

Abstract
Spinal cord injury (SCI) is one kind of severe trauma for central nervous system. Myelin debris clearance and axon regeneration are essential for nerve regeneration after SCI. Metformin, a glucose-lowering drug, has been demonstrated to promote the locomotor functional recovery after SCI. In this study, we investigated the role and molecular mechanism of metformin on myelin preservation in a rat SCI model. SCI was induced in rats by compression at T9 level using a vascular clip. We showed that administration of metformin (50 mg·kg−1·d−1, ip) for 28 days significantly improved locomotor function in SCI rats. Metformin also ameliorated SCI-induced neuronal apoptosis and promoted axon regeneration in the spinal cord. Using co-immunofluorescence of IBa-1 and MBP, and luxol fasting blue (LFB) staining, we demonstrated that metformin promoted the transformation of M1 to M2 phenotype polarization of microglial cells, then greatly facilitated myelin debris clearance and protected the myelin in SCI rats. Furthermore, metformin ameliorated SCI-induced blockade of autophagic flux in the spinal cord, and enhanced the fusion of autophagosome and lysosome by inhibiting the AMPK-mTOR signaling pathway. Moreover, metformin significantly attenuated inflammatory responses in the spinal cord. In LPS-treated BV2 cells, pretreatment with metformin (2 mM) significantly enhanced autophagy level, suppressed inflammation and cell apoptosis. The protective effects were blocked in the presence of an autophagy inhibitor 3-methyladenine (3-MA, 5 mM), suggesting that the effect of metformin on autophagy in microglial cells is essential for the myelin preservation during nerve recovery. This study reveals a novel therapeutic effect of metformin in SCI recovery by regulating the activation of microglial cells and enhancing its autophagy level.
Keywords: spinal cord injury, metformin, microglial cells, autophagy, nerve regeneration, 3-methyladenine
Introduction
Spinal cord injury (SCI) is one kind of severe trauma of central nervous system (CNS), which leads to the dysfunction of the sensory and autonomic control ability and motility of the SCI patients. Myelin integrity is essential for maintaining the normal physiological function of the CNS [1, 2]. SCI destroys the myelin sheath structures (such as the ranffian junction), exposes the axons, and blocks the nerve conduction at the lesion sites [3]. Then, the oligodendrocytes will be lost and subsequently trigger the loss of the myelin sheath around the axons [3]. Moreover, the presence of myelin-sheath sheet fragments surrounding the lesion sites will also amplify the difficulty of nerve regeneration. Accelerating myelin regeneration and maintaining myelin integrity are crucial during SCI recovery. Accordingly, the timely and effective clearance of myelin fragments and promoting myelin preservation are conducive to nerve repair following SCI.
Autophagy is a common biological phenomenon in mammals to degrade misfolded proteins, damaged organelles, or other components to maintain cellular homeostasis. Accumulating evidences suggest that autophagy is associated with myelin debris clearance [4–6]. Recently, the role of autophagy in the functional recovery after SCI has been widely investigated, however, its role in myelin debris clearance and myelin preservation during SCI recovery is still unclear. It has been reported that autophagic flux is somewhat reduced or blocked after SCI or traumatic brain injury (TBI), which may further lead to a delay in functional recovery, especially that of microglial cells [7, 8]. As the resident immune cells of the CNS, microglial cells exert a key role in the immune response during CNS damage. Under damage or stress conditions, microglial cells can quickly transform into the activated state and then recruit circulating monocytes in the CNS [9, 10]. Promoting the autophagy in microglial cells may be an important regulatory mechanism during nerve recovery. The AMP-activated protein kinase (AMPK), a modulator of cellular energy, is also essential for the regulation of autophagy. AMPK has been reported to activate autophagy and enhance autophagic flux in schwann cells (SCs) by suppressing mammalian target of rapamycin (mTOR) during debris accumulation [11, 12]. Thus, we will further explore the role of autophagy in microglial cells during SCI recovery.
Metformin has been widely prescribed for the treatment of type 2 diabetes and other metabolic syndromes since the 1960s [13]. Metformin ameliorates hyperglycemia by inhibiting hepatic glucose production and increasing peripheral glucose utilization. More importantly, its ability is not limited to lower glucose. Accumulating evidences suggest metformin has effective efficacy in various CNS disorders, including Huntington’s disease, Parkinson’s disease, and ischemic brain injury [14–16]. Recent studies have also shown that metformin treatment improves the locomotor functional recovery after SCI [17, 18], however, the molecular mechanism of its action is not very unclear. Metformin is involved in the regulation of autophagy through activating the AMPK/mTOR signaling pathway [19]. Thus, we hypothesized that the benefit of metformin in the outcome of SCI recovery can be partly attributed to the activation of autophagy in microglial cells.
In current study, we aim to detect the role and molecular mechanism of metformin on myelin preservation using a SCI rat model. We found that after SCI, metformin treatment reverses autophagy level in microglial cells and promotes the transformation of M1 to M2 phenotype polarization of microglial cells, subsequently enhancing the myelin debris clearance, which, in turn, promotes myelin preservation and nerve repair following SCI. Our current study further elucidates the novel mechanism of metformin in promoting nerve repair, and provides a new theoretical basis for the clinical application of metformin in SCI recovery.
Materials and methods
Animals and ethic statement
Female Sprague-Dawley rats (female; 220–250 g) were purchased from the Animal Center of Chinese Academy of Science (Shanghai, China). The animals were housed in an OptiMICE Rotary Experimental Animal Cage System (Cat. No. HH-A-5II, Jiangsu, China) under standard housing conditions, including appropriate temperature (22 °C) and humidity (60%). The mice were subjected to a 12 h light/12 h dark cycle and given access to water and food ad libitum. The study was not pre-registered. No exclusion criteria for animals were pre-determined. No animal was excluded. The time-line diagram was shown in Fig. 1a. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University according strictly to the National Institutes of Health Guide.
Fig. 1. Experimental protocol of Met treatment for SCI rats and BV2 cells.

a Time-line diagram of spinal cord injury, drug treatment and experimental analysis in rat. b Time-line diagram of BV2 cells treated with LPS, drug treatment and experimental analysis. c A table containing the group of each experiment and its sample size. SCI spinal cord injury, 3-MA 3-Methyladenine, Met metformin, BBB Basso, Beattie, and Bresnahan, WB Western blotting, IF immunofluorescence staining, LFB luxol fast blue.
Induction of the rat model of SCI and drug treatment
The rats were randomly selected for surgery. After anesthetizing the rats with 2 (%) pentobarbital sodium (40 mg/kg, Cat. No. 4579/50, R&D), the rats were randomly chosen for a laminectomy at the T9 level exposing the cord beneath without disrupting the dura. Then, the rats were subjected to compression using a vascular clip (15 g forces, Oscar, Suzhou, China) for 1 min. Rats in the sham control group underwent a similar surgical procedure but without impact injury, and did not receive pharmacological treatment. Metformin(Cat. No. 1115-70-4, Sigma, MS, USA) was diluted with normal saline to a final metformin concentration of 20 mg/mL. After surgery, the rats were given metformin (50 mg/kg) immediately via intraperitoneal (i.p.) injection and continued to receive a similar dose of metformin every day until they were sacrificed. The rats in the SCI group received an equivolumetric injection of normal saline at the corresponding time points after injury. The time-line diagram was shown in Fig. 1a. The group and number of animals required for each experiment were shown in Fig. 1c. Postoperative care included the manual emptying of urinary bladder twice daily until the return of bladder function.
Assessment of locomotion functional recovery
The locomotion functional recovery was examined using the Basso-Beattie-Bresnahan (BBB) locomotion rating scale, inclined plane test, and footprint analysis. BBB locomotion rating scale was performed at 1, 3, 7, 14, and 28 d after SCI surgery. To determine the BBB locomotion rating score, rats were laid individually on an open field and allowed to move freely for 5 min. Then, the crawling ability of rats was evaluated according to the BBB scale ranging from 0 (no limb movement or weight support) to 21 (normal locomotion). The inclined plane test was performed in each time point after SCI. Briefly, the rats were subjected to this test in two positions (right side or left side up) using a testing apparatus (i.e., a board covered with a rubber mat containing horizontal ridges spaced 3 mm apart). For each position, the maximum angle at which a rat could retain its position for 5 s without falling was recorded and averaged to obtain a single score for each rat. For footprint analysis, the forepaws and hindpaws of rats were dipped in red and blue dye (non-toxic), respectively, then the animals walked across a narrow box (1 m long and 7 cm wide). The footprints were scanned into images, which were used for analyzing the functional condition of the nerve.
Hematoxylin and eosin (H&E) staining and Nissl staining
Lengthwise paraffin sections (5 μm thickness) obtained from spinal cord segments at 14 d after injury. For H&E staining, the sections were stained with H&E reagent following the manufacturer’s instructions (Cat. No. G1120, 2018, Solarbio, China) and observed using a Nikon ECLPSE 80i microscope (Nikon, Tokyo, Japan). The epicenter cavity area of length wise spinal cord segments was quantified using Image ProPlus software. For Nissl staining, the sections obtained from spinal cord segments 14 d after injury were stained with cresyl violet and Nissl differentiation solutions according to the manufacturer’s instructions (Cat. No. C0117, 2018, Beyotime, China) and observed using a Nikon ECLPSE 80i microscope. The Nissl positive cells were visualized at ×400 magnification, and counted at five randomly selected fields per section using the Image-ProPlus.
Western blotting
The spinal cord segments including the lesion center (length 0.5 cm, n = 3 per group) were dissected at 14 d after injury and snap-frozen at −80 °C. The total proteins were purified using protein extraction reagents and the extracted protein was quantified using BCA assay. Then, equal amounts of protein (50 μg) were separated on an 8 (%) or 12 (%) gel and transferred onto a PVDF membrane (Bio-Rad, Hercules, CA, USA). After blocking with 5 (%) milk in TBS for 45 min at room temperature, the membranes were incubated with the following primary antibodies overnight at 4 °C: Cleaved caspase-3 (1:1000, AF7022), NF-200 (1:1000, Ab4680), Growth associated protein-43 (GAP43, 1:1000, Ab16053), Nogo-A (1:1000, BS 0134r), NgR1 (1:1000, BS 0129r), Myelin basic protein (MBP, 1:1000, Ab62631), p-AMPK (1:1000, CST 2535 s), AMPK (1:1000, Ab32047), Phosphorylated-mammalian target of rapamycin (p-mTOR, 1:1000, CST 5536), mTOR (1:1000, CST 2983), p62 (1:1000, Ab56416), ATG7 (1:1000, 10088-2-AP), 1L-6 (1:1000, sc-57315), Tumor necrosis factor-alpha (TNF-α, 1:1000, 17590-1-AP), CD206 (1:1000, 60143-1-Ig), CD86 (1:1000, 13395-1-AP), Bax (1:1000, CST 2772 S), Bcl-2 (1:1000, AF6139), Interleukin-1beta (IL-1β, 1:1000, 66737-1-Ig), Interleukin-10 (IL-10, 1:1000, 20850-1-AP), and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:10000, AP0063). After washing with TBST (TBS with 0.05 (%) tween 20) for 3 times, the membranes were respectively incubated with the corresponding goat anti-rabbit (A10521, Multi Sciences, Hangzhou, China) or goat anti-mouse secondary antibody (1:3000, A00413, Multi Sciences, Hangzhou, China) for 4 h at room temperature. The bands were detected using an enhanced chemiluminescence (ECL) kit. Signals were visualized using ChemiDocXRS + Imaging System (Cat. No.1708195, Bio-Rad, USA). All experiments were performed in triplicate with independently prepared samples. The densitometric values of the band were determined using Image J software, followed by statistical analysis.
Immunofluorescence staining
Rats (n = 5 per group) were sacrificed at 1 d or 14 d after injury, and perfused with normal saline, and subsequently perfused with 4 (%) paraformaldehyde. The spinal cord segments including the lesion center were collected, and fixed in 4 (%) paraformaldehyde for 24 h and embedded in paraffin. After dewaxing, rehydrating and washing, the 5 μm thick sections were incubated in 3 (%) H2O2 for 15 min at room temperature. Next, the sections were incubated with 5 (%) bovine serum albumin (BSA) for 30 min, and then incubated with rabbit anti-Cleaved caspase-3 (1:400, AF7022), and NeuN (1:400, 66836-1-Ig), NF-200 (1:300, Ab4680), Ionized calcium binding adaptor molecule 1 (IBa-1, 1:300, NB100-1028), MBP (1:300, Ab62631), LC3II (1:300, NB100-2220), LAMP1 (1:300, 65050-1-Ig), CD68 (1:400, Ab53444), CD86 (1:300, 13395-1-AP) and CD206 (1:300, 60143-1-Ig) as the primary antibody at 4 °C overnight. Then, the sections were respectively incubated with Alexa Fluor 488/647 donkey anti-rabbit/mouse secondary antibodies (Abcam) for 2 h. The nuclei were stained with DAPI for 5 min at room temperature. The images were observed using a confocal microscopy. The number of positive cells was visualized at ×400 magnification, and counted in five randomly selected fields per section per sample using the Image ProPlus. Then, the images were quantified by Image J software. The Plot profile in Image J was used to analyze the colocalization of two proteins in typical images. The Scatter Plot in Image J was used to quantify the level of colocalization between MBP and IBa-1. The same indicators in different groups were captured under the same laser intensity.
Luxol fast blue (LFB) staining
LFB staining is used to show the morphological structure and pathological change of nerve myelin sheath. For LFB staining, the lengthwise sections (5-μm thickness) were obtained from spinal cord segments at the injury epicenter for LFB staining. The sections were stained using the Luxol fast blue kits (Cat. No. G1030, JingKeHuaXue, China) and sealed after impregnation at 60 °C for 8–16 h. After washing with distilled water, the sections were immersed in 95 (%) alcohol, 0.05 (%) lithium carbonate solution for more than 10 s, and in 70 (%) alcohol. The sections were washed again with distilled water, and stained with 0.25 (%) tar violet solution for 10 min and treated with 70 (%) alcohol. Last, the sections were rinsed with n-butanol for dehydration, and observed using a Nikon ECLIPSE 80i microscope (Nikon, Tokyo, Japan).
Real-time quantitative PCR
Total RNA was isolated from the spinal cord using TRIzol (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Reverse transcription and quantitative PCR were performed using a High-Capacity cDNA Reverse Transcription Kit (Cat. No. RR047A, TaKaRa, 2018). The IL-6, IL-1β and TNF-α were amplified using 7900HT Fast Real-Time PCR System. A final reaction volume of 10 μL was used for PCR reaction using the SYBR Green PCR Master Mix (Bio-Rad, Hercules, CA, USA). The sequences of primers were listed in Table 1. The relative amount of each gene was normalized to the amount of β-actin.
Table 1.
Sequences of primers used in real-time quantitative polymerase chain reaction.
| Primers name | Primer sequences |
|---|---|
| IL-6 | Forward: 5ʹ-CCA AGA GGT GAG TGC TTC CC-3ʹ |
| Reverse: 5ʹ-CTG TTG TTC AGA CTC TCT CCC T-3ʹ | |
| IL-1 β | Forward: 5ʹ-ACT CCT TAG TCC TCG GCC A-3ʹ |
| Reverse: 5ʹ-CCA TCA GAG GCA AGG AGG AA-3ʹ | |
| TNF-α | Forward: 5ʹ-TGA TCC GCG ACG TGG AA-3ʹ |
| Reverse: 5ʹ-ACC GCC TGG AGT TCT GGA A-3ʹ | |
| β-actin | Forward: 5ʹ-CCG TGA AAA GAT GAC CCA GA-3ʹ |
| Reverse: 5ʹ-TAC GAC CAG AGG CAT ACA G-3ʹ |
Microglial cell line (BV2 cells) culture and treatment
The BV2 murine microglial cells (Cat. No. HTX1876, Otwo Biotech, Shenzhen, China) were cultured in DMEM with 5 (%) fetal bovine serum, 2 mM glutamine, and 1 (%) penicillin-streptomycin, as previously described [20]. BV2 cells incubated with Lipopolysaccharide (LPS) (100 ng/mL) in culture medium were treated with 2 mM metformin with or without 5 mM of 3-methyladenine (3-MA, Cat. No. HY-19312, 2018, MedChem Express) and incubated for 24 h before immunofluorescence staining/Western blotting. The time-line diagram was shown in Fig. 1b, and the group and number of animals required for each experiment were shown in Fig. 1c.
Statistical analysis
All experiments and statistical analyses were performed by investigators who were unaware of the animals’ group. Data were presented as mean ± SEM. Using GraphPad Prism 6, the statistical differences were determined by one-way analysis of variance (ANOVA) and Tukey’s test. The BBB locomotor scores and the inclined plane test scores were analyzed using two-way ANOVA followed by Bonferroni post-hoc comparison test. Image J software was used to quantify the expression of protein during immunofluorescence staining. Statistical significance was accepted when P < 0.05. Each experiment was performed at least 3 times to ensure accuracy, and the tissues from each replicate were from different mice.
Results
Metformin improves locomotor functional recovery after SCI
As shown in Fig. 2a, the BBB scores between the Met-treated SCI group and SCI group were not significantly different at 1, 3 and 7 d after SCI injury. However, the BBB score of the Met-treated SCI rats was significantly higher than that of the vehicle-treated SCI rats at 14 d post injury (P < 0.05, Fig. 2a), and this tendency was increasing obviously after 14 d of SCI injury (P < 0.01, Fig. 2a). Consistent with the result obtained in the BBB rating scale, the value of angle of incline test in SCI rats was markedly increased after treatment with metformin and significantly higher than that in SCI group at the 28 d post SCI injury (P < 0.01, Fig. 2b). Thus, we further performed footprint analyses at 14 d post SCI, and showed that SCI rats exhibits inconsistent coordination and a large number of drags as evidenced by ink streaks extending from both hindlimbs (labeled in red and blue dye, respectively) (Fig. 2c). In contrast, Met-treated SCI rats showed fairly consistent forelimb-hindlimb coordination and little toe dragging (Fig. 2c). Moreover, H&E staining of longitudinal section of spinal cord revealed a significant reduction in the total lesion area of Met-treated SCI rats when compared with that in the SCI group (P < 0.01, Fig. 2d and e).
Fig. 2. Metformin improves locomotor functional recovery after SCI.

a The BBB score in the Sham, SCI and SCI + Met group (n = 8). b Angle of incline in different groups (n = 8). c Footprint analysis results of each group. Both the forepaws and hindpaws of rats were dipped in red and blue dye (n = 8). d H&E staining result for the lengthwise section of spinal cord in each group. Scale bar = 1 mm. e Quantification analysis of the lesion cavity area in H&E staining of spinal cord (n = 5). *P < 0.05, **P < 0.01 vs. SCI group. SCI spinal cord injury, Met metformin, BBB, Basso, Beattie, and Bresnahan, H&E hematoxylin and eosin.
Metformin relieves neuronal apoptosis following SCI
Next, we detected the neuroprotective role of metformin following SCI. The result of Nissl staining showed that the number of Nissl positive cells was significantly decreased at 14 d after SCI compared with that in the sham group, and metformin treatment reversed this trend (P < 0.05, DF = 6, Fig. 3a, b). To further examine the effect of metformin on neuronal apoptosis, the co-immunofluorescence of pro-apoptotic protein (labeled with Cleaved-caspase 3) and NeuN (labeled for neuron) was performed, and we found that the fluorescence intensity of Cleaved-caspase 3 in neuron was significantly increased in the SCI group when compared with that in the sham group (P < 0.05), which was attenuated by metformin treatment (P < 0.01, Fig. 3c, d). In addition, the Western blotting result of Cleaved-caspase 3 was consistent with that of the immunofluorescence result (P < 0.05, DF = 8, Fig. 3e, f). These results suggest that metformin ameliorates SCI-induced neuronal apoptosis.
Fig. 3. Metformin relieves neuronal apoptosis following SCI.

a, b Representative images and quantification of the number of Nissl positive cells in the posterior horn area of the spinal cord in each group at 14 d after SCI (n = 5). Scale bar = 50 μm. c, d Immunofluorescence staining of Cleaved caspase-3 (green) and NeuN (red), and the quantification of the number of NeuN-positive cells in the posterior horn area of the spinal cord in each group at day 14 after SCI (n = 5). Scale bar = 50 μm. e, f Western blotting result and quantification of Cleaved caspase-3 in each group at day 14 after SCI (n = 3). #P < 0.05 vs. Sham group, *P < 0.05, **P < 0.01 vs. SCI group. SCI spinal cord injury, Met metformin.
Metformin promotes axon regeneration after SCI
NF-200, a protein in the cytoplasm of neuron, not only acts as skeletal support, but also transports the nutrients. In our current study, it was found that there were very few surviving neurofilaments (labeled with NF-200) in the epicenter 14 d post SCI (P < 0.001, DF = 8, Fig. 4a, b). Metformin treatment enhanced NF-200 immunoreactivity and promoted neurofilament preservation after SCI (P < 0.05, DF = 8, Fig. 4a, b). Metformin treatment also enhanced GAP43 immunoreactivity in the posterior horn of the spinal cord after SCI (Fig. 4c). Consistent with the result from immunofluorescence study, Western blotting result also showed that the SCI-mediated decreases of NF-200 (P < 0.05, F = 4.13) and GAP43 (a axon membrane protein) (P < 0.05, F = 12.75) expression were significantly reversed by metformin treatment (Fig. 4d–f). Next, we examined the expression of myelin growth-inhibitory molecules (Nogo-A) and its receptors (NgR1), which is important for the remyelination and axon regeneration. Western blotting results showed that metformin treatment significantly ameliorated the SCI-associated elevated expression of Nogo-A (P < 0.001, F = 18.65) and NgR1 (P < 0.01, F = 40.22) (Fig. 4g–i). These results suggest that metformin promotes axon regeneration of rats after SCI.
Fig. 4. Metformin promotes axon regeneration after SCI.

a, b The immunofluorescence staining and quantification of NF-200 (labeled for neurofilaments) (green) in posterior horn area of spinal cord from each group at 14 d after SCI (n = 5). Scale bar = 25 μm. c ICH staining for GAP43 in posterior horn area of spinal cord from each group at 14 d after SCI (n = 5). Scale bar = 25 μm. d–f Western blotting results and quantification of NF-200 and GAP43 (the protein of axon membrane) in the spinal cord from each group at 14 d after SCI (n = 3). g–i Western blotting and quantification of Nogo-A (the myelin growth-inhibitory molecules) and NgR1 (Nogo-A’s receptor) in each group at 14 d after SCI (n = 3). #P < 0.05, ##P < 0.01, ###P < 0.001 vs. Sham group, *P < 0.05, **P < 0.01, ***P < 0.001 vs. SCI group. SCI spinal cord injury, Met metformin.
Metformin enhances microglial cells to phagocytose myelin debris
Myelin integrity is essential for the normal physiological function of the CNS. Enhancing myelin debris clearance and promoting myelin preservation are crucial for SCI recovery. We performed co-staining of IBa-1 (labeled for microglial cells) and MBP (labeled for myelin) to detect whether metformin promotes the activation of microglial cells and expedites myelin debris clearance following SCI. It was shown that microglial cells were activated after SCI with their enlarged vesicles, suggesting that they have a phagocytotic function and participation in the myelin debris clearance (P < 0.05, DF = 6, Fig. 5a–e). After metformin treatment, the ability of microglial cells to engulf myelin fragments was enhanced, and myelin fragments (MBP protein in red) were largely observed in the microglial vesicle, suggesting that microglial cells were phagocytosing the myelin fragments (P < 0.05, DF = 6, Fig. 5a–e). More importantly, the ability of microglial cells to engulf myelin fragments in Met-treated SCI group was better than that in SCI group (P < 0.001, Fig. 5e). The total level of MBP was significantly decreased in the SCI group compared with that in the sham group (P < 0.01), and metformin treatment reversed it (P < 0.05, F = 16, Fig. 5f). Moreover, LFB staining further indicated that metformin significantly ameliorated severe demyelination from the epicenter to the left and right sides in the SCI group (Fig. 5g). These results prove that metformin expedites microglial cells to phagocytize myelin debris, which contributes to myelin preservation following SCI.
Fig. 5. Metformin enhances microglial cells to phagocytose myelin debris.

a Co-staining of IBa-1 (a specific protein of microglial cells) (red) and MBP (labeled for myelin) (green) in posterior horn area of spinal cord from each group at 14 d after SCI (n = 5). Scale bar = 25 μm. b–d Colocalization analysis of IBa-1 and MBP in selected area of spinal cord from each group. e Quantification analysis of MBP and IBa-1 colocalization area in co-staining of IBa-1 and MBP in each group at 14 d after SCI. f Western blotting and quantification analysis of MBP in the spinal cord from each group at 14 d after SCI (n = 3). g Luxol fast blue (LFB)-stained longitudinal sections of spinal cord from each group at 14 d post SCI (n = 5 per group). #P < 0.05, ##P < 0.01 vs. Sham group, *P < 0.05, ***P < 0.001 vs. SCI group. SCI spinal cord injury, Met metformin, MBP myelin basic protein.
Metformin ameliorates SCI-induced blockade of autophagic flux in microglial cells
Autophagy is essential for the phagocytosis of myelin fragments [5, 6]. The level of autophagic flux determines the autophagic fluency during the degradation of misfolded proteins and damaged organelles. Therefore, we further assessed the role of metformin in autophagy and autophagic flux during SCI recovery. Results of the Western blotting analysis showed that SCI triggers the decrease in p-AMPK/AMPK (P < 0.01, F = 21.22) and elevation in p-mTOR/mTOR (P < 0.05, F = 14.72) in the spinal cord, whereas metformin reversed these occurrences (P < 0.01) (Fig. 6a, b). Next, we tested the expression of p62 and ATG7, and observed a significant increase in p62 and a decrease in ATG7 in spinal cord from SCI group compared with those in the sham group (P < 0.01, F = 31.65, Fig. 6c–e). As expected, metformin administration reversed the expression of p62 and ATG7 in the spinal cord from SCI group (P < 0.05, F = 31.65, Fig. 6c–e). A reduction in p62 represents an enhancement of autophagic flux [21, 22]. Thus, we further detected whether metformin plays a role in the fusion of autophagosome and lysosome. It was shown that SCI inhibits the fusion of autophagosome (LC3II, labeled for autophagosome) and lysosome (LAMP1, labeled for lysosome), whereas metformin treatment reverses LC3II expression, and promotes their fusion with colocalization of LAMP1 and LC3II (P < 0.001, DF = 6, Fig. 6f). Moreover, we have performed co-staining of IBa-1 and LC3II, and found that metformin treatment not only activates microglial cells, but also promotes the autophagy level in microglial cells as evidenced with much more colocalization of IBa-1 and LC3II after SCI (Fig. 6g). These results indicate that metformin could activate autophagy in microglial cells and promote the conversion of autophagosome to autolysosome, subsequently ameliorating the SCI-induced autophagic flux impairment.
Fig. 6. Metformin ameliorates SCI-induced blockade of autophagic flux in microglial cells.

a, b Western blotting and quantification analysis of p-AMPK and p-mTOR in the spinal cord from each group at 14 d after SCI (n = 3). c–e Western blotting and quantification analysis of p62 and ATG7 in the spinal cord from each group at 14 d after SCI (n = 3). f Co-staining of LAMP1 (green) and LC3II (red) in posterior horn area of spinal cords from each group at 14 d post SCI (n = 5). Scale bar = 25 μm. g Co-staining of IBa-1 (green) and LC3II (red) in posterior horn area of spinal cord from each group at 14 d post SCI (n = 5). Scale bar = 25 μm. #P < 0.05, ##P < 0.01 vs. Sham group, *P < 0.05, **P < 0.01, ***P < 0.001 vs. SCI group. SCI spinal cord injury, Met metformin.
Metformin ameliorates inflammatory level in spinal cord following SCI
We further investigated the infiltration of macrophages (labeled with CD68) after injury. As shown in Fig. 7a and b, metformin treatment significantly reduced the infiltration of CD68 positive cells (P < 0.01). Next, we examined the mRNA and protein levels of the inflammatory cytokines after SCI. As shown in Fig. 7c–e, the mRNA levels of IL-1β, IL-6 and TNF-α were significantly increased after injury (P < 0.01, DF = 8), and were remarkably reversed by metformin treatment (P < 0.05, DF = 8). Consistent with the results of mRNA, Western blotting analysis also showed that metformin treatment significantly ameliorated the SCI-induced the increases of IL-6 and TNF-α (P < 0.05, Fig. 7f–h). More importantly, metformin inhibited the expression of CD86 (M1, proinflammatory factor) and increased the expression of CD206 (M2, proinflammatory factor) (P < 0.05, DF = 6, Fig. 7i–m). Based on the increased labeling of the M2 phenotypic markers (CD206), the M2:M1 ratio in the spinal cord from SCI group was markedly increased after metformin treatment (Fig. 7l and m).
Fig. 7. Metformin ameliorates inflammatory level in spinal cord following SCI.

a, b Immunofluorescence staining of CD68 and DAPI (blue), and quantification analysis of the number of CD68 (green) positive cells in posterior horn area of the spinal cord in each group at 1 d after SCI (n = 5). Scale bar = 50 μm. c–e mRNA levels of IL-6, IL-1β, and TNF-α from the injured spinal cord in each group at 14 d after SCI (n = 5). f–h Western blotting and quantification analysis of IL-6 and TNF-α in the spinal cord from each group at 14 d after SCI (n = 3). i–k Western blotting and quantification analysis of CD206 (red) and CD86 (green) in each group at 14 d after SCI (n = 3). l Co-staining of CD206 and CD86 in posterior horn area of spinal cord from each group at 14 d after SCI (n = 5). Scale bar = 25 μm. m Quantification analysis of the M2 (CD206):M1 (CD86) ratio in posterior horn area of spinal cord from each group during co-staining of CD206 and CD86. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. Sham group, *P < 0.05, **P < 0.01, ***P < 0.001 vs. SCI group. SCI spinal cord injury, Met metformin.
3-MA treatment reverses the protective effect of metformin on BV2 cells
To further confirm the role of metformin-mediated elevated autophagy in microglial cells during SCI recovery, 3-MA, an autophagy inhibitor, was used to treat BV2 cells (a microglial cells line) in vitro. After 3-MA treatment, metformin-mediated elevated autophagy level was significantly suppressed in BV2 cells as evidenced by an increase in p62 and a decrease in LC3II level (P < 0.001, DF = 8, Fig. 8a–c). In addition, 3-MA treatment dramatically enhanced the expression of Cleaved caspase-3 and Bax (P < 0.01, DF = 8, Fig. 8d–f), and inhibited the expression of Bcl-2 (P < 0.01, DF = 8, Fig. 8d, g). Furthermore, 3-MA reversed the metformin-triggered the transformation of BV2 cells from the M2 phenotype polarization to M1 phenotype polarization by significantly enhancing CD86 expression and suppressing CD206 expression (P < 0.05, Fig. 8h–n). These results confirmed the effect of metformin in promoting autophagy in microglial cells.
Fig. 8. 3-MA treatment reverses the protective effect of metformin on BV2 cells.

a, b Immunofluorescence staining of LC3II (green) in BV2 cells, and quantification of the number of LC3II in selected area after treating metformin with or without 3-MA (n = 5). Scale bar = 50 μm. c Western blotting and quantification analysis of p62 in BV2 cells after treating metformin with or without 3-MA (n = 3). d–n Western blotting and quantification analysis of Cleaved-caspase 3, Bax, Bcl-2, IL-1β, IL-6, IL-10, TNF-α, CD206, and CD86 in BV2 cells after metformin treatment with or without 3-MA (n = 3). ##P < 0.01, ###P < 0.001 vs. CON group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. LPS group; &P < 0.05, &&P < 0.01, &&&P < 0.001 vs. LPS + Met group. LPS lipopolysaccharide, Met metformin, 3-MA 3-Methyladenine.
Discussion
Myelin integrity is essential for maintaining the normal physiological function of CNS. Myelin debris clearance and myelin preservation are crucial for SCI recovery. In this study, we investigated the effect of meformin in modulating the myelin debris clearance and promoting myelin preservation following SCI. Using a rat model of SCI, we observed the following results: (1) Rats treated with metformin after SCI showed a remarkably improvement in locomotor functional recovery; Metformin reduced neuronal apoptosis, and promoted axon regeneration. These findings are consistent with the reported results in previous studies [18, 23, 24]; (2) Metformin administration significantly promoted the myelin debris clearance and myelin preservation after SCI; (3) These protective effects of metformin are not only attributed to the metformin-associated enhancement of autophagic flux in microglial cells through the AMPK/mTOR signaling pathway, but also attributed to the transformation of microglial cells from M1 to M2 phenotype polarization. Our current studies provide a novel mechanistic insight into the positive neuroprotective effect of metformin for SCI recovery.
Metformin, a glucose-lowering agent, is one of the first-line drugs in the treatment of type 2 diabetes mellitus. Except for its indication in type 2 diabetes, the neuroprotective role of metformin in cerebral ischemia [25], peripheral nerve [26], and SCI has been reported [17, 24]. Several studies have demonstrated that metformin ameliorates the SCI-induced locomotor dysfunction by relieving BSCB disruption and promoting axon regeneration. However, there is little known regarding the role of metformin in myelin preservation after SCI. Thus, we hypothesized and then verified that metformin enhances the myelin debris clearance and accelerates nerve regeneration during SCI recovery. In the current study, metformin alone was used for the treatment of SCI in rats, thus, there is no synergistic effect of the metformin during treatment for SCI. Moreover, the rats in sham group were also treated with metformin, and there was no significant difference in behavioral outcomes of rats between Sham + Met and Sham group (Supplementary Fig. 1). Furthermore, our mechanistic study further indicated that metformin has an effect on microglial cells and enhances their autophagic flux, thereby promoting the myelin debris clearance, which is consistent with its role on stroke recovery [27]. Our study further highlighted the unique molecular mechanism underlying the neuroprotective role of metformin during SCI recovery.
Myelin debris originates from the breakdown of damaged myelin. Excessive myelin debris accumulation disturbs the regenerative microenvironment of nerve and hinders remyelination [28]. Thus, it is an urgent need to remove myelin debris after SCI. Microglial cells are a specialized population of macrophages in the CNS and exert an important role in maintaining the homeostasis [29, 30]. After injury, the microglial cells are activated; they proliferate, and migrate to phagocytize the cell fragments at the damaged site and maintain the physiological balance. It is well known that the activated microglial cells are polarized to M1 or M2 phenotype. The polarization of M1 phenotype is associated with inflammation and neurodegeneration, and whereas the polarization of M2 phenotype is associated with phagocytosing debris and tissue repair [31–33]. Accordingly, microglial cells are the key factor to facilitate remyelination during nerve injury. Our findings demonstrated that microglial cells were significantly activated and polarized to M1 phenotypes following SCI. Interestingly, metformin treatment transformed the phenotype polarization of microglial cells from M1 to M2 phenotype polarization after injury both in vivo and in vitro. These results indicate that metformin effectively promotes microglial cells to inhibit inflammation and enhances myelin debris clearance following SCI.
After CNS injury, blood-derived monocytes are massively recruited in the CNS, and then differentiate into macrophages and acquire many characteristics of microglial cells [34]. Macrophages and microglial cells share several structure and function. As a consequence, they are still referred as microglial/macrophages in the neuroscience literature. Accumulating evidences suggest that macrophages also play an important role in the myelin debris clearance and contribute to axon regeneration [35–37]. However, there is litter known regarding the role of metformin on macrophages during phagocytizing the myelin debris. We observed that SCI triggers an increase in CD68 positive cells, and metformin reduces the infiltration of CD68 positive cells at 1 d after SCI, indicating that macrophages may also be involved in the metformin treating for SCI. However, whether this is a long-term collaborative effect or is specific to the period during SCI recovery is still unknown. Accordingly, their roles remain to be further clarified.
Autophagy is an adaptive process in body, which is involved in the turnover of long-lived proteins, cytosolic components, or damaged organelles. In general, sequestosome 1 (SQSTM1/p62) binds to ubiquitinated proteins and the autophagy-related protein 8/LC3, and then forms autophagosomes with the presence of ATG family proteins to transport ubiquitinated proteins to lysosomes for degradation [38]. A growing body of evidences indicate that normal function of autophagy is important for the recovery following SCI [39–41]. Zhou et al’s. work had systematically revealed the dynamic changes of autophagic flux during SCI recovery [8]. Inhibition of autophagy delays the myelin fragmentation clearance around the injured nerve [5]. In addition, metformin has been widely reported to be associated with the regulation of autophagy. However, few studies paid attention on impaired autophagic flux and its functional role in microglial cells for the myelin debris clearance after SCI. Our findings provide a valuable evidence that the impairment in autophagy flux occurs in microglial cells after SCI. Metformin treatment significantly reverses this trend, and promotes the myelin debris clearance and myelin preservation. More interestingly, when the autophagic inhibitor, 3-MA, was used to reverse metformin-induced elevated autophagy level in microglial cells, the apoptosis of microglial cells was suppressed.
Increasing evidences demonstrate that autophagy is involved in the balance of immunity and inflammation during CNS injury, and protects against infectious, autoimmune and inflammatory diseases, suggesting that autophagy and inflammation are two intricately linked processes in CNS [42, 43]. Our previous findings suggest that regulation of autophagy after TBI is an effective strategy to limit the progression of inflammatory cascades [7]. As such, we speculated that there is a cross-talk between autophagy and inflammation in microglial cells after SCI. This hypothesis was supported by the findings in our current study. 3-MA reversed the level of inflammatory factors and promoted the transformation of BV2 cells from the M1 to M2 phenotype polarization.
In summary, our current study demonstrates that metformin treatment reverses the SCI-induced blockade of autophagic flux in microglial cells, then promotes the transformation of microglial cells from M1 to M2 phenotype polarization. Then, metformin subsequently enhanced the myelin debris clearance, consequently promoting myelin preservation and nerve regeneration after SCI (Fig. 9). These findings further elucidate the protective mechanism of metformin underlying SCI recovery.
Fig. 9. Metformin promotes microglial cells to facilitate myelin debris clearance and accelerate nerve repairment after SCI.
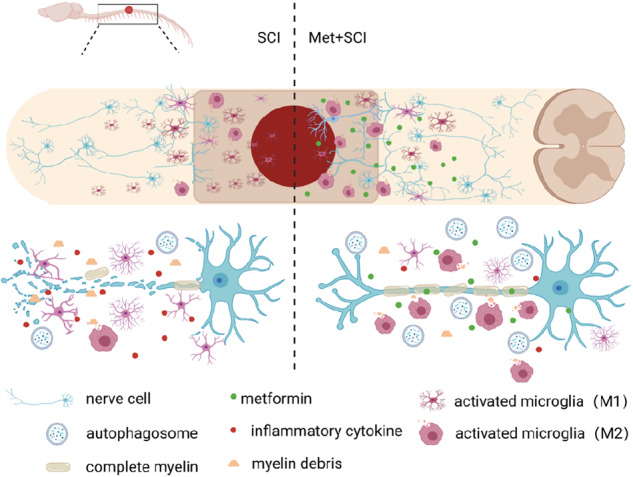
Schematic diagram showing how metformin promotes SCI recovery.
Supplementary information
Acknowledgements
This study was partially supported by National Natural Science Foundation of China (81801233 to YQW, 81802251 to KX), Zhejiang Provincial Natural Science Foundation (LQ18H090011 to YQW and LQ18H150003 to KX), Research Unit of Research and Clinical Translation of Cell Growth Factors and Diseases, Chinese Academy of Medical Science (2019RU010), and Graduate Student Innovation Foundation of Wenzhou University (316202001029).
Author contributions
YQW, JX (Jun Xiong) and ZLH performed research and wrote the manuscript. YY, BNW, JYX, MW, SSZ, SFC, JXZ and KX performed researched. HYZ participated in data analyses and manuscript writing. JX (Jian Xiao) conceived the project, designed the experiments, and wrote the manuscript. All authors have approved the final version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Yan-qing Wu, Jun Xiong, Zi-li He
Contributor Information
Hong-yu Zhang, Email: st_hyz@126.com.
Jian Xiao, Email: xfxj2000@126.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00759-5.
References
- 1.Nave KA. Myelination and support of axonal integrity by glia. Nature. 2010;468:244–52. doi: 10.1038/nature09614. [DOI] [PubMed] [Google Scholar]
- 2.Lüders KA, Nessler S, Kusch K, Patzig J, Jung RB, Möbius W, et al. Maintenance of high proteolipid protein level in adult central nervous system myelin is required to preserve the integrity of myelin and axons. Glia. 2019;67:634–49. doi: 10.1002/glia.23549. [DOI] [PubMed] [Google Scholar]
- 3.Papastefanaki F, Matsas R. From demyelination to remyelination: the road toward therapies for spinal cord injury. Glia. 2015;63:1101–25. doi: 10.1002/glia.22809. [DOI] [PubMed] [Google Scholar]
- 4.Koch JC, Lingor P. The role of autophagy in axonal degeneration of the optic nerve. Exp Eye Res. 2016;144:81–9. doi: 10.1016/j.exer.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 5.Li R, Li D, Wu C, Ye L, Wu Y, Yuan Y, et al. Nerve growth factor activates autophagy in schwann cells to enhance myelin debris clearance and to expedite nerve regeneration. Theranostics. 2020;10:1649–77. doi: 10.7150/thno.40919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yue Z. Regulation of neuronal autophagy in axon: implication of autophagy in axonal function and dysfunction/degeneration. Autophagy. 2007;3:139–41. doi: 10.4161/auto.3602. [DOI] [PubMed] [Google Scholar]
- 7.Zheng Z, Wu Y, Li Z, Ye L, Lu Q, Zhou Y, et al. Valproic acid affects neuronal fate and microglial function via enhancing autophagic flux in mice after traumatic brain injury. J Neurochem. 2020;154:284–300. doi: 10.1111/jnc.14892. [DOI] [PubMed] [Google Scholar]
- 8.Zhou K, Zheng Z, Li Y, Han W, Zhang J, Mao Y, et al. TFE3, a potential therapeutic target for spinal cord injury via augmenting autophagy flux and alleviating ER stress. Theranostics. 2020;10:9280–302. doi: 10.7150/thno.46566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gyoneva S, Davalos D, Biswas D, Swanger SA, Garnier-Amblard E, Loth F, et al. Systemic inflammation regulates microglial responses to tissue damage in vivo. Glia. 2014;62:1345–60. doi: 10.1002/glia.22686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol. 2017;13:420–33. doi: 10.1038/nrneurol.2017.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Tang G, Li Y, Wang Y, Chen X, Gu X, et al. Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. J Neuroinflammation. 2014;11:177. doi: 10.1186/s12974-014-0177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patil SP, Jain PD, Ghumatkar PJ, Tambe R, Sathaye S. Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience. 2014;277:747–54. doi: 10.1016/j.neuroscience.2014.07.046. [DOI] [PubMed] [Google Scholar]
- 16.Vázquez-Manrique RP, Farina F, Cambon K, Dolores Sequedo M, Parker AJ, Millán JM, et al. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum Mol Genet. 2016;25:1043–58. doi: 10.1093/hmg/ddv513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang D, Tang Q, Zheng G, Wang C, Zhou Y, Wu Y, et al. Metformin ameliorates BSCB disruption by inhibiting neutrophil infiltration and MMP-9 expression but not direct TJ proteins expression regulation. J Cell Mol Med. 2017;21:3322–36. doi: 10.1111/jcmm.13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang D, Xuan J, Zheng BB, Zhou YL, Lin Y, Wu YS, et al. Metformin improves functional recovery after spinal cord injury via autophagy flux stimulation. Mol Neurobiol. 2017;54:3327–41. doi: 10.1007/s12035-016-9895-1. [DOI] [PubMed] [Google Scholar]
- 19.Wu H, Ding J, Li S, Lin J, Jiang R, Lin C, et al. Metformin promotes the survival of random-pattern skin flaps by inducing autophagy via the AMPK-mTOR-TFEB signaling pathway. Int J Biol Sci. 2019;15:325–40. doi: 10.7150/ijbs.29009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–37. doi: 10.1016/0165-5728(90)90073-V. [DOI] [PubMed] [Google Scholar]
- 21.Kumsta C, Chang JT, Lee R, Tan EP, Yang Y, Loureiro R, et al. The autophagy receptor p62/SQST-1 promotes proteostasis and longevity in C. elegans by inducing autophagy. Nat Commun. 2019;10:5648. doi: 10.1038/s41467-019-13540-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komatsu M, Kageyama S, Ichimura Y. p62/SQSTM1/A170: physiology and pathology. Pharmacol Res. 2012;66:457–62. doi: 10.1016/j.phrs.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Wang C, Liu C, Gao K, Zhao H, Zhou Z, Shen Z, et al. Metformin preconditioning provide neuroprotection through enhancement of autophagy and suppression of inflammation and apoptosis after spinal cord injury. Biochem Biophys Res Commun. 2016;477:534–40. doi: 10.1016/j.bbrc.2016.05.148. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Zheng Z, Han W, Yuan Y, Li Y, Zhou K, et al. Metformin promotes axon regeneration after spinal cord injury through inhibiting oxidative stress and stabilizing microtubule. Oxid Med Cell Longev. 2020;2020:9741369. doi: 10.1155/2020/9741369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang T, Yu JT, Zhu XC, Wang HF, Tan MS, Cao L, et al. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of AMPK-dependent autophagy. Br J Pharmacol. 2014;171:3146–57. doi: 10.1111/bph.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L, Tian D, Liu C, Yu K, Bai J. Metformin enhances functional recovery of peripheral nerve in rats with sciatic nerve crush injury. Med Sci Monit. 2019;25:10067–76. doi: 10.12659/MSM.918277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin Q, Cheng J, Liu Y, Wu J, Wang X, Wei S, et al. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav, Immun. 2014;40:131–42. doi: 10.1016/j.bbi.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Brosius Lutz A, Chung WS, Sloan SA, Carson GA, Zhou L, Lovelett E, et al. Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc Nat Acad Sci U S A. 2017;114:E8072–E8080. doi: 10.1073/pnas.1710566114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zielasek J, Hartung HP. Molecular mechanisms of microglial activation. Adv Neuroimmunol. 1996;6:191–22. doi: 10.1016/0960-5428(96)00017-4. [DOI] [PubMed] [Google Scholar]
- 30.Gehrmann J, Gold R, Linington C, Lannes VJ, Wekerle H, Kreutzberg GW. Microglial involvement in experimental autoimmune inflammation of the central and peripheral nervous system. Glia. 1993;7:50–9. doi: 10.1002/glia.440070110. [DOI] [PubMed] [Google Scholar]
- 31.Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014;11:98. doi: 10.1186/1742-2094-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73:14–29. doi: 10.1097/NEN.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pischiutta F, Brunelli L, Romele P, Silini A, Sammali E, Paracchini L, et al. Protection of brain injury by amniotic mesenchymal stromal cell-secreted metabolites. Crit Care Med. 2016;44:e1118–e31. doi: 10.1097/CCM.0000000000001864. [DOI] [PubMed] [Google Scholar]
- 34.Bellver LV, Bretheau F, Mailhot B, Vallières N, Lessard M, Janelle ME, et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun. 2019;10:518. doi: 10.1038/s41467-019-08446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuhlmann T, Brück W. Immunoglobulins induce increased myelin debris clearance by mouse macrophages. Neurosci Lett. 1999;275:191–4. doi: 10.1016/S0304-3940(99)00782-X. [DOI] [PubMed] [Google Scholar]
- 36.Kopper TJ, Gensel JC. Myelin as an inflammatory mediator: Myelin interactions with complement, macrophages, and microglia in spinal cord injury. J Neurosci Res. 2018;96:969–77. doi: 10.1002/jnr.24114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grajchen E, Wouters E, van de Haterd B, Haidar M, Hardonnière K, Dierckx T, et al. CD36-mediated uptake of myelin debris by macrophages and microglia reduces neuroinflammation. J Neuroinflammation. 2020;17:224. doi: 10.1186/s12974-020-01899-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang YB, Gong JL, Xing TY, Zheng SP, Ding W. Autophagy protein p62/SQSTM1 is involved in HAMLET-induced cell death by modulating apotosis in U87MG cells. Cell Death Dis. 2013;4:e550. doi: 10.1038/cddis.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhong L, Zhang H, Ding ZF, Li J, Lv JW, Pan ZJ, et al. Erythropoietin-induced autophagy protects against spinal cord injury and improves neurological function via the extracellular-regulated protein kinase signaling pathway. Mol Neurobiol. 2020;57:3993–4006. doi: 10.1007/s12035-020-01997-0. [DOI] [PubMed] [Google Scholar]
- 40.Zhu S, Chen M, Deng L, Zhang J, Ni W, Wang X, et al. The repair and autophagy mechanisms of hypoxia-regulated bFGF-modified primary embryonic neural stem cells in spinal cord injury. Stem Cells Transl Med. 2020;9:603–19. doi: 10.1002/sctm.19-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng Z, Zhou Y, Ye L, Lu Q, Zhang K, Zhang J, et al. Histone deacetylase 6 inhibition restores autophagic flux to promote functional recovery after spinal cord injury. Exp Neurol. 2020;324:113138. doi: 10.1016/j.expneurol.2019.113138. [DOI] [PubMed] [Google Scholar]
- 42.Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol. 2010;189:925–35. doi: 10.1083/jcb.201002021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.She H, He Y, Zhao Y, Mao Z. Release the autophage brake on inflammation: the MAPK14/p38α-ULK1 pedal. Autophagy. 2018;14:1097–8. doi: 10.1080/15548627.2018.1446626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.