

Abstract
Purpose:
The radiosensitivity of the normal intestinal epithelium is the major limiting factor for definitive radiotherapy against abdominal malignancies. Radiosensitizers, which can be used without augmenting radiation toxicity to normal tissue, are still an unmet need. Inhibition of proteosomal degradation is being developed as a major therapeutic strategy for anticancer therapy as cancer cells are more susceptible to proteasomal inhibition-induced cytotoxicity compared to normal cells. Auranofin, a gold-containing anti rheumatoid drug, blocks proteosomal degradation by inhibiting deubiquitinase inhibitors. In the present study we have examined whether Auranofin selectively radiosensitizes colon tumor without promoting radiation toxicity in normal intestine.
Experimental design:
The effect of Auranofin (10 mg/kg i.p.) on the radiation response of subcutaneous CT26 colon tumors and the normal gastrointestinal epithelium was determined using a mouse model of abdominal radiation. The effect of Auranofin was also examined in a paired human colonic organoid system using malignant and non-malignant tissues from the same patient.
Results:
Both in the mouse model of intestinal injury and in the human nonmalignant colon organoid culture, Auranofin pretreatment prevented radiation toxicity and improved survival with the activation of p53/p21-mediated reversible cell cycle arrest. However, in a mouse model of abdominal tumor and in human malignant colonic organoids, Auranofin inhibited malignant tissue growth with inhibition of proteosomal degradation, induction of ER stress/unfolded protein response, and apoptosis.
Conclusion:
Our data suggest that Auranofin is a potential candidate to be considered as a combination therapy with radiation to improve therapeutic efficacy against abdominal malignancies.
Introduction
Radiation-induced intestinal injury is a limiting factor for definitive chemoradiation therapy for abdominal malignancies such as gastric, pancreatic and colorectal cancer. Thus, tumoricidal radiation doses cannot be administered in the treatment of abdominal tumors, resulting in poor survival and early metastatic spread. The radioresistance of abdominal tumors further reduces the efficacy of radiotherapy. Even with advanced intensity modulated radiotherapy (IMRT), significant toxicity in the small bowel was noted which also reduced quality of life in patients. Therefore, a selective radioprotector for normal tissue and/or a radiosensitizer for tumor tissue is needed to improve efficacy of radiotherapy and achieve complete cure. In the present study we have shown for the first time that a repurposed anti-rheumatoid agent auranofin (AUR) sensitizes colon tumors while protecting non-malignant gastrointestinal epithelium.
Intestinal epithelium is a radiosensitive organ due to its high cell renewal rate. Radiation injury to intestine induces a rapid loss of mucosal epithelium, resulting in malabsorption, diarrhea, bacterial influx, and sepsis (collectively, the gastrointestinal or GI syndrome), leading to death. The epithelial response against radiation exposure to the small intestine is regulated by the tumor suppressor protein p53. In some cell types, ionizing radiation-induced activation of p53 promotes cell cycle arrest by inducing the cyclin-dependent kinase (cdk) inhibitor p21 (1–7) which may allow time for repair of DNA damage. p53−/− mice are more susceptible to the GI syndrome when compared to mice containing two copies of the wild-type p53 gene (7–9). Transgenic mice overexpressing p53 in all tissues were protected from the GI syndrome after irradiation. However, pharmacologic induction of p53 signaling and its effect on radioprotection of the GI epithelium has not been studied.
Auranofin a gold-containing compound, has been used clinically to treat rheumatic arthritis since 1985. It has also been reported that AUR has anti-cancer effects (10–13). AUR was recently tested in a Phase II clinical trial of Chronic Lymphocytic Leukemia (http://clinicaltrials.gov/ct2/show/NCT01419691). To date, AUR has been administered orally with only minor GI adverse events reported. To minimize any possibility of GI toxicity and to have maximum control of in situ drug levels, we used an intraperintoneal (i.p.) route throughout this study. We have demonstrated that AUR, a deubiquitinase (DUB) inhibitor, stabilizes p53 by inhibiting deubiquitinase HAUSP7 involved in regulation of p53 stabilization (14). Pre-treatment with AUR induces p53/p21 mediated reversible cell cycle arrest to reduce radiation toxicity in non-malignant GI epithelium and thereby ameliorates the Radiation-Induced Gastro Intestinal Syndrome (RIGS).
According to previous reports, AUR targets thioredoxin reductase, thus inducing the generation of reactive oxygen species (ROS)(15–17). However, a separate set of studies confirms that AUR-mediated apoptosis is associated with inhibition of proteosomal degradation rather than ROS generation. AUR inhibits proteasome-associated DUBs to impair proteosomal degradation and thereby induces cytotoxicity in malignant cells (18). The anticancer effect of AUR has been tested in many preclinical models. It has been shown that AUR promotes cytotoxicity in human chronic leukemia and gastric cancer cells with the induction of lethal endoplasmic reticulum (ER) stress. The hypoxic tumor microenvironment generates oxidative ER stress, resulting in protein misfolding and unfolded protein response (UPR). Inhibition of proteosomal degradation increases the mis-folded or unfolded protein burden. The accumulation of misfolded proteins in the ER produces a stress response, which induces heat shock proteins (HSPs) to initiate cytoprotective measures to correct protein misfolding and restore normal ER function (19–22). These responses are collectively known as unfolded protein response (UPR). If the correction machinery fails to control erroneous protein burden, resulting in prolonged activation of the UPR, the cytoprotective response switches to initiate programmed cell death or apoptosis. Thus, the signaling cascade of UPR involves both cytoprotective and apoptotic/cell death pathways. In this manuscript we have demonstrated that combination treatment of AUR and radiotherapy increases the misfolded protein load beyond the tolerance level and thereby induces cell death with the activation of a UPR downstream apoptotic pathway. We have also demonstrated that in organoids derived from human colonic surgical specimens, AUR protects the non-malignant tissue but sensitizes malignant organoids to radiation. Therefore, this is the first report demonstrating the dual role of a small molecular agent to promote efficacy of radiotherapy both in mice and human tissue.
Materials and Methods:
Animals
Five- to six week-old male C57BL/6 and BALB/c (Jackson Laboratory, Bay Harbor, ME, USA) mice were maintained ad libitum. All studies were performed under the protocols approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center (ACUP Number: 2016–2316).
Cell lines and Human Samples
The murine colorectal carcinoma cell lines CT26 (ATCC CL-2638) and IEC6 (ATCC CRL-1592) were obtained from the American Type Culture Collection [(ATCC), with authentication by short tandem repeat] and maintained in complete medium as described previously (23). Cell lines were routinely characterized in the laboratory based on morphology and gene-expression patterns. Cells were confirmed to be free of mycoplasma contamination using a mycoplasma detection kit (Thermo Fisher Scientific, Waltham, MA, USA). Cells were maintained with RPMI-1640 medium (Corning Cellgro, Manassas, VA, USA) supplemented with 10% heat-inactivated fetal bovine serum (Corning Cellgro) in a 5% CO2 atmosphere at 37°C.
Surgically resected tumor and normal colon from three individual patients were obtained through the University of Kansas Medical Center Biospecimen Repository.
Development of organoid culture from human colon
Human colon tissues were received from the University of Kansas Medical Center Biospecimen Repository (HUS#5929) to perform organoid culture as described previously (24,25). The tissue was scraped and chopped into ~5 mm pieces. The tissue was then washed with cold PBS, and incubated in dissociation buffer for 20 min with agitation at room temperature. The tissue fragments were suspended vigorously with a 10-ml pipette in cold 0.1% BSA in PBS, yielding supernatants enriched in epithelial cells. Samples were passed through 100 μm filters (BD Biosciences, San Jose, CA, USA) to obtain Fraction 1. The same step was repeated three times to get fractions 2, 3 and 4. Fractions 3 and fraction 4 enriched in crypt stem cells were centrifuged at 300 g for 5 min at 4°C and diluted with advanced DMEM/F12 (Invitrogen, Waltham, Massachusetts, USA) containing B27, N2, 1 μM n-Acetylcysteine, 10 mM HEPES, penicillin/streptomycin, and Glutamax (all Invitrogen). Crypts were passed through 100 μm filters (BD Biosciences), and centrifuged at 275 g for 5 min at 4°C and single cells were discarded. Crypts were embedded in extracellular matrix (BD Bioscience) and plated in a pre-warmed 24-well plate. After the matrix/matrigel solidified, Intesticult medium (Stem cell technologies, Inc Vancouver, Canada) with supplements and gentamycin (50ug/ml) was overlaid. Passage was performed once per week. The number of organoids per well was counted by Evos microscope (Thermo Fisher Scientific, Waltham, MA, USA). Images of organoids were acquired using fluorescent (Nikon, 80i) and confocal (Nikon, A1RMP) microscopy. The total number of crypt structures and number of budding crypts were quantified and expressed as a ratio of budding crypts/total crypt structures.
siRNA transfection in human normal colon derived organoid
The single cell suspension from human normal and tumor colon derived organoids was prepared by dissociating the organoid with cell dissociation media (Corning Cellgro) per manufacturer provided protocol. Human p53 and p21 siRNA were purchased from Santa Cruz, Dallas, TX, USA. The transfection of p53 and p21 siRNA and control scrambled RNAs in the normal colon derived organoid cells was carried out using Lipofectamin 2000 (Thermo Fisher Scientific, Waltham, MA, USA) per manufacturer provided protocol. Western blot was done to confirm the silencing of p53 and p21 protein expression.
Cytotoxicity and MTT assay
MTT assay was performed as reported earlier (26). A total of 5000 cells in 100 μl medium per well were plated in 96-well plates and allowed to adhere for 24 h. AUR (Enzo Life Sciences, Farmingdale, NY, USA), was added at indicated concentrations for 2 h, and cultures were rinsed with fresh medium and re-incubated for an additional 24 h. Next, 10 μl of the MTT reagent (5 mg/ml, Promega, Madison, Wisconsin, USA) was added for 2 h, and 150 μl of DMSO was admixed to dissolve the formazan crystals. Absorbance was measured at a wavelength of 540 nm using a spectrophotometer.
Clonogenic assay
Clonogenic assay was performed as reported earlier (27). Cells were seeded in a 6-well plate and treated with 2 μM AUR for 24 h; then X-irradiated at a dose rate of 2.26 Gy/min (XENX, XStrahl, Surrey, UK). After 8 days, cultures were fixed with crystal violet and colonies (> 50 cells) were counted. Surviving fractions (SF) were fitted to the linear quadratic model using GraphPad Prism 5 software (GraphPad Prism Software Inc., La Jolla, CA, USA). The AUR concentration used is similar to the peak concentration of gold that is achieved in humans following chronic treatment with the standard dose of 6 mg daily (28,29).
Isolation of Intestinal Epithelial Cells.
Intestinal epithelial cells were prepared from the jejunum of adult male C57Bl6 mice as previously described (25,30). A catheter was inserted into the intestine of anaesthetized mice through an incision in the most proximal part of duodenum. Another incision was made just proximal to the caecum to perfuse the entire small intestine with ice-cold PBS followed by two times flushing with ice-cold PBS plus 1 mmol/L dithiothreitol (DTT). The entire jejunum was tied at the distal end and filled with isolation citrate buffer (0.9% w/v NaCl, 1.5 mmol/L KCl, 27.0 mmol/L NaCitrate, 8.0 mmol/L KH2PO4 and 5.6 mmol/L Na2HPO4, pH 7.3) followed by incubation at 37°C for 15 min. After incubation, the fluid was emptied from the jejunum and it was further filled with 5 ml ethylene diamine tetra acetic acid (EDTA) buffer (0.9% w/v NaCl, 8 mmol/L KH2PO4, 5.6 mmol/L Na2HPO4, 1.5 mmol/L Na2 EDTA, pH 7.6, plus 0.5 mmol/L DTT and 0.23 mmol/L phenylmethylsulfonyl fluoride (PMSF); Sigma-Aldrich, St. Louis, MO, USA). The jejunum was then physically manipulated and tapped allowing the cells to separate from the interior surface. The jejunum was rinsed twice with 5 ml of EDTA buffer and all the fluid containing epithelial cells was collected, centrifuged at 300 g for 5 min, washed twice with 20 ml of balanced salt solution (BSS) containing 135 mmol/L NaCl, 4.5 mmol/L KCl, 5.6 mmol/L glucose, 0.5 mmol/L MgCl2, 10 mmol/L HEPES and 1.0 mmol/L CaCl2, pH 7.4, and suspended in 2 ml of this solution. The number of viable cells was quantified using an automated cell counter T20 (Biorad, Hercules, CA, USA) by the trypan blue exclusion method.
Irradiation Procedure
Abdominal Irradiation (AIR) or whole body irradiation (WBI) was performed on mice anaesthetized via a continuous flow 1.5 ml/min of 1.5% isoflurane in pure oxygen, using a small animal radiation research platform (XENX, XStrahl, Surrey, UK) as described previously (25). For AIR a 3 cm square area of the mouse abdomen encompassing the GI tract was irradiated (Fig. 1), but shielding the upper thorax, head and neck as well as the lower and upper extremities. This results in significant protection of the bone marrow, while predominantly inducing RIGS. Radiation was delivered to the midline of the abdomen. To ensure homogeneous dose delivery, half of the dose was delivered from the anterior-posterior direction and the other half from the posterior–anterior direction. The total irradiation time to deliver the intended dose was calculated with respect to dose rate, field size for radiation and fractional depth dose to ensure accurate radiation dosimetry.
Figure 1:
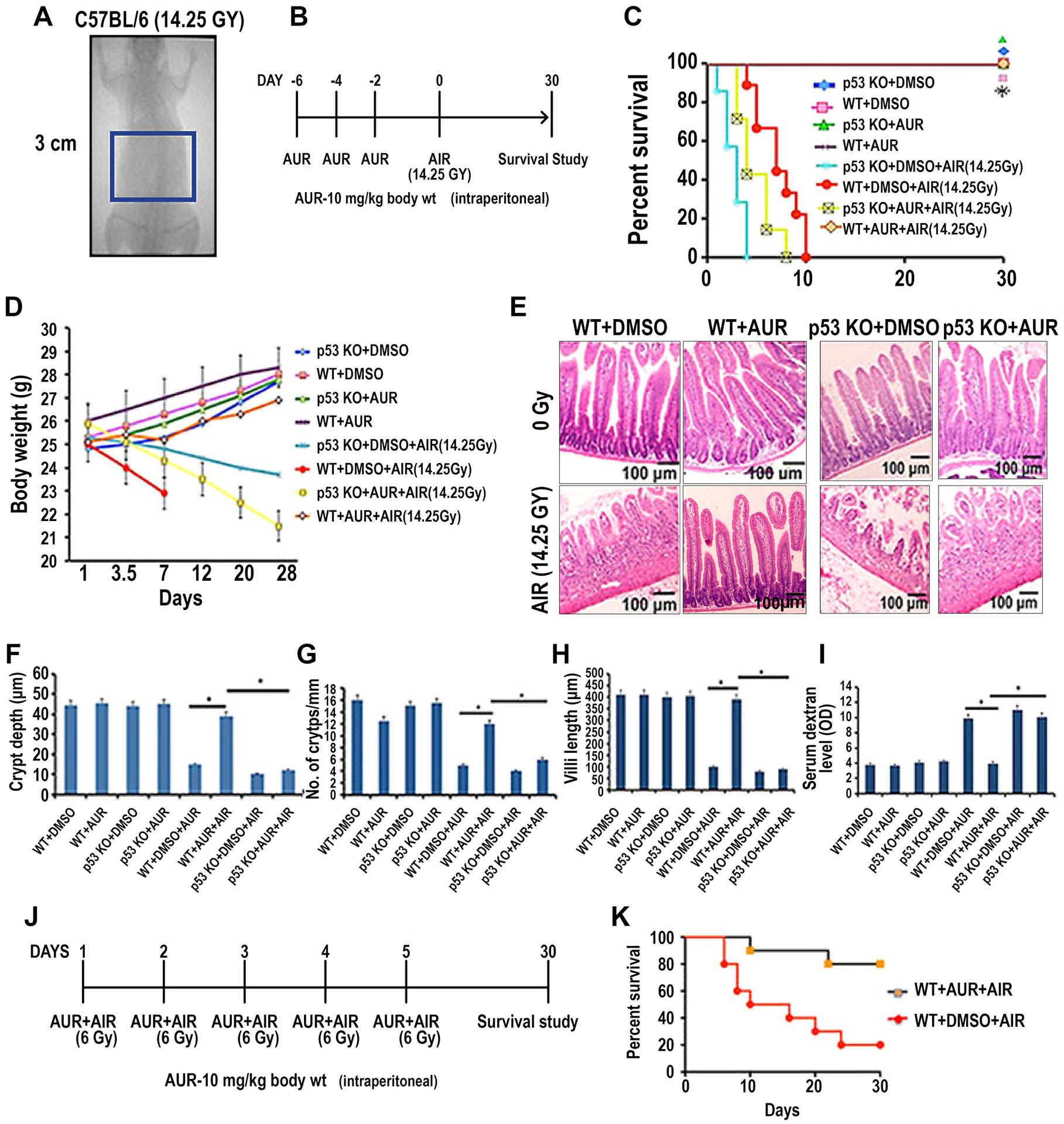
Auranofin treatment protected C57BL/6 mice from radiation-induced lethality. A) Schematic diagram demonstrating AIR exposure field for C57BL/6 mice. A 3 cm square abdominal area of the mouse containing the GI tract was irradiated (irradiation field), thus shielding the upper thorax, head and neck as well as lower and upper extremities, protecting a significant portion of the bone marrow. Mice were treated with auranofin (10 mg/kg i.p.) on days 2, 4 and 6 prior to AIR (14.25 Gy). B) Schematic diagram of the treatment protocol. C) Kaplan–Meier survival analysis. Wild type (WT) male mice receiving auranofin showed 100% survival beyond 30 days post irradiation compared with mice receiving DMSO where 100% of mice died within 10 days after irradiation (p<0.0001, Log-rank (Mantel–Cox) test; n=10 per group). However, auranofin did not inhibit radiation-induced lethality in p53 knockout (KO) mice. D) Body weight of mice at times post irradiation (14.25 Gy AIR). E) Representative H&E stained sections of mouse jejunum (20X). Note, restitution of crypt villus structures in irradiated, auranofin treated WT mice compared with mice receiving only radiation. However, in p53 KO mice auranofin treatment did not protect the intestinal epithelium from radiation damage. F-H) Histograms showing the crypt depth (F), number of crypts/mm (G) and villous length (H) in jejunal sections of WT mice and mice treated with auranofin or AIR or AUR+AIR. Auranofin treatment did not have any effect on the intestine but significant increase in crypt depth (*, p<0.001), crypt count/mm (*, p<0.005) and villi length (*, p<0.001) were observed in the AIR cohort pre-treated with auranofin compared to the AIR only cohort (unpaired t-test, two-tailed) (n=6 per group). AUR treatment in p53 KO mice demonstrated significantly less crypt depth (*, p<0.002), crypt count/mm (*, p<0.006) and villous length (*, p<0.003) compared to WT mice receiving auranofin. I) Histogram demonstrating serum dextran levels in WT and p53KO mice. No significant change in serum dextran level was observed in control and in WT mice treated only with auranofin. Significantly higher serum dextran levels were observed in the AIR group compared to the AIR+AUR group (*, p<0.006) or control (DMSO) group (p<0.004) of WT mice. In p53KO mice AUR pre-treatment did not reduce the serum dextran level following irradiation. J) Schematic diagram of the treatment protocol and fractionated radiation schedule. K) Kaplan–Meier survival analysis of mice receiving fractionated AIR (6 Gy × 5 fraction). Auranofin pre-treatment improved survival in these mice compared to controls (p<0.0002, Log-rank (Mantel–Cox) test; n=10 per group).
Histology
As described previously (25), the intestine was dissected, washed in PBS to remove intestinal contents and the jejunum was fixed in 10% neutral-buffered formalin prior to paraffin embedding. Tissue was routinely processed and 5 μm sections were prepared for haematoxylin and eosin and immunohistochemical staining. All haemotoxylin and eosin (HE) (Fisher Scientific, Pittsburgh, PA, USA) staining was performed at the Pathology Core Facility in the University of Kansas Cancer Center.
Determination of Villi Length and Crypt Depth
Crypt depth was objectively analyzed and quantitfied in a blinded manner from coded digital photographs of crypts from HE-stained slides using ImageJ 1.37 software to measure the height in pixels from the bottom of the crypt to the crypt villus junction. The length from the crypt villus junction to villous tip was measured to determine the villous length. This measurement was recorded in pixel and was converted to length (in μm) using the following conversion factor (1.46 pixels/μm).
Detection of Apoptosis In Situ
Apoptotic cells were detected in situ by performing TUNEL (TdT–mediated digoxigenin labeled dUTP nick end labeling) staining. Briefly, paraffin embedded sections were de-paraffinized, rehydrated through graded alcohol steps and stained using ApopTag Peroxidase In Situ Apoptosis Detection Kit (Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. A total of 60 crypts were examined per animal.
FITC-dextran permeability assay
Mice exposed to AIR and/or pre-treated with AUR were gavaged on day 5 after irradiation with 0.6 mg/kg body weight of a FITC-dextran solution (4,000 kD size, Sigma-Aldrich, St. Louis, MO, USA). Mice were euthanized and serum was obtained with cardiac puncture 4 h after gavage (31). Samples were measured in a 96-well plate using a multi well fluorometer. A standard curve was constructed using mouse serum having increasing amounts of FITC-dextran to determine the serum levels of FITC-dextran in different treatment groups.
BALB/c mice xenograft assay
2×106 CT26 cells in 200 μl PBS was inoculated subcutaneously in the flanks of 6–8 week-old male BALB/c mice. Upon observation of palpable tumors, animals were randomly distributed into treatment groups and treated with either 10% DMSO, Auranofin (i.p., 10 mg/kg/day in 10% DMSO), 12.5 Gy as single fraction AIR or 4 × 4 Gy of fractionated radiation or a combination of Auranofin and radiation as mentioned in the schematic diagrams (Fig. 4A and E). The Auranofin dose of 10 mg/kg has been reported as non-toxic when used alone in preclinical studies (11,32). Auranofin was given three times a week for three weeks and radiation was given twice a week for two weeks. Radiation was started 1 week after the first auranofin injection. Tumors were measured every other day using Vernier calipers. Tumor volumes were determined by the formula: Volume (in cubic millimeters) = W × (L)2/2 (W, width; L, length). When tumor volumes reached 2,000 mm3 or the tumors became necrotic, animals were euthanized (33).
Cell isolation from solid tumor
A single cell suspension from solid tumor was prepared a previously described protocol with little modification (34). Briefly, each tumor was excised from the mouse abdomen without tearing the abdominal cavity. Tumors were transferred to a petri plate containing serum free media and chopped with a razor. Then, the suspension was incubated at 37°C with gentle shaking in 40 μl of Liberase DL stock solution 28 U/ml (Roche), 80 μl of Liberase TL stock solution 14 U/ml (Roche), and 40 μl of DNase I (15 mg/ml stock solution 100X, Sigma-Aldrich, St. Louis, MO, USA) for 30 min. The enzymatic reaction was stopped by adding 2% BSA solution. The suspension was then passed through a 100 μm cell strainer (BD Falcon, Tewksbury MA, USA) and lysed with RBC lysis buffer (Biolegend, San Diego, CA, USA) so as to prepare a single cell suspension. These cells were used for deubiquitinase activity assay, western blot, q-PCR and apoptosis assay.
Deubiquitinase activity assay
Deubiquitinase (DUB) activity was measured according to a protocol reported earlier (35). Briefly, tumor cell lysate and tumor colon organoid cell lysate (5 μg) was dissolved in ice-cold DUB buffer containing 50 mmol/L Tris-HCl (pH 7.5), 250 mmol/L sucrose, 5 mmol/L MgCl2, and 1 mmol/L PMSF, then incubated with Ub-AMC substrate (Enzo Life Sciences, Farmingdale, NY, USA) in a 100 μL reaction volume at 25°C. AMC release generated from the cleaved substrate was temporally recorded with the microplate reader of a fluorescent spectrophotometer.
USP7 Inhibitor assay
USP7 (HAUSP) inhibitor activity of AUR was determined using the USP7 inhibitor screening assay kit (BPS Bioscience) as per manufacturer protocol. The USP7 Inhibitor Screening Assay Kit is designed to measure USP7 activity for screening and profiling of HAUSP7 inhibitor. The USP7 Inhibitor Screening Assay Kit contains fluorogenic, ubiquitinated substrate Ub-AMC, assay buffer, and purified USP7 enzyme. USP7 is incubated with a sample containing assay buffer and Ub-AMC Substrate in presence/absence of HAUSP7 inhibitor and activity was measured by fluorimetric assay on a fluorimeter.
Western blot analysis
Whole cell lysates were prepared in RIPA buffer supplemented with 10 mmol/L β-glycerophosphate, 1 mmol/L sodium orthovanadate, 10 mmol/L NaF, 1 mmol/L PMSF, and 1×Roche Complete Mini Protease Inhibitor Cocktail (Roche, Indianapolis, IN, USA) from crypt cells of BALB/c and C57BL/6 mice, solid tumor of BALB/c mice and normal and tumor colon organoid with different treatments. Cell lysates were subjected to SDS-PAGE, transferred to polyvinylidene difluoride membrane, and immunoblotted with monoclonal primary antibodies and appropriate secondary antibodies (1:10000) Infrared tagged 680RD Goat anti-Mouse IgG and 800CW Goat anti-Rabbit IgG (Li-cor, Lincoln, NE, USA). To detect the level of mono and poly-ubiquitinated proteins or peptides, we used mono- and poly-ubiquitinylated conjugated monoclonal antibody (1:1000) (Enzo Life Sciences, Farmingdale, NY, USA). To determine the p53 and p21 level and UPR signaling molecules in cell lysates, mouse monoclonal antibody to p53 (1:500) (Catalog no. sc6243), p53 (human) (1:200) (Catalog no. sc126), p21(1:1000) (Catalog no. sc397) (Santa Cruz Biotechnology, Dallas, TX, USA); p53 (acetyl K120) (Catalog no. ab78316, Abcam, Cambridge, MA), Tip60 (phospho S86) (Catalog no. ab73207, Abcam, Cambridge, MA); PERK (1:500) (Catalog no. C33E10), p-PERK (1:500) (Catalog no. T980), CHOP (1:1000) (Catalog no. D46F1), PUMA (1:500) (mouse Catalog no. ab9643; Abcam, Cambridge, MA) (human Catalog no. 4976; Cell Signaling Technology, Danvers, MA, USA), BAK (mouse) (1:1000) (Catalog no. 12105; Upstate Biotechnology, Inc. Lake Placid, NY, USA), BAX (mouse) (1:500) (Catalog no. 2772; Santa Cruz Biotechnology, Dallas, TX, USA) and β-Actin (13E5) Rabbit monoclonal antibody (1:1000) (Catalog no. 4970) (Cell Signaling Technology, Danvers, MA, USA) was used.
Human colonic organoids grown in 24-well plates were used for immunoblot analysis. Each 24-well plate(number of organoids 15±3/well) was used for an individual treatment group. For each experiment 30 μg protein was loaded for each sample.
Densitometric analysis of immunoreactive bands was performed using Li-cor image studio lite, version 5.2 (Lincoln, NE, USA). Mean band density was plotted as a histogram after normalization of band density of each protein with band density of β-actin.
Full images of all immunoblots are shown in supplement figure S5–S6.
Cell death assay
Apoptosis in cells from solid tumors and cells from organoids following different treatments was determined by flow cytometry using Annexin V-fluoroisothiocyanate (FITC)/propidium iodide (PI) double staining (Bio-Vision, Milpitas, CA, USA) (36). Briefly, the cells were collected and washed with binding buffer, then incubated in working solution (100 μl binding buffer with 1.0 μl Annexin V-FITC) for 15 min in dark. PI was added just before flow cytometric analysis.
Real Time PCR analysis of UPR target genes
RNA was isolated from mouse intestinal epithelial cells, cells from mouse solid tumor and human malignant colon organoid cells using RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Isolated RNA was subjected to cDNA synthesis using the SuperScriptTM First-Strand Synthesis System (Invitrogen, Grand Island, NY, USA). Real time PCR was performed in a QuantStudio 7 Flex Real-Time time PCR machine (Thermo Fisher Scientific, Waltham, MA, USA) using the ABsolute QPCR SYBER Green Mix (ABgene, Rochester, NY, USA) according to the standard ABgene protocol. To check for primer amplification specificity, a melting curve was generated at the end of the PCR and different samples containing the same primer pair showed matching amplicon melting temperatures. Primers used for real time PCR were XBP1: F 5TGG CCG GGT CTG CTG AGT CCG’ 3’; R 5’ ATC CAT GGG GAG ATG TTC TGG 3’; spliced form of XBP1: F 5’CTG AGT CCG AAT CAG GTG CAG3’; R 5’ATC CAT GGG GAG ATG TTC TGG3’; CHOP: F 5’ ACC AAG GGA GAA CCA GGA AAC G 3’; R 5’ TCA CCA TTC GGT CAA TCA GAG C 3’; GRP78: F 5’TTC CAA GGA ACA CTG TGG TG 3’; R CAA AAG TTC CCA GGA GGT GA 3’; ATF4: F 5’GTT CTC CAG CGA CAA GGC TA 3’ R 5’ATC CTG CTT GCT GTT GTT GG 3’; PMAIP1 (NOXA): F 5’GAG TGC ACC GGA CAT AAC TG3’ R 5’CTCGTCCTTCAAGTCTGCTG3’; BBC3 (PUMA): F 5’GTGTGGAGGAGGAGGAGTG3’ R 5’ TCGATGCTGCTCTTCTTGTC3’; BIM: F 5’GGCCCCTACCTCCCTACA3’ R 5’ GGGGTTTGTGTTGATTTGTCA3’; BAK F 5’ACGCTATGACTCAGAGTTCC3’ R 5’ CTTCGTACCACA-AACTGGCC3’; BAX F 5’ATGGACGGGTCCGGGGAGCA3’ R 5’CCCAGTTGAAGTTGCCGTCA3’; GAPDH: F 5’ TCA GTT GTA GGC AAG CTG CGA CGT 3’ and R 5’ AAG CCA GAG GCT GGT ACC TAG AAC 3’. The internal control gene GAPDH was amplified simultaneously in a separate reaction. Threshold cycle number (CT) of triplicate reactions was determined using the ABI-SDS software and the mean CT of triplicate reactions was determined. The levels of expression of the genes of interest were normalized to GAPDH using the formula 2–ΔDCT, where –ΔDCT = ΔCT (sample) – ΔCT (control) and ΔCT is the CT of the target gene subtracted from the CT of the housekeeping gene GAPDH.
Statistical Analysis
Data from the mouse survival/mortality studies were analyzed by Kaplan-Meier statistics as a function of radiation dose using Graphpad Prism 6.0 software for Mac (25). After genotyping mice were sorted randomly to each experimental and control group. For mouse intestinal histopathological analysis, jejunal sampling regions were chosen at random for digital acquisition for quantitation. Evaluation of digital image data was performed blinded as to treatment. A two-sided student’s t test was used to determine significant differences between experimental cohorts (P < 0.05) with representative standard errors of the mean (SEM).
Results
Auranofin treatment inhibits RIGS
Sensitivity of the intestinal epithelium to radiation is always a concern when using any antineoplastic agent as a radiosensitizer for abdominal radiotherapy. Therefore, we first examined whether auranofin alone or in combination with radiation-induced GI toxicity. In vitro results in IEC6 intestinal epithelial cells demonstrated that auranofin alone does not have any effect on cell survival (Supplementary Fig. S1A). Moreover, IEC6 cells became more resistant to radiation with auranofin pretreatment (Supplementary Fig S1C and S1D). Auranofin treatment, however, reduced the viability of CT26 colon cancer cells (Supplementary Fig. S1A). Clonogenic assay demonstrated that auranofin (2 μmol/L) enhances the radiosensitivity of CT26 cells (Supplementary Fig. S1B). Therefore, the in vitro data indicate that auranofin may be used as a radiosensitizer without promoting toxicity in non-malignant tissue.
Treatment with auranofin in wild type (WT) C57BL/6 male mice did not show any changes in animal health and overall survival. We examined the protective role of AUR against radiation-induced intestinal injury in mice. Mice exposed to 14.25 Gy of abdominal irradiation (AIR) died within 10–15 days of radiation with characteristic signs and symptoms of RIGS, including diarrhea, black stools and weight loss. In contrast, animals that received AUR treatment (3 times, alternate days, 10 mg/kg i.p.) prior to AIR (Fig. 1A–B) had well-formed stools, maintained body weight (Fig 1D) and demonstrated 100% survival beyond 20 days post AIR (p<0.003, Log-rank (Mantel–Cox) test; n=10 per group) (Fig. 1C). This experiment was also repeated in C57BL/6 female mice. Female mice receiving AUR treatment (3 times, alternate days, 10 mg/kg i.p.) prior to AIR demonstrated 90% survival beyond 20 days post AIR (p<0.0003, Log-rank (Mantel–Cox) test; n=10 per group) (supplementary Fig. S2A). Mice rescued from RIGS by auranofin treatment were observed beyond 30 days post radiation. No behavioral changes were noticed, with normal food and water intake.
Histopathological analysis of jejunal sections collected 96 h post AIR showed crypt depletion and villous denudation in untreated WT mice exposed to AIR (25) (Fig. 1E). Treatment with auranofin improved overall crypt villus architecture with an increase in crypt depth (p<0.001) (Fig. 1F), number of crypts (p<0.005) (Fig. 1G) and preserved villous length (p<0.001) (Fig. 1H) following irradiation. Since dextran is unable to cross the GI epithelium unless it is compromised, detection of dextran in the blood is a good indicator of epithelial damage. Blood FITC dextran levels were measured at 4 h after gavage (25). Exposure to AIR significantly increased the serum dextran level compared to un-irradiated control, suggesting increase in epithelial permeability. Treatment with auranofin in irradiated mice significantly reduced the FITC-dextran uptake into the blood (p<0.006) (Fig. 1I). These data indicate restitution of intestinal epithelial integrity by auranofin treatment.
The intestinal epithelium in mice deficient in p53 was more sensitive to higher radiation doses, causing lethal GI syndrome. The presence of p53 in crypt epithelial cells promotes reversible cell cycle arrest after irradiation. However, in p53-deficient epithelium, dysregulated cell proliferation following irradiation results in accelerated death of damaged cells followed by rapid destruction of villi and accelerated lethality. Super p53 mice that contain one extra copy of the wildtype p53 allele, for a total of 3 copies of the p53 genes demonstrated resistance to GI syndrome compared to wildtype mice (37). However, Super p53; p21−/− and wildtype p53; p21−/− did not show any protection from GI syndrome (7). p21 is a cell survival protein which arrests the cell cycle for DNA repair by a p53 dependent pathway. It has been reported that auranofin induces p21 expression with downregulation of Cdk2 and Cdk4 (38). We examined the role of p53 in the radioprotective effect of AUR. Kaplan-Meier survival analysis demonstrated that auranofin could not rescue p53 knockout (KO) mice from radiation-induced toxicity (Fig. 1C–D). Histopathological analysis demonstrated villi denudation (Fig. 1E,H), loss of crypt depth (Fig. 1E,F) and number of crypts (Fig. 1E,G) resulting in an increase in mucosal permeability in these irradiated mice, with/without auranofin treatment (Fig. 1I).
The radioprotective effect of Auranofin was also examined in a fractionated AIR model (39). Mice receiving Auranofin (10 mg/kg i.p.) prior to fractionated AIR exposure (6 Gy × 5 fractions) (Fig1J–K) demonstrated significant improvement in survival compared to AIR controls (p<0.005, Log-rank (Mantel–Cox) test; n=10 per group).
Auranofin pretreatment promotes reversible cell cycle arrest in crypt epithelial cells
To understand the effect of auranofin on crypt cell proliferation, we performed Ki67 staining in sections of jejunum from C57Bl6 mice. Exposure to whole body irradiation (WBI) demonstrated a strong presence of Ki67+ve crypt epithelial cells within the first 4 h. However, a rapid decrease in the number of Ki67+ve cells was observed within 72 h (Fig. 2A–B). Auranofin pre-treatment (10 mg/kg i.p.) reduced cell proliferation prior to radiation which gradually recovers to a higher proliferation level within 3.5 to 4 days post irradiation (Fig. 2A–B) and thereby serves to repair the crypt villus structure in intestinal epithelium (Fig. 2C–E). Auranofin treatment alone also reduced proliferation of un-irradiated crypt cells which then gradually recovers following the auranofin treatment (Fig. 2B). γH2AX staining demonstrated significant reduction in DNA damage accumulation at 4–24 h post irradiation with auranofin treatment compared to irradiated control (Fig. 2F–G). Moreover, TUNEL staining in jejunal sections at 4–24 h post irradiation demonstrated significantly less apoptosis in the auranofin treated group compared to irradiated controls (Fig. 2H–I). AUR also reduces radiation induced mitotic catastrophe (multinucleated structures with multipolar spindles) (40) in mouse crypt cells (Fig 2J–K). These results suggest that auranofin mediated blocking of cell proliferation prior to irradiation inhibits DNA damage and reduce apoptosis within hours after irradiation, which then promotes epithelial regeneration within 3–4 days post-irradiation.
Figure 2:
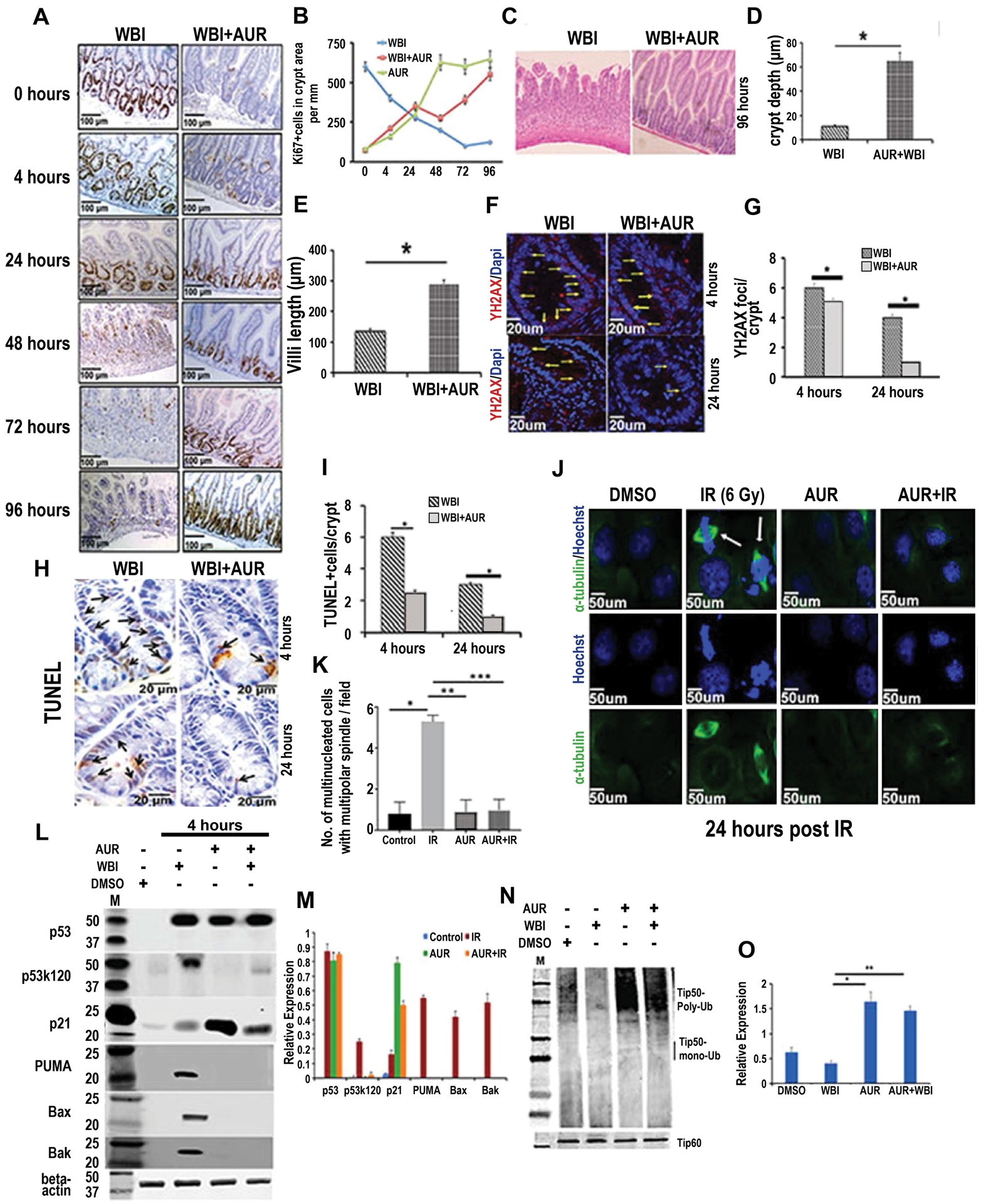
Auranofin modulates crypt cell proliferation kinetics, reduces radiation-induced DNA damage and promotes crypt regeneration. C57BL/6 male mice pretreated with auranofin (10 mg/kg i.p.) or vehicle (DMSO) were exposed to 12.5 Gy WBI. A) Representative images of jejunal sections stained with Ki67. B) Quantification of Ki67 positive cells in jejunum from irradiated mice treated with auranofin or DMSO, as a function of time after irradiation. C) H&E stained jejunal section at 96 h post WBI (n=6 mice per group). D–E) Histogram demonstrating crypt depth and villous length in jejunal sections at 96 h post WBI. Auranofin pretreatment demonstrated higher crypt depth (p<0.0003) and villous length (p<0.0006) compared to WBI control. F) Representative confocal images of γH2AX staining of jejunal sections. G) Histogram demonstrating number of γH2AX positive foci in crypt cells. Auranofin pretreatment reduced number of γH2AX positive foci/crypt compared to WBI control, both at 4 and 24 h post exposure (*p<0.05 and *p<0.003, respectively). H) Representative image of TUNEL staining in jejunal sections. I) Histogram demonstrating number of TUNEL positive cells per crypt. Auranofin pretreatment reduced number of TUNEL positive cells/crypt compared to WBI control, both at 4 and 24 h post exposure (*p<0.008 and p<0.006, respectively). J. Representative confocal microscopic images of α-tubulin and Hoechst staining demonstrate mitotic catastrophe (α-tubulin and Hoechst double positive cells indicated with an arrow). K) Histogram of number of multinucleated cells with multipolar spindle per field (n=3 mice/group). Auranofin pretreatment reduced the number of these cells compared to the IR control (***p<0.0003). Immunoblot demonstrating expression of total p53, acetylated p53 (p53k120), total p21, PUMA, BAX and BAK in crypt cells. M) Histogram demonstrating densitometric analysis of immunoreactive bands representing expression of the above proteins (assessments repeated in 3 mice/group). Significant upregulation of p53k120 (p<7.86E-07), PUMA (p<7.96E-08), BAX (p<6.87E-08), BAK (p<6.92E-08) expression was noted in the WBI group compared to the WBI+AUR group. The WBI group also showed significantly higher expression of these molecules compared to untreated controls [p53k120 (p<8.78E-08), PUMA (p<8.66E-08), BAX (p<8.87E-08), BAK (p<8.32E-08)] or auranofin alone [p53k120 (p<6.78E-06), PUMA (p<7.66E-07), BAX (p<5.87E-07), BAK (p<8.32E-08)]. Expression of p21 was significantly higher for AUR+WBI compared to WBI (p<5.68E-05). N). Immunoblot demonstrating ubiquitinated and total Tip60 in crypt cells. O) Histogram of results from densitometric analysis of immunoreactive bands representing ubiquitinated Tip60. The ubiquitinated form of Tip60 was significantly higher in AUR+WBI compared to WBI mice (p<0.0004).
We have examined the involvement of p53 and p21 in AUR mediated regeneration of crypt epithelium in mice exposed to WBI. Histopathological analysis at 96 h post-irradiation clearly demonstrated that auranofin could not inhibit radiation-induced intestinal epithelial damage (Supplement Fig S2B) in p53 and p21 KO mice. Unlike WT mice, auranofin pre-treatment failed to halt radiation-induced crypt cell proliferation in p53 and p21 knock out mice at 4 h post-irradiation (Supplement Fig S2B–C) resulting in mitotic catastrophe and significant damage to the intestinal epithelium (Supplement Fig S2B).
Cell fate after injury/stress depends on a balance between p53 downstream cell-cycle arrest or proapoptotic signaling (41) (42,43). p53 activates downstream effector genes, such as PUMA and BAX for apoptosis, or a gene expressing cyclin/cdk inhibitor p21 for cell-cycle arrest. In response to radiation-induced DNA damage, acetylation of p53 at k120 switches on the pro-apoptotic pathway in intestinal epithelium (44). A previous report has shown that pharmacological inhibition of p53k120 acetylation blocked radiation-induced apoptosis and PUMA induction without affecting induction of p21 expression. In the mouse model of abdominal irradiation we have also observed upregulation of p53k120 acetylation (Fig 2L–M) resulting in induction of PUMA, BAX, and BAK expression (Fig 2L–M). Pre-treatment with auranofin did not alter the stabilization of p53 but significantly reduced radiation-induced acetylation of p53k120 and expression of PUMA, BAX, and BAK (Fig 2L–M). Auranofin, a potent DUB inhibitor, stabilized p53 by inhibition of HAUSP DUB (14) (Supplement Fig S3A). Absence of HAUSP promoted the ubiquitinated form and thereby proteosomal degradation of MDM2, a negative regulator of p53 levels (14).
Acetylation of p53k120 is mediated by Tip60 acetylatransferase (43). Activity of Tip60 acetyltransferase is modulated by ubiquitination (45). The present study demonstrated that pretreatment with auranofin promoted the ubiquitinated inactive form Tip60 (Fig 2N–O) and thereby reduced the acetylated form of p53k120. The de-acetylated form of p53 shifted the balance from a pro-apoptotic pathway and induced p21 expression (Fig 2L–M). Involvement of Tip60 in acetylation of p53 at k120 and thereby activation of the intrinsic apoptotic pathway was further confirmed by siRNA mediated depletion of Tip60 in IEC6 intestinal epithelial cells. In irradiated IEC6 cells, absence of Tip60 inhibited the acetylation of p53k120 and activation of the apoptotic pathway without affecting p21 expression (supplement Fig S3B–C). Treatment of IEC6 cells with auranofin prior to irradiation also inhibited acetylation of p53k120 and the downstream intrinsic apoptotic pathway and induced p21 expression (supplement Fig S3B–C). Previous studies by other groups have also shown that pharmacological inhibition of Tip60 inhibits acetylation of p53k120 and rescues intestinal stem cells from radiation-induced apoptosis(44). Therefore, these observations, including our present study, clearly suggest that modulation of p53k120 acetylation shifts the balance from a pro-apoptotic pathway and induces p21 expression so as to promote survival of intestinal epithelial cells following irradiation.
Auranofin treatment protects human non-malignant colon derived organoid from radiation.
To determine the role of auranofin in radiation-induced normal tissue toxicity in human colonic epithelium we turned to an ex vivo colonic organoid culture system developed from surgical specimens from human colon (24). The effect of auranofin has been examined in 6 different malignant and non-malignant paired organoids. We have observed interpersonal radiosensitivity differences in both normal and malignant tissue derived organoids (Supplement Fig S4A–B). Following radiation exposure organoids lose their budding crypt structure in a dose dependent manner (Fig 3A–B). The radioprotective role of AUR was examined in non-malignant colon derived organoids. Treatment with auranofin (2 μM) prior to radiation (6 Gy) rescued these organoids from radiation toxicity (Fig 3A) and restored the number of budding crypts to the un-irradiated level as represented by budding crypt/total crypt ratio (Fig 3A–B). No significant difference was noted in organoid morphology or growth between un-irradiated untreated organoids and auranofin treated un-irradiated organoids (Fig 3A, B). Auranofin also promoted cell proliferation of irradiated organoid cells (Fig 3A–D), ATP level (Fig 3C) and inhibited apoptosis (Fig 3A–E) as demonstrated by Ki67 staining, ATP level analysis and cleaved caspase staining, respectively. To analyze the mechanism of action of radioprotective role of auranofin three most radiosensitive organoids (n=3) were further examined.
Figure 3.
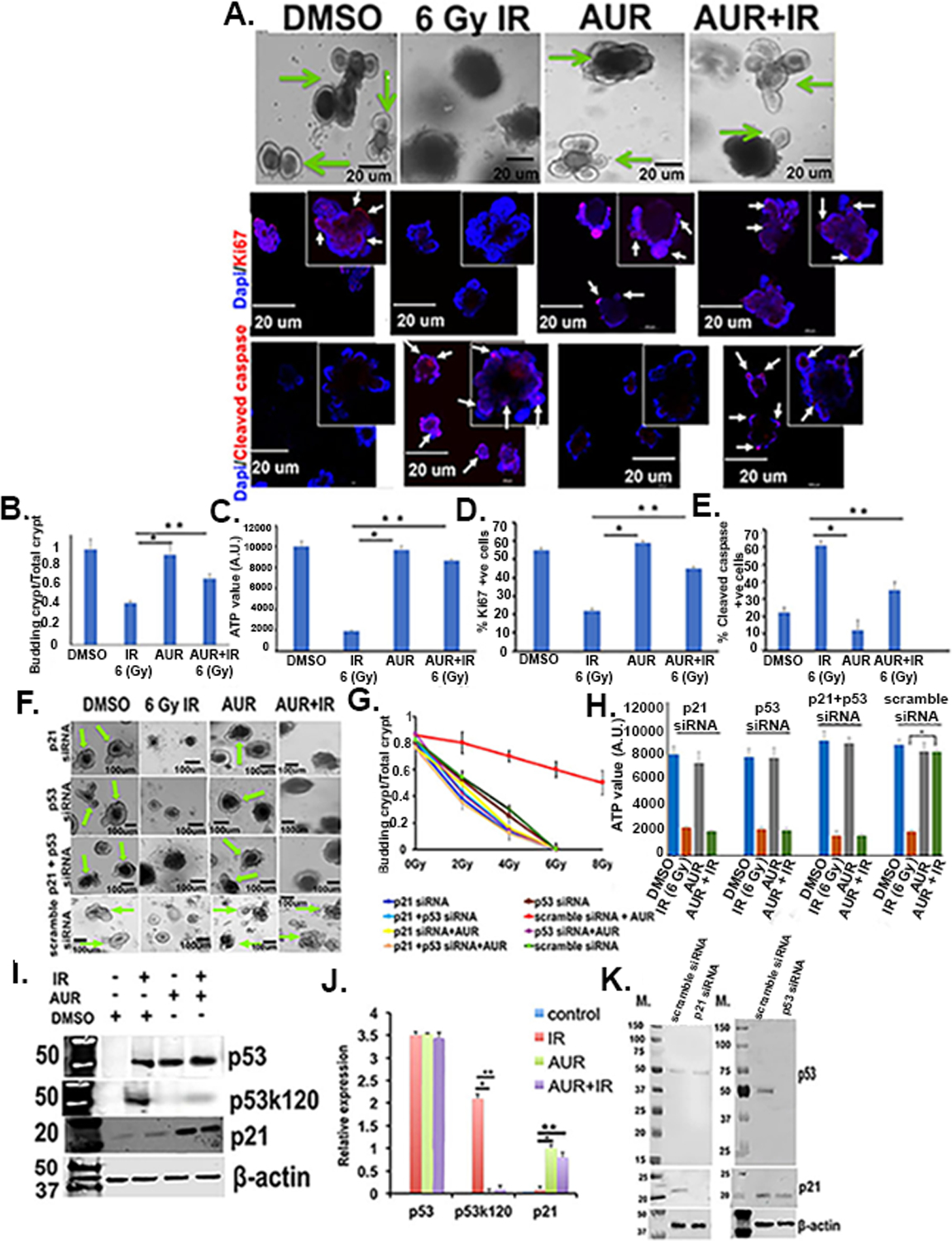
Auranofin protected human normal colon derived organoids from radiation toxicity. Organoids developed from surgical specimens of human non-malignant colon were treated with radiation (2–8 Gy), AUR (2 μmol/L) and the combination of both. Auranofin treatment did not have any toxic effect. Pretreatment with auranofin protected organoids from radiation-induced lethality by inducing crypt formation, compared to the radiation only group. A) Upper panel, Phase contrast microscopic images of organoids withall budding crypts indicated by an arrow). Middle panel: Confocal microscopic images of organoids stained with Ki67 (red color, indicated with arrow). Nucleus stained with DAPI (blue color). Lower panel: Confocal microscopic images of organoids stained with cleaved caspase (red color, indicated with arrow). Nucleus stained with DAPI (blue color). B) Histogram of budding crypt/total crypt ratio. The auranofin and AUR+IR groups had significantly higher budding crypt/total crypt ratio compared irradiated control (*p<0.0002 and **p<0.0006, respectively) (n=6 specimens). C) Histogram of ATP levels in organoids. The auranofin and AUR+IR groups had significantly higher ATP values compared to irradiated controls (*p<0.0004 and **p<0.0005, respectively). D) Histogram demonstrating percent Ki67 positive cells were significantly higher in auranofin and AUR+IR groups compared to irradiated controls (*p<0.0002 and **p<0.0006, respectively). E) Histogram demonstrating percent cleaved caspase positive (apoptotic) cells were significantly higher in irradiated control compared auranofin and AUR+IR treated groups (*p<0.0001 and **p<0.0003, respectively). F) Crypt organoid cells transfected with siRNA for p53, p21, or both did not respond to auranofin treatment. Note loss of budding crypt structures in these organoids even with auranofin treatment. G) Effect of auranofin pre-treatment on irradiated crypt organoid growth. Auranofin treatment improved the budding crypt/total crypt ratio in irradiated organoids compared to irradiated control. Organoids treated with siRNA for p53, p21, or both failed to improve budding crypt to total crypt ratio even with auranofin treatment. Treatment groups: 2 Gy + AUR vs 2 Gy (p<0.0007), 2 Gy +AUR vs 2 Gy + AUR + p53 siRNA (p<1E-04), 2 Gy + AUR vs 2 Gy + AUR + p21 siRNA (p<5.1E-05), 4 Gy + AUR vs 4 Gy (p<0.0003), 4 Gy + AUR vs 4 Gy + AUR + p53 siRNA (p<4.2E-04), 4 Gy + AUR vs 4 Gy + AUR + p21 siRNA (p<5.1E-06), 6 Gy + AUR vs 6 Gy (p<0.0004), 6 Gy + AUR vs 6 Gy + AUR + p53 siRNA (p<3.2E-07), 6 Gy + AUR vs 6 Gy + AUR + p21 siRNA (p<4.1E-06), 8 Gy + AUR vs 8 Gy (p<2.3E-07), 8 Gy + AUR vs 8 Gy + AUR + p53 siRNA (p<6.3E-07), 8 Gy + AUR vs 8 Gy + AUR + p21 siRNA (p<3.3E-08) (n=3 specimens). H) Histogram demonstrating that for irradiated organoids treated with p53 siRNA or p21siRNA or p53+p21 siRNA, auranofin pretreatment did not increase ATP levels. I) Immunoblot of total p53, acetylated p53 (p53k120) and total p21 protein expression. J) Histogram of densitometric analysis of immunoreactive bands representing p53, acetylated p53 (p53k120), total p21. Significant upregulation of p53k120 expression was noted in the IR group compared to auranofin alone (*p<6.4E-07) or AUR+IR group (**p<7.2E-07). Significant upregulation of p21 was noted in AUR alone (**p<0.0008) or AUR+IR group (**p<0.0006). K) Immunoblot of p53 and p21 expression in crypt organoid cells treated with siRNA for p53, p21 or scrambled siRNA. Representative immunoblot images from at least three independent experiments (n=3 specimens) with β-actin loading control.
To determine the involvement of the p53/p21 pathway in the radioprotective role of auranofin, human colonic organoids were first treated with siRNAs targeted to inhibit p53 expression and then treated with auranofin and radiation. A pooled siRNA approach was used (including two to three siRNAs) to inhibit the expression of the gene of interest. Irradiated organoids pretreated with p53 siRNA did not respond to auranofin and demonstrated complete loss of budding crypt like structure upon irradiation (Fig. 3F,G) with significant reduction in ATP level (Fig 3H). These results indicate the involvement of p53 in auranofin-mediated radioprotection of human colonic organoids. siRNA mediated inhibition of p21 also inhibited the auranofin-mediated protection of organoids against radiation (Fig 3F–H) suggesting involvement of p21 in p53-mediated radioprotective effect of auranofin. To further validate these results. expression of both p53 and p21 was inhibited in the same organoids which also showed inhibition of auranofin-mediated radioprotection (Fig 3F–H). We performed immunoblot analysis to determine the transfection efficiency of p53 and p21 siRNA in organoids. Treatment with p53 or p21 siRNAs completely suppressed these target proteins, respectively. Suppression of p21 expression did not show any effect on p53 expression (Fig 3K). However, p53 suppression partially reduced the p21 expression suggesting that p21 is downstream of p53 and therefore regulating the p53-mediated cell survival.
Immunoblot analysis showed significant upregulation of p53 levels in irradiated organoids with/without auranofin treatment (Fig 3I–J). Irradiated organoids in the absence of auranofin treatment demonstrated significant upregulation of the acetylated form of p53 (Fig 3I–J) suggesting activation of a p53 downstream apoptosis pathway. However, in response to auranofin treatment level of acetylated p53 was significantly downregulated (Fig 3I–J) with the upregulation of p21 expression (Fig 3I–J). Involvement of an intrinsic apoptosis pathway in radiation-induced apoptosis in organoids was examined by siRNA-mediated depletion of PUMA. Depletion of PUMA made those organoids more radioresistant. The radioprotective role of auranofin was also maintained in these organoids (Supplementary Fig. S3 D–F).
Therefore, auranofin inhibits a radiation-induced p53 downstream apoptosis pathway and promotes p53/p21 pathway-mediated cell survival.
Pre-treatment with AUR sensitized tumor to radiation and increased therapeutic ratio of abdominal radiation
To determine the role of auranofin on the radiation response of mouse tumors we developed CT26 colon tumors on the abdominal flank region of BALB/c mice by subcutaneous injection of 2×106 CT26 cells (22,30). Mice with palpable subcutaneous abdominal tumors were treated with auranofin (10 mg/kg i.p.) followed by exposure to fractionated abdominal irradiation (AIR) (4 Gy/fraction; 2 fractions/week for two weeks) (Fig 4A). Significant tumor growth delay was observed with combination treatment of AUR+AIR compared to controls (p<0.0006). The median time to achieve tumor size 2000 mm3 in control, AIR, auranofin, and AUR+AIR was 22 days, 29 days, 50 days, and 68 days, respectively.
Figure 4:

Auranofin sensitized tumor and protected intestine from abdominal radiotherapy. A) Schematic diagram of auranofin treatment (10 mg/kg i.p.) and fractionated AIR (4 Gy/fraction) given to BALB/c mice with a CT26 colon tumor implanted in the abdominal flank. B) Mice receiving auranofin prior to AIR exposure demonstrated significant tumor growth retardation compared to DMSO control (p<0.0005), AIR (p<0.003), or auranofin only (p<0.005) treatment (n=10 mice/group). C) Representative picture of CT26 colon tumor at day 25 after first auranofin treatment. D) Immunoblot to detect p53 and p21 protein expression in CT26 colon tumor cells. No significant difference in p53 and p21 protein expression level was observed in auranofin and AIR treatment groups compared to DMSO control. Representative immunoblot images from at least three independent experiments with β-actin loading control. E) Schematic diagram of the treatment protocol and the AIR (12.5 Gy) exposure field for BALB/c mice. A 3 cm square area of the mice containing the GI tract was irradiated (irradiation field), thus shielding the upper thorax, head and neck as well as the lower and upper extremities, protecting a significant portion of the bone marrow. Mice were treated with auranofin (10 mg/kg i.p.) prior to single dose AIR (12.5 Gy). F) Kaplan–Meier survival analysis indicated a significant improvement in survival for the AUR+AIR groupcompared to AIR alone (p<0.0001; log rank (Mantel–Cox) test; n=10 mice per group). G) Body weight of mice at times post AIR. H) Combination treatment of auranofin and single dose AIR resulted in significant reductionof tumor growth compared to the auranofin only (p<0.002) and DMSO control (p<0.001).
CT26 colon tumor cells do not have the Cdnk2a gene (46) responsible for expressing p19ARF. p19ARF functions as a tumor suppressor by inducing p53 stabilization (47). We have observed that expression of p53 and p21 were not altered by radiation and/or auranofin treatment (Fig. 4D). These results suggest that the absence of Cdnk2a made p53 non-responsive to radiation/auranofin treatment and therefore it may not play a significant role in the tumoricidal effect of auranofin.
Radiotherapy with a single large dose is sometimes preferred over fractionated radiation because of better treatment outcome and low risk of tumor recurrence. However, the high radiosensitivity of intestinal epithelium restricts the use of a tumoricidal large single radiation dose against abdominal malignancies. In this study we have examined whether auranofin could be applied with a large single dose of AIR (12.5 Gy) against colon tumors. Mice with palpable subcutaneous abdominal CT26 colon tumor were treated with/without auranofin (10 mg/kg i.p.) followed by exposure to lethal dose abdominal irradiation (12.5 Gy single fraction) (Fig 4E). Mice exposed to AIR without auranofin pre-treatment all died within 15 days of exposure with symptoms of RIGS including loss of body weight (Fig 4G), diarrhea etc. However, auranofin treatment prior to radiation exposure rescued these mice from radiation toxicity along with significant tumor growth retardation compared to un-irradiated controls (Fig 4F, H). The tumor growth curve of untreated AIR mice was not complete due to mortality in all mice within 15 days post exposure. Mice rescued by auranofin continued to survive beyond 30 days post AIR without any sign or symptoms of RIGS.
Therefore, these results demonstrate that administration of auranofin prior to irradiation enhances the radiation response of the colon tumors while providing radioprotection to the normal tissues, including small intestine, thus increasing the therapeutic ratio of abdominal irradiation.
Auranofin treatment induced apoptosis in tumor tissue by inhibiting deubiquitinase (DUB) and increasing unfolded protein response (UPR).
Auranofin inhibits 19S proteasome-associated DUBs to inhibit proteosomal degradation and thereby increases cytotoxicity in malignant tissue (18).To examine the involvement of auranofin mediated inhibition of DUBs in radiosensitization of tumor tissue, DUB activity was measured in the CT26 tumor tissue lysate by ELISA. A significantly lower DUB activity was found in the auranofin treated tumor exposed to radiation compared to irradiated control tumor or untreated tumor (p<0.007 and p<0.002) (Fig. 5A). Treatment with auranofin without radiation also significantly reduced the DUB activity in tumor tissue compared to irradiated or untreated control (p<0.005 and p<0.002) (Fig. 5A). Next, we determined the ubiquitinated protein level in tumor tissue (18). Combination treatment of auranofin and radiation significantly increased the ubiquitinated protein level in tumor tissue compared to irradiated or untreated controls (p<0.005 and p<0.001 respectively) (Fig. 5 B–C). Treatment with auranofin alone also increased the ubiquitinated protein level compared to untreated or irradiated control (p<0.002 and p<0.007) (Fig. 5B–C). Inhibition of proteosomal degradation induces unfolded and misfolded protein burden within cells resulting in initiation of UPR with the induction of endoplasmic reticulum (ER) stress (48). Three major UPR signaling pathways - inositol-requiring enzyme 1α (IRE1α), activating transcription factor 4 (ATF4) and protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) (22,48) - were examined in auranofin-treated tumor tissue. IRE1α, upon activation, splices XBP1 into the active form, producing a potent transcription factor that translocates to the nucleus and activates several UPR target gene expressions including HSPs. ATF4 is a downstream target of PERK which can activate an ER stress downstream pro-apoptotic pathway with induction of the pro-apoptotic protein C/EBP homologous protein (CHOP) (49). Auranofin treatment of tumor prior to irradiation up-regulated sXBP1, GRP78, ATF4 and CHOP mRNA levels compared to irradiated or untreated controls (Fig. 5D). Immunoblot analysis also demonstrated auranofin-mediated upregulation of p-PERK (activated for PERK) and CHOP protein expression which further increased in response to combination treatment (Fig. 5E,F). Since PERK activation attenuates protein synthesis in response to ER stress through the phosphorylation of translation initiation factor eIF2α at serine 51, we determined the levels of phosphorylated eIF2α. Immunohistochemistry of peIF2alpha demonstrated higher peIF2alpha positive tumor cells in auranofin and the combination treatment groups (Fig. 5I). Consistent with these findings, combination treatment of AUR+AIR also increased mRNA levels of UPR downstream pro-apoptotic genes PUMA, PMAIP1, BAK, BAX and BIM (Supplement Table 1) and promoted apoptotic cell death in CT26 tumor cells compared to irradiated controls (Fig. 5G–H, I). The apoptotic cell death was determined in the tumor tissue by detecting the Annexin V/PI positive cells using flow cytometry (Fig. 5G–H) and by using TUNEL staining in tumor sections (Fig 5I). Percent of Annexin V positive CT26 tumor cells was higher in the AUR+AIR group compared to irradiated (p<0.003) or auranofin-treated (p<0.007) or untreated controls (p<0.001) (Fig. 5G–H). TUNEL staining also demonstrated the presence of more apoptotic tumor cells in the combination treatment group (AUR+AIR) compared to irradiated or auranofin-treated groups (Fig 5I). These results indicated that auranofin treatment radiosensitizes the tumor with an increase in apoptotic cell death through activation of a pro-apoptotic ER stress signaling pathway.
Figure 5:

Auranofin inhibited DUB, activated pro-apoptotic pathway of UPR and induced apoptosis in tumor cells. A) DUB activity in CT26 tumor cell lysates was measured by ELISA using the Ub-AMC substrate. Tumor tissue from mice treated with AUR+AIR showed lower DUB activity compared to irradiated (p<0.007) or untreated tumors (p<0.002). Treatment with auranofin without irradiation also reduced DUB activity in tumor tissue compared to irradiated (p<0.005) or untreated controls (p<0.002) (n=6 mice/group). B) Accumulation of ubiquitinated proteins was detected by western blot analysis with a monoclonal mouse primary antibody for ubiquitinated peptides. C) Histogram demonstrating mean band density of immunoreactive bands for ubiquitinated proteins. Combination treatment of auranofin and radiation significantly increased the ubiquitinated protein levels compared to irradiated (**p<0.005) or untreated control (*, p<0.001). Treatment with auranofin alone also increased ubiquitinated protein levels compared to untreated (*p<0.002) or irradiated control (**p<0.007). Representative western blot images from at least three independent experiments with β-actin as loading control (n=3 mice/group). D) qPCR analysis of sXBP1, GRP78, ATF4 and CHOP mRNA in CT26 tumor cells. There were several fold increases in sXBP1, GRP78, ATF4 and CHOP mRNA levels in the AUR+AIR treated group compared to AIR or auranofin treatment groups (n=6 mice/group). E-F) Immunoblot to detect p-PERK, PERK and CHOP expression in CT26 tumor cells. pPERK and CHOP expression were increased in the AUR+AIR treated group compared to control, AIR and auranofin treatment groups (n=3mice/group). G) Flow cytometry plot demonstrating Annexin V-FITC and PI positive apoptotic CT26 tumor cells. H) Histogram demonstrated that AUR+AIR treated tumor had higher numbers of Annexin V-FITC positive apoptotic cells compared to control (***,p<0.001) or AIR (**,p<0.003) or AUR treatment (*,p<0.007) (n=6mice/group). I) Representative images of CT26 colon tumor tissue showing TUNEL staining or immune-histochemical staining to detect peIF2alpha.
Auranofin induced apoptosis in human malignant colonic organoid with the activation of UPR
To determine the effect of auranofin treatment in human colonic malignant tissue we developed primary ex vivo organoid cultures from surgical specimens of malignant colon. For these experiments we used surgical specimens from the same patients considered for the collection of non-malignant tissue. Irradiation of these organoids resulted in loss of budding crypt structures in a dose dependent manner (Fig 6A–B). Similar to their non-malignant counterparts, malignant tissue derived organoids also demonstrated interpersonal differences in radiosensitivity (Supplement Figure S4C). However, radiation-induced damage in these organoids was relatively less compared to non-malignant organoids (Supplement Figure S4A) indicating the radioresistant nature of colonic malignant tissue. Pre-treatment with auranofin radiosensitizes this malignant tissue derived organoids resulting in significant loss of budding crypt like structures with the reduction in budding crypt ratio compared to irradiated controls or untreated controls (Fig 6A–B) (Supplement Figure S4C–D). Ki67+ve proliferative cells (Fig 6A,D) and ATP level (Fig 6A,C) were also significantly decreased in auranofin pre-treated, irradiated organoids compared to irradiated controls.
Figure 6:
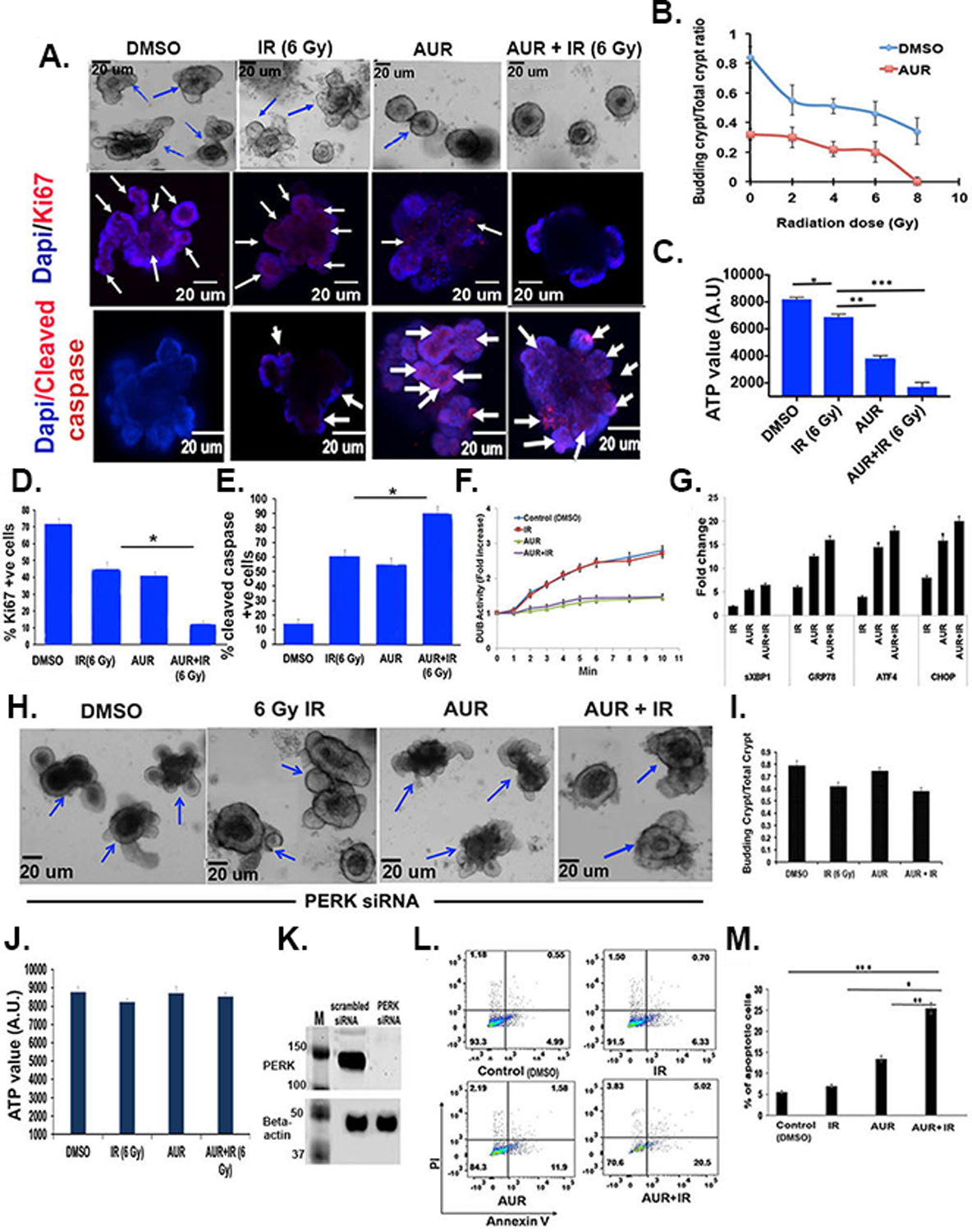
Auranofin treatment sensitized organoids derived from human malignant colon to radiation therapy. A) Images of organoids derived from human malignant colon. Upper panel: Phase contrast microscopic images. Middle panel: Confocal microscopic images of organoids stained with Ki67 (red color, indicated with arrow). Nucleus stained with DAPI (blue color). Lower panel: Confocal microscopic image of organoids stained with cleaved caspase (red color, indicated with arrow). Nucleus stained with DAPI (blue color). B) Budding crypt/total crypt ratio as a function of radiation dose (n=6 specimens). Addition of auranofin resulted in significant reduction of the budding crypt ratio compared to radiation only: 2 Gy+AUR vs 2 Gy+DMSO p<0.0005; 4 Gy + AUR vs 4 Gy + DMSO p<0.0002; 6 Gy + AUR vs 6 Gy + DMSO p<0.0002 and 8 Gy + AUR vs 8 Gy + DMSO p<0.002. C) Histogram demonstrating auranofin pre-treatment significantly reduces ATP levels compared to radiation only (6 Gy vs 6 Gy + AUR ***p<0.0003) (n=3 specimens). D) Histogram demonstrating auranofin pre-treatment significantly reduced Ki67 positive cells compared to radiation only (6 Gy vs 6 Gy + AUR, *p<0.0002) (n=3 specimens). E) Histogram demonstrating auranofin pre-treatment significantly induced cleaved caspase positive apoptotic cells compared to radiation only (6 Gy + AUR vs 6 Gy, *p<0.0004) (n=3 specimens). F) DUB activity over time was assessed in organoid lysate using the Ub-AMC substrate. Significantly lower DUB activity was ovbserved in the AUR plus radiation treated organoid compared to radiation only or untreated control (p<0.008 and p<0.002, respectively). Treatment with auranofin without radiation also significantly reducedDUB activity compared to irradiated or untreated control (p<0.005 and p<0.002, respectively) (n=3 specimens). The experiment was repeated three times, yielding similar results. G) qPCR analysis demonstrated increased mRNA expression of sXBP1, GRP78, ATF4 and CHOP with radiation and radiation plus auranofin. H) Phase contrast images of organoids treated with PERK siRNA. I) Histogram demonstrating budding crypt/total ratio in organoids treated with PERK siRNA. J) Histogram demonstrating ATP level in organoids treated with PERK siRNA. K) Immunoblot demonstrating PERK expression in crypt organoid cells treated with siRNA for PERK or scrambled siRNA. L) Flow cytometry plot demonstrating Annexin V-FITC and PI positive apoptotic cells. M) Histogram demonstrating that AUR+IR treatment resulted in more Annexin V-FITC positive apoptotic cells than for controls (***,p<0.001), radiation only (**, p<0.002), or AUR only (*,p<0.005) (n=3 specimens).
The three most radioresistant organoids were further examined for mechanism of action of auranofin -mediated radioprotection. A significantly lower DUB activity was found in the auranofin treated human tumor colon organoids exposed to irradiation compared to irradiated or untreated control tumor colon organoids (p<0.008 and p<0.002 respectively) (Fig. 6F). Treatment with auranofin without irradiation also significantly reduced DUB activity in human tumor colon organoid compared to irradiated or untreated control (p<0.005 and p<0.002 respectively) (Fig. 6F).
Next we determined the effect of auranofin on UPR in these malignant colon derived organoids. Pre-treatment of auranofin in irradiated organoids up-regulated sXBP1, GRP78, ATF4 and CHOP mRNA levels compared to irradiated organoids (Fig. 6G). Organoids pre-treated with siRNAs targeted to inhibit PERK expression did not show radiosensitization with auranofin treatment (Fig. 6H–J). Absence of PERK in siRNA treated organoids was confirmed by immunoblot (Fig. 6K). However, PERK siRNA treatment in non-malignant colonic organoid did not reverse the radioprotective role of auranofin (supplement Fig S3G–H) suggesting that involvement of PERK is restricted to auranofin mediated radiosensitization of malignant organoids. Consistent with these findings AUR pre-treatment also increased the percent of Annexin-V FITC apoptotic cells in these irradiated organoids compared to irradiated (p<0.002) or AUR only treatment (p<0.005) (Fig. 6L–M). Similar effect was also observed in organoids stained with cleaved caspase (Fig 6A,E). These findings clearly indicate that auranofin pre-treatment radiosensitizes human malignant colonic organoids by inducing a UPR downstream pro-apoptotic signaling pathway.
Discussion:
The current study demonstrates that repurposing of the anti-rheumatoid drug auranofin radioprotects the normal intestinal epithelium but radiosensitizes malignant tissue. In a model of murine CT26 colorectal tumors treated with whole abdominal irradiation, pretreatment with auranofin protected the mice from lethal RIGS, while improving tumor growth suppression. Moreover, colonic organoids developed from surgical specimens of human colon demonstrated similar effects in response to auranofin treatment. The radioprotective ability of auranofin is restricted to organoids developed from non-malignant specimens of human colon. Organoids developed from malignant tissue from the same patient were sensitized by auranofin treatment. This is the first use of paired normal and malignant tissue from the same patient to examine the efficacy of a pharmacological agent for improvement of radiotherapy. This opposite effect of auranofin in irradiated normal and malignant tissue from the same patient is promising for its further use in clinical settings in combination with radiotherapy.
p53 mediated cell cycle arrest and DNA double strand break repair is critical for radioprotection in the intestinal epithelium. Previous reports have shown that selective protection of normal tissue from radiation toxicity could be achieved by pharmacological modulation of cell cycle arrest (50) using inhibitors of cyclin-dependent kinases (CDKs). But a CDK inhibitor could not prevent radiation injury in p53 knock out mice. In the current study, auranofin treatment activated the p53/p21 pathway for DNA check point inhibition, resulting in a reduction of radiation-induced damage in the normal intestinal epithelium. However, combination treatment of auranofin with radiation-induced synthetic lethality in colon tumor through activation of ER stress and ER stress associated pro-apoptotic pathway to induce apoptosis in cancer cells.
It has been reported that p21/p53 deficient mice are more sensitive to RIGS (9). p53 deficient crypt epithelial cells continue to proliferate post-irradiation causing mitotic catastrophe within 72–96 h after irradiation resulting in denudation of villi within 3–5 days post-irradiation. However, the presence of p53 induced reversible growth arrest in crypt epithelial cells within 72 h post-irradiation and thereby prevented mitotic catastrophe. After 72 h post-irradiation these wild-type p53 crypt epithelial cells are proliferating to repair the damage caused by irradiation. p21 plays a critical role in allowing p53 to protect mice from the GI syndrome (7). In response to radiation, the GI epithelium of p21 null mice also undergoes aberrant cell-cycle progression with persistent DNA damage that leads to cell death via mitotic catastrophe. Super p53 mice, which are very resistant to RIGS, become very sensitive with the deletion of the p21 gene (7). In the present study, auranofin pretreatment of WT mice demonstrated initial arrest of crypt cell proliferation which gradually recovered to a higher level resulting in epithelial regeneration within 96 h post-irradiation. However, p53 or p21 knockout mice as well as siRNA mediated inhibition of p53 or p21 in human non-malignant colonic organoids completely abrogated the radioprotective role of auranofin. P53 can also induce apoptosis. In response to radiation injury in the intestine, p53-mediated apoptosis depends on Tip60 acetylatransferase-mediated acetylation of p53k120 (44). Our study in irradiated mouse intestinal epithelium, human non-malignant colon organoids demonstrates clear evidence that auranofin pretreatment inhibits acetylation of p53k120 which reduces radiation-induced apoptosis and activates p21 downstream cell cycle arrest. Therefore, auranofin reduces radiation-induced mitotic catastrophe and promote intestinal epithelial regeneration by modulation of a p53/p21 pathway.
Auranofin inhibits 19S proteasome-associated DUB to block proteosomal degradation (18). In the present study we have shown that auranofin treatment reduced DUB activity and thereby inhibited deubiquitination in irradiated tumor tissue resulting in a significant increase in ubiquitinated protein level. Deubiquitination is a critical step for proteosomal degradation of unfolded and misfolded proteins in cancer cells. Therefore, DUB inhibition increases the unfolded or misfolded protein burden resulting in a UPR. Increase in unfolded or misfolded protein burden beyond tolerance level switches the cytoprotective effect of UPR to the cytotoxic effect by inducing UPR associated apoptosis. Our study demonstrated that the combination of auranofin and radiation-induced synthetic lethality in cancer cells by tipping the balance from the cytoprotective arm of UPR to the PERK-CHOP associated apoptotic arm, resulting in significant reduction of colon tumor growth in mice as well as growth inhibition in human malignant colonic organoids. However, we did not observe any changes in p53/p21 expression in malignant tissue in response to auranofin or radiation suggesting that, unlike normal tissue, the p53/p21 pathway is not playing a central role in the mechanism of action of auranofin.
Cancer cells are more sensitive to proteasome inhibition than are normal cells. In our studies auranofin-mediated proteosomal inhibition increased the radiosensitivity of cancer cells without altering the radiosensitivity of non-malignant cells. Because of cancer cell specific cytotoxicity, proteosomal inhibition is being developed as a major therapeutic target to improve the efficacy of radiation therapy. Moreover, our present data demonstrate that auranofin confers radioprotection to the normal GI epithelium via activation of the p53/p21 pathway while radiosensitizing tumor cells through proteosomal inhibition. Considering the high radiosensitivity of the gastrointestinal epithelium, a pharmacological agent like auranofin should be of great importance in future clinical trials using it in combination therapy with abdominal radiotherapy.
Supplementary Material
Statement of Translational Relevance.
Intestinal injury limits the application of high dose stereotactic radiosurgery (SRS) in abdominal cancers. In the present study we have demonstrated that Auranofin a gold-containing anti rheumatoid agent protects normal intestinal epithelium from radiation injury while radio-sensitizing the colon tumor. Our preclinical study both in mouse tumor model and human colonic surgical specimen derived malignant and non-malignant paired organoid system have clearly shown that Auranofin radio-protects normal tissue, radio-sensitizes malignant tissue and improves the therapeutic ratio for abdominal radiotherapy. This opposite effect of Auranofin in irradiated normal and malignant tissue derived from the same patient is promising for its further use in clinical settings as a combination therapy with radiotherapy. On the basis of our preclinical data, Auranofin, a repurposed drug which has been known for it’s clinical use as anti-rheumatoid agent, is now being considered for a clinical trial as combination therapy with radiation against abdominal malignancy.
Acknowledgements
This work was supported by grants from 1U01AI138323 (S. S.), K01DK096032 (S.S.), ACS IRG Pilot IRG-15-194-07 (B.K.), KUCC support grant P20 168524 (S.S.), KUCC pilot P20 168524 (S.S.).
The authors acknowledge the Biospecimen Repository Core Facility, Histopathology Core Facility and Confocal Microscopy Core Facility at KUMC for their help and support.
Footnotes
“The authors declare no potential conflicts of interest”
Disclosure of Potential Conflicts of Interest
The authors declare no competing financial interests. No potential conflicts of interest were disclosed by any authors.
References:
- 1.Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 1995;377(6549):552–7 doi 10.1038/377552a0 [DOI] [PubMed] [Google Scholar]
- 2.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998;282(5393):1497–501 [DOI] [PubMed] [Google Scholar]
- 3.Castedo M, Perfettini JL, Roumier T, Kroemer G. Cyclin-dependent kinase-1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ 2002;9(12):1287–93 doi 10.1038/sj.cdd.4401130 [DOI] [PubMed] [Google Scholar]
- 4.Komarova EA, Christov K, Faerman AI, Gudkov AV. Different impact of p53 and p21 on the radiation response of mouse tissues. Oncogene 2000;19(33):3791–8 doi 10.1038/sj.onc.1203717 [DOI] [PubMed] [Google Scholar]
- 5.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993;75(4):817–25 [DOI] [PubMed] [Google Scholar]
- 6.Efeyan A, Collado M, Velasco-Miguel S, Serrano M. Genetic dissection of the role of p21Cip1/Waf1 in p53-mediated tumour suppression. Oncogene 2007;26(11):1645–9 doi 10.1038/sj.onc.1209972 [DOI] [PubMed] [Google Scholar]
- 7.Sullivan JM, Jeffords LB, Lee CL, Rodrigues R, Ma Y, Kirsch DG. p21 protects “Super p53” mice from the radiation-induced gastrointestinal syndrome. Radiat Res 2012;177(3):307–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komarova EA, Kondratov RV, Wang K, Christov K, Golovkina TV, Goldblum JR, et al. Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene 2004;23(19):3265–71 doi 10.1038/sj.onc.1207494 [DOI] [PubMed] [Google Scholar]
- 9.Leibowitz BJ, Qiu W, Liu H, Cheng T, Zhang L, Yu J. Uncoupling p53 functions in radiation-induced intestinal damage via PUMA and p21. Mol Cancer Res 2011;9(5):616–25 doi 10.1158/1541-7786.MCR-11-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roder C, Thomson MJ. Auranofin: repurposing an old drug for a golden new age. Drugs R D 2015;15(1):13–20 doi 10.1007/s40268-015-0083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirabelli CK, Johnson RK, Sung CM, Faucette L, Muirhead K, Crooke ST. Evaluation of the in vivo antitumor activity and in vitro cytotoxic properties of auranofin, a coordinated gold compound, in murine tumor models. Cancer Res 1985;45(1):32–9 [PubMed] [Google Scholar]
- 12.Mirabelli CK, Johnson RK, Hill DT, Faucette LF, Girard GR, Kuo GY, et al. Correlation of the in vitro cytotoxic and in vivo antitumor activities of gold(I) coordination complexes. J Med Chem 1986;29(2):218–23 [DOI] [PubMed] [Google Scholar]
- 13.Fiskus W, Saba N, Shen M, Ghias M, Liu J, Gupta SD, et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res 2014;74(9):2520–32 doi 10.1158/0008-5472.CAN-13-2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cummins JM, Vogelstein B. HAUSP is required for p53 destabilization. Cell Cycle 2004;3(6):689–92 [PubMed] [Google Scholar]
- 15.Watson WH, Heilman JM, Hughes LL, Spielberger JC. Thioredoxin reductase-1 knock down does not result in thioredoxin-1 oxidation. Biochem Biophys Res Commun 2008;368(3):832–6 doi 10.1016/j.bbrc.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madeira JM, Gibson DL, Kean WF, Klegeris A. The biological activity of auranofin: implications for novel treatment of diseases. Inflammopharmacology 2012;20(6):297–306 doi 10.1007/s10787-012-0149-1 [DOI] [PubMed] [Google Scholar]
- 17.Casini A, Messori L. Molecular mechanisms and proposed targets for selected anticancer gold compounds. Curr Top Med Chem 2011;11(21):2647–60 [DOI] [PubMed] [Google Scholar]
- 18.Liu N, Li X, Huang H, Zhao C, Liao S, Yang C, et al. Clinically used antirheumatic agent auranofin is a proteasomal deubiquitinase inhibitor and inhibits tumor growth. Oncotarget 2014;5(14):5453–71 doi 10.18632/oncotarget.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007;8(7):519–29 doi 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 20.Back SH, Schroder M, Lee K, Zhang K, Kaufman RJ. ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 2005;35(4):395–416 doi 10.1016/j.ymeth.2005.03.001 [DOI] [PubMed] [Google Scholar]
- 21.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer 2004;4(12):966–77 doi 10.1038/nrc1505 [DOI] [PubMed] [Google Scholar]
- 22.Saha S, Bhanja P, Partanen A, Zhang W, Liu L, Tome W, et al. Low intensity focused ultrasound (LOFU) modulates unfolded protein response and sensitizes prostate cancer to 17AAG. Oncoscience 2014;1(6):434–45 doi 10.18632/oncoscience.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kano A Tumor cell secretion of soluble factor(s) for specific immunosuppression. Sci Rep 2015;5:8913 doi 10.1038/srep08913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011;141(5):1762–72 doi 10.1053/j.gastro.2011.07.050 [DOI] [PubMed] [Google Scholar]
- 25.Saha S, Aranda E, Hayakawa Y, Bhanja P, Atay S, Brodin NP, et al. Macrophage-derived extracellular vesicle-packaged WNTs rescue intestinal stem cells and enhance survival after radiation injury. Nat Commun 2016;7:13096 doi 10.1038/ncomms13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mosmann T Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65(1–2):55–63 [DOI] [PubMed] [Google Scholar]
- 27.Jiang H, De Ridder M, Verovski VN, Sonveaux P, Jordan BF, Law K, et al. Activated macrophages as a novel determinant of tumor cell radioresponse: the role of nitric oxide-mediated inhibition of cellular respiration and oxygen sparing. Int J Radiat Oncol Biol Phys 2010;76(5):1520–7 doi 10.1016/j.ijrobp.2009.10.047 [DOI] [PubMed] [Google Scholar]
- 28.Blocka KL, Paulus HE, Furst DE. Clinical pharmacokinetics of oral and injectable gold compounds. Clin Pharmacokinet 1986;11(2):133–43 doi 10.2165/00003088-198611020-00003 [DOI] [PubMed] [Google Scholar]
- 29.Capparelli EV, Bricker-Ford R, Rogers MJ, McKerrow JH, Reed SL. Phase I Clinical Trial Results of Auranofin, a Novel Antiparasitic Agent. Antimicrob Agents Chemother 2017;61(1) doi 10.1128/AAC.01947-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhanja P, Saha S, Kabarriti R, Liu L, Roy-Chowdhury N, Roy-Chowdhury J, et al. Protective role of R-spondin1, an intestinal stem cell growth factor, against radiation-induced gastrointestinal syndrome in mice. PLoS One 2009;4(11):e8014 doi 10.1371/journal.pone.0008014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest 2004;114(8):1098–106 doi 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kastritis E, Gavriatopoulou M, Kyrtsonis MC, Roussou M, Hadjiharissi E, Symeonidis A, et al. Dexamethasone, rituximab, and cyclophosphamide as primary treatment of Waldenstrom macroglobulinemia: final analysis of a phase 2 study. Blood 2015;126(11):1392–4 doi 10.1182/blood-2015-05-647420 [DOI] [PubMed] [Google Scholar]
- 33.Robinson M, Li B, Ge Y, Ko D, Yendluri S, Harding T, et al. Novel immunocompetent murine tumor model for evaluation of conditionally replication-competent (oncolytic) murine adenoviral vectors. J Virol 2009;83(8):3450–62 doi 10.1128/JVI.02561-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cassetta L, Noy R, Swierczak A, Sugano G, Smith H, Wiechmann L, et al. Isolation of Mouse and Human Tumor-Associated Macrophages. Adv Exp Med Biol 2016;899:211–29 doi 10.1007/978-3-319-26666-4_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med 2011;17(12):1636–40 doi 10.1038/nm.2536 [DOI] [PubMed] [Google Scholar]
- 36.Li X, Liu S, Huang H, Liu N, Zhao C, Liao S, et al. Gambogic acid is a tissue-specific proteasome inhibitor in vitro and in vivo. Cell Rep 2013;3(1):211–22 doi 10.1016/j.celrep.2012.11.023 [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM, et al. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J 2002;21(22):6225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park SJ, Kim M, Kim NH, Oh MK, Cho JK, Jin JY, et al. Auranofin promotes retinoic acid- or dihydroxyvitamin D3-mediated cell differentiation of promyelocytic leukaemia cells by increasing histone acetylation. Br J Pharmacol 2008;154(6):1196–205 doi 10.1038/bjp.2008.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee CL, Oh P, Xu ES, Ma Y, Kim Y, Daniel AR, et al. Blocking Cyclin-Dependent Kinase 4/6 During Single Dose Versus Fractionated Radiation Therapy Leads to Opposite Effects on Acute Gastrointestinal Toxicity in Mice. Int J Radiat Oncol Biol Phys 2018;102(5):1569–76 doi 10.1016/j.ijrobp.2018.07.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cui FM, Sun XJ, Huang CC, Chen Q, He YM, Zhang SM, et al. Inhibition of c-Myc expression accounts for an increase in the number of multinucleated cells in human cervical epithelial cells. Oncol Lett 2017;14(3):2878–86 doi 10.3892/ol.2017.6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vousden KH. Switching from life to death: the Miz-ing link between Myc and p53. Cancer Cell 2002;2(5):351–2 [DOI] [PubMed] [Google Scholar]
- 42.Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 2006;24(6):841–51 doi 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 2006;24(6):827–39 doi 10.1016/j.molcel.2006.11.021 [DOI] [PubMed] [Google Scholar]
- 44.Wang X, Wei L, Cramer JM, Leibowitz BJ, Judge C, Epperly M, et al. Pharmacologically blocking p53-dependent apoptosis protects intestinal stem cells and mice from radiation. Sci Rep 2015;5:8566 doi 10.1038/srep08566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai C, Shi D, Gu W. Negative regulation of the acetyltransferase TIP60-p53 interplay by UHRF1 (ubiquitin-like with PHD and RING finger domains 1). J Biol Chem 2013;288(27):19581–92 doi 10.1074/jbc.M113.476606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castle JC, Loewer M, Boegel S, de Graaf J, Bender C, Tadmor AD, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics 2014;15:190 doi 10.1186/1471-2164-15-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matheu A, Maraver A, Serrano M. The Arf/p53 pathway in cancer and aging. Cancer Res 2008;68(15):6031–4 doi 10.1158/0008-5472.CAN-07-6851 [DOI] [PubMed] [Google Scholar]
- 48.Zou P, Chen M, Ji J, Chen W, Chen X, Ying S, et al. Auranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget 2015;6(34):36505–21 doi 10.18632/oncotarget.5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rao R, Nalluri S, Fiskus W, Savoie A, Buckley KM, Ha K, et al. Role of CAAT/enhancer binding protein homologous protein in panobinostat-mediated potentiation of bortezomib-induced lethal endoplasmic reticulum stress in mantle cell lymphoma cells. Clin Cancer Res 2010;16(19):4742–54 doi 10.1158/1078-0432.CCR-10-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gudkov AV, Komarova EA. Radioprotection: smart games with death. J Clin Invest 2010;120(7):2270–3 doi 10.1172/JCI43794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.