMed] [Google Scholar]
- 11.Miner JH, Sanes JR. Molecular and functional defects in kidneys of mice lacking collagen alpha 3(IV): implications for Alport syndrome. J Cell Biol. 1996;135(5):1403–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu W, Phillips CL, Killen PD, et al. Insertional mutation of the collagen genes Col4a3 and Col4a4 in a mouse model of Alport syndrome. Genomics. 1999;61(2):113–124. [DOI] [PubMed] [Google Scholar]
- 13.Hashikami K, Asahina M, Nozu K, et al. Establishment of X-linked Alport syndrome model mice with a Col4a5 R471X mutation. Biochem Biophys Rep. 2019;17:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cosgrove D, Meehan DT, Grunkemeyer JA, et al. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 1996;10(23):2981–2992. [DOI] [PubMed] [Google Scholar]
- 15.Odiatis C, Savva I, Pieri M, et al. A glycine substitution in the collagenous domain of Col4a3 in mice recapitulates late onset Alport syndrome. Matrix Biol Plus. 2021;9:100053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korstanje R, Caputo CR, Doty RA, et al. A mouse Col4a4 mutation causing Alport glomerulosclerosis with abnormal collagen alpha3alpha4alpha5(IV) trimers. Kidney Int. 2014;85(6):1461–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khoshnoodi J, Cartailler JP, Alvares K, et al. Molecular recognition in the assembly of collagens: terminal noncollagenous domains are key recognition modules in the formation of triple helical protomers. J Biol Chem. 2006;281(50):38117–38121. [DOI] [PubMed] [Google Scholar]
- 18.Boutaud A, Borza DB, Bondar O, et al. Type IV collagen of the glomerular basement membrane. Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J Biol Chem. 2000;275(39):30716–30724. [DOI] [PubMed] [Google Scholar]
- 19.Gross O, Beirowski B, Koepke ML, et al. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003;63(2):438–446. [DOI] [PubMed] [Google Scholar]
- 20.••.Pedchenko V, Bauer R, Pokidysheva EN, et al. A chloride ring is an ancient evolutionary innovation mediating the assembly of the collagen IV scaffold of basement membranes. J Biol Chem. 2019;294(20):7968–7981. [DOI] [PMC free article] [PubMed] [Google Scholar]; First evidence for an evolutionary role of chloride ions in assembly of basement membranes. New thechnology for recombinant production of stable NC1 trimers of collagen IV with controlled composition. Detailed structural and functional role of chloride ions in the collagen IV hexamer assembly.
- 21.••.Boudko SP, Bauer R, Chetyrkin SV, et al. Collagen IV(alpha345) dysfunction in glomerular basement membrane diseases. II. Crystal structure of the alpha345 hexamer. J Biol Chem. 2021;296:100591. [DOI] [PMC free article] [PubMed] [Google Scholar]; First crystal structure of human collagen IVα345 hexamer, allowing the mapping of Alport variants and description of surface features enabling function. It also provides a platform for studyng pathobiology of Alport syndrome and development of new therapy.
- 22.••.Ivanov SV, Bauer R, Pokidysheva EN, Boudko SP. Collagen IV Exploits a Cl- Step Gradient for Scaffold Assembly. Adv Exp Med Biol. 2021;21:129–141. [DOI] [PubMed] [Google Scholar]; Overview of tools to study collagen IV scaffold assembly and role of chloride ions.
- 23.••.Pedchenko V, Boudko SP, Barber M, et al. Collagen IV(alpha345) dysfunction in glomerular basement membrane diseases. III. A functional framework for alpha345 hexamer assembly. J Biol Chem. 2021;296:100592. [DOI] [PMC free article] [PubMed] [Google Scholar]; A recent advance revealing mechanisms that underlie collagen IVα345 assembly and the construction of a prototype molecule for protein replacement therapy in Alport syndrome.
- 24.••.McCall AS, Cummings CF, Bhave G, et al. Bromine is an essential trace element for assembly of collagen IV scaffolds in tissue development and architecture. Cell. 2014;157(6):1380–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]; First evidence for an essential role of bromide ions in the assembly of collagen IV in basement membranes.
- 25.••.Omachi K, Kai H, Roberge M, Miner JH. Aminoglycoside-induced premature termination codon readthrough of COL4A5 nonsense mutations that cause Alport syndrome. bioRxiv. 2021:2021.2006.2011.448099. [Google Scholar]; First testing of small molecule-based premature termination codon readthrough therapy for COL4A5 nonsense mutants as a feasible approach for some patients with AS.
- 26.••.Yamamura T, Horinouchi T, Adachi T, et al. Development of an exon skipping therapy for X-linked Alport syndrome with truncating variants in COL4A5. Nat Commun. 2020;11(1):2777. [DOI] [PMC free article] [PubMed] [Google Scholar]; First report on development of an exon-skipping therapy for severe male X-linked Alport syndrome in mice, which enabled trimer formation, leading to remarkable clinical and pathological improvements including expression of the α5 chain on glomerular and the tubular basement membrane.
- 27.•.Heidet L, Borza DB, Jouin M, et al. A human-mouse chimera of the alpha3alpha4alpha5(IV) collagen protomer rescues the renal phenotype in Col4a3−/− Alport mice. Am J Pathol. 2003;163(4):1633–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]; Provides first proof of principle towards gene therapy for Alport syndrome and supportive evidence for collagen IVα345 scaffold.
- 28.Lin X, Suh JH, Go G, Miner JH. Feasibility of repairing glomerular basement membrane defects in Alport syndrome. J Am Soc Nephrol. 2014;25(4):687–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.••.Daga S, Donati F, Capitani K, et al. New frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells. Eur J Hum Genet. 2020;28(4):480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]; First achievement of a beneficial and stable Alport variant-specific correction in human podocyte-lineage cells using CRISPR/Cas9 genome editing.
- 30.•.Omachi K, Kamura M, Teramoto K, et al. A Split-Luciferase-Based Trimer Formation Assay as a High-throughput Screening Platform for Therapeutics in Alport Syndrome. Cell Chem Biol. 2018;25(5):634–643 e634. [DOI] [PubMed] [Google Scholar]; The first high-throughput screening assay for collagen IVα345 trimer formation as a platform for mutation analysis and potential drug screening.
- 31.•.Tran ML, Genisson Y, Ballereau S, Dehoux C. Second-Generation Pharmacological Chaperones: Beyond Inhibitors. Molecules. 2020;25(14). [DOI] [PMC free article] [PubMed] [Google Scholar]; Overview of a concept of second-generation pharmacological chaperones and recent practical applications of them for several disorders.
- 32.Shin MH, Lim HS. Screening methods for identifying pharmacological chaperones. Mol Biosyst. 2017;13(4):638–647. [DOI] [PubMed] [Google Scholar]
- 33.•.Lin MH, Miller JB, Kikkawa Y, et al. Laminin-521 Protein Therapy for Glomerular Basement Membrane and Podocyte Abnormalities in a Model of Pierson Syndrome. J Am Soc Nephrol. 2018;29(5):1426–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]; Foundational study demonstrating feasibility of delivering and incorporation of functional biological macromolecules to the GBM via blood stream.
- 34.•.Butkowski RJ, Langeveld JP, Wieslander J, et al. Localization of the Goodpasture epitope to a novel chain of basement membrane collagen. J Biol Chem. 1987;262(16):7874–7877. [PubMed] [Google Scholar]; First evidence for the existence of the α3 and α4 chains of collagen IV in the glomerular basement membrane and the α3 chain as the Goodpasture autoantigen.
- 35.•.Saus J, Wieslander J, Langeveld JP, et al. Identification of the Goodpasture antigen as the alpha 3(IV) chain of collagen IV. J Biol Chem. 1988;263(26):13374–13380. [PubMed] [Google Scholar]; Validation of the existence of the α3 chain of collagen IV and its identity as the Goodpasture autoantigen.
- 36.•.Pihlajaniemi T, Pohjolainen ER, Myers JC. Complete primary structure of the triple-helical region and the carboxyl-terminal domain of a new type IV collagen chain, alpha 5(IV). J Biol Chem. 1990;265(23):13758–13766. [PubMed] [Google Scholar]; First evidence for the existence of the α5 chain of collagen IV.
- 37.•.Hostikka SL, Eddy RL, Byers MG, et al. Identification of a distinct type IV collagen alpha chain with restricted kidney distribution and assignment of its gene to the locus of X chromosome-linked Alport syndrome. Proc Natl Acad Sci U S A. 1990;87(4):1606–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]; First evidence for gene locus of the α5 chain of collagen IV and restricted location of the chain in kidney.
- 38.•.Barker DF, Hostikka SL, Zhou J, et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science. 1990;248(4960):1224–1227. [DOI] [PubMed] [Google Scholar]; First evidence linking the gene encoding the α5 chain of collagen IV with Alport syndrome.
- 39.•.Hudson BG. The molecular basis of Goodpasture and Alport syndromes: beacons for the discovery of the collagen IV family. J Am Soc Nephrol. 2004;15(10):2514–2527. [DOI] [PubMed] [Google Scholar]; A review of the stragtegies and evidence for the discovery of collagen IV as a family of six alpha chains and a molecular commonality between Goodpasture and Alport diseases.
- 40.•.Gunwar S, Saus J, Noelken ME, Hudson BG. Glomerular basement membrane. Identification of a fourth chain, alpha 4, of type IV collagen. J Biol Chem. 1990;265(10):5466–5469. [PubMed] [Google Scholar]; Validation of the existence of the α4 chain of collagen IV.
- 41.•.Hudson BG, Reeders ST, Tryggvason K. Type IV collagen: structure, gene organization, and role in human diseases. Molecular basis of Goodpasture and Alport syndromes and diffuse leiomyomatosis. J Biol Chem. 1993;268(35):26033–26036. [PubMed] [Google Scholar]; A succinct review of foundational discoveries of collagen IV that underlie Alport and Goodpasture diseases.