

Abstract
IL-10 can be considered an important agent in the resolution of inflammation. Originally named “cytokine synthesis inhibitory factor” for its ability to inhibit IFN-γ and IL-2 production in Th2 cells, it is secreted by monocytes, macrophages, mast cells, T and B lymphocytes, and dendritic cells (DCs). IL-10 production and release by monocytic cells in response to allergic challenge is upregulated by TNF-α, and by negative feedback regulation of itself. However, it is also secreted by T regulatory cells (Tregs), under the control of IL-2. Importantly in the context of asthma, IL-10 inhibits eosinophilia, by suppression of IL-5 and GM-CSF, by direct effects on eosinophil apoptosis, and effects on cell proliferation through down-regulation of IL-1. A number of its cytokine suppressive characteristics are now thought to occur through its upregulation of suppressor of cytokine signaling (SOCS)-3. IL-10 is also a suppressor of nitric oxide (NO) production, which may have ramifications for its role in airway inflammatory diseases. Initial clinical trials have demonstrated relative safety and few clinically adverse events at doses of recombinant human IL-10 below 50 μg/kg, with mixed success in treatment of patients with inflammatory bowel disease and psoriasis. However, both steroid therapy and allergen specific immunotherapy are known to elevate endogenous IL-10 levels, which may account for their efficacy, suggesting that further study of IL-10 as a target for treatment of airway inflammatory diseases such as asthma and COPD is warranted.
Keywords: Allergy, antigen-presenting cells, asthma, T regulatory cells
INTRODUCTION
Interleukin (IL)-10 is an important “anti-inflammatory” cytokine, due to its effects in downregulating pro-inflammatory Th2 cytokine production, and its potential to promote resolution of inflammation. In this context, we elucidate the role of IL-10 as an anti-inflammatory factor, through brief review of its molecular, functional, and immunologic effects, and we review its potential relevance to clinical medicine through its clinical applications in treating allergic and autoimmune diseases. We place specific focus throughout on the potential role of IL-10 as an airway anti-inflammatory factor, which may have important ramifications for the diseases of asthma and chronic obstructive pulmonary disease (COPD).
STRUCTURE AND CHARACTERISTICS OF IL-10
IL-10, in both purified and recombinant forms, has three polypeptides of approximate molecular weight between 17, 19, and 21 kD, as determined by gel electrophoresis analysis [1]. This structural heterogeneity is due, in large part, to the glycosylation binding to the amino acid residue. The superfamily of IL-10 and IFN-γ has the same conserved helix C region based on conserved amino acid sequences, with IL-10 existing in a dimeric form in its native state [2]. The IL-10 family includes IL-19, IL-20, IL-22, IL-24, IL-26 (AK 155), IL-28, and IL-29 [3]. As a family, these cytokines all have unique heterodimer receptor complexes, and they share a partial homology in their amino acid sequences, structural similarities in interferon receptors, and display anti-viral activity [3].
Consistent with its structure, IL-10 binds as a dimer to IL-10 membrane receptors on human lymphoma and mouse mast cell lines [4]. The soluble extracellular portion of the membrane receptor has been reported to be glycosylated, having a mass of 35,000–45,000; however, in vitro studies in solution indicated that IL-10 and its receptor form a single complex made up of two IL-10 dimers and four receptor monomers, suggesting that IL-10 may bind to its receptors in a unique and cooperative way, in vivo [5]. In fact, there are two IL-10 receptors: IL-10R1, which is the high-affinity receptor, and IL-10R2, which is the low-affinity receptor. The activity of IL-10 as a cell-signaling initiator is considered to be ultimately dependent on binding to both receptors sequentially to form a complex, first to IL-10R1, and then to IL-10R2 [6,7]. Particularly for human neutrophils, in vitro studies have indicated that the upregulation of suppressor of cytokine signaling (SOCS)-3 mRNA, by IL-10, is dependent on the level of expression of IL-10R1 [8]. However, mouse studies also have suggested that the relative level of expression of IL-10R1 can determine whether IL-10 produces immunostimulative or immunosuppressive effects [9], which may account for some of the varied anti-inflammatory and pro-inflammatory responses to IL-10 reported in some experiments. In all, the necessity for IL-10 protein-dual receptor complex formation is common to other cytokines in the same family, including viral IL-10, IL-22, IL-26, IL-28, and IL-29 [6], suggesting a conservation of receptor signaling mechanism within the IL-10 cytokine family.
IMMUNOLOGIC SOURCES AND EFFECTS OF IL-10
IL-10 was first described as “cytokine synthesis inhibition factor”, produced by mouse Th2 cells, because it inhibited cytokine production by T helper 1 cells [10, 11]; specifically it was an inhibitor of IFN-γ and IL-2 production in Th2 cells [10]. Further investigations showed that in humans, IL-10 is produced by both Th1 and Th2 lymphocytes and inhibits both Th1 and Th2 mediated inflammation [11]. This cytokine-inhibitory property of IL-10 also has been demonstrated for other peripheral blood cells [12]. The direct effects of IL-10 on Th1 and Th2 states appear to be mainly due to inhibition of IL-2 production and cell proliferation [13], as well as CD28 tyrosine phosphorylation in signaling [14].
IL-10 is produced by a variety of specific cells in mammals, including macrophages, monocytes, mast cells, T and B lymphocytes, and dendritic cells; in humans, monocytes, macrophages, and B cells are the major lymphocytic sources [15]. Although IL-10 stimulates proliferation of some specific immune cells, such as B cells and mast cells, and can facilitate immunoglobulin synthesis [16], as alluded to above, its most prominent and potent activities relate to functional suppression of inflammatory cells (Fig. 1). IL-10 RNA has been found in stimulated Th2 lymphocytes of CD4+ cell lines, and in mast cells and conventional B cells (Ly1−(CD5−) B cell) [17], consistent with its known immunologic functions. Furthermore, IL-10 has been shown to decrease TNF-α production in human cord-blood derived mast cells [18], potentially further closing the loop of understanding regarding IL-10 message induction, protein expression, and cell signaling response. Due to the importance of the mast cell associated with human lung allergic mechanisms [19], it would seem to be of some importance that an IL-10 mechanism be present in those cells to decrease histamine and pro-inflammatory cytokine production, as part of the resolution of the airway inflammatory response.
Figure 1.
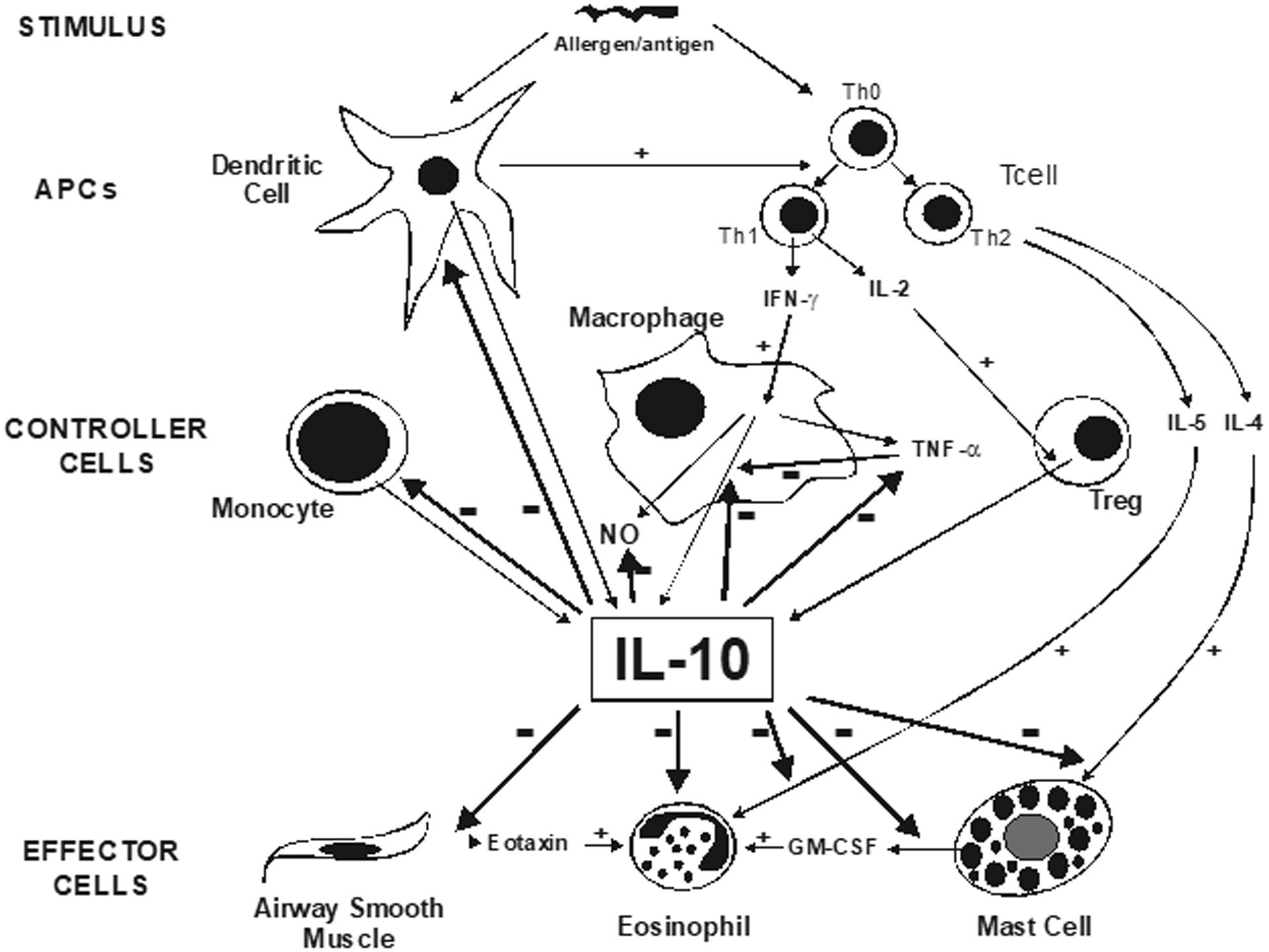
Regulation of IL-10 and its cytokine modulatory effects.
A primary mechanism by which IL-10 inhibits Th1 and Th2 cytokine production is considered to be inhibition of co-stimulators provided by molecules on antigen presenting cells (APC) for T helper cell activation. The co-stimulators, including B7–1 (CD80) and B7–2 (CD86), are inhibited by IL-10, therefore inhibiting macrophage co-stimulatory activity [20,21]. In the absence of B7, T lymphocytes do not respond to antigen, and do not proliferate and produce cytokines; they also become non-responsive to stimuli, and irreversibly tolerant [20]. Thus, expression of IL-10 by APCs represents an established pathway for induction of antigen-specific tolerance, including tolerance to allergens. In the case of T helper cells, T helper cell activation requires co-stimulators provided by molecules on APCs, and is sequentially followed by cytokine production.
However, IL-10 has also been shown to an important factor in induction of the expression of SOCS-3 mRNA in human monocytes [22]. Experiments in LPS-stimulated mouse macrophages have demonstrated the importance of SOCS-3 in the suppression of pro-inflammatory cytokine signaling, such that SOCS-3 transfected cells released less nitric oxide, TNF-α, IL-6, and GM-CSF [23]. Recent evidence has shown that increased expression of SOCS-3 in mouse macrophages can diminish IL-10-dependent tyrosine phosphorylation of signal transducer and activator of transcription (STAT)-3 [23], an important factor in the transcription of cytokine-responsive genes. Consistent with those findings, it also has been shown, in human monocytes in vitro, that IL-10 inhibits expression of IFN-γ-induced genes IP-10 and ICAM-1, and does so by suppression of assembly of STAT factors on those genes, specifically by preventing IFN-γ-associated tyrosine phosphorylation of STAT-1 [24], thereby diminishing IFN-γ-promoted pro-inflammatory mechanisms. However, it bears consideration that with the increasing importance assigned to neutrophils in airway inflammation and remodeling in asthma and COPD [25–27], that IL-10 has been observed to act without triggering IFN-γ-associated STAT1 and STAT3 tyrosine phosphorylation in human neutrophils, but rather can induce SOCS-3 mRNA expression in a STAT-independent manner [28], suggesting a cell-specificity to the specific mechanism by which IL-10 upmodulates SOCS-3. Therefore, IL-10-driven upregulation of SOCS-3, and SOCS-3 attendant suppression of pro-inflammatory cytokines and their effects, constitute another potentially important pathway through which IL-10 can exert its anti-inflammatory effects.
Consistent with its anti-inflammatory characteristics, IL-10 inhibits eosinophil survival, by multiple mechanisms. One mechanism is through inhibition of IL-5, a pro-inflammatory cytokine that induces eosinophilic inflammation, through its effects on eosinophil survival and sustenance [29]. IL-10 also inhibits resting T cells producing IL-5, by interfering with B7-CD28 APC dependent signals [30], which may favor limiting eosinophil recruitment. It is known that survival of eosinophils is prolonged by IL-3, GM-CSF, TNF-α and bacterial lipopolysaccharide (LPS) [31]; specifically, LPS promotes release of GM-CSF and TNF-α by eosinophils, but IL-10 suppresses GM-CSF and TNF-α release [32], thereby representing another eosinophilia-suppressing mechanism. Finally, IL-10 has been reported to effect eosinophils directly, by accelerating eosinophil apoptosis [33].
IL-10 may also exert effects on the IL-1 axis, which is important in the cell proliferative facet of the inflammatory response. For example, IL-10 has been reported to inhibit IL-1receptor(R) gene expression in human monocytes, specifically through decreased phosphorylation of STAT-6 [34], which would presumably have the effect of decreasing IL-1R and its attendant promotion of cell proliferation. Additional evidence in mouse macrophages has suggested that IL-10 may also upregulate the specific IL-1R antagonist, potentially in association with, or as a result of, upregulation of SOCS-3 [23]. The suppression of these IL-1-associated cell proliferative activities may be significant in modulation of the airway remodeling response thought to be important in chronic asthma and COPD.
Dendritic cells (DCs) are important APCs in the differentiation of naïve T cells to either Th-1 or Th-2 cells, and are considered to be the primary APC of the immune system [35].They have received increased attention in recent years, because they are thought to play an important role in allergic airway inflammation [36], in part due to their ability to produce IL-10 [37], and the fact that their accumulation in the large airways has been associated with chronic eosinophilic airway inflammation [38]. In fact, there are two primary populations of DCs within the lung, airway DCs within the epithelium of the conducting airways, and parenchymal DCs [39], each serving a locale-specific APC function in the lung. The proximity of DCs to the airway epithelium allows them to serve an antigen sentinel function, while those in the parenchyma may be more closely associated with mast cells [35]. As such, IL-10-secreting DCs are in a position to have strong immunomodulatory effects on other cells in the lung, as well as themselves, which include inhibition of DC maturation [40], and the induction of immune tolerance [41]. However, at the present time, the overall physiological effects of IL-10 secreted by pulmonary DCs on airway contractility and inflammation remain controversial, and await further study [35].
IMMUNOLOGIC REGULATION OF IL-10
A classical scheme of IL-10 control involves negative feedback regulation, which includes IL-10, itself (Fig. 1). For example, in macrophages and monocytes, a major inducer of IL-10 expression and release is TNF-α [42], which acts to simultaneously promote pro-inflammatory events in other cells, while acting to self-upregulate IL-10, as the anti-inflammatory agent which eventually leads to the resolution of the inflammatory response [43]. Once induced, IL-10 feeds back to suppress TNF-α release, while simultaneously suppressing its own release [44]. Thus, this scheme provides a tidy mechanism that takes advantage of the anti-inflammatory cytokine-suppressive characteristics of IL-10, through which IL-10 expression and release is modulated upward and then downward, as part of inflammation resolution.
However, another important focus of IL-10 regulation are the regulatory T cells (Tregs), which are an important T helper cell subtype in innate and adaptive immunity, being naturally developed in the thymus and induced by TGF-β1 [45–48]. Importantly, Treg cells constitute 5–10% of the CD4+ cells in healthy adult humans, expressing CD4 and CD25 constitutively (the α-chain of the IL-2 receptor) [49]. Although Tregs do not produce IL-2, they do compete for IL-2 secreted by responder T cells, and with IL-2 binding become primed to produce IL-10 [50]. Recent in vitro evidence in a Treg subtype suggests that IL-2 enhances production of IL-10 through synergistic activation of STAT-5 and NF-κB [51]. Thus, IL-2 is an essential growth factor for IL-10 production [49], and the Treg cell-associated IL-2-dependent mechanism of IL-10 modulation can be considered a key initiating element of the inflammatory resolution process (Fig. 1), which has a basis in the overlap of innate and adaptive immune responses.
A subset of Treg cells are the inducible type 1 Treg (Tr1) cells. Tr1 cells are peripherally-derived regulatory T cells with upregulated CD25, and significant capacity for IL-10 production, which can be exploited in treatment of atopy (Table 1). For example, allergen immunotherapy is a form of treatment aimed at decreasing sensitivity to allergens, and IL-10 and Treg cells are considered to play a major role in this treatment. After allergen-specific immunotherapy (SIT) is initiated, IL-10 and TGF-β. are produced in an autocrine fashion, and peripheral T-cell tolerance is induced, through upregulation of IL-10 release by Tr1 cells. Examples include grass pollen immunotherapy [52] and bee venom immunotherapy [53], both resultant of induction of increased IL-10 production by CD4+CD25+T cells.
Table 1.
CD4+ T cells with regulatory function associated with IL-10.
| Regulatory T cell | Regulation | Characteristic |
|---|---|---|
| Naturally- occuring Treg | Cell contact, cytokines (IL-10?) | CD4+CD25+ foxP3+ Thymus and possibly peripheral origin Suppresses auto-immunity Mediates self-tolerance |
| Tr1 | IL-10 | Peripheral origin Suppresses auto-immunity Mechanism of immunotherapy |
| Th3 | TGF-β | Peripheral origin Suppresses auto-immunity in gastrointestinal tract |
IL-10: ROLE IN AIRWAY INFLAMMATION AND HYPERRESPONSIVENESS
In vitro studies in human airway cells have constituted a starting point for more focused airway investigations of IL-10 in other models [54,55]. For example, consistent with its anti-inflammatory properties on lymphocytic cells outlined above, IL-10 has been shown to suppress eotaxin in human airway smooth muscle cells [56], suggesting a role modulating the paracrine pro-inflammatory function of that important airway effector cell. Mouse models of allergic airway inflammation have been helpful in confirming the role of IL-10 as an anti-inflammatory cytokine. Transfer of ovalbumin-specific CD4+CD25+ Tcells to ovalbumin-sensitized mice decreased airway eosinophilia and Th2 cytokine (IL-5 and IL-13) production within the lung, which was found to be IL-10-dependent [57], further confirming its importance and potential Tcell specificity related to allergen responses. However, the sources of IL-10 regulation, and its specific effects and targets, have not been universally determined in animal models of airway inflammation. For example, in some studies, IL-10-deficient mice demonstrated increased eosinophilic airway inflammation with sensitization and challenge [58,59], while other studies of those mice showed decreased eosinophilic airway inflammation [60,61]. The discrepancies in these findings may be model-dependent, such that longer-term models of allergic airway inflammation may be necessary to better determine the source of the effects of IL-10, and its relevance to human airway inflammatory diseases. However, those differences also could be due to the relative expression of IL-10R1 in those various models [9], as alluded to above, but which has not yet been quantified and compared.
The role of IL-10 in airway inflammation-associated airway hyperresponsiveness (AHR) remains controversial, despite recent research. Based on the anti-inflammatory properties of IL-10 outlined above, it might be expected that a lack of IL-10 would result in AHR, associated with unresolved inflammation. In fact, the reported effects of IL-10 on AHR have been variable, being decreased, unchanged, or increased, and may be dependent on the model employed. However, paradoxically decreased AHR with a lack of IL-10 has been reported in several mouse models of airway inflammation, as has re-establishment of AHR with administration of IL-10 [58–65]. Furthermore, Wilson et al. [66] reported that the reduced pathological responses in IL-10 knockout mice were associated with increased production of IL-13Rα2. They created IL-10 and IL-13Rα2 double knockout mice, and these mice developed more AHR and goblet hyperplasia following allergen exposure in the lung, compared to single IL-10 knockout mice. These data would suggest that IL-13Ra2 and IL-10 appear to be required to suppress Th2-dependent inflammation and prevent development of downstream immunopathologies; however, their interaction remains to be sufficiently elucidated in human airway inflammatory diseases.
Some evidence suggests that IL-10 potentiates other factors to increase airway smooth muscle contractility [59], possibly through IL-5 [61], or regulation of surfactant protein D [67]. Interestingly, the recent work of Koya, et al. [68] has shown that IL-10-treated dendritic cells can suppress expression of CD11c, CD80, CD86, IL-12 and Th2 cytokines in vitro, while decreasing BAL eosinophilia and AHR in vivo. These findings suggest a link between allergic airway inflammation and AHR, on which IL-10 can possibly exert a modulatory influence.
IL-10 AND NITRIC OXIDE
The association between IL-10 and nitric oxide (NO) has been established mainly through in vitro studies in cell culture systems, in which IL-10 has been reported to be a natural suppressor of nitric oxide synthase (NOS) activity, and therefore NO production [69]. However, while exhaled NO has been reported to be elevated in asthmatics, and reduced with corticosteroid treatment [70], it has been assumed that elevated NO in airway inflammatory diseases may be an indicator of ongoing inflammation that is quelled due to the anti-inflammatory properties of steroids [71]. Furthermore, given the findings that decreased IL-10 message and protein production have been reported in asthmatics [72], it is reasonable to postulate that the elevated exhaled NO in asthmatics may be due to deficiency in IL-10 production, essentially constituting removal of a normally-present brake on NO production. While this association in humans remains unproven, there is reason to speculate that a down-regulation of IL-10 may be a function of physiologic feedback mechanisms called into play in the setting of asthma.
One potential physiologic mechanism relates to the property of NO as a bronchodilator, through its actions on airway smooth muscle relaxation and suppression of airway hyperresponsiveness [73, 74], As mentioned above, findings from several laboratories in IL-10 knockout mice have indicated airway hypores-ponsiveness in response to methacholine [58–60], which was opposite of the originally-expected results, in a system missing a major inflammatory down-regulator. While the expected increases in recruited leukocytes with induction of airway inflammation were observed in the absence of IL-10, one group has linked the airway hyporesponsiveness to increased exhaled NO, which was reversed by administration of either NOS inhibitors, or reconstitution of IL-10 [58, 65]. Admittedly, mouse models of airway inflammation cannot be considered to precisely extrapolate to human asthma; however, the core of the association between IL-10 and NO may be similar, such that a down-regulation of IL-10 in asthma may constitute a mechanism to defend airway caliber through production of NO. Further research is needed to test this hypothesis and better determine the mechanism and potential function behind the reduced IL-10 production and elevated airway NO levels associated with asthma.
AIRWAY INFLAMMATORY DISEASES POTENTIALLY INVOLVING IL-10
Asthma
Asthma is a disease of the airways that occurs in approximately 7% of the world population [75], the same percent of Americans (20 million) [76], and is associated with approximately 5,000 deaths, annually. Progress has been made in the understanding of the disease in recent years, and specifically the role of cytokines in asthma has been the focus of intense study, due to the recognition that airway inflammation associated with leukocyte activity is an important component of the disease [77]. As such, asthma has been characterized as a disease associated with recruitment, accumulation, and activation of eosinophils in the lung [78]. Furthermore, the bronchial tissue and bronchoalveolar lavage fluid (BALF) from asthmatics have activated CD4+ T cells, eosinophils and mast cells, and increased Th2 cytokines, such as IL-4 and IL-5, have been measured in the BALF from asthmatic patients [79]. Mast cells also generate tumor necrosis factor-α (TNF-α), which leads to leukocyte recruitment, adhesion, activation and survival [80], as well as modulation of IL-10 production, as outlined above.
Healthy people express IL-10 constitutively in their respiratory tract, produced by immature dendritic cells and mononuclear phagocytic cells. This expression of IL-10 can help induce and maintain tolerance to allergens or aerosols. However, in asthmatic patients, IL-10 production is diminished, which may lead to unresolved airway inflammation, as IL-10 is known to inhibit dendritic cell maturation, eosinophil survival and IL-4-induced IgE synthesis [72]. The origins and mechanism behind this lack of induction of IL-10 in cells from asthma patients remains poorly understood, and suggests a potential area for future investigation into the role of IL-10 in asthma.
One important attribute of asthma is that acute exacerbations can be precipitated due to neutrophil infiltration together with eosinophil degranulation. Grissel et al. [81] reported that IL-10 mRNA was significantly increased in sputum from respiratory virus-induced acute asthma patients. Interestingly, increased IL-10 mRNA was not observed in either virus infection without asthma, acute asthma without virus infection, or in stable asthma, and the IL-10 mRNA levels were subsequently reduced on recovery from the acute exacerbation. These findings suggest that IL-10 may play a particularly significant role in virally-associated asthma. Further study is need to better define the role of IL-10 in asthma originating or associated with different infectious and non-infectious triggering agents.
COPD
Similar to asthma, chronic airway inflammation is a key feature of COPD. However, in contrast to asthma, the inflammation is predominantly associated with neutrophils [27], with cigarette smoking being an important causative factor in the development of the disease [82]. However, COPD accounts for over 762,000 hospitalizations, and over 124,000 deaths per year [83], and therefore has garnered renewed attention over the past ten years. Some antiinflammatory drugs that were originally formulated for asthma treatment have also been utilized in treating COPD, thus the connection to the potential anti-inflammatory role of IL-10 may be important in the setting of this disease, as well as asthma.
Since IL-10 is considered to be an inhibitory cytokine of airway inflammation, less IL-10 production might be expected in COPD patients, who can be typically non-atopic and likely have lower baseline inflammation, than that associated with allergic asthma. However, like asthma, this expectation also might be based on the potential for dysfunctional IL-10 upregulation, which may hinder the inflammation resolution response [72]. In support of this expectation, Takanashi, et al. showed that in sputum, both COPD and asthma patients have significantly less IL-10-positive cells compared to healthy nonsmokers [84]. Thus, overall, both asthma and COPD may be associated with a hindered inflammation resolution response, due to downregulation of IL-10, and it might be expected that therapies that upregulate its production could be useful.
CLINICAL ADMINISTRATION OF IL-10
Given that IL-10 directly suppresses the production of cytokines in human T cells [11], as well as indirectly, in an APC-associated manner [20,21], there has been consideration of IL-10 as a therapeutic target for airway inflammatory disease treatment. Furthermore, due to the importance and overlap between innate and adaptive immunity represented by IL-2-modulated Treg production of IL-10, it would seem that upregulation of IL-10-secretion activity may represent a therapeutic potential for airway allergic diseases inducing peripheral tolerance [85]. However, to date, there have been no clinical experiments or trials of IL-10 administration in humans for the treatment of airway inflammatory disease.
Initial studies of administration of recombinant human (rh)IL-10 in humans demonstrated few clinical adverse events at subcutaneous injection doses below 50 μg/kg, while significantly suppressing IL-1β and TNF-α production in whole blood samples ex vivo [86]. Interestingly, a comparison of subcutaneous and intravenous administration routes for IL-10 administration indicated that the subcutaneous route appears to be more effective in suppressing those cytokines [87]. Subsequently, a single intravenous low dose of rhIL-10(10 μg/kg) was studied, the results of which suggested that administered IL-10 transiently altered both lymphocyte trafficking and the antigen presentation of monocytes [88]. In all, these initial studies suggest that low doses of IL-10 can be tolerated and may offer potential therapeutic utility for inflammatory diseases.
There have also been trials of administration of recombinant human IL-10, as Tenovil® (Schering-Plough, Kenilworth, NJ), for other inflammatory diseases including Crohn’s disease and psoriasis, which may shed some light as to its promise for airway disease remission. Inflammatory bowel disease (IBD) is thought to stem from a chronic gastrointestinal tract inflammation caused by immune disregulation, manifested as Crohn’s disease and ulcerative colitis, associated with Th1- and Th2-associated inflammation, respectively. Consistent with these characteristics, IL-10 knockout mice are known to develop spontaneous enteric colitis with aging [89], and IL-10 administration and gene therapy has been shown to reduce gut inflammation [90–92]. However, reduction of gut inflammation with direct administration of IL-10 to humans has been equivocal to mildly efficacious [93–95], indicating further study is necessary before consideration can be given to lung therapy.
Administration of IL-10 for psoriasis, a Th1-associated disease, resulted in temporary clinical improvement in patients with moderate to severe disease [96]. However, it should be noted that injected IL-10, for the cases of treatment for psoriasis [96] and for Crohn’s disease [89], was associated with unmeasurable to minor improvement, similar to the transient remission of established colitis with injected IL-10, in IL-10 knockout mice [97]. These data suggest that the local and consistent availability of IL-10 to the target tissue will be important in elucidating its true potential to treat persistent inflammation in the lung associated with diseases such as asthma and COPD.
Although direct IL-10 therapy for allergic airway disease remains to be determined by future study, it is known that some asthmatic patients fail to respond to inhaled and oral glucocorticoid therapy, and are thus grouped as glucocorticoid-resistant (SR, derived from “steroid resistant”) asthmatics [98]. It has been shown that the CD4+ T cells from SR asthmatics fail to induce IL-10 synthesis, following in vitro stimulation in the presence of dexamethasone, unlike glucocorticoid-sensitive asthmatics (SS, derived from “steroid sensitive”) that respond to steroid treatment [99]. Interestingly, dexamethasone by itself does not enhance secretion of IL-10 by CD4+ T cells from SR asthmatics, but co-administration of vitamin D3 with dexamethasone significantly enhances IL-10 synthesis to levels observed in cells from SS asthmatics cultured with dexamethasone alone [100]. Moreover, pretreatment of SR asthmatic CD4+ T cells with IL-10 upregulated glucocorticoid-receptor expression, suggesting another potentially important anti-inflammatory effect of IL-10, on steroid receptor metabolism.
Finally, IL-10 is induced and secreted prominently by allergen specific immunotherapy (SIT) [101]. SIT is known to reduce eosinophil and basophil recruitment in mucosal tissue [102], due to inhibition of IgE-dependent mast cell activation, and inhibition of eosinophil cytokine production and survival. However, as indicated in Fig. (2), SIT is effective, in large part, because it induces Tregs and dendritic cells which produce IL-10 [103]. IL-10 not only generates tolerance in T cells but also inhibits antigen-specific IgE production and increases specific lgG4 and IgA [104]. While in practice for approximately a century, SIT is currently indicated for patients with IgE-mediated diseases, such as allergic rhinitis due to sensitivity to known allergens, including pollen, molds, animal dander and insect venom. Typically, SIT administration is subcutaneous and continued for 3–5 years, through phased increases to a maintenance dose, unless adverse reactions occur. Most importantly, after discontinuation of SIT, beneficial effects have been reported to endure for at least 3 years [105, 106]. Furthermore, in pediatric populations, SIT has demonstrated protective effects from onset of asthma, as well as inhibition of allergic progression and new sensitizations [107]. Thus, these findings offer some theoretical potential for development of future lung-specific SIT-like treatments for airway inflammatory diseases.
Figure 2.
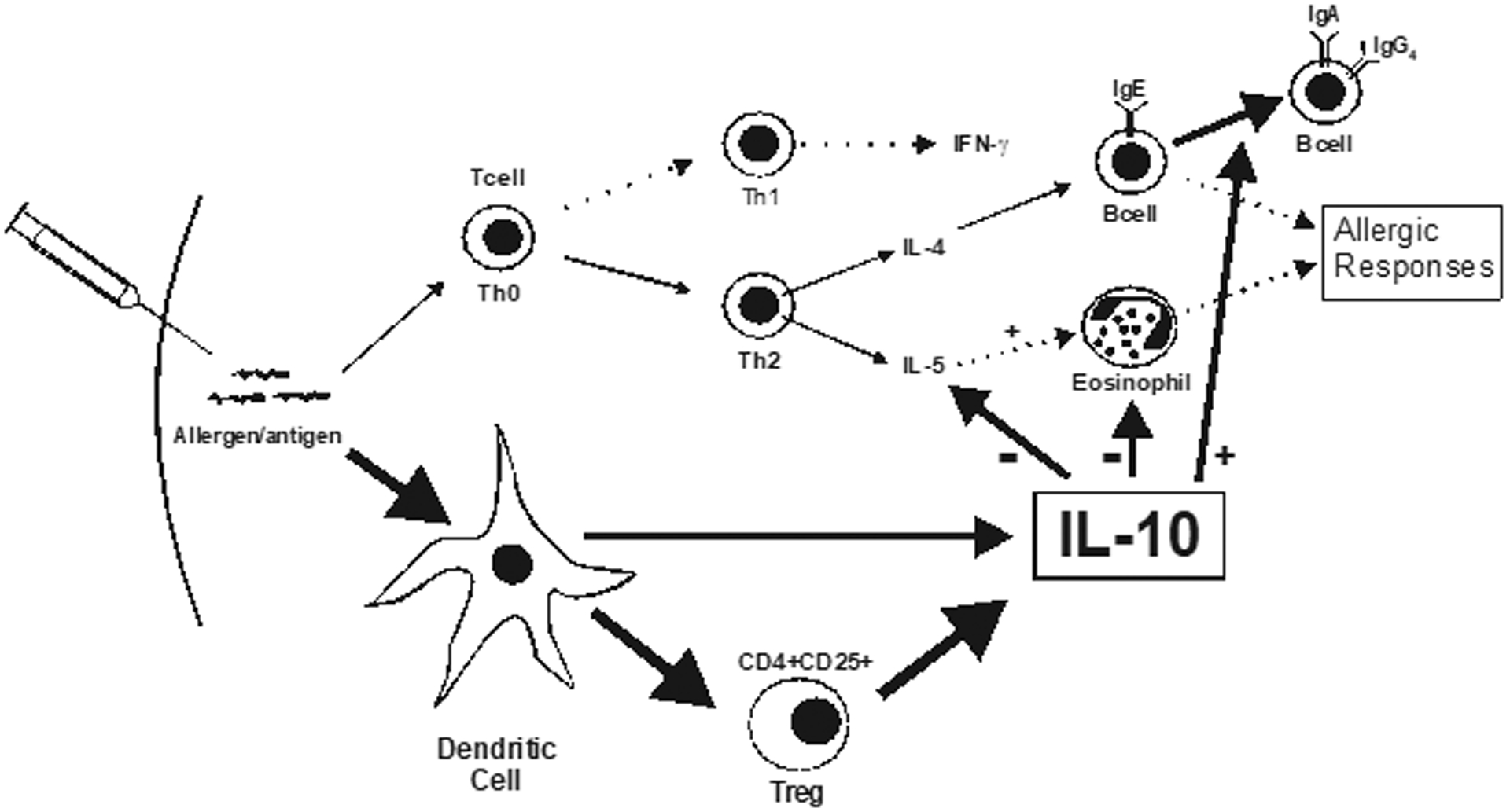
IL-10-associated mechanisms promoted with immunotherapy. (adapted from ref. 88).
SUMMARY
IL-10 is a potent anti-inflammatory and immunosuppressive cytokine that mediates its major immunosuppressive function by inhibiting cytokine production and APC function by macrophages, monocytes, and DCs. IL-10 also regulates effector responses associated with established allergic and asthmatic disease, including inhibition of cytokine production by Th2 cells, as well as mast cell and eosinophil function. IL-10 can be thought of as an “off-switch” that promotes the resolution of inflammation, and is therefore an important cytokine to understand with regard to its mechanisms and regulation. With continued study, IL-10 may hold some promise as a therapeutic target for the treatment of inflammatory airway diseases such as asthma and COPD.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the assistance of William J. Calhoun, M.D., in the preparation of this article.
REFERENCES
- [1].Vieira P, de Waal-Malefyt R, Dang MN, Johnson KE, Kastelein R, Fiorentino DF, deVries JE, Roncarolo MG, Mosmann TR and Moore KW (1991) Proc. Natl. Acad. Sci. USA, 88, 1172–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pieper U, Eswar N, Davis F, Madhusudhan MS, Rossi A, Marti-Renom MA, Karchin R, Webb B, Eramian D, Shen M-Y, Kelly L, Melo F and Sali A (2006) Nucleic Acids Res, 34, D291–D295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Conti P, Kempuraj D, Frydas S, Kandere K, Boucher W, Letourneau R, Madhappan B, Sagimoto K, Christodoulou S and Theoharides TC (2003) Immunol. Lett, 88,171–4. [DOI] [PubMed] [Google Scholar]
- [4].Tan JC, Indelicato SR, Narsula SK, Zavodny PJ and Chou CC (1993) J. Biol. Chem, 268, 21053–9. [PubMed] [Google Scholar]
- [5].Tan JC, Braun S, Rong H, DiGiacomo R, Dolphin E, Baldwin S, Narula SK, Zavodny PJ and Chou CC (1995) J. Biol. Chem, 270, 12906–11. [DOI] [PubMed] [Google Scholar]
- [6].Yoon SI, Logsdon NJ, Sheikh F, Donnelly RP and Walter MR (2006) J. Biol. Chem, 281, 35088–96. [DOI] [PubMed] [Google Scholar]
- [7].Josephson K, Logsdon NJ and Walter MR (2001) Immunity, 15, 35–46. [DOI] [PubMed] [Google Scholar]
- [8].Crepaldi L Gasperini S, Lapinet JA, Calzetti F Pinardi C, Liu Y, Zurawski S, se Waal Malefyt R, Moore KW and Cassatella MA (2001) J. Immunol, 167, 2312–22. [DOI] [PubMed] [Google Scholar]
- [9].Ding Y, Qin L, Zamarin D, Kotenko SV, Pestka S, Moore KW and Bromberg JS (2001) J. Immunol, 167, 6884–92. [DOI] [PubMed] [Google Scholar]
- [10].Fiorentino DF, Bond MW and Mosmann TR (1989) J. Exp. Med, 170, 2081–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Del Prete G, De Carli M, Almerigogna F, Giudizi MG, Biagiotti R and Romagnani S (1993) J. Immunol, 150, 353–60. [PubMed] [Google Scholar]
- [12].de Waal Malefyt R, Abrams J, Bennett B, Figdor CG and de Vries JE (1991) J. Exp. Med, 174,1209–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].de Waal Malefyt R, Yssel H and de Vries JE (1993) J. Immunol, 150, 4754–4765. [PubMed] [Google Scholar]
- [14].Akdis CA, Joss A, Akdis M, Faith A and Blaser K (2002) FASEBJ, 14, 1666–1668. [DOI] [PubMed] [Google Scholar]
- [15].Wanidworanun C and Strober W (1993) J. Immunol, 151, 6853–61. [PubMed] [Google Scholar]
- [16].Moore KW, de Waal Malefyt R, Coffman RL and O’Garra A (2001) Annu. Rev. Immunol, 19, 683–765. [DOI] [PubMed] [Google Scholar]
- [17].Gieni RS, Umetsu DT and DeKruyff RH (1997) Cell Immunol, 175,164–70. [DOI] [PubMed] [Google Scholar]
- [18].Royer B, Varadaradjalou S, Saas P, Guillosson JJ, Kantelip JP and Arock M (2001) Clin. Exp. Allergy, 31, 694–704. [DOI] [PubMed] [Google Scholar]
- [19].Holgate ST, Benyon RC, Howarth PH, Agius R, Hardy C, Robinson C, Durham SR, Kay AB and Church MK (1985) Int. Arch. Allergy Appl. Immunol, 77, 47–56. [DOI] [PubMed] [Google Scholar]
- [20].Ding L, Linsley PS, Huang L-Y, Germain RN and Shevach IEM (1993) J. Immunol, 151, 1224–1234. [PubMed] [Google Scholar]
- [21].Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW and O’Garra A (1991) J. Immunol, 146, 3444–3451. [PubMed] [Google Scholar]
- [22].Williams L, Jarai G, Smith A and Finan P (2002) J. Leukocyte Biol, 72, 800–809. [PubMed] [Google Scholar]
- [23].Berlato C, Cassatella MA, Kinjyo I, Gatto L, Yoshimura A and Bazzoni F (2002) J. Immunol, 168, 6404–11 [DOI] [PubMed] [Google Scholar]
- [24].Ito S, Ansari P, Sakatsume M, Dickensheets H, Vazquez N, Donnelly RP, Lamer AC and Finbloom DS (1999) Blood, 93, 1456–63. [PubMed] [Google Scholar]
- [25].Ennis M (2003) Curr. Allergy Asthma Rep, 3, 159–65. [DOI] [PubMed] [Google Scholar]
- [26].Macdowell AL and Peters SP (2007) Curr. Allergy Asthma Rep, 7, 464–8. [DOI] [PubMed] [Google Scholar]
- [27].Barnes PJ (2000) Chest, 117, 105–45. [Google Scholar]
- [28].Cassatella MA, Gasperini S, Bovolenta C, Calzetti F, Vollebregt M, Scapini P, Marchi M, Suzuki R, Suzuki A, Yoshimura A (1999) Blood, 94, 2880–9. [PubMed] [Google Scholar]
- [29].Pretolani M (1997) Immunol. Today, 18, 277–280. [DOI] [PubMed] [Google Scholar]
- [30].Schandene L, Alonso-Vega C, Willems F, Gerard C, Delvaux A, Velu T, Devos R, de Boer M and Goldman M (1994) J. Immunol, 152, 4368–74. [PubMed] [Google Scholar]
- [31].Rossi AG, Ward C, Murray J, Martin MC, Fujihara S, Sransfield I and Haslett C, (2000) Resp. Med, 94, 1259–1262. [Google Scholar]
- [32].Grutz G (2005) J. Leukoc. Biol, 77, 3–15. [DOI] [PubMed] [Google Scholar]
- [33].Takanashi S, Nonaka R, Xing Z, O’Byrne P, Dolovich J and Jordana M (1994) J. Exp. Med, 180, 711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dickensheets HL and Donnelly RP (1997) J. Immunol, 159, 6226–33. [PubMed] [Google Scholar]
- [35].Bharawaj AS, Bewtr AK and Agrawal DK (2007) Can. J. Physiol. Pharmacol, 85, 686–99. [DOI] [PubMed] [Google Scholar]
- [36].Van Rijt LS, Vos N, Hijdra D, De V, Hoogsteden HC and Lambrecht BN (2003) J. Immunol, 171, 3372–78. [DOI] [PubMed] [Google Scholar]
- [37].Akbari O, DeKruyff RH and Umetsu DT (2001) Nat. Immunol, 2, 725–31. [DOI] [PubMed] [Google Scholar]
- [38].Schon-Hegrad MA, Oliver J, McMenamin PG and Holt PG (1991) J. Exp. Med, 173, 1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Holt PG and Stumbles PA (2000) J. Aerosol. Med, 13, 361–67. [DOI] [PubMed] [Google Scholar]
- [40].De Smedt T, Van Mechelen M, De Becker G, Urbain J, Leo O and Moser M (1997) Eur. J. Immunol, 27, 1229–35. [DOI] [PubMed] [Google Scholar]
- [41].Akbari O, Freeman GJ, Meyer EH, Greenfield EA, Chang TT, Sharpe AH, Berry G, DeKruyff RH and Umetsu DT. (2002) Nat. Med, 8, 1024–32. [DOI] [PubMed] [Google Scholar]
- [42].Wandiworanun C and Strober W (1993) J. Immunol, 151, 6853–6861. [PubMed] [Google Scholar]
- [43].Zuany-Amorim C, Hailiaee S, Ledue D, Dumarey C, Huerre M, Vargaftig BB and Pretolani M (1995) J. Clin. Investig, 95, 2644–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE (1991) J. Exp. Med, 174, 1209–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sakaguchi S (2000) Cell, 101, 455–458. [DOI] [PubMed] [Google Scholar]
- [46].Levings MK, Bacchetta R, Schulz U and Roncarolo MG. (2002) Int. Arch. Allergy Immunol, 129, 263–76. [DOI] [PubMed] [Google Scholar]
- [47].Shevach E (2004) Nat. Rev. Immunol, 2, 389–400. [DOI] [PubMed] [Google Scholar]
- [48].Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ and Powrie F (2003) J. Exp. Med, 197, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kumanogoh A, Wang X, Lee I, Watanabe C, Kamanaka M, Shi W, Yoshida K, Sato T, Habu S, Itoh M, Sakaguchi N, Sakaguchi S and Kikutani H (2001) J. Immunol, 166, 353–360. [DOI] [PubMed] [Google Scholar]
- [50].de la Rosa M, Rutz S, Dorninger H and Scheffold A (2004) Eur. J. Immunol, 34, 2480–8. [DOI] [PubMed] [Google Scholar]
- [51].Tsuji-Takayama K, Suzuki M, Yamamoto M, Harashima A, Okochi A, Otani T, Inoue T, Sugimoto A, Motoda R, Yamasaki F, Nakamura S and Kibata M (2008) Exp. Hematol, 36, 181–92. [DOI] [PubMed] [Google Scholar]
- [52].Francis JN, Till SJ and Durham SR (2003) J. Allergy Clin. Immunol, 111, 1255–1261. [DOI] [PubMed] [Google Scholar]
- [53].Akdis CA, Blesken T, Akdis M, Wuthrich B and Blaser K (1998) J. Clin. Invest, 102, 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Borish L (1998) J. Allergy Clin. Immunol, 101, 293–7. [DOI] [PubMed] [Google Scholar]
- [55].Hawrylowicz CM and O’garra A (2005) Nat. Rev. Immunol, 5, 271–83. [DOI] [PubMed] [Google Scholar]
- [56].Chung KF, Patel HJ, Fadlon EJ, Rousell J, Haddad EB, Jose PJ, Mitchell J and Belvisi M Sr. J. Pharmacol, 127, 1145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kearley J, Barker JE, Robinson DS and Lloyd CM (2004) J. Exp. Med, 202, 1539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ameredes BT, Zamora R, Sethi JM, Liu HL, Kohut LK, Gligonic AL, Choi AMK and Calhoun WJ (2005) J. Immunol, 175, 1206–1213. [DOI] [PubMed] [Google Scholar]
- [59].Justice JP, Shibata Y, Sur S, Mustafa J, Fan M and Van Scott MR (2001) Am. J. Physiol. Lung Cell Mol. Physiol, 280, L363–L368. [DOI] [PubMed] [Google Scholar]
- [60].Yang X, Wang S, Fan Y and Han X (2000) Eur. J. Immunol, 30, 382–391. [DOI] [PubMed] [Google Scholar]
- [61].Oh JW, Seroogy CM, Meyer EH, Akbari O, Berry G, Fathman CG, Dekruyff RH and Umetsu DT (2002) J. Allergy Clin. Immunol, 110,460–468. [DOI] [PubMed] [Google Scholar]
- [62].Grunstein MM, Hakonarson H, Leiter J, Chen M, Whelan R, Grunstein JS and Chuang S (2001) Am. J. Physiol. Lung Cell Mol. Physiol, 281, L1130–7. [DOI] [PubMed] [Google Scholar]
- [63].Makela MJ, Kanehiro A, Borish L, Dakhama A, Loader J, Joetham A, Xing Z, Jordana M, Larsen GL and Gelfand EW (2000) Proc. Natl. Acad. Sci. USA, 97, 6007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Stampfli MR, Cwiartka M, Gajewska BU, Alvarez D, Rifz SA, Inman MD, Xing Z and Jordana M, Am. J. Respir. Cell Mol. Biol, 21, 586–96. [DOI] [PubMed] [Google Scholar]
- [65].Ameredes BT, Sethi JM, Liu HL, Choi AM and Calhoun WJ (2005) Am. J. Physiol. Lung Cell Mol. Physiol, 288, L868–73. [DOI] [PubMed] [Google Scholar]
- [66].Wilson MS, Elnekave E, Mentink-Kane MM, Hodges MG, Pesce JT, Ramalingam TR, Thompson RW, Kamanaka M, Flavell RA, Keane-Myers A, Cheever AW and Wynn TA (2007) J. Clin. Invest, 117, 2941–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Takeda K, Miyahara N, Rha YH, Taube C, Yang ES, Joetham A, Kodama T, Balhorn AM, Dakhama A, Duez C, Evans AJ, Voelker DR and Gelfand EW (2003) Am. J. Respir. Crit. Care Med, 168, 783–9. [DOI] [PubMed] [Google Scholar]
- [68].Koya T, Matsuda H, Takeda K, Matsubara S, Miyahara N, Balhom A, Dakhama A and Gelfand EW (2007) J. Allergy Clin. Immunol, 119,1241–1250. [DOI] [PubMed] [Google Scholar]
- [69].Cuhna FQ, Moncada S and Liew FY (1992) Biochem. Biophys. Res. Commun, 182, 1155–59. [DOI] [PubMed] [Google Scholar]
- [70].Kharitonov SA, Yates DH and Barnes PJ (1996) Am. J. Respir. Crit. Care Med, 153, 454–7. [DOI] [PubMed] [Google Scholar]
- [71].Kharitonov SA and Barnes PJ (2000) Eur. Respir. J, 16, 781–92. [DOI] [PubMed] [Google Scholar]
- [72].Borish L, Aarons A, Rumbyrt J, Cvietusa P, Negri J and Wenzel S (1996) J. Allergy Clin. Immunol, 97, 1288–96. [DOI] [PubMed] [Google Scholar]
- [73].Folkerts G. and Nijkamp FP (2006) Curr. Phanri. Des, 12, 3221–32. [DOI] [PubMed] [Google Scholar]
- [74].Janssen LJ, Premji M, Lu-Chao H, Cox G and Keshavjee S (2000) Am. J. Physiol. Lung Cell Mol. Physiol, 278, L899–905. [DOI] [PubMed] [Google Scholar]
- [75].Centers for Disease Control. Surveillance for Asthma. MMWR; March 29, 2002/51 (SS01); 1–13. [Google Scholar]
- [76].American Lung Association. Epidemiology & Statistics Unit, Research and Program Services. Trends in Asthma Morbidity and Mortality May 2005. [Google Scholar]
- [77].Nakajima H and Takatsu K (2007) Int. Arch. Allergy Immunol, 142,265–273. [DOI] [PubMed] [Google Scholar]
- [78].Busse WW, Nagata M and Sedgwick JB (1996) Eur. Respir. J Suppl. 132s–135s. [PubMed] [Google Scholar]
- [79].Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR and Kay AB (1992) N. Engl. J. Med, 326, 298–304. [DOI] [PubMed] [Google Scholar]
- [80].Tracey KJ and Cerami A (1993) Annu. Rev. Cell Biol, 9, 317–43. [DOI] [PubMed] [Google Scholar]
- [81].Grissell TV, Powell H, Shafren DR, Boyle M.j, Hensley MJ, Jones PD, Whitehead BF and Gibson PG (2005) Am. J. Respir. Crit. Care Med, 172, 433–439. [DOI] [PubMed] [Google Scholar]
- [82].Tetley TD (2005) Curr. Drug Targets Inflamm. Allergy, 4, 607–18. [DOI] [PubMed] [Google Scholar]
- [83].CDC Fast Facts A-Z. Vital Health Statistics, 2003. [Google Scholar]
- [84].Takanashi S, Hasegawa Y, Kanehira Y, Yamamoto K, Fujimoto K, Satoh K and Okamura K (1999) Eur. Respir. J, 14, 309–14. [DOI] [PubMed] [Google Scholar]
- [85].Sakaguchi S (2004) Annu. Rev. Immunol, 22, 531–562. [DOI] [PubMed] [Google Scholar]
- [86].Huhn RD, Radwanski E, Gallo J, Affrime MB, Sabo R, Gonyo G, Monge A and Cutler DL (1997) Clin. Pharmacol. Ther, 62, 171–80. [DOI] [PubMed] [Google Scholar]
- [87].Radwanski E, Chakraborty A, Van Wart S, Huhn RD, Cutler DL, Affrime MB and Jusko WJ (1998) Pharm. Res, 15, 1895–901. [DOI] [PubMed] [Google Scholar]
- [88].Huhn RD, Pennline K, Radwanski E, Clarke L, Sabo R and Cutler DL (1999) Immunopharmacology, 41, 109–17. [DOI] [PubMed] [Google Scholar]
- [89].Kuhn R, Lohler J, Roanoke D, Rajewsky K and Muller W (1993) Cell, 75, 263–274. [DOI] [PubMed] [Google Scholar]
- [90].Nakase H, Okazaki K, Tabata Y, Ozeki M, Watanabe N, Ohana M, Uose S, Uchida K, Nishi T, Mastuura M, Tamaki H, Itoh T, Kawanami C and Chiba T (2002) J. Pharmacol. Exp. Ther, 301, 59–65. [DOI] [PubMed] [Google Scholar]
- [91].Lindsay JO, Ciesielski CJ, Scheinin T, Hodgson HJ and Brennan FM (2001) J. Immunol, 166, 7625–33. [DOI] [PubMed] [Google Scholar]
- [92].Lindsay JO, Ciesielski CJ, Scheinin T, Brennan FM and Hodgson HJ (2003) Gut, 52, 33–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Colombel JF, Rutgeerts P, Malchow H, Jacyna M, Nielsen OH, Rask-Madsen J, Van Deventer S, Ferguson A, Desreumaux P, Forbes A, Geboes K, Melani L and Cohard M (2001) Gut, 49, 42–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Fedorak RN, Gangl A, Elson CO, Rutgeerts P, Schreiber S, Wild G, Hanauer SB, Cohard M, LeBeaut A and Feagan B (2000) Gastroenterology, 119,1473–82. [DOI] [PubMed] [Google Scholar]
- [95].van Deventer SJ, Elson CO and Fedorak RN (1997) Gastroenterology, 113, 383–9. [DOI] [PubMed] [Google Scholar]
- [96].Kimball AB, Kawamura T, Tejura K, Boss C, Hancox AR, Vogel JC, Steingberg SM, Turner ML and Blauvelt A (2002) Arch. Dermatol, 138, 1341–1346. [DOI] [PubMed] [Google Scholar]
- [97].Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, Thompson-Snipes L, Leach MW and Rennick D (1996) J. Clin. Invest, 98, 1010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Leung DY, de Castro M, Szefler SJ and Chrousos GP (1998) Ann. N.Y. Acad. Sci, 840, 735–746. [DOI] [PubMed] [Google Scholar]
- [99].Hawrylowicz C, Richards D, Loke TK, Corrigan C and Lee T (2002) J. Allergy Clin. Immunol, 109, 369–370. [DOI] [PubMed] [Google Scholar]
- [100].Xystrakis E, Kusumakar S, Boswell S, Peek E, Urry Z, Richards DF, Adikibi T, Pridgeon C, Dallman M, Loke TK, Robinson DS, Barrat FJ, O’Garra A, Lavender P, Lee TH, Corrigan C and Hawrylowicz CM (2006) J. Clin. Invest, 116, 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Till SJ, Francis JN, Nouri-Aria K and Durham SR (2004) J. Allergy Clin. Immunol, 113, 1025–34. [DOI] [PubMed] [Google Scholar]
- [102].Wilson DR, Irani AM, Walker SM, Jacobson MR, Mackay IS, Schwartz LB and Durham SR (2001) Clin. Exp. Allergy, 31, 1705–1713. [DOI] [PubMed] [Google Scholar]
- [103].Akdis M and Akdis CA (2007) J. Allergy Clin. Immunol, 119, 780–9. [DOI] [PubMed] [Google Scholar]
- [104].Jutel M, Akdis M, Budak F, Aebischer-Casaulta C, Wrzyszcz M, Blaser K and Akdis CA (2003) Eur. J. Immunol, 33, 1205–1214. [DOI] [PubMed] [Google Scholar]
- [105].Durham SR, Walker SM, Varga EM, Jacobson MR, O’Brien F, Noble W, Till SJ, Hamid QA and Nouri-Aria KT (1999) 341,468–475. [DOI] [PubMed] [Google Scholar]
- [106].Golden DB, Kwiterovich KA, Kagey-Sobotka A, Valentine MD and Lichtenstein LM (1996) J. Allergy Clin. Immunol, 97, 579–587. [DOI] [PubMed] [Google Scholar]
- [107].Moller C, Dreborg S, Ferdousi HA, Halken S, Host A, Jacobson L, Koivikko A, Koller DY, Niggemann B, Norberg LA, Urbanek R, Valovirta E and Wahn U (2002) J. Allergy Clin. Immunol, 109, 251–256. [DOI] [PubMed] [Google Scholar]