

Abstract
Macrophage migration inhibitory factor (MIF) is a pluripotent pro-inflammatory cytokine and is related to acute and chronic inflammatory responses, immune disorders, tumors, and other diseases. In this study, an integrated virtual screening strategy and bioassays were used to search for potent MIF inhibitors. Twelve compounds with better bioactivity than the prototypical MIF-inhibitor ISO-1 (IC50 = 14.41 μM) were identified by an in vitro enzymatic activity assay. Structural analysis revealed that these inhibitors have novel structural scaffolds. Compound 11 was then chosen for further characterization in vitro, and it exhibited marked anti-inflammatory efficacy in LPS-activated BV-2 microglial cells by suppressing the activation of nuclear factor kappa B (NF-κB) and mitogen-activated protein kinases (MAPKs). Our findings suggest that MIF may be involved in the regulation of microglial inflammatory activation and that small-molecule MIF inhibitors may serve as promising therapeutic agents for neuroinflammatory diseases.
Keywords: macrophage migration inhibitory factor, virtual screening, tautomerase assay, naive Bayesian classification, neuroinflammation
Introduction
Neurodegenerative disorders, including Parkinson’s disease (PD) and Alzheimer’s disease (AD), are characterized by progressive neuronal dysfunction and loss in the brain and can result in cognitive and motor dysfunction. Microglia-mediated neuroinflammation is a common feature and is linked to the pathogenesis of neurodegenerative diseases [1]. As resident innate immune cells in the central nervous system (CNS), microglia are closely associated with neurodegenerative progression by adjusting their morphological and functional phenotypes response to pathophysiological brain injuries [2, 3]. Overactivated microglia produce several inflammatory mediators, including interleukin (IL)-6, tumor necrosis factor (TNF)-α, IL-1β, chemokines, nitric oxide (NO), and reactive oxygen species (ROS), which can induce neuronal damage or death [4]. Accordingly, the development of novel small-molecule drugs targeting microglial activation may be an important method to treat inflammatory-related CNS diseases.
MIF, a multifunctional protein, can act as a cytokine-, enzyme-, endocrine-, and chaperone-like molecule. As a cytokine, MIF is present in various cell types, especially in the endocrine and nervous systems, which are linked to the pathogenesis of many inflammatory and immune disorders [5]. MIF mRNA and protein have been found in both glial cells and neurons [6, 7]. Abnormal expression of MIF has been found in the progression of multiple neuroinflammation-related diseases, such as stroke [8], multiple sclerosis (MS) [9], AD [10], and PD [11]. MIF can catalyze the tautomerization of D-dopachrome [12] and phenylpyruvate tautomerase [13] and has thiol-protein oxidoreductase activity, in addition to its physiological and pathophysiological activities [14]. MIF is a homotrimer, and its enzymatic active sites are located at the binding interface of any two adjacent monomers. D-dopachrome tautomerase (DDT), the homolog of MIF, is also known as MIF2 and has an overlapping functional spectrum [15]. In terms of tautomerase enzymatic activity, DDT is ten times weaker than MIF and can also activate the CD74 receptor.
Targeting the tautomerase activity of MIF is an effective strategy to discover small-molecule MIF inhibitors [16, 17]. To date, many small-molecule inhibitors of MIF tautomerase activity have been found through high-throughput virtual screening [18, 19]. However, MIF inhibitors, which have high activity and good druggability and have been definitively characterized in relevant disease models, remain limited. Herein, an integrated virtual screening strategy based on multiple MIF crystal structures and a naive Bayesian classification (NBC) model were used to discover novel MIF inhibitors. A total of 12 novel compounds with potent tautomerase activity against MIF were discovered, four of which had remarkable anti-inflammatory efficacy in BV-2 microglial cells. These compounds could also markedly suppress the LPS-induced expression of pro-inflammatory cytokines, while their anti-neuroinflammatory effects were abolished when MIF was knocked down by siRNA. Moreover, the most potent compound, compound 11, could inhibit the neurotoxicity of LPS-activated conditioned medium towards HT-22 neuroblastoma cells by reducing inflammatory cytokines and ROS.
Materials and methods
Preparation of representative MIF-ligand structures and datasets
The MIF crystal structure complex cannot characterize the structural flexibility of this protein upon ligand binding. In addition, due to the diversity of ligand chemical structures, the binding pockets in different crystal structures of the same target are different [20]. Therefore, integrated virtual screening protocols based on multiple MIF complex structures were used as described in our previous studies (Fig. 1) [21, 22]. The crystal structures of 25 MIF-inhibitor complexes with high resolutions were retrieved from the RCSB Protein Data Bank (PDB) [23]. A three-step structural clustering protocol was then used to select representative MIF complex structures: (1) the residues within 10 Å of each cocrystallized ligand were aligned by the STAMP algorithm in VMD [24]; (2) the root-mean-square deviation (RMSD) values were used to generate a phylogenetic tree by using the create phylogenetic tree module in VMD. A validation dataset consisting of known MIF inhibitors and noninhibitors was selected to assess the “screening power” of these eight structures of MIF. A total of 257 known inhibitors of MIF were screened from the BindingDB database with experimental biological activity [25], while 10,000 noninhibitors were selected from the ChemBridge library via the Find Diverse Molecules protocol in Discovery Studio 3.5 (DS3.5).
Fig. 1. Workflow of virtual screening.

It includes machine learning, MIF complex structures and bioassays.
Molecular docking procedure
The crystal structures of the MIF-inhibitor complexes were used as the initial templates for virtual screening. The Protein Preparation Wizard module in Schrödinger was used to prepare each MIF complex. The ligand situated in the active site between chains A and B was maintained. The complex was then refined with the OPLS force field [26]. Pro1, an important nucleophilic residue, was protonated according to experimental and theoretical results [27]. The Glide module in Schrödinger was used for molecular docking calculations [28]. The tautomers and protonation states of the ligands were generated at pH =7.4 by LigPrep in Schrödinger and then docked to the binding site of MIF by Glide in extra precision (XP) scoring mode. The bounding box was centered on the cocrystallized ligand.
Integrated virtual screening through naive Bayesian classification
According to our previous studies, the integrated virtual screening protocol developed by NBC based on multiple receptor structures can yield better performance than docking-based VS according to any single rigid conformation [21, 29]. Here, the eight docking scores for each ligand were used as the independent variables (X), and 0/1 served as the dependent variable (Y): 0 represents a noninhibitor and 1 represents an inhibitor. The Create Bayesian Model protocol in DS3.5 was used to develop the classifiers to differentiate the known inhibitors from noninhibitors. The following measures were calculated to evaluate the classification performance of each NBC classifier: true negative (TN), false negative (FN), true positive (TP), false positive (FP), global accuracy (GA), sensitivity (SE), specificity (SP), and Matthews correlation coefficient (C) (Eq. (1)).
| 1 |
In addition, the area (AUC) of the receiver operating characteristic (ROC) curve was used to estimate the classification capability.
The compounds in the ChemBridge and ChemDiv libraries were processed by the LigPrep module in Schrödinger. Then, the Glide module with XP scoring was used to dock these compounds in the binding sites of eight MIF structures, and the 1000 top-ranked compounds were then rescored by the best Bayesian classifier. The REOS rules were used to filter the compounds with harmful or otherwise undesirable moieties [30]. The remaining 753 structures were then clustered into 70 groups by the Find Diverse Molecule protocol in DS3.5. Finally, 63 compounds (purity >95%, Supplementary Table S1) were chosen and purchased for subsequent experimental analysis.
MIF tautomerase activity measurement
MIF tautomerase activity was measured according to a previous study [31, 32]. Briefly, equal volumes of 6 mM l-3,4-dihydroxyphenylalanine methyl ester hydrochloride (Sigma-Aldrich, USA) and 12 mM sodium metaperiodate (Sigma-Aldrich, USA) were used to prepare fresh l-dopachrome. Different concentrations of potential inhibitors (ChemBridge or ChemDiv) and ISO-1 (Merch, Germany) were dissolved in DMSO before being incubated with 120 nM recombinant human MIF (hMIF) (Novoprotein, China) for 30 min on ice, and then 30 μL of fresh l-dopachrome was added to a 96-well plate containing hMIF with or without the compounds in 10 mM potassium phosphate buffer and 0.5 mM EDTA, pH 6.2. The inhibitory effect of each compound on the tautomerase activity of MIF was measured by a microplate reader (Infinite M1000 Pro, Tecan, Switzerland) by measuring the absorbance at 475 nm every 10 s for 3 min at 37 °C. For the inhibitory pattern assay, various concentrations of the compounds were added to the reaction buffer containing 120 nM hMIF and incubated for 30 min prior to the addition of different concentrations (1–6 mM) of l-dopachrome methyl ester. Tautomerase activity was assessed using nonlinear regression analysis by Prism 6 (GraphPad Prism). Ki (the inhibition constant) was determined by nonlinear regression against the competitive equation using Prism 8.
Cell culture and siRNA transfection
BV-2 microglial cells and HT-22 neuroblastoma cells were cultured in DMEM with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin in an incubator at 37 °C with 5% CO2. For the siRNA transfection assay, BV-2 microglial cells were plated in 24-well plates (2 × 104 cells per well) and then treated with MIF siRNA (GenePharma, China). The sequence was 5′-UUCUCCGAACGUGUCACGUTT-3′, and Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, USA) was added and incubated for 36–48 h according to the manufacturer’s instructions and our previous report [7]. The level of MIF RNA knockdown was assessed using real-time quantitative PCR.
Culture of primary astrocytes and microglia
Primary astrocytes and microglia were obtained from the cerebral cortex of newborn mice as described previously [33, 34]. Briefly, murine cerebral cortices were harvested, and the meninges were removed and placed in serum-free DMEM. The tissue was digested by papain (2 mg/mL) for 20 min at 37 °C with gentle agitation. Digestion was stopped with 10% FBS in DMEM/F12, and the cells were passed through a 20 μm cell strainer. Then, the cells were centrifuged for 3 min, and the cell suspension was plated on poly-d-lysine-coated T-75 flasks and cultured at 37 °C and 5% CO2. After being cultured for ~2 weeks, microglial cells were isolated by shaking for ~2 h at 150 r/min, and astrocytes were obtained by continuing shaking for ~6–12 h. All mouse experimental procedures conformed to the National Institutes of Health guidelines and were approved by the Animal Welfare and Research Ethics Committee of Xiangyang No. 1 People’s Hospital.
NO measurement
NO release in the cell supernatant was determined by Griess reagent (0.2% N-(1-naphthyl)-ethylenediamine dihydrochloride and 2% sulfanilamide in 5% H3PO4) as previously described [31]. BV-2 microglial cells were plated in 96-well plates (2 × 104 cells per well) and then pretreated with compounds (1–50 µM) for 30 min prior to the addition of LPS (0.2 µg/mL) for 24 h. Then, 50 µL of culture supernatant was transferred to a new 96-well plate with an equal volume of Griess reagent and incubated for 3 min, avoiding direct light and bubbles. The optical density (OD) was determined by a microplate reader at 570 nm. A standard curve was prepared with sodium nitrite to calculate the concentration of NO.
Cell viability assay
Cell viability was evaluated by a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) (Solarbio, China) assay. BV-2 microglial cells were plated in 96-well plates (2 × 104 cells per well) and then primed with compounds (1–50 µM) for 30 min prior to LPS (0.2 µg/mL) stimulation for ~24 h. The cell culture medium was removed, and 30 µL of MTT was added and incubated for ~2–4 h. Then, the formazan dye was dissolved in 100 µL of DMSO. The OD value was determined by a microplate reader at 540 nm.
Drug affinity responsive target stability (DARTS)
After reaching ~80%–85% confluence, BV-2 microglial cells were lysed in M-PER Reagent (Thermo Scientific, USA) on ice for 10 min as described previously [35]. Then, the samples were centrifuged for 10 min at 18,000×g at 4 °C, and the supernatant was transferred to a new 1.5 mL tube. After measuring the concentration with a BCA kit (Bio-Rad, USA), the protein was diluted to 5 g/L by precooled 10× TNC [500 mmol/L Tris-HCl (pH 8.0), 500 mmol/L NaCl, 100 mmol/L CaCl2]. Then, the compounds and samples (containing 250 μg of protein) were mixed for ~1 h on ice and 20 min at 37 °C prior to the addition of pronase (1:1000, Roche, Germany) for 15 min at room temperature. The digestion reaction was stopped by the addition of a protease inhibitor cocktail (Roche, Germany) on ice for 15 min. The samples were processed with 5× SDS-PAGE after being heated at 70 °C for 10 min.
Enzyme-linked immunosorbent assay (ELISA)
BV-2 microglial cells were plated in 96-well plates (2 × 104 cells per well) and then exposed to compounds (1–25 µM) for 30 min prior to LPS (0.2 µg/mL) stimulation for ~24 h. The cell supernatant was collected and stored at −80 °C to avoid repeated freezing. The level of TNF-α and IL-6 in the cell supernatant was measured by ELISA kits according to the manufacturer’s instructions (R&D Systems, USA). The OD was determined by a microplate reader set to 540 or 570 nm.
RNA extraction and quantitative real-time polymerase chain reaction (qPCR)
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instructions and our previous report [34]. After measuring the RNA concentration using a NanoDrop2000, 1 μg of total RNA was used to prepare complementary DNA (cDNA) according to the protocol of the M-MLV reverse transcriptase kit (Promega, USA). qPCR analysis of cDNA was performed using SYBR Green PCR master mix (Promega, USA), and amplification was performed using a 7500 Real-Time PCR System (ThermoFisher Scientific, USA). The relative gene expression was normalized to GAPDH, and the sequences of the cDNA primers for qPCR are shown in Supplementary Table S2.
Immunofluorescence (IF)
A total of 5000 cells per well were plated on sterile poly-d-lysine-coated coverslips in 24-well plates. After pretreatment with compounds (25 µM) for 30 min prior to the addition of LPS (0.2 µg/mL) for 1 h, the medium was removed, and the cells were fixed using precooled methanol for 20 min and blocked with 3% bovine serum albumin (BSA) plus 1% Triton X-100 at room temperature for a minimum of 1 h. The coverslips were incubated with p65 antibodies (1:200, Cell Signaling Technology, USA) overnight at 4 °C. After being washed with PBST (PBS containing 0.05% Tween) three times for ~10 min each, Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:500, Invitrogen, USA) was added and incubated for at least 90 min at room temperature in the dark. Hoechst (0.2 mg/mL, 1:10,000) was added and incubated for 15 min at 37 °C to visualize the nuclei. The coverslips were imaged by fluorescence microscopy.
Western blotting
BV-2 microglial cells were harvested and lysed using RIPA lysis buffer (Beyotime, China) containing protease and phosphatase inhibitors (Roche, Germany) for 30 min on ice. The protein samples were normalized and separated by 10%–12% SDS polyacrylamide gel electrophoresis, transferred to PVDF membranes (Millipore, MA), and blocked with 5% milk for 1 h [36] before being treated with the indicated primary antibodies against MIF (1:800, Sigma-Aldrich, USA), COX-2 (1:500, Absin, China), iNOS (1:500, Absin, China), IκBα (1ː800, Proteintech, China), phospho-p65 (1:1000, Cell Signaling Technology, USA), p65 (1:1000, Cell Signaling Technology, USA), phospho-ERK (1:1000, Cell Signaling Technology, USA), ERK (1:1000, Cell Signaling Technology, USA), phospho-p38 (1:800, Cell Signaling Technology, USA), p38 (1:1000, Cell Signaling Technology, USA), phospho-JNK (1:1000, Cell Signaling Technology, USA), JNK (1:1000, Cell Signaling Technology, USA) and α-tubulin (1:5000, Sigma-Aldrich, USA) at 4 °C overnight. After being washed with TBST 3 times for ~10 min each, the membranes were probed with matched IgG polyclonal secondary antibodies (Sigma, USA) for ~90 min. Finally, the protein expression was determined by enhanced chemiluminescence (ECL) (Bio-Rad, USA) with a ChemiDoc XRS (Bio-Rad, USA).
ROS measurement
ROS production in BV-2 microglial cells was determined using CellROX Oxidative Stress Reagent (ThermoFisher Scientific, USA) according to the manufacturer’s instructions. Cells were stained with CellROX reagent and Hoechst for 30 min at 37 °C. Then, the cells were collected using 0.25% trypsin and identified by flow cytometry.
Conditioned media (CM) of microglia/neuron cocultures
The microglial CM/neuron co-culture system was used as previously described [37]. Briefly, BV-2 microglial cells were cultured in 96-well plates and pretreated with compound 11 (1–25 μM) for 30 min prior to the addition of LPS (0.2 µg/mL) or vehicle for 6 h. Then, the culture medium was discarded, and a fresh medium was added. After 24 h of incubation, the resulting CM was collected and used to treat HT-22 cells for an additional 24 h. Then, HT-22 cell viability was measured using the MTT assay.
Statistical analysis
Data analysis was performed by using GraphPad Prism 6.0 software. The data are representative of three independent experiments performed in triplicate, and the values are expressed as the mean ± SD of three replicates. One-way or two-way ANOVA was used for multiple-group comparisons. *P < 0.05 was considered statistically significant.
Results and discussion
Naive Bayesian classifiers based on multiple MIF complexes
In structure-based virtual screening approaches, semiflexible molecular docking is usually chosen to ensure computational efficiency, while the receptor and ligands are treated with rigidity and flexibility, respectively. Because protein flexibility and chemical structure diversity are not considered, the prediction results of molecular docking based on different crystal structures of the same target are quite different. Accordingly, a molecular docking method based on multiple typical receptor conformations of a specified target may be a desirable comprise. Our previous studies suggested that the NBC model could integrate the prediction results of different scoring functions and exhibited the best prediction power [21, 22]. According to the structural clustering results, multiple MIF crystal structures can be divided into eight clusters (Supplementary Fig. S1). Through a comprehensive assessment of resolution, scoring power and discrimination power, eight complexes were ultimately selected (1CA7 [38], 2OOH [39], 3B9S [40], 3CE4 [41], 3IJJ [42], 3JSG [43], 3SMC [44], and 4F2K [45]). Scoring power and discrimination power are two important indicators of molecular docking [46]. As reported in our previous studies [47], the RMSD value between the docking pose and crystallographic structure was calculated, and RMSD ≤ 2.0 Å (docking power) can be considered successful docking. As shown in Supplementary Table S3, all RMSD values were <2 Å, suggesting that the Glide docking with the XP scoring mode reproduces the near-native conformations of the crystallographic ligands in the eight MIF complexes. The discrimination power was evaluated by the t-test (P-value), which can assess the significance of the difference between the mean values of the distributions of the docking scores for the known inhibitors and noninhibitors. As shown in Supplementary Table S3 and Supplementary Fig. S2, in terms of the relatively low P-values (<10−24), molecular docking can effectively distinguish known inhibitors and noninhibitors for the eight MIF complex structures. The predictions for different complexes also exhibit variable sampling powers (Supplementary Fig. S2). 1CA7 has the best discrimination power (P-value = 3.01 × 10−68), whereas that of 3SMC is only 1.64 × 10−24. The eight representative MIF complexes and receptor-ligand interactions are shown in Supplementary Fig. S3.
The eight MIF complexes met the requirements of RMSD ≤ 2.0 Å and P-value ≤ 10−20 with XP scoring mode (Supplementary Table S2 and Supplementary Fig. S2). The docking scores for the molecules in the validation dataset were used to generate the data matrix to build the NBC models (Supplementary Fig. S2). For the compounds that could not be docked into the binding site of an MIF protein, penalty values of 20 were assigned. As shown in Table 1, by combining the docking scores for the eight MIF protein structures, the AUC of the classifier achieved 0.832, suggesting that the VS strategy based on multiple MIF conformations may serve as a powerful tool to identify potent MIF inhibitors.
Table 1.
Performances of naive Bayesian classifiers via docking scores as descriptors.
| Descriptors | TP | FN | FP | TN | SE | SP | GA | C | AUC |
|---|---|---|---|---|---|---|---|---|---|
| 1CA7_XP | 165 | 72 | 5883 | 24117 | 0.696 | 0.804 | 0.803 | 0.110 | 0.737 |
| 2OOH_XP | 174 | 63 | 8898 | 21102 | 0.734 | 0.703 | 0.704 | 0.084 | 0.710 |
| 3B9S_XP | 184 | 53 | 8888 | 21112 | 0.776 | 0.704 | 0.704 | 0.092 | 0.730 |
| 3CE4_XP | 177 | 60 | 8895 | 21105 | 0.747 | 0.704 | 0.704 | 0.087 | 0.740 |
| 3IJJ_XP | 179 | 58 | 8893 | 21107 | 0.755 | 0.704 | 0.704 | 0.088 | 0.744 |
| 3JSG_XP | 170 | 67 | 11926 | 18074 | 0.717 | 0.602 | 0.603 | 0.058 | 0.648 |
| 3SMC_XP | 177 | 60 | 11919 | 18081 | 0.747 | 0.603 | 0.604 | 0.063 | 0.651 |
| 4F2K_XP | 190 | 47 | 12690 | 17310 | 0.802 | 0.577 | 0.579 | 0.068 | 0.677 |
| Combined_Scores | 177 | 60 | 6251 | 23749 | 0.747 | 0.792 | 0.791 | 0.116 | 0.832 |
Structural analyses and binding mechanisms of the active compounds
ISO-1, the prototypical MIF inhibitor, was chosen as the reference control. As shown in Fig. 2 and Table 2, twelve hits were more potent than ISO-1, with IC50 values less than 14.4 μM. Compound 1 showed the most potent inhibitory activity (IC50 = 1.22 μM), whereas compound 12 exhibited the weakest inhibitory activity (IC50 = 14.00 μM). The twelve active molecules have diverse structures and novel scaffolds (Fig. 2). The FCFP4 fingerprints identified with the fingerprints protocol were used to estimate the novelty of these compounds with reference to known MIF inhibitors deposited in the BindingDB database. Except for compounds 2, 7, and 8, the other compounds exhibited relatively low Tanimoto similarities (0.22–0.31, Table 2). It was also noted that although the potency of MIF inhibition may not be very high, our data provided structural novelty and diversity of MIF inhibitors that will be new potential resources for structure–activity assays to search for selective MIF inhibitors. In addition, the blood–brain barrier (BBB) is a complicated physical and biochemical interface composed of blood capillary endothelial cells, and BBB penetration of compounds is important for effective CNS-acting drugs. Here, we predicted the BBB coefficient and other drug-likeness properties of these inhibitors by Qikprop [48], which can evaluate pharmaceutically relevant properties and provide ranges with those of 95% of known drugs. As shown in Table 2, the logBB values were in the range of −3.0 to 1.2, indicating that these novel MIF inhibitors were predicted to cross the BBB.
Fig. 2. Chemical structures of compounds.

They include ISO-1 and the 12 active compounds identified by machine learning-based virtual screening with multiple MIF structures and enzyme-based assays.
Table 2.
Experimentally determined half-maximal inhibitory concentrations (IC50) of MIF tautomerase activity (TIC50) and NO production of compounds with TIC50 < 14.41 μM in LPS-activated BV-2 microglia cells (NIC50).
| No. | TIC50 (μM)a | NIC50 (μM)b | MWc | logPd | logSe | PCacof | logBBg | Similarityh |
|---|---|---|---|---|---|---|---|---|
| ISO-1 | 14.41 ± 1.59 | 100.03 ± 2.00 | 235.24 | 1.08 | −2.88 | 228.56 | −1.05 | |
| 1 | 1.22 ± 0.09 | 65.59 ± 1.82 | 312.38 | 1.55 | −3.74 | 326.82 | −1.27 | 0.25 |
| 2 | 2.45 ± 0.39 | 349.40 | 3.76 | −5.56 | 888.85 | −0.34 | 0.63 | |
| 3 | 5.16 ± 0.71 | 68.22 ± 1.84 | 402.78 | 3.44 | −5.42 | 459.27 | −0.41 | 0.22 |
| 4 | 7.30 ± 0.86 | 55.75 ± 1.75 | 438.56 | 4.73 | −6.49 | 493.57 | −1.19 | 0.22 |
| 5 | 7.55 ± 0.88 | 35.40 ± 1.55 | 459.89 | 4.01 | −5.02 | 484.48 | −1.09 | 0.30 |
| 6 | 7.57 ± 0.88 | 113.32 ± 2.05 | 314.29 | 2.42 | −3.86 | 886.96 | −0.26 | 0.29 |
| 7 | 8.34 ± 0.92 | 59.20 ± 1.77 | 263.25 | 2.38 | −3.16 | 902.02 | −0.14 | 0.49 |
| 8 | 8.91 ± 0.95 | 55.18 ± 1.74 | 363.39 | 3.85 | −5.69 | 486.32 | −1.07 | 0.47 |
| 9 | 9.49 ± 0.98 | 28.32 ± 1.45 | 338.38 | 3.59 | −4.92 | 394.41 | −1.34 | 0.28 |
| 10 | 10.80 ± 1.03 | 290.68 | 3.32 | −4.70 | 914.35 | −0.06 | 0.31 | |
| 11 | 11.95 ± 0.98 | 23.84 ± 1.38 | 291.37 | 3.06 | −4.40 | 896.71 | −0.57 | 0.23 |
| 12 | 14.00 ± 1.15 | 27.80 ± 1.58 | 285.73 | 3.18 | −4.34 | 915.71 | −0.36 | 0.23 |
aDopachrome tautomerase assay.
bNitric oxide (NO) measurement.
cMolecular weight (130.0–725.0).
dPredicted octanol/water partition coefficient (−2.0 to 6.5).
ePredicted aqueous solubility, S in mol/L (−6.5 to 0.5).
fPredicted Caco-2 cell permeability in nm/s (500 great).
gPredicted brain/blood partition coefficient (−3.0 to 1.2).
hTanimoto similarity indices based on the FCFP_4 fingerprints for each inhibitor with the known MIF inhibitors.
These inhibitors exhibited similar binding poses in the active site of MIF predicted by Glide docking. Although compound 1 had the most potent MIF tautomerase activity (IC50 = 1.22 μM), its cell cytostatic activity was only 65.59 μM. Compound 1 is a unique chemotype that possesses benzenesulfonyl and two amide groups. Compound 4 has two amide groups and showed relatively low MIF tautomerase inhibitory activity (IC50 = 7.30 μM), while its cell cytostatic activity was 55.75 μM. Five compounds (2, 5, 6, 7, and 8) are coumarin derivatives, and compound 10 is a chromen-4-one analog, which is structurally similar to the inhibitors reported by a previous study [49]. The binding structures of the four inhibitors with the strongest cell cytostatic activity (5, 9, 11, and 12) were predicted by molecular docking, and they were predicted to be located in the hydrophobic pocket formed by the side chains of Pro1, Met2, Ile64, Val106, and Phe113 (Fig. 3). As shown in Supplementary Figs. S3a and S4a, the oxygen atoms of the coumarin ring of compound 5 form H-bond interactions with Lys32 and Ile64, which is consistent with a previous study [50]. The phenyl of the coumarin ring interacts with Tyr95 to form an aryl-aryl group and can also form cation-π interactions with Pro1. The chlorophenyl of compound 5 also forms an aryl-aryl interaction with Phe113. As shown in Supplementary Figs. S3b and S4b, the phenolic ring of compound 9 forms a typical H-bond with Asn97, and the phenolic moiety is considered a key structural feature of the activity of phenolic hydrazones [50]. The carbonyl oxygen forms two bifurcated H-bonds of Ile64 and Lys32, and the two different phenyl groups of compound 5 form aryl-aryl interactions with Phe113 and Tyr95. The carbonyl oxygen of compound 11 also forms bifurcated H-bonds with Ile64 and Lys32 (Supplementary Figs. S3c and S4c). The nitrogen atom of the amide group forms a salt-bridge interaction with Pro1. The phenyl and thiazole rings of compound 11 also form aryl-aryl interactions with Tyr95 and Tyr36, respectively. As shown in Supplementary Figs. S3d and S4d, the amino group of compound 12 forms a H-bond with Asn97, and the carbonyl oxygen can also form a H-bond with Lys32. Ile64 forms a bifurcated H-bond with the two oxygen atoms of compound 12. The detailed 2D interaction patterns between MIF and compounds 5, 9, 11, or 12 are shown in Supplementary Fig. S4. Currently, most MIF tautomerase inhibitors have been identified through screening efforts using d-dopachrome methyl ester or p-hydroxyphenyl pyruvic acid as substrates. Pro1 has a pKa of 5.6, ~4 pH units lower than the pKa of proline amide, and is the most nucleophilic residue of MIF. Many reported MIF inhibitors have unsaturated functional groups and tend to undergo addition reactions [51]. Therefore, a mix of covalent and noncovalent MIF inhibitors may be commonplace. NAPQI, a well-known covalent inhibitor of MIF, was confirmed by mass spectrometry experiments in 2002 [52]. However, a subsequent article confirmed that only approximately one-third of MIF was covalently complexed to NAPQI, while it yielded 96% inhibition. The structure of MIF cocrystallized with NAPQI reveals that the NAPQI undergoes a chemical alteration forming an acetaminophen dimer (bi-APAP) and binds noncovalently to MIF at the mouth of the active site, and kinetic data indicate that NAPQI inhibits MIF both covalently and noncovalently [41]. In the present study, some MIF inhibitors (1, 3, 4, 5, 9, 11, and 12) have carbonyl (R–C=O) groups, which may be good electrophilic centers for Pro1. However, we found that most of the groups connected to the carbonyl group were electron-donating groups by analyzing their chemical structures and included several strong electron-donating groups (–NHR, –OR), which reduce the electrophilicity of the carbonyl carbon atoms to a certain extent. In addition, based on careful investigations of the MIF-related PDB database and the literature, we hypothesize that these inhibitors are mainly bound with MIF in a noncovalent form.
Fig. 3. The 3D presentation of the interactions.

MIF binding pockets for 5 (a), 9 (b), 11(c) and 12 (d).
Cell-based analysis of the four strongest MIF tautomerase activity inhibitors
MIF is mainly produced and stored in immune cells such as macrophages and T cells in response to multiple stimuli and causes an immune response by inducing itself and other pro-inflammatory cytokines, such as IL-6, TNF-α, and IL-1β [53–55]. Microglia are resident cells in the brain that contribute to neurodegenerative progression by regulating the expression of various neurotoxic mediators [2]. Our previous study confirmed that MIF is largely present in brain cells, including microglia and astrocytes [7]. Furthermore, a few studies reported that an inhibitor with MIF tautomerase activity could inhibit LPS-induced macrophage/microglial activation [56, 57]. Thus, we examined the effects of MIF tautomerase inhibitors on activated microglial cells. Excessive secretion of NO is a known hallmark of inflammation in activated microglial cells. Thus, compounds with potent tautomerase activities (IC50 < 14.41 μM) were chosen, and their effects on NO production were examined in LPS-stimulated BV-2 microglial cells. The results revealed that all compounds could strongly attenuate NO release in LPS-primed BV-2 microglial cells. Among the tested compounds, compounds 5, 9, 11, and 12 exhibited more potency than ISO-1 (Table 2 and Fig. 4a). MTT assays were used to examine compound cytotoxicity and excluded the reduction in NO caused by cell deactivation. We found that except for compounds 2, 6, and 10, no compounds exhibited significant cytotoxicity at the tested concentrations (Fig. 4b). iNOS and COX-2 are known to be key pro-inflammatory enzymes that contribute to the synthesis of NO and prostaglandin E2 (PGE-2), respectively, in the brain [58]. Thus, we tested the effect of MIF tautomerase activity inhibitors on the expression of iNOS and COX-2 at the protein level in LPS-primed BV-2 microglial cells. Similar to the effect on NO, compounds 5, 9, 11, and 12 obviously decreased the expression of iNOS and COX-2 at the protein level (Fig. 4c).
Fig. 4. Effects of the compounds on the production of NO, cell viability, protein levels of iNOS, and COX-2 in LPS-primed BV-2 microglial cells.
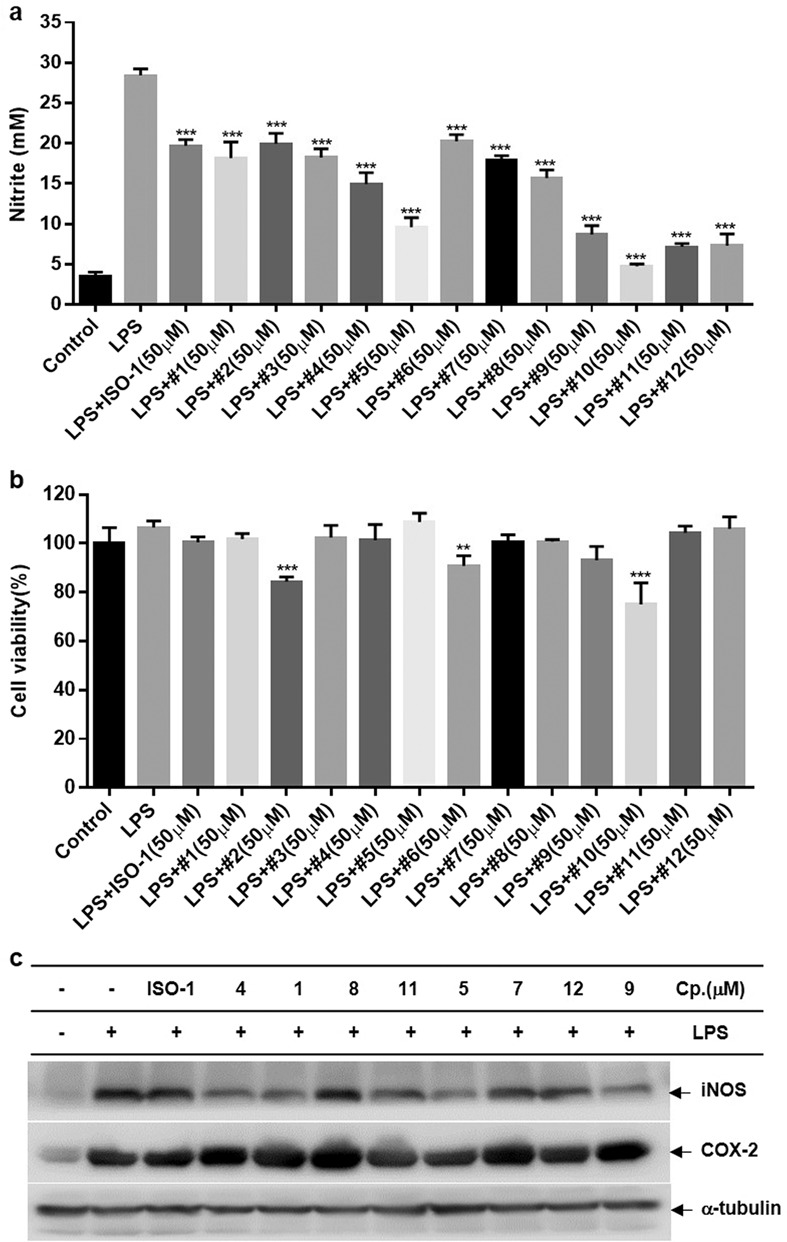
Cells were exposed to the compounds (50 μM) for 30 min prior to LPS (0.2 µg/mL) stimulation for 24 h. a The release of NO in cell culture medium was evaluated by Griess reagent. b Cell viability was measured using an MTT reagent. c The expression of iNOS and COX-2 at the protein level was examined by Western blotting. The expression of α-tubulin was used as an internal control. **P < 0.01, ***P < 0.001 compared to the LPS-alone group.
Since compounds 5, 9, 11, and 12 strongly inhibited microglial NO production without significant cytotoxicity, the anti-inflammatory effects of these compounds were further elucidated. We found inhibitory effects of the MIF inhibitors on NO release in LPS-activated primary microglia and primary astrocyte cultures (Fig. 5a, b), suggesting that NO inhibition by MIF inhibitors was not unique to BV-2 microglial cells. Pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β are induced by activated microglial cells and linked to neuronal injuries during the progression of CNS diseases [59]. Thus, we examined the role of these compounds on LPS-induced IL-6, TNF-α, and IL-1β expression in activated microglial cells. As shown in Fig. 5c–e, all four compounds could significantly decrease the mRNA expression of these cytokines. The results demonstrated that small-molecule inhibitors of MIF tautomerase activity could markedly decrease LPS-induced microglial inflammatory activation.
Fig. 5. Effects of compounds 5, 9, 11, and 12 on the production of NO and the mRNA expression of pro-inflammatory factors in glial cells.

Cells were pretreated with 25 μM compounds 5, 9, 11, and 12 for 30 min prior to LPS or LPS + INF-γ stimulation. a, b After 24 h of LPS (0.2 µg/mL) or LPS+INF-γ (50 U) stimulation, the levels of NO in the supernatants of primary microglial cells (a) and primary astrocytes (b) were determined by Griess reagent. c–e RT-qPCR analysis of TNF-α (c), IL-1β (d), and IL-6 (e) mRNA levels after LPS stimulation for 6 h. *P < 0.05, **P < 0.01, ***P < 0.001 compared to the LPS-alone group.
DARTS is a technology for identifying and validating the protein targets of small molecules. This methodology relies on the fact that the binding of small molecules may stabilize the target protein against proteolysis [35]. We verified compound-MIF interactions using DARTS. The results showed that compounds 5, 9, 11, and 12 markedly blocked the degradation of MIF protein (Fig. 6a). To further elucidate whether the anti-inflammatory effects of compounds 5, 9, 11, and 12 are dependent on MIF expression, we next examined the effects of these four compounds in MIF knockdown BV-2 microglial cells. As shown in Fig. 6b–d, consistent with previous results, LPS-induced iNOS expression was significantly impaired in MIF knockdown BV-2 microglial cells. Furthermore, the inhibitory effects of compounds on LPS-induced iNOS gene expression were abolished in MIF knockdown BV-2 microglial cells, clearly demonstrating that the anti-inflammatory effects of compounds 5, 9, 11, and 12 were dependent on MIF.
Fig. 6. The anti-inflammatory effects of MIF tautomerase inhibitors were dependent on MIF expression.

a Lysates of BV-2 microglial cells were collected by M-PER lysis and incubated with compounds 5, 9, 11, and 12 (50 μM) for 1 h. Then, pronase E was added and incubated for 15 min, and each reaction was stopped by the addition of a protease inhibitor cocktail. The protein level of MIF was determined by Western blotting. b–d BV-2 microglial cells were treated with MIF siRNA or scrambled siRNA (NC) for 36–48 h and then primed with LPS (0.2 µg/mL) with or without compounds 5, 9, 11, and 12 (50 μM) for 6 h. mRNA expression of MIF (b) and iNOS (c, d) was examined by RT-qPCR. ***P < 0.001 compared to the LPS-alone group.
Since compound 11 exhibited the most bioactivity, we focused on this compound in the following experiments. First, we analyzed the competitive type of compound 11 and determined the inhibition constant. The Lineweaver–Burk plots of the dose–response curves showed that compound 11 was a competitive inhibitor, and the value of the inhibition constant (Ki) was 7.949 ± 0.8378 μM (Fig. 7a, b).
Fig. 7. Competitive inhibition pattern and the inhibition of MIF-mediated abrogation of glucocorticoid activity by compound 11.

a, b Nonlinear regression analysis and Lineweaver−Burk plot showing the inhibitory constant (Ki) and type of inhibition. c, d Primary microglial cells were pretreated for 30 min with dexamethasone (DEX; 0.1 μM) and recombinant MIF (0.25 μM) in the presence or absence of the compounds (5-50 μM) prior to LPS (0.2 μg/mL) stimulation. At 24 h after LPS stimulation, the levels of NO in the supernatants were measured using Griess reagents (c), and the levels of IL-6 were measured using ELISA (d). *P < 0.05, ***P < 0.001 compared with the LPS + DEX + MIF group.
It is well-known that MIF itself can override the anti-inflammatory properties of glucocorticoids [31]. Therefore, we determined whether compound 11 could inhibit the immunoregulatory effects of MIF on primary microglial cells. As expected, recombinant MIF abrogated the inhibitory effect of dexamethasone on the production of NO and IL-6 in LPS-activated primary microglial cells, and compound 11 significantly inhibited the counter-regulation of MIF on glucocorticoid-mediated immunosuppression in a dose-dependent manner (Fig. 7c, d).
Next, BV-2 microglial cells were used to test the anti-inflammatory effect of compound 11. A dose–response analysis of compound 11 on NO release was performed in LPS-stimulated BV-2 microglial cells (Fig. 8a, b). The results demonstrated that compound 11 markedly decreased LPS-induced NO production in BV-2 microglial cells in a dose-dependent manner. In addition, compound 11 also dose-dependently decreased iNOS, COX-2, TNF-α, IL-6, and IL-1β mRNA and protein levels (Fig. 8c–k) in LPS-activated BV-2 microglial cells. Overactivated microglia can also produce ROS, which is related to neuroinflammation and neurodegeneration. Thus, we evaluated the effect of compound 11 on ROS generation in LPS-primed BV-2 microglial cells. Similar to the above results, compound 11 (25 μM) significantly impaired intracellular ROS production in LPS-stimulated BV-2 microglial cells (Fig. 8l). These data further confirmed that compound 11 induced a strong anti-inflammatory effect on activated microglial cells.
Fig. 8. Effects of compound 11 on the generation of pro-inflammatory factors in LPS-primed BV-2 microglial cells.

Cells were pretreated with compound 11 (1–50 μM) for 0.5 h prior to LPS (0.2 µg/mL) stimulation. a, b After 24 h of LPS stimulation, NO levels were evaluated by Griess reagent (a), and cell viability was determined by MTT assays (b). c, d The production of IL-6 (c) and TNF-α (d) in supernatants after LPS stimulation for 24 h was evaluated by ELISA. e–j The mRNA expression of iNOS, COX-2, TNF-α, IL-6, IL-1β, and MIF after LPS treatment for 6 h was assessed by RT-qPCR. k Western blot analysis of iNOS, COX-2, MIF, and α-tubulin protein expression in lysates of LPS-primed BV-2 microglial cells at 16 h (k, left). The expression of α-tubulin was used as an internal control, and the relative expression of MIF, iNOS, and COX-2 was quantified by densitometric analysis (k, right). l After 16 h of LPS stimulation, the production of intracellular ROS was measured by flow cytometry. *P < 0.05, **P < 0.01, ***P < 0.001 compared with the LPS-alone group.
The transcription factor NF-κB mediates certain cellular signal transduction pathways that contribute to the production of pro-inflammatory cytokines, including COX-2, iNOS, IL-6, IL-1β, and TNF-α. Inflammatory stimuli such as LPS can induce IKKβ activation, phosphorylation of the p65 subunit of NF-κB and IκBα degradation, ultimately resulting in nuclear translocation of p65 [60]. We explored the effects of compound 11 on NF-κB signals in LPS-primed BV-2 microglial cells. As shown in Fig. 9a, b, compound 11 significantly suppressed LPS-induced p65 phosphorylation, IκBα degradation, and the nuclear translocation of p65 in BV-2 microglial cells. Further results from the immunofluorescence assay revealed that p65 translocated into the nucleus after LPS stimulation for ~60 min, and this effect was significantly inhibited by compound 11 pretreatment (Fig. 9c). The MAPKs signaling cascade is also thought to regulate the expression of pro-inflammatory factors in activated microglial cells [61]. The role of compound 11 in the activation of MAPKs was assessed by Western blot analysis. Compound 11 inhibited LPS-induced phosphorylation of the MAPKs p38, JNK, and ERK (Fig. 9d). These results indicated that the anti-inflammatory effects of compound 11 were achieved by inhibiting the activation of NF-κB and MAPKs signaling in LPS-primed BV-2 microglial cells.
Fig. 9. Compound 11 inhibits LPS-induced BV-2 microglial cells activation via NF-κB and MAPK signaling.

BV-2 microglial cells were primed with compound 11 (25 μM) for 30 min before LPS stimulation. a Western blot analysis of phospho-p65, p65, IκBα, and α-tubulin protein levels in LPS-primed BV-2 microglial cells at 20 min (a, left). The relative expression of phospho-p65 and IκBα was quantified by densitometric analysis (a, right). b Western blot analysis of p65 and Histone H2B in the nuclear fraction and Western blot analysis of p65 and GAPDH in the cytoplasm of LPS-primed BV-2 microglial cells at 1 h (b, left). Histone H2B and GAPDH were used as internal controls. The relative expression of p65 in the nucleus and cytoplasm was quantified by densitometric analysis (b, right). c After 1 h of LPS stimulation, NF-κB p65 subunit localization was visualized by immunofluorescence analysis, and all cell nuclei were counterstained with Hoechst. The scale bar represents 10 μm (c, left). The ratio of cells with p65 nuclear translocation was calculated by counting at least 100 cells in a coverslip (c, right). d Western blot analysis of MAPKs (p38, JNK, ERK) in the lysates of LPS-primed BV-2 microglial cells at 1 h (d, left). Total p38, JNK, and ERK were used as internal controls, and the relative phosphorylation level of p38, JNK, and ERK was quantified by densitometric analysis (d, right). **P < 0.01, ***P < 0.001 compared with the LPS-alone group.
Overactivated microglia can produce many inflammatory cytokines that trigger neuronal damage or amplify neurodegeneration [62]. Indeed, anti-inflammatory compounds, which inhibit microglial activation, have been suggested to exert a neuroprotective effect in vitro and in vivo [63, 64]. To examine the potential neuroprotective effect of compound 11 in vitro, we used a microglial conditioned media/neuron co-culture system. As shown in Fig. 10, the addition of CM from LPS-primed BV-2 microglial cells decreased HT-22 neuroblastoma cell viability, and compound 11 (25 μM) markedly attenuated the decrease in the viability of HT-22 neuroblastoma cells. Although these co-culture systems cannot completely mimic the condition of neurodegenerative diseases, they partially reflect the neuroinflammatory microenvironments in which activated microglia induce surrounding neuronal cell death by producing neurotoxic factors. These results indicate that compound 11 may be neuroprotective by suppressing microglial pro-inflammatory activation.
Fig. 10. Neuroprotective effect of compound 11 on CM from a microglia/neuroblastoma co-culture model.
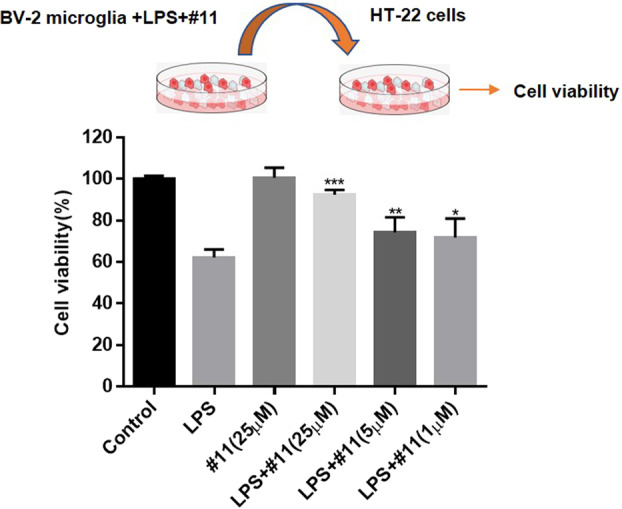
BV-2 microglial cells were pretreated with or without compound 11 (1–25 μM) and then stimulated with LPS (0.2 µg/mL) for 6 h. The culture media was discarded, and fresh medium was added. After 24 h of incubation, the culture supernatants were collected and then added to HT-22 neuronal cells as described in the Methods. After co-culture for 24 h, cell viability was determined using an MTT reagent. *P < 0.05, **P < 0.01, ***P < 0.001 compared with the LPS-alone group.
Conclusion
In this study, an integrated virtual screening strategy was used to identify novel MIF inhibitors. Tautomerase activity experiments confirmed that 12 molecules had stronger inhibitory activity than ISO-1, including 9 hits with IC50 values below 10 μM. Their bioactivities were measured by assessing the production of NO in LPS-activated BV-2 microglial cells. Among them, compounds 5, 9, 11, and 12 significantly inhibited NO production without inducing cytotoxicity at a dose of 50 μM. These compounds, especially compound 11, the most potent compound, also markedly decreased LPS-induced the production of pro-inflammatory cytokines, including IL-6, TNF-α, iNOS, COX-2, and IL-1β in microglia cells. The strong anti-inflammatory effects of these compounds were abolished when MIF was knocked down by siRNA. Moreover, the anti-inflammatory effect of compound 11 in LPS-activated microglial cells was linked to the suppression of the activation of NF-κB and MAPKs pathways. Additionally, compound 11 attenuated the neurotoxicity of microglial CM towards HT-22 neuroblastoma cells. These results indicate that MIF may be involved in the regulation of neuroinflammatory responses and that small-molecule MIF inhibitors may serve as promising therapeutic agents for neuroinflammatory diseases.
Supplementary information
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81703496, 81973334, 31970909, 21173156, 81803430, 81773702, and 81373382), Natural Science Foundation of Hubei Province (2020CFB543), and Natural Science Foundation of Jiangsu Province (BE2019650) and the Priority Academic Program Development of the Jiangsu Higher Education Institutes (PAPD), National Center for International Research (2017B01012), and Inner Mongolia Natural Science Foundation (2019MS08197).
Author contributions
LTZ, XCZ, and TJH designed the research. Yu Zhang, LX, and Yao Zhang designed the experiments. Yu Zhang, LX, and Yao Zhang conducted virtual screening and biological assays. JP, PQW, ST, HTL, and BWG helped to perform part of biological assays. Yu Zhang, LX, Yao Zhang, LTZ, XCZ, and TJH wrote the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Yu Zhang, Lei Xu, Yao Zhang
Contributor Information
Ting-jun Hou, Email: tjhou@zju.edu.cn.
Xue-chu Zhen, Email: zhenxuechu@suda.edun.cn.
Long-Tai Zheng, Email: zhenglongtai@suda.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00753-x.
References
- 1.Amor S, Peferoen LAN, Vogel DYS, Breur M, Der Valk PV, Baker D, et al. Inflammation in neurodegenerative diseases- an update. Immunology. 2014;142:151–66. doi: 10.1111/imm.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Block ML, Zecca L, Hong J. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 3.Dong XH, Zhen XC. Glial pathology in bipolar disorder: potential therapeutic implications. Neurosci Ther. 2015;21:393–7. doi: 10.1111/cns.12390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perry VH, Nicoll JAR, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 5.Xu L, Li Y, Sun H, Zhen X, Qiao C, Tian S, et al. Current developments of macrophage migration inhibitory factor (MIF) inhibitors. Drug Discov Today. 2013;18:592–600. doi: 10.1016/j.drudis.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 6.Zhang W, Li L, Wang J, An L, Hu X, Xie J, et al. Expression of macrophage migration inhibitory factor in the mouse neocortex and posterior piriform cortices during postnatal development. Cell Mol Neurobiol. 2014;34:1183–97. doi: 10.1007/s10571-014-0094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Gu R, Jia J, Hou T, Zheng LT, Zhen X. Inhibition of macrophage migration inhibitory factor (MIF) tautomerase activity suppresses microglia-mediated inflammatory responses. Clin Exp Pharmacol Physiol. 2016;43:1134–44. doi: 10.1111/1440-1681.12647. [DOI] [PubMed] [Google Scholar]
- 8.Zis O, Zhang S, Dorovini-Zis K, Wang L, Song W. Hypoxia signaling regulates macrophage migration inhibitory factor (MIF) expression in stroke. Mol Neurobiol. 2015;51:155–67. doi: 10.1007/s12035-014-8727-4. [DOI] [PubMed] [Google Scholar]
- 9.Benedek G, Meza-Romero R, Jordan K, Zhang Y, Nguyen H, Kent G, et al. MIF and D-DT are potential disease severity modifiers in male MS subjects. Proc Natl Acad Sci USA. 2017;114:E8421–E9. doi: 10.1073/pnas.1712288114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang S, Zhao J, Zhang Y, Zhang Y, Cai F, Wang L, et al. Upregulation of MIF as a defense mechanism and a biomarker of Alzheimer’s disease. Alzheimers Res Ther. 2019;11:1–12. doi: 10.1186/s13195-018-0453-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicoletti A, Fagone P, Donzuso G, Mangano K, Dibilio V, Caponnetto S, et al. Parkinson’s disease is associated with increased serum levels of macrophage migration inhibitory factor. Cytokine. 2011;55:165–7. doi: 10.1016/j.cyto.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 12.Sugimoto H, Taniguchi M, Nakagawa A, Tanaka I, Suzuki M, Nishihira J. Crystal structure of human D-dopachrome tautomerase, a homologue of macrophage migration inhibitory factor, at 1.54 Å resolution. Biochemistry. 1999;38:3268–79. doi: 10.1021/bi982184o. [DOI] [PubMed] [Google Scholar]
- 13.Rosengren E, Åman P, Thelin S, Hansson C, Ahlfors S, Björk P, et al. The macrophage migration inhibitory factor MIF is a phenylpyruvate tautomerase. FEBS Lett. 1997;417:85–8. doi: 10.1016/S0014-5793(97)01261-1. [DOI] [PubMed] [Google Scholar]
- 14.Kleemann R, Kapurniotu A, Frank RW, Gessner A, Mischke R, Flieger O, et al. Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol-protein oxidoreductase. J Mol Biol. 1998;280:85–102. doi: 10.1006/jmbi.1998.1864. [DOI] [PubMed] [Google Scholar]
- 15.Gunther S, Fagone P, Jalce G, Atanasov AG, Guignabert C, Nicoletti F. Role of MIF and D-DT in immune-inflammatory, autoimmune, and chronic respiratory diseases: from pathogenic factors to therapeutic targets. Drug Discov Today. 2019;24:428–39. doi: 10.1016/j.drudis.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Garai J, Lorand T. Macrophage migration inhibitory factor (MIF) tautomerase inhibitors as potential novel anti-inflammatory agents: current developments. Curr Med Chem. 2009;16:1091–114. doi: 10.2174/092986709787581842. [DOI] [PubMed] [Google Scholar]
- 17.Trivediparmar V, Jorgensen WL. Advances and insights for small molecule inhibition of macrophage migration inhibitory factor. J Med Chem. 2018;61:8104–19. doi: 10.1021/acs.jmedchem.8b00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kok T, Wasiel AA, Cool RH, Melgert BN, Poelarends GJ, Dekker FJ. Small-molecule inhibitors of macrophage migration inhibitory factor (MIF) as an emerging class of therapeutics for immune disorders. Drug Discov Today. 2018;23:1910–8. doi: 10.1016/j.drudis.2018.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cisneros JA, Robertson MJ, Valhondo M, Jorgensen WL. A fluorescence polarization assay for binding to macrophage migration inhibitory factor and crystal structures for somplexes of two potent inhibitors. J Am Chem Soc. 2016;138:8630–8. doi: 10.1021/jacs.6b04910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanrikulu Y, Kruger B, Proschak E. The holistic integration of virtual screening in drug discovery. Drug Discov Today. 2013;18:358–64. doi: 10.1016/j.drudis.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Tian S, Sun H, Li Y, Pan P, Li D, Hou T. Development and eEvaluation of an integrated virtual screening strategy by combining molecular docking and pharmacophore searching based on multiple protein structures. J Chem Inf Model. 2013;53:2743–56. doi: 10.1021/ci400382r. [DOI] [PubMed] [Google Scholar]
- 22.Tian S, Sun H, Pan P, Li D, Zhen X, Li Y, et al. Assessing an ensemble docking-based virtual screening strategy for kinase targets by considering protein flexibility. J Chem Inf Model. 2014;54:2664–79. doi: 10.1021/ci500414b. [DOI] [PubMed] [Google Scholar]
- 23.Berman HM, Westbrook JD, Feng Z, Gilliland GL, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 25.Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007;35:198–201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaminski GA, Friesner RA, Rives JT, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105:6474–87. doi: 10.1021/jp003919d. [DOI] [Google Scholar]
- 27.Soares TA, Goodsell DS, Ferreira R, Olson AJ, Briggs JM. Ionization state and molecular docking studies for the macrophage migration inhibitory factor: the role of lysine 32 in the catalytic mechanism. J Mol Recognit. 2000;13:146–56. doi: 10.1002/1099-1352(200005/06)13:3<146::AID-JMR497>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 28.Maestro, version 10.3 (Schrödinger, LLC, New York, NY, 2015).
- 29.Tian S, Wang X, Li L, Zhang X, Li Y, Zhu F, et al. Discovery of novel and selective adenosine A2A receptor antagonists for treating parkinson’s disease through comparative structure-based virtual screening. J Chem Inf Model. 2017;57:1474–87. doi: 10.1021/acs.jcim.7b00188. [DOI] [PubMed] [Google Scholar]
- 30.Walters WP, Stahl MT, Murcko MA. Virtual screening: an overview. Drug Discov Today. 1998;3:160–78. doi: 10.1016/S1359-6446(97)01163-X. [DOI] [Google Scholar]
- 31.Xu L, Zhang Y, Zheng LT, Qiao C, Li Y, Li D, et al. Discovery of novel inhibitors targeting the macrophage migration inhibitory factor via structure-based virtual screening and bioassays. J Med Chem. 2014;57:3737–45. doi: 10.1021/jm401908w. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Xu L, Zhang Z, Zhang Z, Zheng LT, Li D, et al. Structure-activity relationships and anti-inflammatory activities of N-carbamothioylformamide analogues as MIF tautomerase inhibitors. J Chem Inf Model. 2015;55:1994–2004. doi: 10.1021/acs.jcim.5b00445. [DOI] [PubMed] [Google Scholar]
- 33.Han CJ, Zheng JY, Sun L, Yang HC, Cao ZQ, Zhang XH, et al. The oncometabolite 2-hydroxyglutarate inhibits microglial activation via the AMPK/mTOR/NF-kappaB pathway. Acta Pharmacol Sin. 2019;40:1292–302. doi: 10.1038/s41401-019-0225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tao L, Zhang F, Hao L, Wu J, Jia J, Liu JY, et al. 1-O-tigloyl-1-O-deacetyl-nimbolinin B inhibits LPS-stimulated inflammatory responses by suppressing NF-kappaB and JNK activation in microglia cells. J Pharm Sci. 2014;125:364–74. doi: 10.1254/jphs.14025FP. [DOI] [PubMed] [Google Scholar]
- 35.Pai MY, Lomenick B, Hwang H, Schiestl R, McBride W, Loo JA, et al. Drug affinity responsive target stability (DARTS) for small-molecule target identification. Methods Mol Biol. 2015;1263:287–98. doi: 10.1007/978-1-4939-2269-7_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo CH, Cao T, Zheng LT, Waddington JL, Zhen XC. Development and characterization of an inducible Dicer conditional knockout mouse model of Parkinson’s disease: validation of the antiparkinsonian effects of a sigma-1 receptor agonist and dihydromyricetin. Acta Pharmacol Sin. 2020;41:499–507. doi: 10.1038/s41401-020-0379-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu ZX, Wu J, Zheng JY, Ma HK, Zhang HJ, et al. Design, synthesis and evaluation of a series of non-steroidal anti-inflammatory drug conjugates as novel neuroinflammatory inhibitors. Int Immunopharmacol. 2015;25:528–37. doi: 10.1016/j.intimp.2015.02.033. [DOI] [PubMed] [Google Scholar]
- 38.Lubetsky JB, Swope M, Dealwis C, Blake P, Lolis E. Pro-1 of macrophage migration inhibitory factor functions as a catalytic base in the phenylpyruvate tautomerase activity. Biochemistry. 1999;38:7346–54. doi: 10.1021/bi990306m. [DOI] [PubMed] [Google Scholar]
- 39.Crichlow GV, Cheng KF, Dabideen D, Ochani M, Aljabari B, Pavlov VA, et al. Alternative chemical modifications reverse the binding orientation of a pharmacophore scaffold in the active site of macrophage migration inhibitory factor. J Biol Chem. 2007;282:23089–95. doi: 10.1074/jbc.M701825200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winner M, Meier J, Zierow S, Rendon BE, Crichlow GV, Riggs R, et al. A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer Res. 2008;68:7253–7. doi: 10.1158/0008-5472.CAN-07-6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crichlow GV, Lubetsky JB, Leng L, Bucala R, Lolis EJ. Structural and kinetic analyses of macrophage migration inhibitory factor active site interactions. Biochemistry. 2009;48:132–9. doi: 10.1021/bi8014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho Y, Crichlow GV, Vermeire JJ, Leng L, Du X, Hodsdon ME, et al. Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc Natl Acad Sci USA. 2010;107:11313–8. doi: 10.1073/pnas.1002716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mclean LR, Zhang Y, Li H, Li Z, Lukasczyk U, Choi Y, et al. Discovery of covalent inhibitors for MIF tautomerase via cocrystal structures with phantom hits from virtual screening. Bioorg Med Chem Lett. 2009;19:6717–20. doi: 10.1016/j.bmcl.2009.09.106. [DOI] [PubMed] [Google Scholar]
- 44.Crichlow GV, Fan C, Keeler C, Hodsdon ME, Lolis E. Structural interactions dictate the kinetics of macrophage migration inhibitory factor inhibition by different cancer-preventive isothiocyanates. Biochemistry. 2012;51:7506–14. doi: 10.1021/bi3005494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tyndall JDA, Lue H, Rutledge MT, Bernhagen J, Hampton MB, Wilbanks SM. Macrophage migration inhibitory factor covalently complexed with phenethyl isothiocyanate. Acta Crystallogr. 2012;68:999–1002. doi: 10.1107/S1744309112030552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider G. Virtual screening: an endless staircase? Nat Rev Drug Discov. 2010;9:273–6. doi: 10.1038/nrd3139. [DOI] [PubMed] [Google Scholar]
- 47.Zhou W, Duan M, Fu W, Pang J, Tang Q, Sun H, et al. Discovery of novel androgen receptor ligands by structure-based virtual screening and bioassays. Genomics Proteomics Bioinformatics. 2018;16:416–27. doi: 10.1016/j.gpb.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dahlgren MK, Garcia AB, Hare AA, Tiradorives J, Leng L, Bucala R, et al. Virtual screening and optimization yield low-nanomolar inhibitors of the tautomerase sctivity of Plasmodium falciparum macrophage migration inhibitory factor. J Med Chem. 2012;55:10148–59. doi: 10.1021/jm301269s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Orita M, Yamamoto S, Katayama N, Aoki M, Takayama K, Yamagiwa Y, et al. Coumarin and chromen-4-one analogues as tautomerase inhibitors of macrophage migration inhibitory factor: discovery and X-ray crystallography. J Med Chem. 2001;44:540–7. doi: 10.1021/jm000386o. [DOI] [PubMed] [Google Scholar]
- 50.Dabideen D, Cheng KF, Aljabari B, Miller EJ, Pavlov VA, Alabed Y. Phenolic hydrazones are potent inhibitors of macrophage migration inhibitory factor proinflammatory activity and survival improving agents in sepsis. J Med Chem. 2007;50:1993–7. doi: 10.1021/jm061477+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cisneros JA, Robertson MJ, Valhondo M, Jorgensen WL. Irregularities in enzyme assays: The case of macrophage migration inhibitory factor. Bioorg Med Chem Lett. 2016;26:2764–7. doi: 10.1016/j.bmcl.2016.04.074. [DOI] [PubMed] [Google Scholar]
- 52.Senter PD, Al-Abed Y, Metz CN, Benigni F, Mitchell RA, Chesney J, et al. Inhibition of macrophage migration inhibitory factor (MIF) tautomerase and biological activities by acetaminophen metabolites. Proc Natl Acad Sci USA. 2002;99:144–9. doi: 10.1073/pnas.011569399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med. 1994;179:1895–902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calandra T, Bucala R. Macrophage migration inhibitory factor (MIF): a glucocorticoid counter-regulator within the immune system. Crit Rev Immunol. 1997;17:77–88. doi: 10.1615/CritRevImmunol.v17.i1.30. [DOI] [PubMed] [Google Scholar]
- 55.Bloom J, Sun S, Al-Abed Y. MIF, a controversial cytokine: a review of structural features, challenges, and opportunities for drug development. Expert Opin Ther Targets. 2016;20:1463–75. doi: 10.1080/14728222.2016.1251582. [DOI] [PubMed] [Google Scholar]
- 56.Lubetsky JB, Dios A, Han J, Aljabari B, Ruzsicska B, Mitchell R, et al. The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J Biol Chem. 2002;277:24976–82. doi: 10.1074/jbc.M203220200. [DOI] [PubMed] [Google Scholar]
- 57.Al-Abed Y, Dabideen D, Aljabari B, Valster A, Messmer D, Ochani M, et al. ISO-1 binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. J Biol Chem. 2005;280:36541–4. doi: 10.1074/jbc.C500243200. [DOI] [PubMed] [Google Scholar]
- 58.Stollg G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol. 1999;58:233–47. doi: 10.1016/S0301-0082(98)00083-5. [DOI] [PubMed] [Google Scholar]
- 59.Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87:10–20. doi: 10.1016/j.brainresbull.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park J, Min JS, Kim B, Chae UB, Yun JW, Choi MS, et al. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neurosci Lett. 2015;584:191–6. doi: 10.1016/j.neulet.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 62.Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7:354–65. doi: 10.1016/j.nurt.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu XL, Lin J, Lu XY, Feng JH, Zhang XQ, Wang H, et al. Synthesis and biological evaluation of clovamide analogues as potent anti-neuroinflammatory agents in vitro and in vivo. Eur J Med Chem. 2018;151:261–71. doi: 10.1016/j.ejmech.2018.03.081. [DOI] [PubMed] [Google Scholar]
- 64.He P, Yan S, Zheng J, Gao Y, Zhang S, Liu Z, et al. Eriodictyol attenuates LPS-induced neuroinflammation, amyloidogenesis, and cognitive impairments via the inhibition of NF-κB in male C57BL/6J mice and BV2 microglial cells. J Agric Food Chem. 2018;66:10205–14. doi: 10.1021/acs.jafc.8b03731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.