

Metformin is the first-line therapy for the treatment of individuals with type 2 diabetes, yet its precise mechanism of action remains unclear. Ma and colleagues now identify a membrane protein PEN2 as a novel target of metformin at low concentrations which specifically activates lysosomal AMP-activated protein kinase (AMPK) via AMP-independent mechanism, leading to therapeutic benefits.
The biguanide drug metformin is currently recommended as the first-line oral glucose-lowering agent in most clinical guidelines on the management of type 2 diabetes. The mechanisms underlying its clinical benefits are complex and still not fully understood. While metformin has pleotropic effects on glucose metabolism, it has been proposed that its major glucose-lowering effect is mostly mediated through inhibition of hepatic glucose production.1 Yet not all of its effects can be explained by this mechanism, and there is growing evidence of an important role for the gut. Early studies have shown that metformin targets hepatic mitochondrion and modestly reduces ATP synthesis through inhibiting complex I of the respiratory chain. This leads to increases in AMP levels, activation of the cellular energy sensor AMP-activated protein kinase (AMPK), and inhibition of other AMP-regulated enzymes (fructose 1,6-bisphosphatase, adenylate cyclase) involved in hepatic gluconeogenesis. Even though these AMPK-dependent and -independent mechanisms have been robustly studied in preclinical models and shown to be involved in glucose-lowering and insulin-sensitizing effects of metformin,2,3 clinical relevance of these observations has been questioned due to the use of relatively high, likely supra-therapeutic, metformin concentrations.1
Lin and colleagues previously reported that AMPK can be activated by low concentrations of metformin, through a different mechanism not involving AMP but involving the translocation of AXIN in complex with the upstream kinase LKB1 to the lysosomal surface via docking onto the Ragulator/vacuolar-type ATPase (v-ATPase) complex (Fig. 1).4 They speculated that low-dose metformin specifically activates lysosomal AMPK, and v-ATPase might act as an sensor/effector of metformin through binding with specific subunit(s). In a recent study published in Nature by Ma et al.,5 building on their previous work, the authors explored the mechanism by which low-dose metformin activates lysosomal AMPK via AMP-independent mechanism and the resulting therapeutic benefits. After having observed that low-dose metformin inhibited the v-ATPase on the lysosome, the authors undertook an affinity-based approach in which they used a photoactive metformin probe to “fish out” metformin-binding proteins from extracts of purified lysosomes. This resulted in identification of 113 candidate proteins that were individually knocked down by RNAi-mediated gene silencing in cells. Remarkably, depletion of presenilin enhancer 2 (PEN2, a subunit of γ-secretase complex6), but not others, specifically rendered the cells insensitive to metformin-induced activation of AMPK and inhibition of v-ATPase. In line with this, genetic knockout of PEN2 resulted in abrogation of low-dose metformin-induced AMPK activation in mouse primary hepatocytes. In vitro biophysics analysis revealed that the binding affinity of metformin to PEN2 is within clinically relevant intracellular concentrations of metformin. They next studied how metformin binding causes PEN2 to intersect with and inhibit v-ATPase. Among several metformin-regulated PEN2-interacting proteins identified, the authors were particularly interested in ATP6AP1, an accessory protein of v-ATPase. Bioinformatics search for functional domains coupled with mutagenesis studies identified that the transmembrane domain of ATP6AP1 was responsible for PEN2 binding and regulation of v-ATPase activity. In line with this, deletion of ATP6AP1 led to constitutive activation of AMPK. Finally, the authors demonstrated that the PEN2-ATP6AP1 pathway is distinct from the glucose/fructose-1,6-bisphosphate (FBP)-sensing aldolase pathway,7 but the two pathways converge on v-ATPase to control lysosomal AMPK activation independently of AMP (Fig. 1).
Fig. 1. Schematic diagram illustrating the regulation of AMPK at lysosomal and non-lysosomal (e.g., cytoplasm, mitochondrion, nucleus) compartments in response to glucose starvation (low glucose) and low or high intracellular concentrations of metformin.
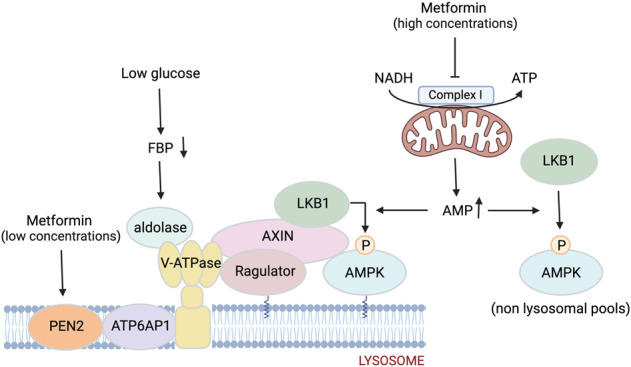
Low glucose availability results in removal of FBP from aldolase leading to the formation of a supercomplex involving the v-ATPase, Ragulator, AXIN, LKB1, and AMPK, thereby triggering phosphorylation/activation of AMPK (via LKB1). Metformin at low concentrations binds to PEN2, and the metformin-bound PEN2 is recruited to v-ATPase via interacting with the transmembrane domain of ATP6AP1, leading to inhibition of v-ATPase and activation of the lysosomal AMPK. Metformin at high concentrations inhibits complex I of the mitochondrial respiratory chain, leading to global AMPK activation through reduced ATP and increased levels of AMP. The illustration is created with BioRender.com.
To explore physiological and therapeutic implications of the PEN2-ATP6AP1-AMPK axis in mediating the effects of metformin, the authors generated intestine- and liver-specific PEN2-knockout mice. Intestine-specific PEN2-knockout mice exhibited impaired postprandial glucose-lowering response to metformin (supplied in drinking water with the dose optimized to keep the plasma metformin levels within clinically relevant concentrations), which was associated with loss or blunting of elevation of plasma GLP-1 and insulin levels stimulated by oral glucose administration in the metformin-treated mice. Notably, these observations were largely phenocopied in intestine-specific AMPK-knockout mice, suggesting that metformin exerts glucose-lowering effect through the intestinal PEN2-AMPK dependent mechanism. In sharp contrast to this observation, a recent study reported that the intestine-specific AMPK-knockout mice exhibit similar metformin-induced glucose lowering and glucose-induced GLP-1 secretion compared to control animals.8 The reason for this discrepancy between the two studies is unknown, but differences in timing/duration and dose of metformin treatment might have effects. Nevertheless, whether PEN2-ATP6P1-AMPK axis mediates glucose lowering through GLP-1 requires further investigation, as a previous study reported that the acute glucoregulatory actions of metformin are independent of GLP-1 receptor.9 In addition, to address the therapeutic role for intestinal PEN2-AMPK axis, it is important to study the effect of metformin under diabetic/hyperglycemic conditions. Liver-specific PEN2 knockout led to blunted improvements of hepatic triglyceride levels and glucose tolerance in high-fat diet-induced obese mice chronically treated with metformin. This is consistent with a previous study that the mice bearing a knockin mutation at the AMPK phosphorylation site of ACC1 and ACC2, key regulators of fatty acid synthesis and oxidation, respectively, were resistant to the lipid- and glucose-lowering effect of chronic metformin treatment.10 Intriguingly, the authors showed that knockdown of PEN2 in C. elegans resulted in abrogation of metformin-induced extension of lifespan, extending previous data showing that v-ATPase-mediated AMPK activation contributes to the lifespan extension effect of metformin in worms.11 These results imply that allosteric AMPK activators that stimulate AMPK independently of AMP would mimic the effects of low-dose metformin. Indeed, small-molecule AMPK activators have been shown to be effective in reversing elevated blood glucose in rodents and non-human primates; however, there are remaining safety issues, such as the potential for global/chronic AMPK activation to promote cardiac hypertrophy.12
Collectively, the study suggests that low dose of metformin specifically activates lysosomal AMPK through a novel PEN2-ATP6AP1-v-ATPase pathway, leading to therapeutic benefits in preclinical models. These findings have several important implications. Metformin elicits gastrointestinal side effects, which is a common cause for treatment discontinuation. The PEN2-ATP6AP1 axis may offer potential targets as substitutes for metformin. It would be of interest to obtain crystal structure of the metformin–PEN2 complex, which would readily inform specific candidate compounds for screening. However, remaining questions include: (1) clarification of the mechanism by which low-dose metformin specifically regulates “lysosomal PEN2-ATP6AP1 axis”, leading to inhibition of v-ATPase, and (2) addressing whether chronic inhibition of v-ATPase would affect lysosomal function (e.g., autophagy) through altering lysosomal pH.
Finally, Ma et al. are careful by choosing low doses of metformin throughout their cellular and preclinical studies. However, are these doses clinically relevant? Effects of metformin depend on route of administration, cellular uptake, and distribution of the drug. The distribution depends on specific organic cation transporter proteins that are organ and species specific. Therefore, future human studies exploring the biodistribution, and kinetic as well as interindividual differences (e.g., by using 11C-labeled metformin)13 will be attractive to assist the investigations performed in preclinical models. Given that metformin shows promise for the treatment of several other age-related chronic diseases beyond type 2 diabetes, its mechanisms of action remain important topic.
Competing interests
The authors declare no competing interests.
References
- 1.LaMoia TE, Shulman GI. Endocr. Rev. 2021;42:77–96. doi: 10.1210/endrev/bnaa023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foretz M, Guigas B, Viollet B. Nat. Rev. Endocrinol. 2019;15:569–589. doi: 10.1038/s41574-019-0242-2. [DOI] [PubMed] [Google Scholar]
- 3.Hunter RW, et al. Nat. Med. 2018;24:1395–1406. doi: 10.1038/s41591-018-0159-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang CS, et al. Cell Metab. 2016;24:521–522. doi: 10.1016/j.cmet.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Ma T, et al. Nature. 2022;603:159–165. doi: 10.1038/s41586-022-04431-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai T, Tomita T. Semin. Cell Dev. Biol. 2020;105:102–109. doi: 10.1016/j.semcdb.2020.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Zhang CS, et al. Nature. 2017;548:112–116. doi: 10.1038/nature23275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olivier S. Mol Metab. 2021;47:101183. doi: 10.1016/j.molmet.2021.101183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maida A, Lamont BJ, Cao X, Drucker DJ. Diabetologia. 2011;54:339–349. doi: 10.1007/s00125-010-1937-z. [DOI] [PubMed] [Google Scholar]
- 10.Fullerton MD, et al. Nat. Med. 2013;19:1649–1654. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Cell Metab. 2016;23:1060–1065. doi: 10.1016/j.cmet.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinberg GR, Carling D. Nat. Rev. Drug Discov. 2019;18:527–551. doi: 10.1038/s41573-019-0019-2. [DOI] [PubMed] [Google Scholar]
- 13.Sundelin E, Jensen JB, Jakobsen S, Gormsen LC, Jessen N. J. Clin. Endocrinol. Metab. 2020;105:3374–3383. doi: 10.1210/clinem/dgaa332. [DOI] [PubMed] [Google Scholar]