

Abstract
A number of emerging studies in field of immune metabolism have indicated that cellular metabolic reprograming serves as a major administrator in maintaining the viability and functions of both tumor cells and immune cells. As one of the most important immunosuppressive cells in tumor stroma, myeloid-derived suppressor cells (MDSCs) dynamically orchestrate their metabolic pathways in response to the complicated tumor microenvironment (TME), a process that consequently limits the therapeutic effectiveness of anti-cancer treatment modalities. In this context, the metabolic vulnerabilities of MDSCs could be exploited as a novel immune metabolic checkpoint upon which to intervene for promoting the efficacy of immunotherapy. Here, we have discussed about recent studies highlighting the important roles of the metabolic reprograming and the core molecular pathways involved in tumor-infiltrating MDSCs. In addition, we have also summarized the state-of-the-art strategies that are currently being employed to target MDSC metabolism and improve the efficacy of antineoplastic immunotherapy.
Keywords: myeloid-derived suppressor cells, tumor microenvironment, metabolic reprograming, cancer immunotherapy, metabolic intervention
Introduction
Tumorigenesis relies on the alternations in cellular metabolism to meet the demands of unbridled growth and metastasis, a process called “metabolic reprogramming” as a recently revisited hallmark of cancer development, which further sculpts the commonly acidic, hypoxic, and hypo-nutrient tumor microenvironment (TME) [1]. TME is an intricate and dynamic structure consisting of multiple immunosuppressive signals and diverse cells that participate in heterotypic interactions with one another [2]. The immunosuppressive signals within TME include, but are not limited to, the expression of suppressive ligands, soluble mediators and metabolites [3]. These signals facilitate tumor progression in plenty of ways: [4] sustaining tumor proliferative signaling, shielding tumor from host immunity, fostering chemotherapy resistance, rendering tumor-infiltrating immune cells to immunosuppressive status and recruiting the immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs) to primarily evade anti-tumor immune responses, among others (Fig. 1).
Fig. 1. Metabolic interactions of MDSCs with TME.

Tumor-derived factors VEGF, CCL, MIF, GM-CSF, G-CSF, M-CSF and S100A8/9 mobilize MDSCs from the bone marrow to tumor sites. Disorganized tumor vasculature brings about inadequate oxygen and glucose for energy supply. Hypoxic tumor cells secrete elevated lactate through glycolysis and consequently contribute to the acidic TME. During tumor progression, the stressed cells release ATP to avoid excessive damage. Tumor stromal adipocytes and neoplastic fat tissues increase the extracellular liberation of fatty acids and cholesterol. MDSCs produce multifarious immunosuppressive mediators to remarkably impair T cell functions. VEGF, vascular endothelial growth factor; CCL chemokine (C-C motif) ligand, GM-CSF granulocyte-macrophage colony-stimulating factor, G-CSF granulocyte colony-stimulating factor, M-CSF macrophage colony-stimulating factor, MIF macrophage migration inhibitory factor, NO nitric oxide, IDO indoleamine 2,3-dioxygenase, Arg-1 arginase-1, iNOS inducible nitric oxide synthase, ROS reactive oxygen species, GLUT glucose transporter, MCT monocarboxylate transporter, LDH lactate dehydrogenase.
The field of cancer metabolism has become a topic of emerging interest in the past decade, while the potential roles of MDSC metabolic pathways in the context of cancer have been investigated recently [5]. MDSCs dynamically reprogram the physiologically metabolic ways to meet their bioenergetic and biosynthetic needs during the various stages of tumorigenesis, thereby adapting to the alterations in TME, such as hypoxia and neo-vascularization [6]. For this reason, it should be further illustrated that how the complex TME manipulates the fate of MDSCs in a metabolic fashion. The aim of this review is to summarize the ways that TME effectively fuels metabolic rewiring in regulation of MDSCs, and to highlight the various opportunities for targeting metabolic vulnerabilities of MDSCs in order to form the basis for novel antineoplastic strategies.
MDSCs
MDSCs are immature cells originating from the bone marrow [7]. As one of the most important immunosuppressive cells in tumor milieu, they possess certain similarities with tumor cells related to the mechanisms of metabolism and adaptive survival. MDSCs belong to a heterogeneous innate cell population and comprise two different subsets: the monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs; also named granulocytic MDSCs, G-MDSCs). MDSCs are known to express multifarious surface biomarkers. The M-MDSCs and PMN-MDSCs from mice display the phenotype of CD11b+ Ly6G+Ly6Chigh and CD11b+ Ly6G−Ly6Clow, respectively [8]. In addition, the human MDSCs are approximately classified as CD33+ HLA-DRlow/−, with M-MDSCs defined as CD11b+CD33+CD14+CD15−HLA-DRlow/− and PMN-MDSCs defined as CD11b+CD33+CD14−CD15+HLA-DRlow/− [9]. Simultaneously, a small group of early-stage MDSCs (e-MDSCs) in human displays the phenotype of Lin− (CD3, CD14, CD15, CD19, CD56) CD33+HLA-DR − [10]. In the pathological conditions like cancer, MDSCs are effectively mobilized from bone marrow to the tumor sites by selective chemokines (such as CCL-2, CCL-5, etc.) and tumor-derived factors (containing GM-CSF, G-CSF, M-CSF, VEGF, S100A8/9, interleukin-1β, etc.) [11]. Thereafter, the transcription factors STAT3/5, CCAAT-enhancer-binding proteins β (C/EBPβ) and nuclear factor kappa-B (NF-κB) drive MDSC expansion, accumulation and immunosuppressive functions [7].
More importantly, MIF derived from several immune and tumor cells confreres advantages for the recruitment, survival and immunosuppressive functions of MDSCs in TME [12–16]. More recently, a second member of the cytokine MIF family, the D-dopachrome tautomerase (DDT, also known as MIF2), also possesses potentiality in regulation of MDSCs [17]. In addition, several MIF inhibitors, including sulforaphane and ibudilast, have shown excellent anti-glioblastoma activity in different preclinical studies through reducing the expansion and functions of circulating MDSCs, thereby ultimately enhancing anti-tumor responses of immune effector cells [15, 18].
The immunosuppressive potencies of MDSCs mediated either through cell-surface receptors and/or the release of short-lived soluble mediators have been well-recognized. MDSCs release multiple oncogenic mediators, typically containing ROS, iNOS, Arg-1, IDO, prostaglandin E-2 (PGE-2), NADPH oxidase-2 (NOX2) and transforming growth factor-β (TGF-β), deplete key amino acids (L-arginine, L-cysteine, etc.), express immune checkpoint including programmed cell death-ligand 1 (PD-L1) and also recruit other tolerant immune cells such as regulatory T cells (Tregs) to significantly impair T cell-mediated immune responses [19, 20]. Furthermore, they also influence the tumor angiogenesis by producing VEGF, basic fibroblast growth factor (bFGF), prokineticin 2 (Bv8), MMP-9, etc [10].
Metabolism is a determinant that effectively shapes the differentiation, functional plasticity, and survival of immune cells [21]. Correspondingly, MDSCs evolve rapid and effective metabolic strategies to maintain immunosuppressive activity and adapt to the drastic changes under a variety of stressed conditions. The metabolic reprograming controls the availability of nutrients and oxygen, as well as the critical receptor signaling events to manipulate the fate of MDSCs. It has become evident that metabolic alternations encompass all the stages of MDSC-TME interactions: (1) In the well-established amino acid metabolism, MDSCs deprive L-arginine via releasing frequently over-expressed metabolic enzymes Arg-1 and iNOS, leading to the deficiency or alteration of TCR-ζ chain (i.e., nitration/nitrosylation) and inhibition of T cell proliferation [22, 23]. Moreover, MDSCs also produce rate-limiting enzyme, IDO, which catalyzes tryptophan along the kynurenine pathway and produces immune-toxic metabolites to induce T cell cycle arrest and stimulate the expansion of Treg cells [24]. (2) The hypoxia and low pH condition of TME contributes to switching the metabolic pathways of MDSCs from oxidative phosphorylation (OXPHOS) to glycolysis, and thus make them compete with the immune effector cells for the limited glucose [25]. (3) Lipid accumulation in TME renders MDSCs to undergo metabolic reprogramming from glycolysis to fatty acid oxidation (FAO), and to use oxidized lipids as the major energy source to potentiate their immunosuppressive activity [26]. Multiple metabolic mechanisms converge to meet the increased requirements of MDSCs: rapid ATP synthesis to maintain energetic status, increased production of immune-toxic metabolites for enhancing their immunosuppressive activity, and dynamic modulation of the metabolic pathways to reduce the unfavorable impact of TME.
Metabolic interactions of MDSCs with TME
Tumor cells are proficient in utilizing the nutrients preferentially assigned to the metabolic pathways that contribute to cellular oncogenic properties [5]. In fact, competition for essential nutrients between enormously proliferative cancer cells and intertumoral cells drastically imparts profound effects on the fates of both innate and adaptive immune cells [27]. Metabolic reprogramming and its underlying signaling events of MDSCs are directly modulated by the complex TME (Figs. 2 and 3). A better understanding of this process may be conducive for development and optimization of novel anti-cancer strategies based on drug repositioning and innovation.
Fig. 2. An overview of cellular metabolic reprograming in MDSCs.

This diagram depicts several important metabolic pathways affected in MDSCs in response to the complex TME. G6P glucose-6-phosphate, MPO myeloperoxidase, MPC mitochondrial pyruvate carrier, PKM pyruvate kinase M, LDL low-density lipoprotein.
Fig. 3. An overview of key metabolic pathways and their associated signaling events in MDSCs during metabolic alternations.
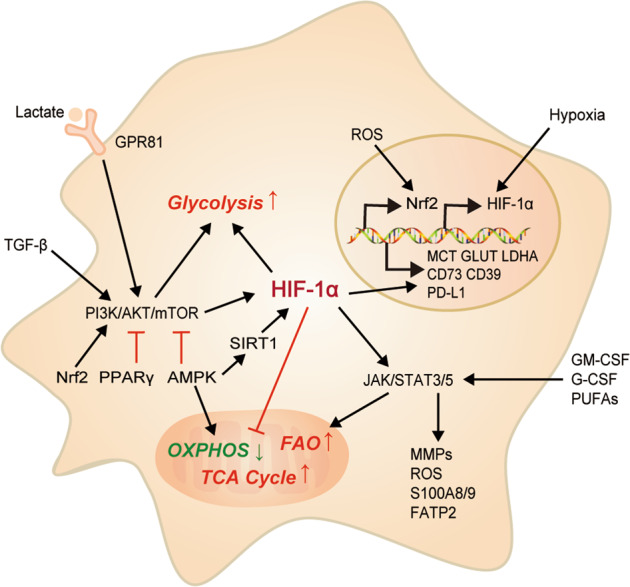
HIF-1α accumulation induced by hypoxia in MDSCs manipulates the transcription of multiple metabolism-related genes. PI3K/Akt/mTOR pathway and the activated HIF-1α facilitate glycolysis, and the AMPK pathway drives glycolysis towards OXPHOS. JAK/STAT3/5 axis promotes FAO via upregulating the expression of FATP2. PI3K phosphatidylinositol 3-kinase, AKT protein kinase B, mTOR mammalian target of rapamycin, JAK janus kinase, STAT 3/5 signal transducer and activator of transcription 3/5, Nrf2 nuclear factor E2-related factor 2, PPARγ peroxisome proliferator-activated receptor γ, AMPK adenosine 5ʹ-monophosphate (AMP)-activated protein kinase, SIRT1 sirtuin 1, MMPs matrix metalloproteinases.
Deregulated uptake of glucose
Glycolysis is a typical metabolic change with an increasing reliance on glucose uptake, thereby supplying numerous metabolites as anabolic precursors and rapidly generating energy [28]. Myeloid cells up-regulate their glycolytic genes when they are confronted with tumor-derived factors and have the greatest capacity to take up intertumoral glucose across a range of cancer models [29, 30]. Besides, the metabolic signature of immune cell is highly anabolic and provokes a striking increase in glucose uptake especially during the periods of proliferation and/or maturation [31]. Although less efficient in generating ATP and more dependent on the requirements for glucose, glycolysis allows for effective disposal of excessive carbon and regeneration of NAD+ while preserving the mitochondrial enzymatic activity for anabolic processes [1]. Recent study reveals that the immature MDSCs share an ability to acquire heavy glucose utilization within TME, exhibiting the flexibility of metabolism [32]. Correspondingly, as dynamic metabolic flux analysis reveals that the maturation of MDSCs is pertaining to the high glycolytic flux and tricarboxylic acid cycle (TCA) activity, while the pentose phosphate pathway (PPP) and OXPHOS activity are remaining at a low level to ensure anabolic precursors synthesis and NADPH production [25]. NADPH provides reducing power for a wide variety of biosynthetic reactions and also helps retain the cellular redox capacity. A prominently increased consumption of glucose by MDSCs generates energy-rich nucleotides and carbon intermediates to support their immunosuppressive mechanisms, with 95% ATP production obtained by glycolysis [25]. Compared with splenic M-MDSCs, tumor-invasive M-MDSCs exhibit stronger suppressive activity coupled with more glucose uptake and lower oxygen consumption rate (OCR), similar to the Warburg effect of cancer cells [33]. In addition, the up-regulation of glycolytic catabolism leads to the production of phosphoenolpyruvate (PEP), as a vital antioxidant agent able to prevent the surplus ROS overproduced by MDSCs, which protects MDSCs from apoptosis [29]. Glycolysis restriction impairs the suppressive potencies of intertumoral MDSCs. For instance, 2-deoxyglucose (2-DG), an inhibitor of the glycolytic pathway, significantly inhibits M-MDSC differentiation from their precursors in TME [34]. Besides, recent research reveals that GLUT3 knockdown by siRNA significantly triggers apoptosis and reduces glucose uptake in PMN-MDSCs, suggesting that the survival of PMN-MDSCs is dependent on both the glucose uptake and GLUT3 overexpression [35].
Mechanistically, mTOR and Nrf2 function as the master regulators of glycolic reprograming in MDSCs. mTOR phosphorylation level has been found to be elevated in tumor M-MDSCs, and mTOR blockade by rapamycin decreases the glycolysis, thereby impairing the suppressive activity of these cells and consequently impeding the tumor growth [33]. Besides, Tuo et al. found that methionine enkephalin (MENK), an endogenous opioid peptide, reduced glycolysis and decreased ROS production of MDSCs through PI3K/AKT/mTOR pathway, thus preventing the development of colon carcinoma [36]. Hence, mTOR signaling is central to enhancing immunosuppressive functions of MDSCs via glycolysis regulation. Nrf2 activated by ROS drives the expression of antioxidant genes and protects cells from oxidative damage. Although the dysregulated ROS levels are toxic to the majority of cells, MDSCs can survive despite their elevated release of ROS, which is regulated by Nrf2 [37]. Nrf2 by increasing H2O2 production of MDSCs potentiates their suppressive activity, as well as increasing the quantity of tumor-infiltrating MDSCs through reducing their oxidative stress and rate of apoptosis. In mouse model with a constitutively active form of Nrf2, the activated Nrf2 was demonstrated to regulate activation and balance between glycolysis and mitochondrial metabolism of MDSCs, thus ultimately increasing the expansion of highly suppressive MDSCs [38]. Concurrently, a high mTOR phosphorylation in Nrf2-induced MDSCs was detected [38]. Rapamycin inhibits the Nrf2-induced MDSC generation, as a direct mTOR activation by Nrf2 has been shown in MDSCs [38]. As discussed above, these investigations clearly demonstrate that mTOR and Nrf2 regulate the diverse suppressive potencies of MDSCs in a glycolytic fashion, and maintain a steady-state level of circulating MDSCs in tumor-bearing individuals.
Hence, restraining deregulated glucose uptake of MDSCs in TME may provide a potential strategy to overcome cancer. Of note, strengthening immune responses via balancing the glycolysis and mitochondrial OXPHOS of MDSCs should be a more reliable strategy than exclusive glycolysis inhibition [39].
Immunosuppressive role of elevated lactate
High transcriptional activity of hypoxia-inducible factor-1α (HIF-1α), Myc, and p53 in cancer cells up-regulates the expression of glycolytic genes to enhance glucose uptake and then provides ATP in a faster way [40]. Along glycolytic pathway, pyruvate dehydrogenase (PDH) catalyzes pyruvate into Acetyl-CoA, the substrates of TCA cycle. Nevertheless, in most cancers, pyruvate dehydrogenase kinase (PDK) is activated by HIF-1α and Myc thus causing the selective repression of PDH [40]. As a result, pyruvate fails to enter the TCA cycle and finally gets converted into large amounts of lactate. Subsequently, excessive lactate (up to 40 mM) produced by tumor cells is excreted from cytoplasm into extracellular environment by MCT, which leads to the TME with associated low pH (6.0~6.5) [41, 42]. But glycolysis is not the sole pathway responsible for producing lactate, as it has been found in MDSC cell line (MSC-1) that L-glutamine serves as another potential source of carbon which leads to an increased lactate production through glutaminolysis-TCA cycle-NADP+-dependent malic enzyme axis [43]. Metabolically, metabotropic glutamate receptor (mGluR) 2/3 transports glutamine from TME into MDSCs [44]. Thereafter, glutamine is metabolized by glutaminase (GLS) to glutamate, which replenishes the TCA cycle after conversion by glutamate dehydrogenase (GLUD1) to α-ketoglutarate (α-KG). And α-KG is subsequently metabolized to malate along TCA cycle, thereby supporting lactate production, a process called anaplerosis [1].
In terms of immunosuppressive mechanism, circulating lactate in TME has been reported that lactate acts as a “signaling molecule”, binding to the G-protein coupled receptor 81 (GPR81) via mTOR/HIF-1α/STAT3 pathway to activate MDSCs [45]. And the endocellular lactate generated in MDSCs through glycolysis elevates their expression of PD-L1 which interacts with cytotoxic T lymphocyte-antigen associated protein 4 (CTLA4) and ultimately impairs T cell-mediated immunity [46]. Moreover, lactate is an essential mediator that significantly facilitates the recruitment of MDSCs within tumor regions. Previous study has found that exogenous lactate increases the frequency of MDSCs generated from mouse bone marrow cells with GM-CSF and IL-6 in vitro [46]. Moreover, the depletion of glucose levels using a ketogenic diet to lower lactate production by glycolytic tumor cells results in smaller tumors, decreases MDSC frequency and improves effectiveness of anti-tumor immune responses [46]. Experimental evidence has also demonstrated that lactate and hypoxia act as synergistic cues to stimulate VEGF and Arg-1 expression in tumor-associated macrophages (TAMs), which induce revascularization of the nutrient-deprived tumor regions and promote tumor progression, as well as the recruitment of MDSCs [47]. Furthermore, in two mouse models of triple-negative breast cancer (TNBC), the knockout of lactate dehydrogenase A (LDHA) which catalyzes pyruvate into lactate was observed to attenuate tumor G-CSF and GM-CSF expression and decrease the number of MDSCs in spleen and tumor [48]. Consequently, lactate in TME contributes to MDSC-induced immune suppression. Inhibiting superfluous lactate production may be an effective way to normalize TME and then impair the immunosuppressive activity of MDSCs to enhance tumor killing.
Lactate as the end product of glycolysis is often considered a waste metabolite but also a fuel for oxidative cells during conditions of nutrient depletion. It has been well established that the poorly vascularized cancerous tissues contain normoxic and hypoxic zones, and it has become increasingly clear that cancer cell metabolism is heterogeneous. MCTs exert essential roles in lactate shuttling between oxygenated and hypoxic regions of tumors. Tumor cells adapt to acidotic TME with the cooperativity that exists between lactate transporters (MCT1 and MCT4) in regulating the lactate levels [49]. Under the anoxic condition, MCT4 up-regulated as a direct target of HIF-1α also increases the expression of various invasion-related genes in cancer cells, such as VEGF, CD147 and MMP2/9, which recruit MDSCs and also deteriorate TME [50]. Hypoxic cancer cells generally obtain energy via glycolysis and subsequently release lactate of toxic levels through MCT4. As lactate excreted by glycolytic cancer cells enters into the aerobic cancer cells through MCT1, these cells are able to preferentially utilize lactate and derive as much as 7.5-fold higher ATP relative to aerobic glycolysis through Krebs cycle and OXPHOS, thereby saving glucose for hypoxic tumor cells, a process known as “metabolic symbiosis” [51, 52].
However, it is still to be ascertained whether superfluous lactate as active participants in intracellular energy metabolism could also be transported into MDSCs by MCT1 and/ or released into extracellular environment by MCT4. Moreover, there are still remaining gaps in knowledge concerning whether MDSCs also use lactate to fuel mitochondrial respiration. Myeloid cells located in tumor margins are considered to be less hypoxic than the center of tumor mass [45]. It is likely that the balance between lactate-generating glycolysis and OXPHOS is dependent on the degree of hypoxia in TME [53]. However, whether the phenotypes, pro-tumor effects and metabolic ways of MDSCs in tumor normoxic and hypoxic regions are heterogeneous and how abundant lactate there affects this process still remain largely elusive.
Hypoxia as a major regulator
A consequence of a lack of vascular oxygen delivery in growing tumors leads to the emergence of hypoxic areas. Oxygen deficit (oxygen tensions <10 mmHg) discovered in most human cancers is a primary driver profiting immunosuppressive TME and contributes to the activation of generous oncogenic transcription factors in tumor cells, which encourage ultimately malignant progression, escape from immunosurveillance, angiogenesis, invasiveness and metastasis [54]. Hypoxia mainly relies on the transcription factor HIF-1α which functions as a master regulator to balance oxygen supply and requirement in mammalian cells [55].
MDSCs accumulate in hypoxic area of tumors where they highly express HIF-1α. Meanwhile, frequent HIF-1α accumulation in MDSCs causes the up-regulation of the PD-L1, because of the direct binding of HIF-1α to a transcriptionally active hypoxia-response element (HRE) in the PD-L1 proximal promoter [56, 57]. Moreover, hypoxia via HIF-1α redirects MDSCs differentiation towards TAMs, thereby dramatically raising their NO production and Arg-1 activity [58]. As such, hypoxia and HIF-1α are found to be primarily responsible for the effects observed in TME on MDSC recruitment, differentiation and functions.
The metabolic shift of tumor MDSCs from OXPHOS to glycolysis is mainly dominated by HIF-1α [59]. Mechanistically, in hypoxia state, PI3K/mTOR signaling stimulates HIF-1α-medicated transcriptional program of glycolysis related genes, resulting in the increased expression of glucose transporters and glycolytic enzymes like LDHA, as well as the decrease of mitochondrial oxygen consumption [45]. PI3K and mTOR inhibitors markedly reduce the expression of HIF-1α in MDSCs [45]. In contrast to HIF-1α, AMPK drives the transformation from glycolysis to OXPHOS through inhibiting PI3K/Akt/mTOR signaling pathway during glucose metabolism [60]. Except for AMPK, SIRT1 also acts as a key metabolic sensor to interfere glycolysis and has shown negative regulatory effects on mTOR but positive regulatory effects on HIF-1α [61, 62]. Besides, SIRT1 is directly regulated by AMPK. MDSCs in TME could be divided into M1-MDSCs associated with tumor cell attack or M2-MDSCs associated with tumor cell protection [63]. SIRT1 targets the mTOR/HIF-1α glycolytic pathway to reprogram the glycolytic activity of M2-MDSCs and instruct their differentiation, whereas limiting the glycolytic activity of M1-MDSCs. The underlying mechanism of these reported subtle metabolic differences between M1 and M2-MDSCs needs to be clarified. As a whole, these studies show that hypoxia coordinating with HIF-1α dominates the metabolic reprogramming pathways of MDSCs, thereby manipulating their fate in TME. Re-educating TME by targeting hypoxia and impairing HIF-1α signaling probably lead to a more effective and durable anti-tumor response in patients with cancer.
Metabolism of extracellular ATP
Adenosine is an ATP-derived nucleoside. Intracellular adenosine is mostly used for energy metabolism and methionine cycle, whereas emerging evidences have identified extracellular adenosine as an important pro-tumor immunomodulatory factor in TME [64, 65]. The conditions associated with immunological stress, including inflammation, trauma, hypoxia or ischemia, lead to necrotic tumor cells death with concomitant ATP release [66]. At same time, the damaged or stressed cells would decrease the ratio of ATP/adenosine and mediate an increase in extracellular hydrolysis of ATP into adenosine, thereby protecting themselves from excessive damage [67]. In cancer lesions, accumulative extracellular adenosine in TME (up to 100 Μm) induces strong immune suppression and supports tumor to evade immune destruction via A2 adenosine receptors (A2AR and A2BR), which are the most abundant adenosine receptors in human and mice [68]. The adenosine enhances the immunosuppressive activity of intra-tumoral MDSCs via A2BR [69]. Furthermore, adenosine signaling may ameliorate energetic status through metabolizing the stored substances. Previous report has indicated that the treatment with A2AR agonists substantially increased energy expenditure and improved glucose tolerance in mice fed a high-fat diet [70]. Similarly, tumor or immune cells possibly also benefit from adenosine-mediated energy metabolism as another aggravating immunosuppression mechanism, which is yet to be investigated in detail.
Surface ectonucleotidases, CD39 (ENTPDase1) and CD73 (ecto-5ʹ-nucleotidase), sustain a delicate balance between the proinflammatory ATP and anti-inflammatory adenosine in TME [71, 72]. Extracellular ATP and ADP are hydrolyzed to 5′-AMP by CD39, which is then processed to adenosine by CD73 [73]. Notably, a critical role of adenosine-rich TME is emerging in the modulation of MDSC expansion and differentiation. Study in Lewis lung carcinoma mouse model reports that the generation of adenosine promotes the preferential expansion and facilitates immunosuppressive activity of the PMN-MDSCs expressing CD73 at high levels in vitro, coupled with rendering myeloid progenitors (MPs) expressing A2BR to differentiate into tolerogenic phenotypes [74]. Activation of A2BR by its selective agonist Bay60-6583 in melanoma mouse model shows increased STAT3 phosphorylation and MDSCs accumulation, subsequently enhancing tumor VEGF expression and vessel density [67]. Furthermore, TGF-β signaling directly induces the generation of CD39/CD73 myeloid cells in tumors [75]. Mechanistically, Li et al. further confirmed that TGF-β derived from TME triggered the phosphorylation of mTOR and then activated HIF-1α, thereby inducing CD39/CD73 expression on MDSCs [76]. Moreover, TGF-β also facilitates the maturation of MDSCs into tumorigenic terminally differentiated myeloid mononuclear cells (TDMMCs) characterized with the higher levels of CD39/CD73 expression and VEGF secretion, which forms a vicious circle [75]. Aside from regulating the accumulation of MDSCs, extracellular CD39/CD73-adenosine pathway in TME indiscriminately dampens anti-tumor immune responses by hampering the cytotoxicity of T cells through A2AR and activating immune suppressive cells such Treg cells, and consequently facilitating tumor growth [68, 77]. Furthermore, Ryzhov et al. further demonstrated that the ability of PMN-MDSCs to suppress CD3/CD28-induced T cell proliferation was significantly enhanced in the presence of the CD73 substrate 5ʹ-AMP [74]. Hence, CD39/CD73 by catalyzing adenosine production confers immunosuppressive functions to MDSCs.
Taken together, the aforementioned findings provide ample evidences for that MDSCs potentiate their suppressive activity via extracellular ATP metabolism. Extracellular adenosine may represent a potentially therapeutic window for cancer patients, especially those receiving immune therapy. But the intracellular molecular mechanisms and associated signaling events that drive this process are far from clear and require further investigations.
Increased demand for fatty acids
Systemic dysregulation of fatty acid (FAs) metabolism supports cancer-related immune dysfunctions, which is among one of the most prominent metabolic alternations in cancer [27, 78]. The extracellular release of free FAs in TME mostly from stromal adipocytes and neoplastic fat tissue is increased, which causes cancer-associated MDSCs to alter their main energetic source from glucose towards FAs, since mitochondrial FAs oxidation produces more than twice as much ATP per mole as oxidation of glucose and generates high levels of ATP and acetyl-CoA which participates in cholesterol biosynthesis [26, 79]. Such a property may contribute to the selective survival advantage of MDSCs over other immune cells, since they have to compete with the cancer cells for rare glucose in nutrient-deficient TME. Therefore, in tumor-bearing mice on a high fat diet, obesity increases the frequency of MDSCs and enhances their capability thus limiting the activation of tumor-reactive CD8+ T cells [80]. Correspondingly, MDSCs up-regulate the expression of lipid transport receptors, including fatty acid translocase (CD36), macrophage scavenger receptor 1 (Msr1) and fatty acid transport protein (FATP), and take up substantial amounts of exogenous lipids from TME, concomitant with an increased mitochondrial mass and up-regulation of key FAO enzymes including carnitine palmitoyltransferase (CPT), peroxisome proliferator-activated receptor gamma coactivator 1β (PGC1β), acyl CoA dehydrogenase (ACADM), and 3-hydroxyacyl-CoA dehydrogenase (HADHA) [81]. Study in several tumor mice models shows that tumor-derived growth cytokines (G-CSF and GM-CSF) and the subsequent STAT3/5 signaling induce MDSCs to up-regulate the expression of lipid transport receptors, coupled with the resulting increase in the uptake of lipids with high concentrations in TME [82]. Moreover, the overexpression of FATP2 in PMN-MDSCs is controlled by GM-CSF through the activation of the STAT5 transcription [83]. FATP2, exclusively up-regulated by MDSCs both in human and mouse, mediates the suppressive activity involved in the uptake of arachidonic acid and the synthesis of PGE-2 by cyclooxygenase-2 (COX-2) [84]. And the genetic depletion of FAs translocase FATP2 and CD36 represses the activation of oxidative metabolism and immunosuppressive capacity of tumor-infiltrating MDSCs, thereby resulting in a T cell-dependent delay in tumor growth [82, 83].
In addition, the supplementation of exogenous unsaturated free FAs, in particular sodium oleate, induces suppressive phenotype in the myeloid suppressor cell line MSC-2 and diminishes the anti-tumor T cell response, paralleled with increased intracellular lipid droplets formation which positively correlates with the NO production [85]. Yan et al. further contended that polyunsaturated fatty acids (PUFAs) mediated MDSC immunosuppressive effects through JAK/STAT3 signaling which regulated the expression of genes related to MDSC expansion, such as S100A8/9, and increased ROS production [86]. Recent study also suggests that MPO-driven lipid peroxidation in PMN-MDSCs might serve as a possible non-cell autonomous mechanism of restraining the antigen cross-presentation by DCs [87]. Besides, Hossain et al. suggested that FAO inhibition repressed the immunosuppressive potency and overall metabolic activity of MDSCs, conspicuously delayed tumor progress in a T cell-dependent manner and significantly decreased the production of G-CSF, GM-CSF and IL-6 thus weakening the immunosuppressive circumstance [81].
Interestingly, MDSCs in different phenotypes may be inclined to possess distinct metabolic patterns. Indeed, M-MDSCs prefer FAO rather than glycolysis to fuel ATP generation, whereas PMN-MDSCs prefer glycolysis and OXPHOS [88]. However, compared PMN-MDSCs, M-MDSCs exhibit a stronger suppressive capacity mainly by producing NO, TGF-β and IL-10, which indirectly reflects the regulation of FAO on the MDSC functions [89]. In summary, abundant FAs in TME are not merely metabolic fuels, but also critical signaling molecules manipulating the activation of MDSCs, which partly interprets the failure of cancer therapies and poor clinical outcomes caused by obesity and high-calorie diets.
Intracellular cholesterol overload
It is well accepted that malignant tumor cells require excess cholesterol and its intermediates to maintain a high level of proliferation [90]. Similar to tumor cells, MDSCs alter their cholesterol profile in TME. Condamine et al. demonstrated that lectin-type oxidized LDL receptor-1 (LOX-1) functioned as a novel proprietary biomarker of human PMN-MDSC with robust immunosuppressive activity and up-regulation of endoplasmic reticulum (ER) stress [91]. Moreover, TME produces ROS, inflammatory cytokines and oxLDL which elevate the surface LOX-1 expression in MDSCs. Lysosomal acid lipase (LAL) cleaves cholesteryl esters and triglycerides in lysosomes to generate free fatty acids and cholesterol [91, 92]. PPARγ serves as the nuclear receptor triggered by FAs derived from LAL enzymatic action. Thereafter, endocellular cholesterol is primarily metabolized by the enzyme CYP7A1 in a traditional bile acid synthesis pathway, concurrently developing into 27-hydoxycholesterol (27-OHC) produced by the enzyme CYP27A1, the primary metabolite of cholesterol. 27-OHC is the most abundant oxysterol in the circulation and acts as the important ligand activating at least liver X receptor (LXR) and endogenous selective estrogen receptor in vivo [93]. Abnormal amounts of cellular cholesterol and its derivatives (i.e., oxysterols) directly activate LXR, initiating the process of reverse excess cholesterol transport through ATP-binding cassette transporters A1 (ABCA1) and G1 (ABCG1), both transporters of intracellular lipids [94]. They promote the efflux of cholesterol onto high-density lipoprotein (HDL) particles or onto the lipid-poor form of apolipoprotein A1 (apoA1), which finally transport cholesterol from peripheral tissues back to the liver for reuse or excrete them into bile in form of bile acids [95].
Preclinical studies tend to more consistently support the tumor facilitation triggered by cholesterol imbalance in TME during tumor onset [96]. Noteworthy, excessive cholesterol deposit in tumor infiltrating MDSCs could be likely mediator of this effect. In melanoma tumor mouse model with myeloid-specific loss of ABCA1, Daryoush et al. showed that the blockade of cholesterol efflux pathways dramatically lessened tumor volume which correlated closely with decreased frequency of MDSCs, suggesting the excessive intracellular cholesterol removal was critically involved in the survival of tumor MDSCs [97]. Besides, recent study finds that 27-OHC promotes the differentiation of M-MDSCs and ovarian tumor-bearing mice treated with 27-OHC display increased M-MDSCs within their tumors [98]. Tumors fail to thrive in mice lacking CYP27A1, which could be likely attributed to the improper differentiation and/or functions of M-MDSCs [98]. Intriguingly, however, CYP27A1 expression is considered as a poor prognostic in terms of overall survival in patients with incipient ovarian cancer, while its expression serves as a good prognostic for patients at late-stage disease [98]. On the basis of this, one might speculate that 27-OHC promotes the increased abundance of M-MDSCs only during the early stage of ovarian cancer, whereas the anti-proliferative effects of 27-OHC could be more obvious after a suitable TME has already been created. But its idiographic action of mechanism needs to be addressed. Because of this, the impaired cholesterol homeostasis and the expansive frequency of MDSCs may be responsible for the specific forepart diagnostic indexes for ovarian cancer.
Although there are few studies about the possible influence on the inhibitory activity of MDSCs based on their cholesterol metabolism. LAL deficiency causes the expansion of MDSCs and loss of T cells [92]. MDSCs from LAL−/− mice directly stimulate B16 melanoma cells not only for in vitro proliferation, but also promote both in vivo growth and metastatic dissemination, the remarkable tumor-promoting capacity is found to be mediated, at least in part, through hyperactivation of the mTOR pathway [99]. Mechanistically, LAL−/− MDSCs overuse glycolysis to compensate the energy deficit, which leads to an increased ATP production, ROS over-production and oxidative stress, as well as the impairment of mitochondrial membrane potential and functions triggered by mTOR overactivation [100]. However, 9-hydroxyoctadecadienoic acid (9-HODE), a LAL downstream metabolic derivative, acts as hormonal ligands for PPARγ and then effectively reverses the aforementioned mTOR overactivation and ROS overproduction, thus leading to the dysfunction of tumor MDSCs [101].
These findings underline the linkage between cholesterol metabolic homeostasis and MDSC fate. Collectively, rectifying cholesterol metabolic dysregulation of MDSCs in TME may be a novel immunotherapeutic paradigm to control the development of cancer.
Metabolic intervention in MDSCs to improve anti-tumor therapy
Nowadays, cancer immunotherapy has emerged as a revolutionary therapeutic approach that may be able to potentially overcome the suppressive TME. However, to date, the therapeutic effectiveness has been limited to a minority of patients and cancer types, and the response rates remain low due to undefined suppression mechanisms [102]. Besides, conventional antineoplastic agents also exhibit limitations. Since the immunosuppressive cross-talk between MDSCs and TME may cause multifaceted downstream effects to blunt the effectiveness of anti-tumor therapy, immunotherapy based on selective targeting MDSCs is an attractive option. Investigational treatment strategies targeting MDSCs at present have mainly been attempted to dampen MDSC development, expansion and functions, differentiate them into more mature cells or destroy MDSCs [103]. TME shares features of complex and erratic ecosystems, therefore, the requirement for a tight nutrient balance might be a vulnerability of MDSCs upon which to intervene for boosting tumor-specific immunity. Aforementioned observations have led to a major interest in characterizing the immune-microenvironment in cancer bearers and MDSC metabolism-targeting drugs appear to be promising tools for providing a novel anti-cancer immunotherapy.
Hypoglycemic drugs
Metformin is the most broadly prescribed type 2 diabetes drug and currently under investigation for its antineoplastic activity and underlying mechanisms, which have received great attention [104]. Metformin actually activates AMPK and inhibits mTOR [105]. Several studies have reported that metformin exerts its anti-tumor efficacy through targeting MDSCs in tumor bearers. A study in ovarian cancer patients shows that metformin induces the reduced presence of circulating CD39+CD73+MDSCs and increased presence of circulating CD8+ T cells with enhanced anti-tumor activity, thereby prolonging overall survival in the diabetic patients with ovarian cancer [106]. Mechanistically, metformin triggers activation of AMPKα and subsequently suppresses HIF-1α, thereby dampening immunosuppression of CD39/CD73-dependent MDSCs in cancer [106]. Another recent study also finds that metformin is able to decrease the accumulation and impair the suppressive capacity of PMN-MDSCs, delay tumor progression and elicit Th1 and CTL responses in colon cancer mouse model [107]. Since metformin also reduces STAT3 phosphorylation levels and then down-modulates the production of downstream molecules such as ROS and Arg-1 in PMN-MDSCs [107]. Furthermore, experimental evidence in esophageal squamous cell carcinoma (ESCC) indicates that the treatment with metformin reduces MDSC migration and accumulation in cancer patients [108]. The underlying mechanisms prove that metformin enhances AMPK phosphorylation followed by the inhibition of NF-κB, induces dachshund homologue 1 (DACH1) expression and subsequently inhibits CXCL1 secretion both in ESCC cells and tumor xenografts thus abating chemotaxis in tumor MDSCs [108]. However, limited information is available related to the metabolic intervention of metformin to achieve its antineoplastic activity. Uehara et al. demonstrated that metformin treatment in osteosarcoma mouse model decreased basal respiration and OCR/extracellular acidification rate (ECAR) ratio of tumor MDSCs, which in turn reduced ROS production and proton leakage in these cells [109]. Metabolically, metformin redirected the metabolism of MDSCs via lowering their OXPHOS while relatively elevating glycolysis, along with down-regulating FAO, thereby pushing the TME to a state that inhibited the tumor growth [109].
Aside from metformin, other type 2 diabetes medicines such as phenformin and rosiglitazone have shown analogous anti-cancer activity. Phenformin selectively diminishes PMN-MDSC accumulation in the spleens and tumors of melanoma mouse model and decreases expressed levels of Arg-1 and S100A8/9 in MDSCs [110]. Co-treatment with phenformin and anti-programmed cell death protein 1 (PD-1) antibody creates synergistic effects in inducing CD8+ T cell infiltration in TME and reducing tumor growth [110]. As mentioned above, PPARγ activation in MDSCs dampens their immunosuppressive potencies. Previous study in pancreatic cancer mouse model showed that PPARγ agonist rosiglitazone in combination with gemcitabine limited MDSC accumulation and enhanced T cell-modulated immunity to increase tumor destruction [111]. These observations give rise to the possibility of immunotherapy involving hypoglycemic agents as promising approaches to selectively target MDSCs. Further studies on the metabolic regulation of hypoglycemic drugs on MDSCs will shed light to increase treatment options for certain cancers that are refractory to conventional therapies.
Drugs targeting lactate
The development of drugs targeting critical proteins (LDHA, PDK, and MCT, among others) during lactate metabolism has shown broad prospects in anti-tumor preclinical studies. Dichloroacetate (DCA), an inhibitor of PDK and promoter of PDH, is used clinically to cure patients with lactic academia [112]. DCA acts on both tumor and immune cells to suppress glycolysis and then decrease the lactate production to improve the immunosuppressive TME modulated by lactate [112]. Oncolytic viro-immunotherapy holds significant promise for cancer treatment, as it robustly triggers the host immune activation. Recent study suggests that DCA targeting aerobic glycolysis significantly reduces lactate release, MDSC infiltration, STAT3 activation and IDO1 up-regulation in oncolytic Newcastle disease virus (NDV) -treated HCC bearing mice to alleviate immune negative feedbacks [113]. Moreover, a series of blockers with a higher affinity for MCTs recently have been used as promising therapeutic avenue in oncology [114–116]. Meanwhile, given the TME with low pH caused by circulating lactate may impair the uptake of chemotherapeutic drugs, curtailing or neutralizing the acidification of the TME should be explored as a potential strategy to increase the sensitivity of tumor cells to cytotoxic agents [117]. For instance, proton pump inhibitors (PPIs) or bicarbonate therapy in combination with chemotherapy and/or immunotherapy has been reported to significantly improve drug sensitivity and anti-tumor responses in different pre-clinical models, encompassing clinical cures in some subjects [118, 119]. As described above, lactate from TME imposes essential advancement in cancer promotion. But few studies investigate that lactate inhibitors or pH regulators exhibit their antineoplastic activity through targeting tumor-conditioned MDSCs, which may establish unexpected therapeutic windows and also open new options for “re-purposing” existing metabolic drugs.
Drugs targeting hypoxia
Tumor hypoxia and its associated immune negative feedbacks (angiogenesis, induction of PD-L1 expression both on MDSCs and cancer cells, extracellular lactate and adenosine accumulation, among others) are serious impediments that improve the overall survival of patients with cancer and reduce the chemoresistance or radiotherapy sensitivity. Targeted modulation of tumor hypoxia might convert patient resistance to immunotherapy into those that receive clinical benefits. Beyond its antihyperglycemic effect, metformin is also capable of blocking the complex 1 in mitochondrial respiratory chain, a key complex in electron transfer during FAO [120]. It works like an O2 economizer, alleviating intratumoral hypoxia through reducing oxygen consumption of tumor cells in vitro and in vivo. Metformin plus PD-1 blockade therapy result in improved T-cell functions and clearance of highly aggressive tumor [120]. Recently, Zuo et al. exhibited a novel strategy for selectively clearing tumor MDSCs. They showed novel mesoporous silica nanoparticles (MSNs) with CeO2 as the gatekeeper that selectively delivered the photosensitizer IR780 and metformin, and then controlled their release once arriving at the tumor sites [121]. After that the engagement of metformin significantly inhibited mitochondrial respiration and CeO2 improved the generation of O2 [121]. Consequently, MSNs consequently reversed MDSC-mediated immunosuppression, attenuated MDSC recruitment and reduced the expression of PD-L1 on MDSCs through remodeling hypoxic TME [121]. Besides, in contrast to the normal counterparts, respiratory hyperoxia therapy in TNBC mouse model leads to reoxygenation of lung TME and then decrease the presence of MDSCs and the expression of PD-L1 both in the primary tumor and metastatic lung, suggesting that hypoxia tumor tissue could be impaired by this treatment modality [122].
Decursin is an active compound extracted from the roots of Angelica gigas and has been shown to have potent antiangiogenic activity [123]. Ge et al. revealed its underlying mechanism that Decursin inhibited HIF-1 activation and promoted its degradation under hypoxia condition, consequently reducing the tumor hypoxic area and restraining HIF-1α and PD-L1 expression, paralleled with a significant reduction in Arg-1 production by MDSCs [123]. In addition, Thymosin alpha-1 (TA) has been reported to inhibit tumor growth as an immunomodulator. Study in non-small cell lung carcinoma (NSCLC) shows that TA down-regulates the expression of HIF-1α further to suppress the production of VEGF in tumor cells thus decreasing M-MDSCs accumulation and migration in TME, and promotes the apoptosis of M-MDSCs by reducing the B-cell lymphoma-2 (Bcl-2)/ BCL-2 associated X (BAX) ratio [124]. Based on the above facts, it is conceivable that the high frequency of MDSCs and the elevated expression of PD-L1 could be regarded as the specific biomarkers of hypoxia in cancer. Although antiangiogenic therapy has been thought to hold extensive potentiality for eradicating tumors, the antiangiogenic agents sunitinib and bevacizumab increase the population of human breast cancer stem cells by inducing intratumor hypoxia, which in turn leads to the delayed cancer therapy progress primarily mediated by HIF-1α [125]. Hence, HIF-1 inhibitors may impose synergistic effects with antiangiogenic agents to reverse the above highlighted negative consequences. Coincident with this perspective, study in a murine model of breast cancer indicates that the combination therapy of sunitinib with HIF-1 inhibitor acriflavine evidently inhibits tumoral angiogenesis and increases intratumor necrosis and apoptosis, followed by a significant reduction of MDSC accumulation in the spleen, indicating the reversion of systemic immunosuppression [126]. HIF-1 inhibitors by reducing tumor hypoxia and targeting MDSCs may pave the way to foster entirely novel immunotherapy opportunities.
Drugs targeting extracellular adenosine
Inhibition of ATP-degrading enzymes ubiquitously expressed on MDSCs or blocking surface adenosine receptors exhibit anti-tumoral efficacies in different preclinical cancer models. Multiple studies have shown that drugs targeting the cell-surface enzyme CD73 and CD39 reduce the growth and metastasis of primary tumors through degrading peritumoral adenosine [127–129]. Recently, a novel series of CD73 antibody developed by Jin et al., Ab001/Ab002 and humanized version Hu001/Hu002, exhibited potent CD73 inhibition properties and efficiently protected effector T lymphocyte functions from adenosine-imposed toxicity, followed by the conspicuous reduction of MDSCs in patients with cancer [130]. However, there still remains lack of studies on metabolic drugs interfering with the CD73/CD39-adenosine pathway in tumor MDSCs, a characteristic functional hallmark of these cells. Owing to the immunosuppressive effects of ectocytic adenosine catalyzed by CD73 and CD39, targeting the downstream A2AR and A2BR also has high therapeutic potential. PSB1115, a selective A2BR antagonist, reduces tumor angiogenesis and MDSCs accumulation in melanoma mice, consequently leading to a significant delay in the melanoma growth and enhanced effectiveness of anti-VEGF treatment [67]. Not alone, PSB1115 targeting the accumulation and immunosuppressive activity of MDSCs in TME effectively increases the frequency of CD8 + T cells and natural killer T (NKT) cells, resulting in the reinforced anti-tumor immune responses and melanoma growth delay [131]. These observations indicate that extracellular adenosine inhibitors could further be investigated as adjuvants in the treatment of cancer.
Drugs targeting FAs and therapeutic FAs
Interrupting the mechanisms of FAs uptake from MDSCs might be an effective immune metabolic checkpoint. The selective pharmacological inhibition of FATP2 by small molecule inhibitor lipofermata abrogates the suppressive activity of FATP2-overexpressed PMN-MDSCs and substantially inhibits tumor progression in four tested tumor models [83]. In combination with immune checkpoint inhibitors like anti-CTLA4 antibody, lipofermata blocks tumor progression in mice due to its highly selective targeting of MDSCs in TME [83]. In addition, recent study on lipofermata shows that it decreases FAs accumulation, reduces ROS release and impairs the functions of MDSCs, consequently decreasing tumor burden [132]. More importantly, the combination therapy between anti-PD-L1 antibody and lipofermata blocks suppressive activity of MDSCs and reinforces T-cell effective ability for the production of IFN-γ and TNF-α [132]. These findings indicate that FAO as a metabolic target spot on tumor MDSCs can improve the efficacy of cancer therapy. Similarly, combination treatment of adoptive cell transfer (ACT) with etomoxir blocking the rate-limiting enzyme CTP1 in the FAO cycle drastically inhibits tumor development in the LLC mouse model [81]. This effect is paralleled with the decreased immunosuppressive Arg-1 and ROS release by MDSCs, as well as tumor-derived factors related to MDSC expansion (i.e., G-CSF, GM-CSF, IL-6, IL-10) [81].
As for the FAs, ω−6 PUFAs intake has been implicated in mechanisms of tumor incidence, metastasis and poor outcomes, while the anti-inflammatory ω−3 PUFAs can counter tumor growth and ω−6 PUFA associated inflammation [133]. Docosahexaenoic acid (DHA) is one of the most bioactive FAs belonging to ω−3 PUFAs family and harbors both anti-cancer and anti-inflammatory properties. Since DHA improves the anti-cancer properties of 5-fluorouracil (5-FU) through repressing 5-FU-induced IL-1β secretion by MDSCs in lymphoma mouse model [134]. Mechanistically, DHA inhibits the 5-FU-induced ROS production and then represses the ROS-induced activation of JNK mediating IL-1β secretion [134]. Another signaling pathway shows that DHA in a β-arrestin-2-dependent way inhibits the activity of NLRP3 inflammasome and phosphorylation of JNK in tumor MDSCs [134]. In addition, anticonvulsant drug valproic acid (VPA) is a short-chain fatty acid, showing additional activity including anti-cancer and immunoregulation by inhibition of histone deacetylases. In the same lymphoma mouse model, Xie et al. indicated that VPA decreased the proportion of PMN-MDSCs and impaired their ability to stimulate tumor progression in a dose-dependent manner in vivo [135]. Mechanistically, VPA dramatically down-regulated the immunosuppression-related genes expression such as Arg-1, PD-L1 and TLR4 of MDSCs and thereby recovered immune responses in TME. Of relevance, the modification of FAs intake may be a preventive or therapeutic approach to regulate MDSC-induced immune repression and remains an attractive, but little studied intervention strategy.
LXR agonists
Emerging evidences show that manipulating cholesterol metabolism reshapes the immunological landscape and reinvigorates anti-tumor immunity [136]. GW3965, a class of selective LXR agonists, exerts conspicuous efficacy of cancer immunotherapy through transcriptional induction of stromal apolipoprotein-E (apoE), impeding melanoma cells invasiveness, endothelial recruitment and tumor progression [137]. Mechanistically, the LXR transcriptional target apoE binds with LRP8 receptors expressed on MDSCs to achieve these antineoplastic effects [137]. Besides, a more potent LXR agonist, RGX-104 representing the first MDSC-targeting therapeutic agent, markedly reduces MDSC abundance and enhances the CTL responses both in murine models and clinical patients with metastatic melanoma, as well as rescuing the efficacy of anti-PD-1 therapy in the poorly immunogenic resistant model [138]. Recent research also finds the improved radiosensitivity of NSCLC mediated by RGX-104, which greatly promotes MDSC apoptosis and thereafter augments anti-tumor immune response in TME [139]. Immunotherapy targeting cholesterol metabolism pathway of tumor MDSCs is a significant but overlooked therapeutic window in neoplastic diseases, which remains to be extensively investigated.
Perspectives and prospects
Metabolic reprogramming of MDSCs is triggered by stimuli originated from TME and the bioenergetic needs, which allows them to utilize adaptive metabolic pathways to sustain their viability in metabolically unfavorable conditions. In this review, we have presented a comprehensive overview about the intricate interplay of TME and MDSCs, and analyzed the essentially signaling events involved in the process. A number of studies on MDSCs metabolism have expanded our understanding of the mechanisms and functional consequences of metabolic alternations during the multistep development of cancer and, accordingly, novel targets for therapeutic approaches aimed at metabolic programming to enhance cancer immunotherapy. However, a further systematic profiling of MDSCs metabolic pathways should focus on several important issues such as: (1) What are the divergent metabolic traits between PMN-MDSCs and M-MDSCs in various diseases. (2) How the variable metabolic pathways of MDSCs temporally and spatially alter during the various stages of tumorigenesis. (3) Since nutrient uptake is strictly modulated by growth factor signaling, how the tumor-derived cytokines and growth factors regulate the metabolic switches of MDSCs [5]. (4) How aforementioned metabolic alternations influence the epigenetic states of MDSCs and then govern their functional activity, among others. Increasing evidences suggest that the metabolic reprograming of tumor and immune cells could be responsible for the failure of anti-tumor immunity [27]. Based on this scenario, we have summarized the medicines targeting metabolic reprograming of MDSCs and provided new train of thoughts for developing efficacious anti-cancer modalities, further highlighting the therapeutic potentiality of metabolic interventions as modulators of anti-cancer immune responses. Predictably, regulating the metabolic pathways of MDSCs in TME may allow for more precise and targeted strategies to treat human malignancies.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82071749).
Author contributions
QL designed and drafted the manuscript; MX designed and edited the manuscript.
Competing interests
The authors declare no competing interests.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00776-4.
References
- 1.Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer. 2020;20:516–31. doi: 10.1038/s41568-020-0273-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001–12.e5. doi: 10.1016/j.cmet.2021.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joshi RS, Kanugula SS, Sudhir S, Pereira MP, Jain S, Aghi MK. The role of cancer-associated fibroblasts in tumor progression. Cancers. 2021;13:1399. doi: 10.3390/cancers13061399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erin N, Grahovac J, Brozovic A, Efferth T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist Updat. 2020;53:100715. doi: 10.1016/j.drup.2020.100715. [DOI] [PubMed] [Google Scholar]
- 7.Dysthe M, Parihar R. Myeloid-derived suppressor cells in the tumor microenvironment. Adv Exp Med Biol. 2020;1224:117–40. doi: 10.1007/978-3-030-35723-8_8. [DOI] [PubMed] [Google Scholar]
- 8.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–44. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 9.Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells. 2020;9:561. doi: 10.3390/cells9030561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5:3–8. doi: 10.1158/2326-6066.CIR-16-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Y, Wu T, Shao S, Shi B, Zhao Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology. 2016;5:e1004983. doi: 10.1080/2162402X.2015.1004983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavalli E, Mazzon E, Mammana S, Basile MS, Lombardo SD, Mangano K, et al. Overexpression of macrophage migration inhibitory factor and its homologue d-dopachrome tautomerase as negative prognostic factor in neuroblastoma. Brain Sci. 2019;9:284. doi: 10.3390/brainsci9100284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mangano K, Mazzon E, Basile MS, Di Marco R, Bramanti P, Mammana S, et al. Pathogenic role for macrophage migration inhibitory factor in glioblastoma and its targeting with specific inhibitors as novel tailored therapeutic approach. Oncotarget. 2018;9:17951–70. doi: 10.18632/oncotarget.24885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavalli E, Ciurleo R, Petralia MC, Fagone P, Bella R, Mangano K, et al. Emerging role of the macrophage migration inhibitory factor family of cytokines in neuroblastoma. Pathogenic effectors and novel therapeutic targets? Molecules. 2020;25:1194. doi: 10.3390/molecules25051194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alban TJ, Bayik D, Otvos B, Rabljenovic A, Leng L, Jia-Shiun L, et al. Glioblastoma myeloid-derived suppressor cell subsets express differential macrophage migration inhibitory factor receptor profiles that can be targeted to reduce immune suppression. Front Immunol. 2020;11:1191. doi: 10.3389/fimmu.2020.01191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Ye YL, Li MX, Ye SB, Huang WR, Cai TT, et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene. 2017;36:2095–104. doi: 10.1038/onc.2016.367. [DOI] [PubMed] [Google Scholar]
- 17.Günther S, Fagone P, Jalce G, Atanasov AG, Guignabert C, Nicoletti F. Role of MIF and D-DT in immune-inflammatory, autoimmune, and chronic respiratory diseases: from pathogenic factors to therapeutic targets. Drug Discov Today. 2019;24:428–39. doi: 10.1016/j.drudis.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Kumar R, de Mooij T, Peterson TE, Kaptzan T, Johnson AJ, Daniels DJ, et al. Modulating glioma-mediated myeloid-derived suppressor cell development with sulforaphane. PLoS One. 2017;12:e0179012. doi: 10.1371/journal.pone.0179012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells-an overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology. 2015;4:e954829. doi: 10.4161/21624011.2014.954829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang CH, Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol. 2016;17:364–8. doi: 10.1038/ni.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamada KJ, Heim CE, Aldrich AL, Gries CM, Staudacher AG, Kielian T. Arginase-1 expression in myeloid cells regulates staphylococcus aureus planktonic but not biofilm infection. Infect Immunity. 2018;86:e00206–18. doi: 10.1128/IAI.00206-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammami I, Chen J, Murschel F, Bronte V, De Crescenzo G, Jolicoeur M. Immunosuppressive activity enhances central carbon metabolism and bioenergetics in myeloid-derived suppressor cells in vitro models. BMC Cell Biol. 2012;13:18. doi: 10.1186/1471-2121-13-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190:3783–97. doi: 10.4049/jimmunol.1201449. [DOI] [PubMed] [Google Scholar]
- 25.Goffaux G, Hammami I, Jolicoeur M. A dynamic metabolic flux analysis of myeloid-derived suppressor cells confirms immunosuppression-related metabolic plasticity. Sci Rep. 2017;7:9850. doi: 10.1038/s41598-017-10464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan D, Adeshakin AO, Xu M, Afolabi LO, Zhang G, Chen YH, et al. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front Immunol. 2019;10:1399. doi: 10.3389/fimmu.2019.01399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bleve A, Durante B, Sica A, Consonni FM. Lipid metabolism and cancer immunotherapy: immunosuppressive myeloid cells at the crossroad. Int J Mol Sci. 2020;21:5845. doi: 10.3390/ijms21165845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong Y, Tu R, Liu H, Qing G. Regulation of cancer cell metabolism: oncogenic MYC in the driver’s seat. Signal Transduct Target Ther. 2020;5:124. doi: 10.1038/s41392-020-00235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jian SL, Chen WW, Su YC, Su YW, Chuang TH, Hsu SC, et al. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. 2017;8:e2779. doi: 10.1038/cddis.2017.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature. 2021;593:282–8. doi: 10.1038/s41586-021-03442-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–52. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 32.Rice CM, Davies LC, Subleski JJ, Maio N, Gonzalez-Cotto M, Andrews C, et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat Commun. 2018;9:5099. doi: 10.1038/s41467-018-07505-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng Y, Yang J, Luo F, Qian J, Liu R, Zhang D, et al. mTOR-mediated glycolysis contributes to the enhanced suppressive function of murine tumor-infiltrating monocytic myeloid-derived suppressor cells. Cancer Immunol Immunother. 2018;67:1355–64. doi: 10.1007/s00262-018-2177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu T, Zhao Y, Wang H, Li Y, Shao L, Wang R, et al. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci Rep. 2016;6:20250. doi: 10.1038/srep20250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu C, Fu Z, Jiang C, Xia C, Zhang Y, Gu X, et al. CD205+ polymorphonuclear myeloid-derived suppressor cells suppress antitumor immunity by overexpressing GLUT3. Cancer Sci. 2021;112:1011–25. doi: 10.1111/cas.14783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuo Y, Zhang Z, Tian C, Hu Q, Xie R, Yang J, et al. Anti-inflammatory and metabolic reprogramming effects of MENK produce antitumor response in CT26 tumor-bearing mice. J Leukoc Biol. 2020;108:215–28. doi: 10.1002/JLB.3MA0120-578R. [DOI] [PubMed] [Google Scholar]
- 37.Beury DW, Carter KA, Nelson C, Sinha P, Hanson E, Nyandjo M, et al. Myeloid-derived suppressor cell survival and function are regulated by the transcription factor Nrf2. J Immunol. 2016;196:3470–8. doi: 10.4049/jimmunol.1501785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohl K, Fragoulis A, Klemm P, Baumeister J, Klock W, Verjans E, et al. Nrf2 is a central regulator of metabolic reprogramming of myeloid-derived suppressor cells in steady state and sepsis. Front Immunol. 2018;9:1552. doi: 10.3389/fimmu.2018.01552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia L, Gao Y, Zhou T, Zhao XL, Hu HY, Chen DW, et al. Enhanced response to PD-L1 silencing by modulation of TME via balancing glucose metabolism and robust co-delivery of siRNA/Resveratrol with dual-responsive polyplexes. Biomaterials. 2021;271:120711. doi: 10.1016/j.biomaterials.2021.120711. [DOI] [PubMed] [Google Scholar]
- 40.Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cell Mol life Sci. 2008;65:3981–99. doi: 10.1007/s00018-008-8224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Multhoff G, Vaupel P. Hypoxia compromises anti-cancer immune responses. Adv Exp Med Biol. 2020;1232:131–43. doi: 10.1007/978-3-030-34461-0_18. [DOI] [PubMed] [Google Scholar]
- 42.Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, et al. Targeting lactate dehydrogenase–a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014;19:795–809. doi: 10.1016/j.cmet.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hammami I, Chen J, Bronte V, DeCrescenzo G, Jolicoeur M. L-glutamine is a key parameter in the immunosuppression phenomenon. Biochemical Biophys Res Commun. 2012;425:724–9. doi: 10.1016/j.bbrc.2012.07.139. [DOI] [PubMed] [Google Scholar]
- 44.Morikawa N, Tachibana M, Ago Y, Goda H, Sakurai F, Mizuguchi H. LY341495, an mGluR2/3 antagonist, regulates the immunosuppressive function of myeloid-derived suppressor cells and inhibits melanoma tumor growth. Biol Pharm Bull. 2018;41:1866–9. doi: 10.1248/bpb.b18-00055. [DOI] [PubMed] [Google Scholar]
- 45.Yang X, Lu Y, Hang J, Zhang J, Zhang T, Huo Y, et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the radioresistance of pancreatic. Cancer Cancer Immunol Res. 2020;8:1440–51. doi: 10.1158/2326-6066.CIR-20-0111. [DOI] [PubMed] [Google Scholar]
- 46.Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191:1486–95. doi: 10.4049/jimmunol.1202702. [DOI] [PubMed] [Google Scholar]
- 47.Carmona-Fontaine C, Deforet M, Akkari L, Thompson CB, Joyce JA, Xavier JB. Metabolic origins of spatial organization in the tumor microenvironment. Proc Natl Acad Sci USA. 2017;114:2934–9. doi: 10.1073/pnas.1700600114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab. 2018;28:87–103.e6. doi: 10.1016/j.cmet.2018.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDonald PC, Chafe SC, Dedhar S. Overcoming hypoxia-mediated tumor progression: combinatorial approaches targeting pH regulation, angiogenesis and immune dysfunction. Front Cell Dev Biol. 2016;4:27. doi: 10.3389/fcell.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun Q, Hu LL, Fu Q. MCT4 promotes cell proliferation and invasion of castration-resistant prostate cancer PC-3 cell line. EXCLI J. 2019;18:187–94. doi: 10.17179/excli2018-1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hayes C, Donohoe CL, Davern M, Donlon NE. The oncogenic and clinical implications of lactate induced immunosuppression in the tumour microenvironment. Cancer Lett. 2021;500:75–86. doi: 10.1016/j.canlet.2020.12.021. [DOI] [PubMed] [Google Scholar]
- 52.Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–42. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–84. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18:157. doi: 10.1186/s12943-019-1089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123:3664–71. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74:665–74. doi: 10.1158/0008-5472.CAN-13-0992. [DOI] [PubMed] [Google Scholar]
- 57.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–90. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–53. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.LaGory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol. 2016;18:356–65. doi: 10.1038/ncb3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hardie DG. AMPK–sensing energy while talking to other signaling pathways. Cell Metab. 2014;20:939–52. doi: 10.1016/j.cmet.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Back JH, Rezvani HR, Zhu Y, Guyonnet-Duperat V, Athar M, Ratner D, et al. Cancer cell survival following DNA damage-mediated premature senescence is regulated by mammalian target of rapamycin (mTOR)-dependent Inhibition of sirtuin 1. J Biol Chem. 2011;286:19100–8. doi: 10.1074/jbc.M111.240598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu Q, Dong L, Li Y, Liu GSIRT1. and HIF1α signaling in metabolism and immune responses. Cancer Lett. 2018;418:20–6. doi: 10.1016/j.canlet.2017.12.035. [DOI] [PubMed] [Google Scholar]
- 63.Liu G, Bi Y, Shen B, Yang H, Zhang Y, Wang X, et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1α-dependent glycolysis. Cancer Res. 2014;74:727–37. doi: 10.1158/0008-5472.CAN-13-2584. [DOI] [PubMed] [Google Scholar]
- 64.Ohta A, Ohta A, Madasu M, Kini R, Subramanian M, Goel N, et al. A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J Immunol. 2009;183:5487–93. doi: 10.4049/jimmunol.0901247. [DOI] [PubMed] [Google Scholar]
- 65.Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA. 2006;103:13132–7. doi: 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Montalbán Del Barrio I, Penski C, Schlahsa L, Stein RG, Diessner J, Wöckel A, et al. Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages - a self-amplifying, CD39- and CD73-dependent mechanism for tumor immune escape. J Immunother Cancer. 2016;4:49. doi: 10.1186/s40425-016-0154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget. 2015;6:27478–89. doi: 10.18632/oncotarget.4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muller-Haegele S, Muller L, Whiteside TL. Immunoregulatory activity of adenosine and its role in human cancer progression. Expert Rev Clin Immunol. 2014;10:897–914. doi: 10.1586/1744666X.2014.915739. [DOI] [PubMed] [Google Scholar]
- 69.Morello S, Miele L. Targeting the adenosine A2b receptor in the tumor microenvironment overcomes local immunosuppression by myeloid-derived suppressor cells. Oncoimmunology. 2014;3:e27989. doi: 10.4161/onci.27989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gnad T, Scheibler S, von Kügelgen I, Scheele C, Kilić A, Glöde A, et al. Adenosine activates brown adipose tissue and recruits beige adipocytes via A2A receptors. Nature. 2014;516:395–9. doi: 10.1038/nature13816. [DOI] [PubMed] [Google Scholar]
- 71.Morello S, Pinto A, Blandizzi C, Antonioli L. Myeloid cells in the tumor microenvironment: role of adenosine. Oncoimmunology. 2016;5:e1108515. doi: 10.1080/2162402X.2015.1108515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stagg J, Thompson LF, Dwyer KM. Ectonucleotidases in cancer and inflammation. J Biomed Biotechnol. 2012;2012:951423. doi: 10.1155/2012/951423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8:517. doi: 10.1038/s41467-017-00530-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol. 2011;187:6120–9. doi: 10.4049/jimmunol.1101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ryzhov SV, Pickup MW, Chytil A, Gorska AE, Zhang Q, Owens P, et al. Role of TGF-β signaling in generation of CD39+CD73+ myeloid cells in tumors. J Immunol. 2014;193:3155–64. doi: 10.4049/jimmunol.1400578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology. 2017;6:e1320011. doi: 10.1080/2162402X.2017.1320011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014;74:7250–9. doi: 10.1158/0008-5472.CAN-13-3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 2020;31:62–76. doi: 10.1016/j.cmet.2019.11.010. [DOI] [PubMed] [Google Scholar]
- 79.Kleinfeld AM, Okada C. Free fatty acid release from human breast cancer tissue inhibits cytotoxic T-lymphocyte-mediated killing. J lipid Res. 2005;46:1983–90. doi: 10.1194/jlr.M500151-JLR200. [DOI] [PubMed] [Google Scholar]
- 80.Clements VK, Long T, Long R, Figley C, Smith DMC, Ostrand-Rosenberg S. Frontline science: high fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J Leukoc Biol. 2018;103:395–407. doi: 10.1002/JLB.4HI0517-210R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3:1236–47. doi: 10.1158/2326-6066.CIR-15-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. 2017;6:e1344804. doi: 10.1080/2162402X.2017.1344804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569:73–8. doi: 10.1038/s41586-019-1118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kuroda H, Mabuchi S, Yokoi E, Komura N, Kozasa K, Matsumoto Y, et al. Prostaglandin E2 produced by myeloid-derived suppressive cells induces cancer stem cells in uterine cervical cancer. Oncotarget. 2018;9:36317–30. doi: 10.18632/oncotarget.26347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu H, Weidinger C, Schmidt F, Keye J, Friedrich M, Yerinde C, et al. Oleate but not stearate induces the regulatory phenotype of myeloid suppressor cells. Sci Rep. 2017;7:7498. doi: 10.1038/s41598-017-07685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yan D, Yang Q, Shi M, Zhong L, Wu C, Meng T, et al. Polyunsaturated fatty acids promote the expansion of myeloid-derived suppressor cells by activating the JAK/STAT3 pathway. Eur J Immunol. 2013;43:2943–55. doi: 10.1002/eji.201343472. [DOI] [PubMed] [Google Scholar]
- 87.Ugolini A, Tyurin VA, Tyurina YY, Tcyganov EN, Donthireddy L, Kagan VE, et al. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI insight. 2020;5:e138581. doi: 10.1172/jci.insight.138581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dai H, Xu H, Wang S, Ma J. Connections between metabolism and epigenetic modification in MDSCs. Int J Mol Sci. 2020;21:7356. doi: 10.3390/ijms21197356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zheng W, Song H, Luo Z, Wu H, Chen L, Wang Y, et al. Acetylcholine ameliorates colitis by promoting IL-10 secretion of monocytic myeloid-derived suppressor cells through the nAChR/ERK pathway. Proc Natl Acad Sci USA. 2021;118:e2017762118. doi: 10.1073/pnas.2017762118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cruz PM, Mo H, McConathy WJ, Sabnis N, Lacko AG. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. Front Pharmacol. 2013;4:119. doi: 10.3389/fphar.2013.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016;1:aaf8943. doi: 10.1126/sciimmunol.aaf8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Qu P, Yan C, Blum JS, Kapur R, Du H. Myeloid-specific expression of human lysosomal acid lipase corrects malformation and malfunction of myeloid-derived suppressor cells in lal−/− mice. J Immunol. 2011;187:3854–66. doi: 10.4049/jimmunol.1003358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Karuna R, Holleboom AG, Motazacker MM, Kuivenhoven JA, Frikke-Schmidt R, Tybjaerg-Hansen A, et al. Plasma levels of 27-hydroxycholesterol in humans and mice with monogenic disturbances of high density lipoprotein metabolism. Atherosclerosis. 2011;214:448–55. doi: 10.1016/j.atherosclerosis.2010.10.042. [DOI] [PubMed] [Google Scholar]
- 94.Tall AR, Yvan-Charvet LCholesterol. inflammation and innate immunity. Nat Rev Immunol. 2015;15:104–16. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang NHDL. ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–75. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 96.Kuzu OF, Noory MA, Robertson GP. The role of cholesterol in cancer. Cancer Res. 2016;76:2063–70. doi: 10.1158/0008-5472.CAN-15-2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zamanian-Daryoush M, Lindner DJ, DiDonato JA, Wagner M, Buffa J, Rayman P, et al. Myeloid-specific genetic ablation of ATP-binding cassette transporter ABCA1 is protective against cancer. Oncotarget. 2017;8:71965–80. doi: 10.18632/oncotarget.18666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.He S, Ma L, Baek AE, Vardanyan A, Vembar V, Chen JJ, et al. Host CYP27A1 expression is essential for ovarian cancer progression. Endocr-Relat Cancer. 2019;26:659–75. doi: 10.1530/ERC-18-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao T, Du H, Ding X, Walls K, Yan C. Activation of mTOR pathway in myeloid-derived suppressor cells stimulates cancer cell proliferation and metastasis in lal−/− mice. Oncogene. 2015;34:1938–48. doi: 10.1038/onc.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ding X, Zhang W, Zhao T, Yan C, Du H. Rab7 GTPase controls lipid metabolic signaling in myeloid-derived suppressor cells. Oncotarget. 2017;8:30123–37. doi: 10.18632/oncotarget.16280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhao T, Du H, Blum JS, Yan C. Critical role of PPARγ in myeloid-derived suppressor cell-stimulated cancer cell proliferation and metastasis. Oncotarget. 2016;7:1529–43. doi: 10.18632/oncotarget.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–23. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Anani W, Shurin MR. Targeting myeloid-derived suppressor cells in cancer. Adv Exp Med Biol. 2017;1036:105–28. doi: 10.1007/978-3-319-67577-0_8. [DOI] [PubMed] [Google Scholar]
- 104.Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr-Relat Cancer. 2010;17:351–60. doi: 10.1677/ERC-09-0252. [DOI] [PubMed] [Google Scholar]
- 105.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li L, Wang L, Li J, Fan Z, Yang L, Zhang Z, et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 2018;78:1779–91. doi: 10.1158/0008-5472.CAN-17-2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu P, Yin K, Tang X, Tian J, Zhang Y, Ma J, et al. Metformin inhibits the function of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Biomed Pharmacother. 2019;120:109458. doi: 10.1016/j.biopha.2019.109458. [DOI] [PubMed] [Google Scholar]
- 108.Qin G, Lian J, Huang L, Zhao Q, Liu S, Zhang Z, et al. Metformin blocks myeloid-derived suppressor cell accumulation through AMPK-DACH1-CXCL1 axis. Oncoimmunology. 2018;7:e1442167. doi: 10.1080/2162402X.2018.1442167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Uehara T, Eikawa S, Nishida M, Kunisada Y, Yoshida A, Fujiwara T, et al. Metformin induces CD11b+-cell-mediated growth inhibition of an osteosarcoma: implications for metabolic reprogramming of myeloid cells and anti-tumor effects. Int Immunol. 2019;31:187–98. doi: 10.1093/intimm/dxy079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim SH, Li M, Trousil S, Zhang Y, Pasca di Magliano M, Swanson KD, et al. Phenformin inhibits myeloid-derived suppressor cells and enhances the anti-tumor activity of PD-1 blockade in melanoma. J Investig Dermatol. 2017;137:1740–8. doi: 10.1016/j.jid.2017.03.033. [DOI] [PubMed] [Google Scholar]
- 111.Bunt SK, Mohr AM, Bailey JM, Grandgenett PM, Hollingsworth MA. Rosiglitazone and Gemcitabine in combination reduces immune suppression and modulates T cell populations in pancreatic cancer. Cancer Immunol Immunother. 2013;62:225–36. doi: 10.1007/s00262-012-1324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, et al. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer. 2013;133:1107–18. doi: 10.1002/ijc.28114. [DOI] [PubMed] [Google Scholar]
- 113.Meng G, Li B, Chen A, Zheng M, Xu T, Zhang H, et al. Targeting aerobic glycolysis by dichloroacetate improves Newcastle disease virus-mediated viro-immunotherapy in hepatocellular carcinoma. Br J Cancer. 2020;122:111–20. doi: 10.1038/s41416-019-0639-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pucino V, Cucchi D, Mauro C. Lactate transporters as therapeutic targets in cancer and inflammatory diseases. Expert Opin Therapeutic Targets. 2018;22:735–43. doi: 10.1080/14728222.2018.1511706. [DOI] [PubMed] [Google Scholar]
- 115.Guan X, Rodriguez-Cruz V, Morris ME. Cellular uptake of MCT1 inhibitors AR-C155858 and AZD3965 and their effects on MCT-mediated transport of L-lactate in murine 4T1 breast tumor cancer cells. AAPS J. 2019;21:13. doi: 10.1208/s12248-018-0279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Polański R, Hodgkinson CL, Fusi A, Nonaka D, Priest L, Kelly P, et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res. 2014;20:926–37. doi: 10.1158/1078-0432.CCR-13-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Raghunand N, He X, van Sluis R, Mahoney B, Baggett B, Taylor CW, et al. Enhancement of chemotherapy by manipulation of tumour pH. Br J Cancer. 1999;80:1005–11. doi: 10.1038/sj.bjc.6690455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Luciani F, Spada M, De Milito A, Molinari A, Rivoltini L, Montinaro A, et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst. 2004;96:1702–13. doi: 10.1093/jnci/djh305. [DOI] [PubMed] [Google Scholar]
- 119.Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016;76:1381–90. doi: 10.1158/0008-5472.CAN-15-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res. 2017;5:9–16. doi: 10.1158/2326-6066.CIR-16-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zuo H, Hou Y, Yu Y, Li Z, Liu H, Liu C, et al. Circumventing myeloid-derived suppressor cell-mediated immunosuppression using an oxygen-generated and -economized nanoplatform. ACS Appl Mater interfaces. 2020;12:55723–36. doi: 10.1021/acsami.0c18180. [DOI] [PubMed] [Google Scholar]
- 122.Qian X, Zhang Q, Shao N, Shan Z, Cheang T, Zhang Z, et al. Respiratory hyperoxia reverses immunosuppression by regulating myeloid-derived suppressor cells and PD-L1 expression in a triple-negative breast cancer mouse model. Am J Cancer Res. 2019;9:529–45. [PMC free article] [PubMed] [Google Scholar]
- 123.Ge Y, Yoon SH, Jang H, Jeong JH, Lee YM. Decursin promotes HIF-1α proteasomal degradation and immune responses in hypoxic tumour microenvironment. Phytomedicine. 2020;78:153318. doi: 10.1016/j.phymed.2020.153318. [DOI] [PubMed] [Google Scholar]
- 124.Yang Z, Guo J, Cui K, Du Y, Zhao H, Zhu L, et al. Thymosin alpha-1 blocks the accumulation of myeloid suppressor cells in NSCLC by inhibiting VEGF production. Biomed Pharmacother. 2020;131:110740. doi: 10.1016/j.biopha.2020.110740. [DOI] [PubMed] [Google Scholar]
- 125.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. 2012;109:2784–9. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yin T, He S, Shen G, Wang Y. HIF-1 dimerization inhibitor acriflavine enhances antitumor activity of sunitinib in breast cancer model. Oncol Res. 2014;22:139–45. doi: 10.3727/096504014X13983417587366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Terp MG, Olesen KA, Arnspang EC, Lund RR, Lagerholm BC, Ditzel HJ, et al. Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J Immunol. 2013;191:4165–73. doi: 10.4049/jimmunol.1301274. [DOI] [PubMed] [Google Scholar]
- 128.Allard D, Allard B, Stagg J. On the mechanism of anti-CD39 immune checkpoint therapy. J Immunother Cancer. 2020;8:e000186. doi: 10.1136/jitc-2019-000186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bastid J, Regairaz A, Bonnefoy N, Déjou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res. 2015;3:254–65. doi: 10.1158/2326-6066.CIR-14-0018. [DOI] [PubMed] [Google Scholar]
- 130.Jin R, Liu L, Xing Y, Meng T, Ma L, Pei J, et al. Dual mechanisms of novel CD73-targeted antibody and antibody-drug conjugate in inhibiting lung tumor growth and promoting antitumor immune-effector function. Mol Cancer Ther. 2020;19:2340–52. doi: 10.1158/1535-7163.MCT-20-0076. [DOI] [PubMed] [Google Scholar]
- 131.Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia. 2013;15:1400–9. doi: 10.1593/neo.131748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Adeshakin AO, Liu W, Adeshakin FO, Afolabi LO, Zhang M, Zhang G, et al. Regulation of ROS in myeloid-derived suppressor cells through targeting fatty acid transport protein 2 enhanced anti-PD-L1 tumor immunotherapy. Cell Immunol. 2021;362:104286. doi: 10.1016/j.cellimm.2021.104286. [DOI] [PubMed] [Google Scholar]
- 133.Khadge S, Sharp JG, McGuire TR, Thiele GM, Talmadge JE. Lipid inflammatory mediators in cancer progression and therapy. Adv Exp Med Biol. 2017;1036:145–56. doi: 10.1007/978-3-319-67577-0_10. [DOI] [PubMed] [Google Scholar]
- 134.Dumont A, de Rosny C, Kieu TL, Perrey S, Berger H, Fluckiger A, et al. Docosahexaenoic acid inhibits both NLRP3 inflammasome assembly and JNK-mediated mature IL-1β secretion in 5-fluorouracil-treated MDSC: implication in cancer treatment. Cell Death Dis. 2019;10:485. doi: 10.1038/s41419-019-1723-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xie Z, Ago Y, Okada N, Tachibana M. Valproic acid attenuates immunosuppressive function of myeloid-derived suppressor cells. J Pharmacol Sci. 2018;137:359–65. doi: 10.1016/j.jphs.2018.06.014. [DOI] [PubMed] [Google Scholar]
- 136.Huang B, Song BL, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab. 2020;2:132–41. doi: 10.1038/s42255-020-0174-0. [DOI] [PubMed] [Google Scholar]
- 137.Pencheva N, Buss CG, Posada J, Merghoub T, Tavazoie SF. Broad-spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell. 2014;156:986–1001. doi: 10.1016/j.cell.2014.01.038. [DOI] [PubMed] [Google Scholar]
- 138.Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, et al. LXR/ApoE activation restricts innate immune suppression in cancer. Cell. 2018;172:825–40.e18. doi: 10.1016/j.cell.2017.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liang H, Shen X. LXR activation radiosensitizes non-small cell lung cancer by restricting myeloid-derived suppressor cells. Biochem Biophys Res Commun. 2020;528:330–5. doi: 10.1016/j.bbrc.2020.04.137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.