

Abstract
Background
Cobalamin C (cblC), a vitamin B12 processing protein, plays a crucial role in metabolism for the conversion of homocysteine to methionine and methylmalonyl-CoA to succinyl-CoA. CblC deficiency, an inborn error of cobalamin processing, is a rare cause of atypical hemolytic-uremic syndrome (aHUS) and results in hyperhomocysteinemia and methylmalonic aciduria. Both substances are thought to contribute to thrombotic microangiopathy (TMA) in cblC deficiency patients. However, the roles of homocysteine and methylmalonic acid (MMA) in these patients remain unclear. We want to shed more light on the contributions of homocysteine and MMA levels as contributing factors for thrombotic microangiopathy (TMA)/aHUS by a follow-up of a cblC deficiency patient over six years.
Case Diagnosis
A 27-day-old Hispanic female presented with abnormal C3-carnitine on her newborn screen, poor feeding, decreased activity, and oligouria. She was diagnosed with cblC deficiency after laboratory results revealed elevated serum homocysteine, and serum MMA along with genetic testing showing a homozygous pathogenic frameshift variant in MMACHC. The patient developed aHUS and acute kidney injury (AKI), which resolved after appropriate therapy. Over six years, she continued to have normal kidney function with no thrombocytopenia despite persistently elevated homocysteine and MMA levels.
Conclusion
We question the roles of homocysteine and MMA as causative of aHUS/TMA in cblC deficiency as they remained elevated during follow-up but did not result in aHUS/TMA or AKI. Hyperhomocysteinemia and/or MMA caused by other metabolic diseases do not result in aHUS/TMA or AKI. This suggests that other nephrotoxic factors may trigger aHUS/TMA in cblC patients.
Introduction
Hemolytic uremic syndrome (HUS) is characterized by hemolytic anemia, thrombocytopenia, kidney failure, and thrombotic microangiopathy (TMA) [1]. Approximately 90% of HUS cases are associated with diarrhea due to Shiga-toxin-producing Escherichia coli, and the remaining 10% are caused by rare compliment and metabolic abnormalities and are summarized as atypical HUS (aHUS) [1]. One metabolic disease causing aHUS is cobalamin C (cblC) deficiency, an autosomal recessive disorder caused by biallelic loss-of-function variants in the MMACHC gene, causing a mortality of up to 44% [2]. The significance of MMACHC for development has also been demonstrated in zebrafish and mouse models [3, 4]. Deficiency of cblC results in impaired cobalamin processing with subsequent accumulation of homocysteine and methylmalonic acid (MMA) [2]. Hyperhomocysteinemia contributes to endothelial damage, due to reducing nitric oxide, increasing oxidative stress, stimulating smooth cell proliferation, and altering elastic wall properties [5]. MMA levels are thought to cause kidney injury by impairing mitochondrial metabolism and raise with kidney disease [2]. MMA has been associated with chronic kidney disease (CKD) in patients with classical methylmalonic aciduria [6]. We report a child with cblC deficiency and infantile aHUS followed for over six years. While the diagnosis of cblC deficiency was confirmed by genetic sequencing, we question the role of elevated homocysteine and MMA as the true cause for aHUS/TMA in the etiology of cblC-deficiency-mediated aHUS, given her persistently elevated homocysteine and MMA levels years after her initial aHUS/TMA episode.
Case Report
A 27-day-old Hispanic female with abnormal C3-carnitine on her newborn screen presented with one day of poor feeding, tachypnea, decreased activity, decreased urine output, and diarrhea without vomiting or fever. She was born at term with a weight of 2.892 kg. Pregnancy and delivery were uncomplicated. There was no family history of kidney disease. She was listless, dehydrated, and mottled. Her weight was 3.12 kg, she had a pulse rate of 142 beats/minute, respiratory rate of 66 per minute, and blood pressure of 97/67 mmHg. Her laboratory results revealed pancytopenia with a hemoglobin (Hgb) of 8.4 g/dL (10-18 g/dL), a white blood cell count of 3,000/mm3 (5-19.5 thousands/mm3), and platelets of 5,000/mm3 (150–600/mm3) with schistocytes (2+) on her peripheral smear. She had a creatinine of 0.6 mg/dL and blood urea nitrogen (BUN) was 41 mg/dL consistent with AKI. She also had an elevated C3-carnitine of 15.4μM (< 0.56 μM). Extended metabolic work-up revealed: vitamin B-12 was 1,092 (180 – 914) pg/mL, serum homocysteine was severely elevated with 179 (3.2-10.7) μmol/l, and serum MMA was 164.35 (0.00 - 0.40) μmol/L (Figure 1A). Urine for organic acids revealed elevated MMA and methylcitrate levels. Haptoglobin was low at 8 mg/dL (22-164), lactate dehydrogenase (LDH) was elevated with 518 (129-376) units/L confirming TMA. She decompensated, required intubation, and developed cardiac failure which was treated with milrinone drip. Urinalysis reported 50 red blood cells per high power field (RBC/HPF), and 3+ proteinuria. A kidney sonogram revealed increased kidney echogenicity. She developed oliguria (0.71 mL/kg/hr). We suspected aHUS due to cblC deficiency and started intramuscular hydroxycobalamin, betaine, levocarnitine, and folic acid. Her urine output improved to > 2 mL/kg/hr after receiving blood transfusion, diuretics, and the initiation of cobalamin therapy. However, the following day her creatinine increased from 0.6 to 0.8 mg/dL and she became hypertensive (121/80 mmHg). As her urine output improved, her hemolysis resolved and her kidney function normalized (creatinine 0.2 mg/dL) (Figure 1A). Her hypertension was controlled with atenolol. By the 12th day of hospitalization, the homocysteine levels declined to 25.6 μmol/l (Figure 1A). At discharge, all medications except hydroxycobalamin injections twice weekly were discontinued. However, after 5 weeks of follow-up, her homocysteine levels rose again (113 μmol/L) (Figure 1B). Betaine was restarted and her homocysteine levels improved. MMACHC sequencing revealed a previously reported homozygous deletion of four nucleotides, resulting in a frame shift (c.328_331delAACC; p.N110Dfs*13) [7]. Despite persistently elevated serum homocysteine and MMA levels over the next six years, she continues to have age-appropriate kidney function and no thrombocytopenia to this day (Figure 1B).
Figure 1.
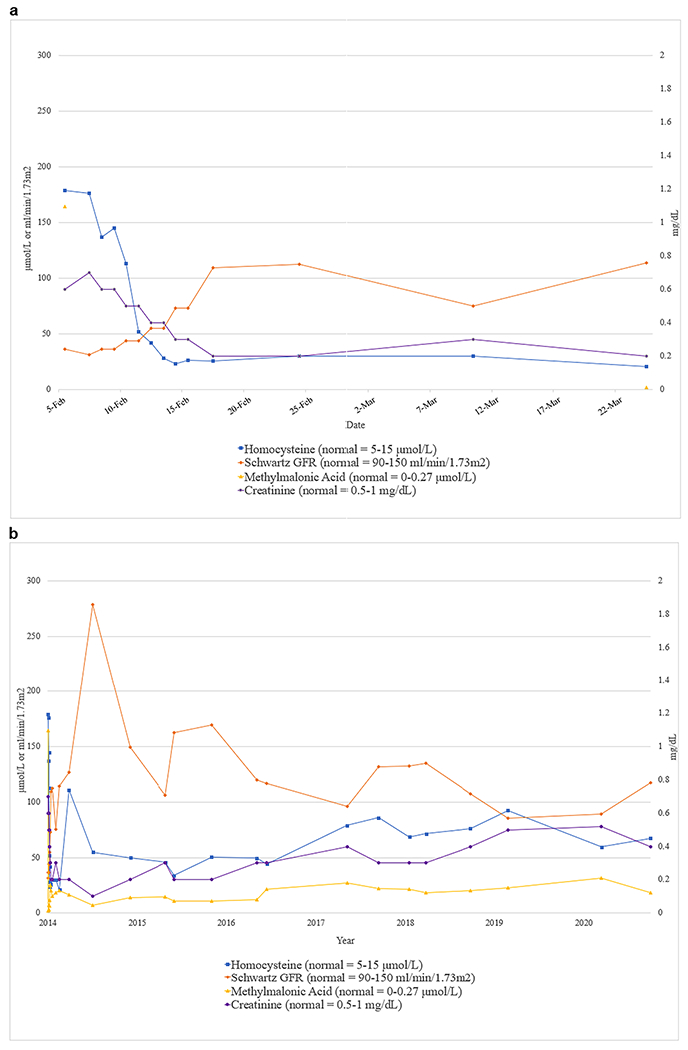
a. Values for serum homocysteine, MMA, GFR (Schwartz) and serum creatinine during the patient’s initial hospitalization are shown. Reference ranges are shown in parentheses
b. Course of serum homocysteine, MMA, GFR (Schwartz) and serum creatinine in the patient over the six years of follow-up. Reference ranges are shown in parentheses
Discussion
CblC deficiency is a disorder of the vitamin B12 pathway but serum vitamin B12 levels are frequently normal or even elevated [2]. The therapeutic effects are only achieved with hydroxycobalamin but not with cyanocobalamin [2]. We question the causative role of homocysteine and MMA in cblC deficiency patients with aHUS/TMA based on the following observations. Only about 7% of cblC-deficient patients are published to develop TMA and aHUS [2]. Patients with the identical mutation as in our patient did not develop kidney involvement [7]. Some publications propose an above threshold level for homocysteine and/or MMA for development of aHUS/TMA. However, Beck et al described that half of the cases of 36 cblC patients with aHUS had intermediate homocysteine levels (30-100 μmol/L) whereas the other half had severe homocysteinemia with levels > 100 μmol/L at presentation [2]. Our patient had severely elevated homocysteine levels > 100 μmol/L at presentation and intermediate homocysteine levels (ranged from 49.4 to 113 μmol/L, mostly in the 40-90 μmol/L range) during the six years of follow-up. Unlike the 50% of patients in the study with intermediate homocysteine levels she did not develop any new episodes of TMA during follow-up [2]. Her MMA levels also remained consistently elevated (ranged from 2.82 to 43.94 μmol/L, mostly in the 20-30 μmol/L range) (Figure 1B). Despite persistently four- to seven-fold elevated homocysteine levels, and 50- to 75-fold elevated MMA levels, her creatinine and glomerular filtration rate (GFR) remained stable. An interesting thought is that perhaps there may exist an age-dependent threshold level, similar to younger patients being more sensitive to ACE inhibitors, rendering younger patients more susceptible to TMA/aHUS with less severe MMA and/or homocysteine elevations. There are even a few cases of asymptomatic MMACHC cases with similarly elevated homocysteine levels but who have not developed symptoms or aHUS [2]. Finally, there are also several other cobalamin defects such as cblD, cblF, and cblJ that are also associated with elevated homocysteine and MMA but do not cause aHUS/TMA [2, 8].
Defects in absorbing vitamin B12 due to mutations in TCN2 (causing Transcobalamin II deficiency) or Cubilin (causing Imerslund–Gräsbeck) also result in homocysteinemia and methylmalonic aciduria. However, in Transcobalamin II deficiency no kidney involvement is known and in Imerslund–Gräsbeck syndrome kidney involvement is characterized by proteinuria but normal kidney function and no TMA [9]. Pernicious anemia also leads to elevated homocysteine and MMA levels. While pernicious anemia is quite common in the general population with an incidence of 0.1% it rarely causes kidney disease or TMA [10]. Moreover, resection of the ileum (the primary site of vitamin B12 absorption), does not typically result in TMA.
Among the homocystinurias, gene mutations in MTRR (causing cblE disorder) or MTR (causing cblG disorder) can rarely cause TMA [2, 8, 11–12]. However, most isolated homocystinurias (without increased MMA) with even higher homocysteine levels due to classic homocystinuria, methylenetetrahydrofolate reductase (MTHFR) deficiency, or cysthatione beta-synthase (CBS) deficiency do not cause aHUS/TMA [2]. While these elevated homocysteine levels in these diseases overlap with the levels seen in our patient, they typically do not result in aHUS/TMA. This points to possibly other contributing factors causing aHUS/TMA besides homocysteine or MMA.
For HUS and complement-mediated aHUS, multiple hits are frequently required to cause TMA such as Shiga-toxin plus variants in genes regulating the complement cascade or anti-FH autoantibodies, infections, or environmental causes [13]. Our patient had no concern for any infection with negative viral stool and sputum cultures and negative bacterial blood and urine cultures. For cblC deficiency there is a notion that it takes at least two or more metabolic factors to develop aHUS/TMA in cobalamin-related disorders. In addition to homocysteinemia either MMA, methionine deficiency, or methylenetetrahydrofolate dehydrogenase deficiency (MTHFD1) may be the second causative factor [2]. TMA caused by nutritional vitamin B12 deficiency, MMACHC, and MTHFD1 are all characterized by increased homocysteine, MMA, and low methionine [2]. However, in our patient chronic elevation of both homocysteine and MMA did not result in any new episodes of TMA. Very little is known about additional environmental factors or infections triggering cblC-deficiency mediated aHUS.
The dissociation between elevated homocysteine and/or MMA levels and the inconsistent and infrequent association with TMA suggest that other mechanisms may be involved. During development, MMACHC is expressed in mesonephric mesenchyme and the endothelium of vessel and the heart [14]. Zebrafish and mouse models of cblC deficiency are also consistent with a developmental role of MMACHC in CNS and eye development causing increased embryonic lethality [3, 4]. Many of these findings are similar to human characteristics in a large Chinese cohort of cblC deficiency patients ranging from hydrocephalus, developmental delay, visual impairment, and seizures [15]. This suggests developmental disturbances contributing to these conditions in addition to the metabolic abnormalities. Alternatively, the elevated homocysteine and MMA levels could be secondary due to impaired kidney function on top of the inherited metabolic disease. Kidney failure patients also have frequently elevated homocysteine which serves as a predictor of cardiovascular disease. More research is warranted in determining the etiology of aHUS/TMA in cblC deficiency.
Conflict of interest
The authors have no conflicts of interest to declare. MTW is supported by Children’s Clinical Research Advisory Committee (CCRAC), Department of Defense (W81XWH1910205), and the NIH (P30 DK079328-11, R01DK119631).
References
- 1.Noris M, Remuzzi G (2005) Hemolytic uremic syndrome. J Am Soc Nephrol 16:1035–1050 [DOI] [PubMed] [Google Scholar]
- 2.Beck BB, van Spronson F, Diepstra A, Berger RMF et al. (2017) Renal thrombotic microangiopathy in patients with cblC defect: review of an under-recognized entity. Pediatr Nephrol 32:733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sloan JL, Achilly NP, Arnold ML, Catlett JL et al. (2020) The vitamin B12 processing enzyme, mmachc, is essential for zebrafish survival, growth, and retinal morphology. Hum Mol Genet 29:2109–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chern T, Achilleos A, Tong X, Hsu CW et al. (2020) Mouse models to study the pathophysiology of combined methylmalonic acidemia and homocystinuria, cblC type. Dev Biol 468:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Guldener C, Stehouwer CD (2000) Hyperhomocysteinemia, vascular pathology, and endothelial dysfunction. Semin Thromb Hemost 26:281–289 [DOI] [PubMed] [Google Scholar]
- 6.Morath MA, Okun JG, Muller IB, Sauer SW et al. (2008) Neurodegeneration and chronic renal failure in methylmalonic aciduria. J Inherit Metab Dis 31:35–43 [DOI] [PubMed] [Google Scholar]
- 7.Lerner-Ellis JP, Tirone JC, Pawelek PD, Dore C et al. (2006) Identification of the gene responsible for methylmalonic aciduria and homocysteinuria, cblC type. Nat Genet 38:93–100 [DOI] [PubMed] [Google Scholar]
- 8.Sloan JL, Carrillo N, Adams D, Venditti CP et al. (2008) Disorders of Intracellular Cobalamin Metabolism. In: Adam MP, Ardinger HH, Pagon RA et al. (eds). GeneReviews® [Internet]. University of Washington, Seattle; 1993–2021. Available from https://www.ncbi.nlm.nih.gov/books/NBK1328/ [PubMed] [Google Scholar]
- 9.Bedin M, Boyer O, Servais A, Li Y et al. (2020) Human C-terminal CUBN variants associate with chronic proteinuria and normal renal function. J Clin Invest 130:335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Podder S, Cervates J, Dey BR (2015) Association of acquired thrombotic thrombocytopaenic purpura in a patient with pernicious anemia. BMJ Case Rep 2015:bcr2015211989. 10.1136/bcr-2015-211989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Labrune P, Zittoun J, Duvaltier I, Trioche P et al. (1999) Haemolytic uraemic syndrome and pulmonary hypertension in a patient with methionine synthase deficiency. Eur J Pediatr 158:734–739 [DOI] [PubMed] [Google Scholar]
- 12.Paul EA, Guttenberg M, Kaplan P, Watkins D et al. (2013) Atypical glomerulopathy associated with the cblE inborn error of vitamin B12 metabolism. Pediatr Nephrol 28:1135–1139 [DOI] [PubMed] [Google Scholar]
- 13.Frimat M, Boudhabhay I, Roumenina LT (2019) Hemolysis Derived Products Toxicity and Endothelium: Model of the Second Hit. Toxins 11:660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonsino E, Ogier H, Mercier JC, Devictor et al. (1985) [Congenital anomaly of the metabolism of vitamin B12. Histopathological study] Arch Anat Cytol Pathol 33:98–99 [PubMed] [Google Scholar]
- 15.He R, Zhang H, Kang L, Li H et al. (2020) Analysis of 70 patients with hydrocephalus due to cobalamin C deficiency. Neurology 95:e3129–e3137 [DOI] [PubMed] [Google Scholar]