

Abstract
INTRODUCTION:
Chronic traumatic encephalopathy (CTE) is a neurodegenerative tauopathy associated with repetitive head impacts (RHI) typically sustained by contact sport athletes. Post-translation modifications to tau in CTE have not been well delineated or compared to Alzheimer disease (AD).
METHODS:
We measured phosphorylated tau epitopes within dorsolateral frontal cortex from post-mortem brains with neither CTE nor AD (n=108), CTE (n=109), AD (n=223), and both CTE and AD (n=33).
RESULTS:
Levels of p-tau202, p-tau231, and p-tau396 were significantly increased in CTE. Total years of RHI exposure was significantly associated with increased p-tau202 levels (p=0.001), but not p-tau396. Instead, p-tau396 was most closely related to Aβ1–42 levels (p<0.001). The p-tau202:p-tau396 ratio was significantly increased in early and late CTE compared to AD.
DISCUSSION:
In frontal cortex p-tau202 is the most upregulated p-tau species in CTE, while p-tau396 is most increased in AD. p-tau202 and p-tau396 measurements may aid in developing biomarkers for disease.
Keywords: Alzheimer Disease / pathology*, Amyloid beta-Peptides, Brain / pathology, Chronic Traumatic Encephalopathy / pathology*, Football / statistics & numerical data*, Humans, Male, Microtubule-Associated Proteins, tau Proteins, Tauopathies
1. BACKGROUND
CTE is a neurodegenerative disease which has been associated with repetitive head impacts (RHI) sustained in a wide variety of contact sports, such as American football, ice hockey, rugby, wrestling, and association football (soccer) [1–5]. Some of the more common clinical features observed in participants with CTE include impulsivity, aggression, depression, impaired memory, and executive dysfunction [5–7]. Currently, CTE can only be diagnosed after death. The neuropathologic criteria that defines CTE is the perivascular accumulation of abnormal hyperphosphorylated tau (p-tau) in neurons primarily and astroglia found at the depths of cortical sulci [8]. Multiple other pathologies, such as amyloid-beta (Aβ) [9], Lewy bodies, and TDP-43 may also frequently occur in CTE, but the presence of p-tau is the defining feature. The tau epitopes involved in the progression of CTE have not yet been delineated.
Alzheimer’s disease (AD), in contrast to CTE, is characterized by the combined presence of Aβ deposits in the form of neuritic plaques (NP) and tau-immunoreactive neurofibrillary tangles (NFT), progressing in a stereotypical pattern first involving the medial temporal lobe [10] [11]. The tau phosphorylation sites involved in the progression of AD have been extensively studied, especially in the temporal cortex, with approximately 30 serine/threonine residues associated with physiological functions [12] [13] [14] [15] [16]. Some of the more commonly recognized phosphorylation sites include Thr181 (p-tau181), Ser202 (p-tau202), Thr231 (p-tau231), and Ser396 (p-tau396). Phosphorylation at p-tau231 has been shown to be an early event in AD [17], although the temporal phosphorylation patterns of p-tau202 and p-tau396 remain controversial. Some studies have found that phosphorylation of p-tau396 is a later event which succeeds phosphorylation of p-tau202 [18] [19], while others suggest that phosphorylation of p-tau396 precedes p-tau202 [20], or occurs in parallel [21]. Distinctions between the type and pattern of tau pathology in CTE versus AD are still being established [10] [22–24].
Here we set out to determine which p-tau epitopes are most involved at different stages of CTE and compare to different stages of AD. Specifically, we tested the four aforementioned epitopes which are commonly recognized in AD: p-tau181, p-tau202, p-tau231, and p-tau396, as well as levels of total Aβ (Aβ1–38, Aβ1–40, and Aβ1–42) in participants with neither CTE nor AD, CTE, AD, and both CTE and AD. Using total years of playing contact sports as a proxy for cumulative RHI exposure [3,9,25–28],we further tested the hypothesis that total years of play would predict increased phosphorylation of CTE relevant tau epitopes. In doing so, we not only have systematically delineated four p-tau epitopes at varying stages of both CTE and AD, but have also evaluated any potential relationships between specific phosphorylation sites and type of neurodegenerative disease.
2. METHODS
2.1. Participants
The study goal was to examine pathologic groups including CTE, AD, both, and neither. Participants were selected from three different study groups that included Understanding Neurologic Injury and Traumatic Encephalopathy (UNITE) as part of the Veteran’s Affairs-Boston University-Concussion Legacy Foundation (VA-BU-CLF), the Boston University’s Alzheimer Disease Research Center (BU ADRC), and Framingham Heart Study (FHS) brain banks. The original study sample included 607 autopsy participants with fresh frozen brain tissue received prior to 10/8/19. Participants were excluded if they had one or more of the following comorbidities: frontotemporal lobar degeneration (both FTLD-TDP and FTLD-tau, including PSP and CBD) (FTLD, n=74), motor neuron disease (MND, n=74), or neocortical Lewy body disease (n=43) resulting in a total of 473 participants. In order to increase the generalizability of our findings, we did not exclude age-related pathologies from our groups, such as primary age-related tauopathy (PART) or limbic-predominant age-related TDP-43 encephalopathy (LATE). Of the 473 brains, 210 were part of the UNITE study, which requires a history of exposure to RHI, including football, ice hockey, rugby, wrestling, and boxing [29]. An additional 179 participants were available from the FHS, a longitudinal and community-based study [30]. The remaining 84 brains were part of the BU ADRC, which recruits control and Alzheimer disease participants during life, as previously described [31] [32]. Total years of all contact sports participation as well as duration of American football was determined from next of kin after death in the UNITE and FHS groups [27]. History of contact sports was unknown in the BU ADRC brain donors. All consents for research participation and brain donation were provided by next of kin. Institutional Review Boards of the Bedford VA Healthcare System and Boston University Medical Center approved all study protocols.
2.2. Pathological Assessment
All brains included in this study were assessed for neuropathological changes of CTE, AD, and other neurodegenerative disorders by using previously published procedures and selection criteria [29,33]. Specifically, brains were hemisected and pathological assessment was performed on the fixed hemibrain; the opposing hemibrain was frozen for biochemical protein quantification. CTE was diagnosed using previously established consensus criteria from the National Institute of Neurological Disorders and Stroke consensus [8]. CTE staging was performed as previously described [3,34]. AD was diagnosed using National Institute of Aging Reagan criteria, including low, intermediate, or high probability of dementia caused by AD [35]. NIA-Reagan criteria was used instead of the more recent NIA-AA AD criteria since the majority of cases were previously evaluated with NIA-Reagan and recent studies suggest the Consortium to Establish a Registry for Alzheimer Disease (CERAD) neuritic plaque score and Braak neurofibrillary tangle stage utilized by NIA-Reagan are the best predictors of cognitive impairment in AD [36].
Using CTE staging and the NIA-Reagan criteria, participants were separated into six different pathologic groups: No CTE/No AD (controls), Low CTE (stages I and II), High CTE (stages III and IV), Low AD, AD (intermediate or high), and both CTE & AD. Participants with no evidence of CTE, no evidence of intermediate or high AD, and CERAD scores of zero were labeled as the “No CTE/No AD” group and used as our control group. Other pathologies such as vascular disease, non-neocortical LBD, and limbic TDP-43 pathology were not selected for. Participants having a CTE Stage of I or II and no evidence of intermediate or high AD were determined to represent early stage disease, and thus were termed “Low CTE.” Conversely, those with a CTE Stage of III or IV and no evidence of AD were determined to have late stage disease, and were labeled as “High CTE.” Low and high stage CTE also correlated with recently suggested consensus CTE staging criteria [34]. Participants who had no evidence of CTE and no evidence of AD, but had a CERAD score of one or greater and a Braak Score greater than zero were placed in the “Low AD” group. Those with a CTE Stage of zero and evidence of AD were classified as “AD.” Lastly, participants having both CTE and threshold criteria for intermediate or high AD were termed “CTE & AD.”
2.3. Measurements of pathological proteins
Frozen tissue from the gyral crest of the dorsolateral prefrontal cortex was weighed and placed on dry ice. Brain tissue was homogenized in 5:1 volume of freshly prepared, ice cold 5M Guanidine Hydrochloride in Tris-buffered saline (20 mM Tris-HCl, 150 mM NaCl, pH 7.4), which contained 1:100 Halt protease inhibitor cocktail (Thermo Fischer Scientific, Waltham, MA) and 1:100 Phosphatase inhibitor cocktail 2 & 3 (Sigma-Aldrich, St. Louis, MO) as previously reported [37]. The homogenate was then shaked (regular rocker) overnight at room temperature. The lysate was diluted with 1% Blocker A (Meso Scale Discovery (MSD), Rockville, Maryland, #R93BA-4) in wash buffer according to specific immunoassays. 1:4000 for Aβ1–38, Aβ1–40, and Aβ1–42 (MSD #K15200E-2), and 1:300 for p-tau181, p-tau202 (MSD custom kit), total tau and p-tau231 (MSD #K15121D-2). Samples were centrifuged at 17,000g and 4°C for 15 minutes. The supernatant was subsequently applied to the immunoassays, and the original homogenate was aliquoted and stored at −80 °C.
To capture tau phosphorylated at Thr residue 181, antibody AT270 was used. The detecting antibody was the biotinylated HT7 that recognizes residue 159–163 of tau (Thermo Scientific, Rockford, IL). To measure p-tau396, a rabbit monoclonal antibody against p-tau396 (Abcam, ab156623) was used as the capturing antibody, and HT7 was used as a detecting antibody. Sulfo-tag conjugated streptavidin secondary antibody was used for signal detection by the MSD platform. MSD SECTOR Imager 2400 was used to measure p-tau396 levels. Internal calibrators of p-tau and tau were used (MSD). p-tau levels were measured in arbitrary units, which may or may not be related among the different epitopes. Standards with known concentrations were used for Aβ, and all standards and samples were run in duplicate. Measurements were made using the multi-detection SPECTOR 2400 Imager (MSD).
2.4. Statistical analysis
Statistical analysis was performed using SPSS 26.0 (IBM Corp, Armonk, NY) and Prism v7 and v8 (Graph-Pad Software, La Jolla, CA). The ROUT method [38] was used to detect outliers by directly controlling for false discoveries, with a maximum False Discovery Rate set to Q=1%. Outliers were removed from each amyloid-beta or p-tau group studied, across pathologic disease groups, but were retained for linear regressions which utilized rank-based normalization.
Relative amounts of Aβ1–38, Aβ1–40, Aβ1–42, p-tau181, p-tau202, p-tau231, p-tau396 were computed by dividing each data point by the mean of the control group. An analysis of covariance (ANCOVA) was used to adjust for age at death and compare differences of each analyte between CTE pathology groups and, separately, AD pathology groups. The p-tau202:p-tau396 ratio was compared between CTE and AD pathology groups. Statistical significance was determined at p<0.05. Tau epitope and Aβ levels, as well as total years of contact sports play underwent rank-based normalization for regression analyses [39]. Multiple linear regression analyses were used to evaluate associations between p-tau and predictors total years of playing contact sports and Aβ1–42 levels, adjusting for age at death and sex. Two separate regression models were run, in order to separately examine CTE (excluding AD) and AD (excluding CTE).
3. RESULTS
3.1. Participant groups
The participants were separated into six different pathologic groups based on both the presence or absence and level of severity of CTE and AD pathology. The groups differed significantly in age at death, sex, and RHI, as seen in Table 1. In order from youngest to oldest, the Low CTE, control, High CTE, and CTE & AD groups all were younger in comparison to the AD and Low AD groups. Low CTE, High CTE, and CTE & AD groups had a much higher composition of men than the other pathologic groups, as well as a greater likelihood of playing contact sports for a longer period of time.
Table 1.
Demographic, clinical, and pathological measures between pathology groups
| no CTE/no AD (control) | low CTE | high CTE | low AD | AD | CTE and AD | p value | |
|---|---|---|---|---|---|---|---|
| Sample size (n) | 108 | 37 | 72 | 93 | 130 | 33 | |
| Cohort | |||||||
| UNITE | 42 (38.9%) | 35 (94.6%) | 71 (98.6%) | 9 (9.7%) | 21 (16.2%) | 32 (97.0%) | <0.001b |
| FHS | 52 (48.1%) | 1 (2.7%) | 0 (0%) | 66 (71.0%) | 59 (45.4%) | 1 (3.0%) | <0.001b |
| BUADC | 14 (13.0%) | 1 (2.7%) | 1 (1.4%) | 18 (19.4%) | 50 (38.5%) | 0 (0%) | <0.001b |
| Age at death (S.E.M) | 70.2 (2.5) | 53.7 (2.9) | 73.6 (1.2) | 86.1 (1.0) | 83.5 (0.8) | 75.6 (1.5) | <0.001a |
| Sex m/f (%male) | 67/41 (62.0%) | 36/1 (97.3%) | 71/1 (98.6%) | 52/41 (55.9%) | 71/59 (54.6%) | 33/0 (100%) | <0.001b |
| RHI y/n (%exposed) | 41/31 (56.9%) | 34/0 (100%) | 70/0 (100%) | 10/44 (18.5%) | 23/41 (35.9%) | 31/0 (100%) | <0.001b |
| Total years* (S.E.M) | 6.6 (1.2) | 15.1 (1.9) | 17.0 (0.7) | 1.3 (0.5) | 3.2 (0.7) | 15.9 (1.8) | <0.001a |
| Sport y/n (%played) | |||||||
| football | 33/39 (45.8%) | 33/1 (97.1%) | 69/1 (98.6%) | 9/45 (16.7%) | 19/45 (29.7%) | 30/2 (93.8%) | <0.001b |
| hockey | 5/67 (6.9%) | 2/32 (5.9%) | 5/65 (7.1%) | 1/53 (1.9%) | 3/61 (4.7%) | 4/28 (12.5%) | 0.492b |
| rugby | 1/71 (1.4%) | 4/30 (11.8%) | 4/66 (6.1%) | 0/54 (0%) | 1/63 (1.6%) | 1/31 (3.1%) | 0.036b |
| wrestling | 2/70 (2.8%) | 3/31 (8.8%) | 11/59 (15.7%) | 0/54 (0%) | 1/63 (1.6%) | 1/31 (3.1%) | 0.001b |
| boxing | 0/72 (0%) | 2/32 (5.9%) | 7/63 (10.0%) | 1/53 (1.9%) | 1/63 (1.6%) | 1/31 (3.1%) | 0.029b |
| Other† | 11/61 (15.3%) | 4/30 (11.8%) | 2/68 (2.9%) | 2/52 (3.7%) | 1/63 (1.6%) | 2/30 (6.3%) | 0.011b |
| Braak Stage (n) | |||||||
| Stage 0 | 38 (35.2%) | 14 (38.9%) | 2 (2.9%) | 0 (0%) | 0 (0.7%) | 0 (0%) | |
| Stage I | 20 (18.5%) | 6 (16.7%) | 0 (0%) | 17 (18.5%) | 0 (0.7%) | 0 (0%) | |
| Stage II | 33 (30.6%) | 11 (30.5%) | 9 (13.2%) | 32 (34.8%) | 0 (0.7%) | 0 (0%) | |
| Stage III | 15 (13.9%) | 2 (5.6%) | 40 (58.8%) | 33 (35.9%) | 7 (5.6%) | 0 (0%) | |
| Stage IV | 2 (1.9%) | 3 (8.3%) | 11 (16.2%) | 6 (6.5%) | 19 (15.2%) | 3 (9.1%) | |
| Stage V | 0 (0%) | 0 (0%) | 4 (5.9%) | 2 (2.2%) | 31 (24.8%) | 8 (24.2%) | |
| Stage VI | 0 (0%) | 0 (0%) | 2 (2.9%) | 2 (2.2%) | 68 (54.4%) | 22 (66.7%) | |
| CTE Stage n (%) | |||||||
| Stage I | 20 (51.4%) | 0 (0%) | 1 (3.0%) | ||||
| Stage II | 17 (45.9%) | 0 (0%) | 2 (6.1%) | ||||
| Stage III | 0 (0%) | 38 (52.8%) | 7 (21.2%) | ||||
| Stage IV | 0 (0%) | 34 (47.2%) | 23 (69.7%) | ||||
| CERAD Score n (%) | |||||||
| 0 | 108 (100%) | 35 (94.6%) | 40 (56.3%) | 0 (0%) | 0 (0.7%) | 0 (0%) | |
| 1 | 0 (0%) | 2 (5.4%) | 29 (40.8%) | 77 (82.8%) | 40 (32.0%) | 7 (21.2%) | |
| 2 | 0 (0%) | 0 (0%) | 2 (2.8%) | 16 (17.2%) | 49 (39.2%) | 19 (57.6%) | |
| 3 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 36 (28.8%) | 7 (21.2%) | |
| Thal Phase n (%) | |||||||
| 0 | 44 (69.8%) | 26 (70.3%) | 15 (21.1%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| 1 | 6 (9.5%) | 5 (13.5%) | 13 (18.3%) | 2 (6.5%) | 0 (0%) | 0 (0%) | |
| 2 | 2 (3.2%) | 0 (0%) | 7 (9.9%) | 3 (9.7%) | 1 (1.9%) | 0 (0%) | |
| 3 | 10 (15.9%) | 4 (10.8%) | 11 (15.5%) | 13 (41.9%) | 4 (7.7%) | 2 (6.2%) | |
| 4 | 1 (1.6%) | 2 (5.4%) | 15 (21.1%) | 5 (16.1%) | 14 (26.9%) | 6 (18.8%) | |
| 5 | 0 (0%) | 0 (0%) | 10 (14.1%) | 8 (25.8%) | 33 (63.5%) | 24 (75.0%) |
Data are presented as mean (S.E.M.) years for age at death and total years and as # yes/# no (%) unless otherwise indicated.
CTE chronic traumatic encephalopathy, AD Alzheimer disease, RHI repetitive head impacts
Total years of contact sports participation primarily available from UNITE and FHS groups. Includes participants who played multiple sports.
includes soccer, lacrosse, mixed martial arts, karate, equestrian, professional wrestling, and martial arts
ANOVA
χ2 test for proportions between all pathology groups
3.2. Beta-amyloid levels are differentially altered in CTE and AD
3.2.1. Changes in CTE (Figure 1A, 1B, 1C, 1D)
Fig. 1. Beta-amyloid levels in frontal cortex of low and high stage CTE.
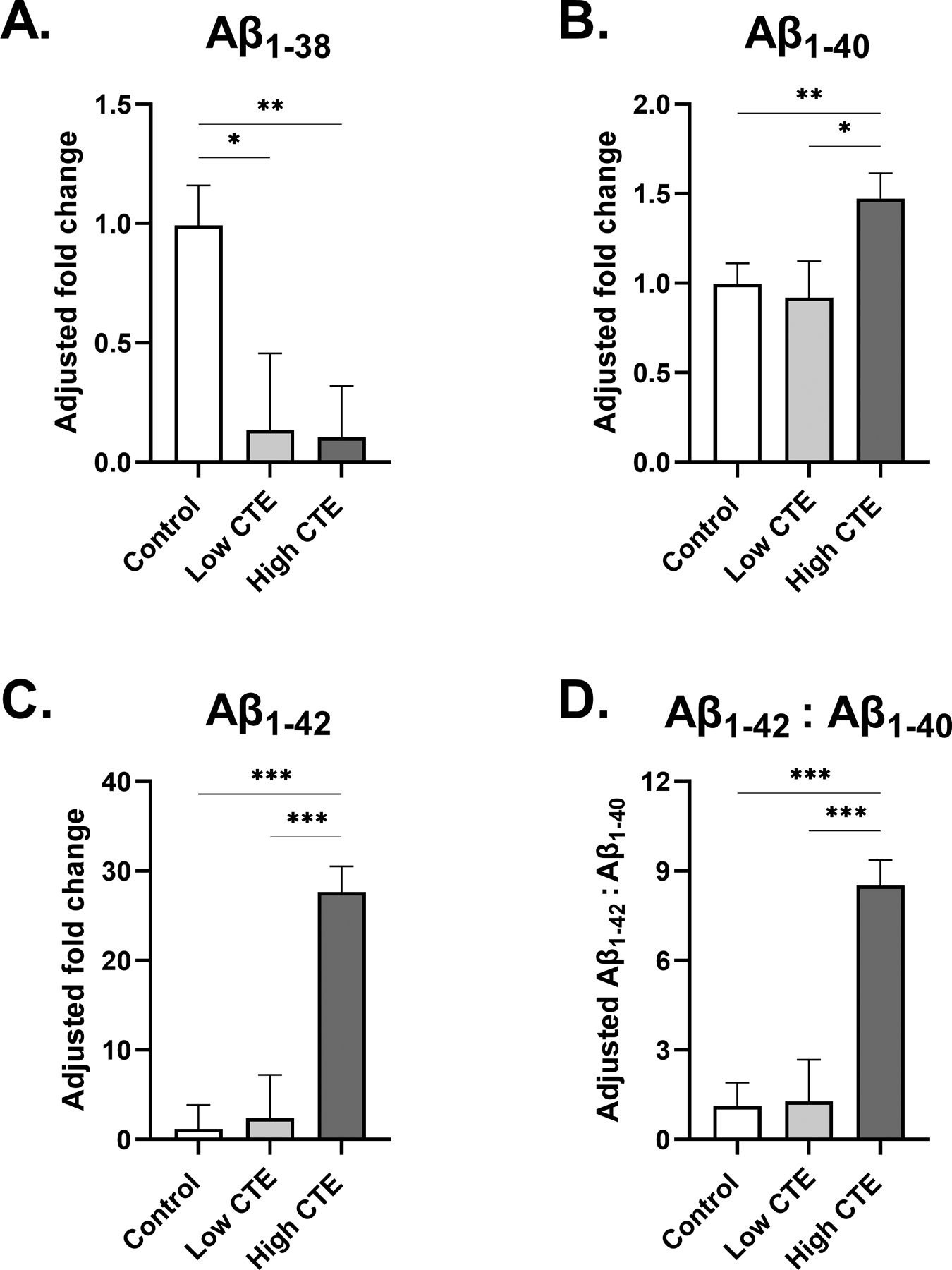
(A-C) Estimated marginal mean and SEM of fold change from control is displayed. (A) Participants with low and high stage CTE displayed significantly decreased levels of Aβ1–38 in comparison to control. (B) Aβ1–40 levels in participants with High CTE were slightly increased in comparison to both control and Low CTE groups. (C) Like Aβ1–40, no significant difference in Aβ1–42 levels was observed between participants of control and Low CTE groups. Participants with High CTE displayed a highly significant, 28-fold increase in Aβ1–42 levels in comparison to the control. (D) Estimated marginal mean and SEM of Aβ1–42:Aβ1–40 ratio is displayed. The High CTE group had significantly increased Aβ1–42:Aβ1–40 levels compared to both control and Low CTE groups. For (A-D) *p<0.05, **p<0.01, ***p<0.001, ANCOVA adjusting for age at death.
Both Low CTE and High CTE groups had significantly lower levels of Aβ1–38 compared to the control group (p=0.019 and p=0.001, respectively). Aβ1–40 fold change to control was significantly increased in High CTE (1.5-fold change from control, SEM=0.14, p=0.009), while no differences were observed between the control and Low CTE groups. Similarly, although there was no significant fold change of Aβ1–42 between control and Low CTE groups, Aβ1–42 was greatly increased in High CTE, statistically significant from Low CTE and control groups (28-fold change from control, SEM=2.9, p<0.001). It is evident that while Aβ1–38 was a low abundant species in CTE, Aβ1–40, and Aβ1–42 in particular, were the main Aβ species in High CTE. To further examine the relationship between the two, the Aβ1–42:Aβ1–40 ratio was computed, and a significant increase was observed in High CTE compared to the Low CTE and control groups (8.5-fold change from control, SEM=0.86, p<0.0001). Lastly, a secondary analysis was performed to compare beta-amyloid levels between our High CTE and CTE & AD participants (data not shown). As expected, CTE & AD participants exhibited significantly higher Aβ1–38, Aβ1–40, and Aβ1–42 levels (p<0.001).
3.2.2. Changes in AD (Figure 2A, 2B, 2C, 2D)
Fig. 2. Beta-amyloid levels in frontal cortex of low and high stage AD.
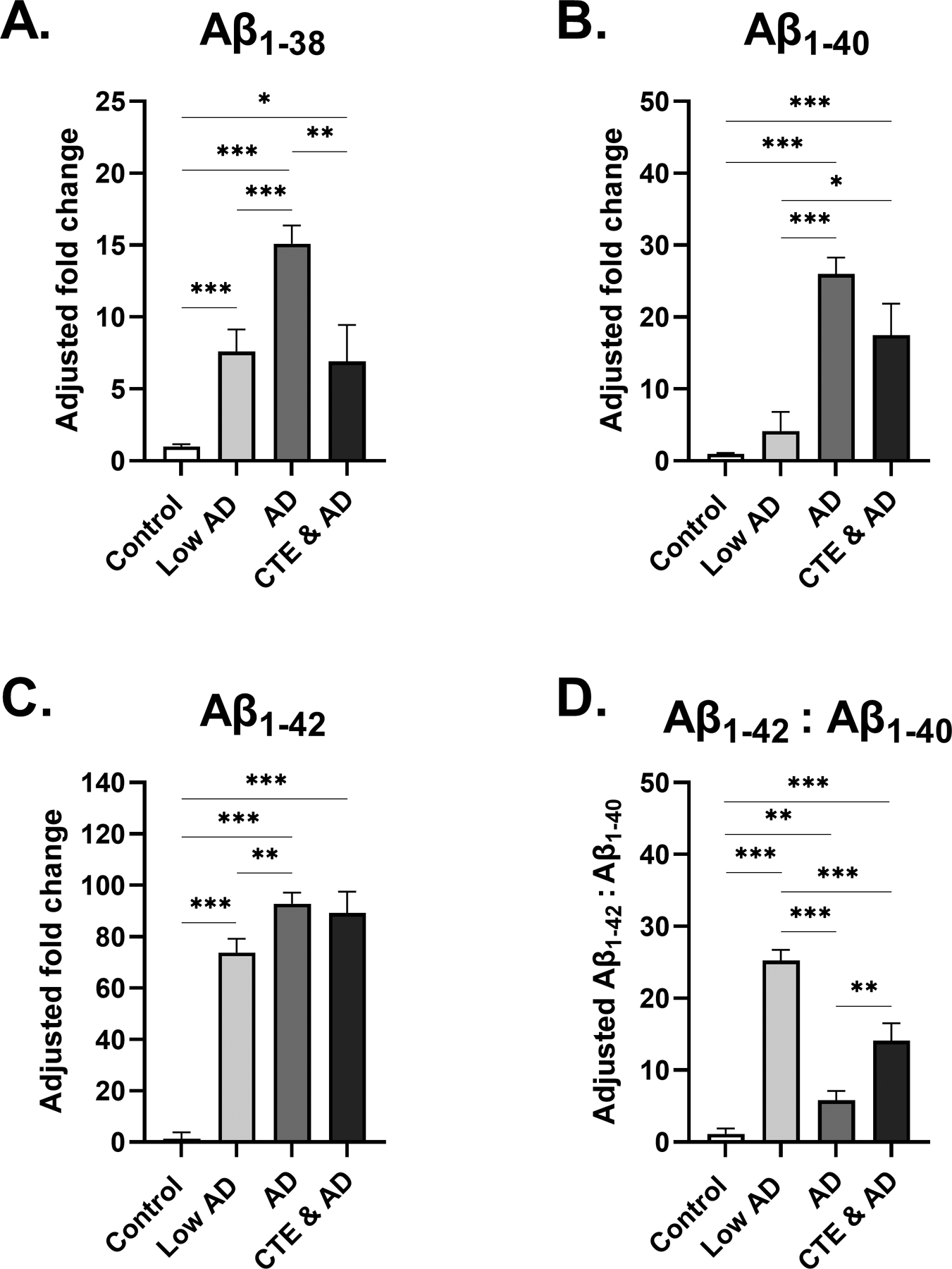
(A-C) Estimated marginal mean and SEM of fold change from control is displayed. (A) Participants with AD displayed significantly increased Aβ1–38 in comparison to control, Low AD, and CTE & AD groups. (B) In participants with AD, Aβ1–40 fold change to control was significantly increased from the control and Low AD group. (C) Aβ1–42 fold change to control was increased even further in participants with Low AD, AD, and CTE & AD, with Aβ1–42 displaying a 93-fold increase with respect to control. (D) Estimated marginal mean and SEM of Aβ1–42:Aβ1–40 ratio is displayed. Aβ1–42 was increased relative to Aβ1–40 in all three groups. However, the greatest increase in Aβ1–42:Aβ1–40 levels was observed in the Low AD group, which was highly significant from controls, AD, and CTE & AD groups. For (A-D) *p<0.05, **p<0.01, ***p<0.001, ANCOVA adjusting for age at death.
It is evident that Low AD, AD, and CTE & AD groups all had much higher levels of all forms of amyloid-beta (Aβ1–38, Aβ1–40, and Aβ1–42) compared to the control and CTE groups. As expected, levels of each Aβ species increased from Low AD to AD (Aβ1–38 p<0.001, Aβ1–40 p<0.001, Aβ1–42 p=0.006). The Low AD group also displayed significantly increased levels of Aβ1–38 (p<0.001) and Aβ1–42 (p<0.0001) compared to controls. However, in AD, the largest fold increase from control was observed in Aβ1–42 (93-fold change from control, SEM=4.3, p<0.0001). In Low AD, the ratio of Aβ1–42:Aβ1–40 displayed a 25-fold increase in Aβ1–42 levels with respect to Aβ1–40 levels, highly significant from both the AD (p<0.001) and CTE & AD groups (p<0.001). Additionally, although significantly less than the increases observed in Low AD and CTE & AD groups, Aβ1–42 was increased 5.8-fold relative to Aβ1–40 in AD (SEM=1.3).
3.3. Tau epitopes are differentially altered in CTE and AD
3.3.1. Changes in CTE (Figure 3A, 3B, 3C, 3D)
Fig 3: Phosphorylated tau levels in frontal cortex of low and high stage CTE.
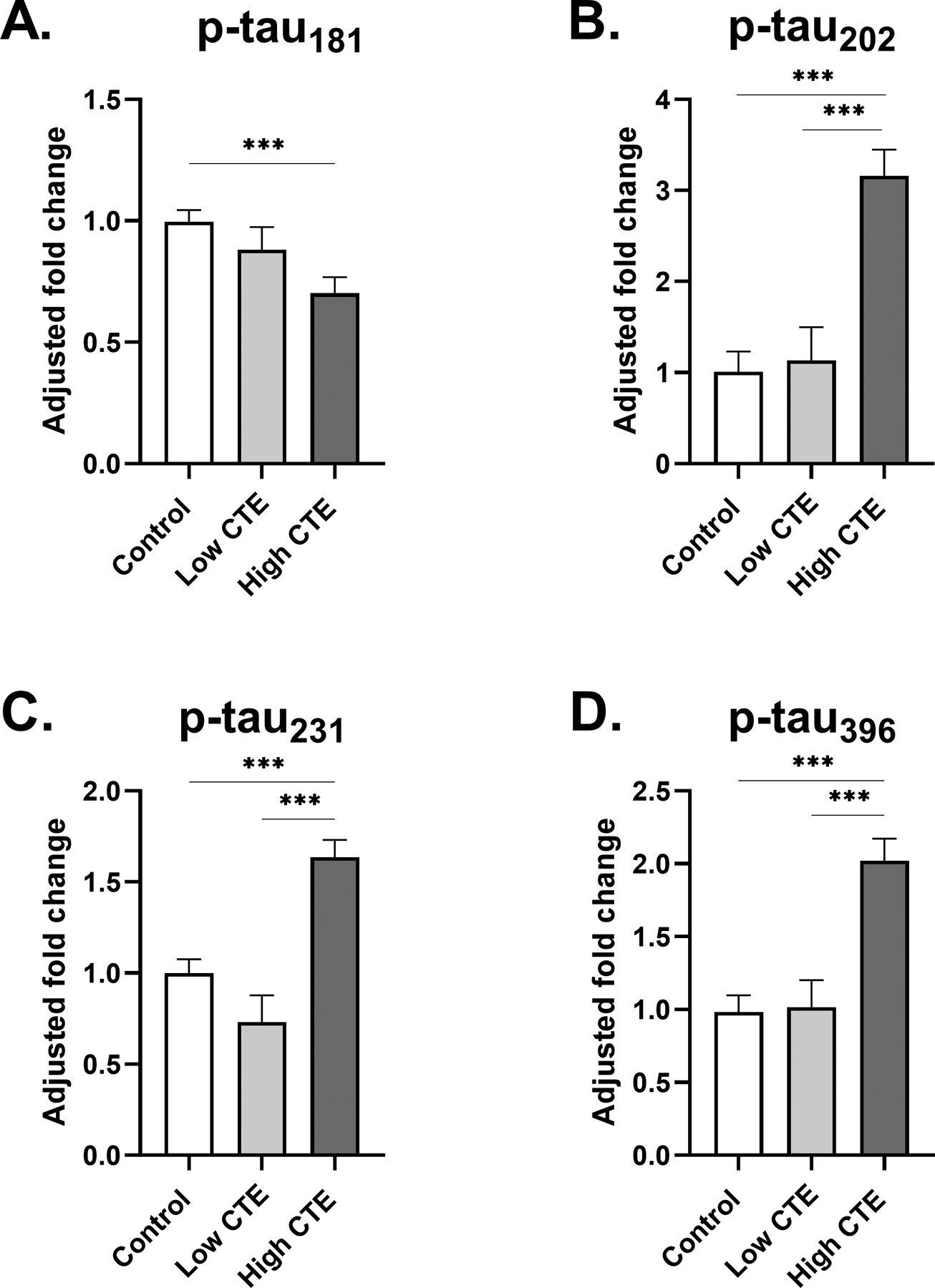
(A-D) Estimated marginal mean and SEM of fold change from control is displayed. (A) A significant fold decrease of p-tau181 with respect to control was observed in participants with High CTE. (B) A large 3.2-fold increase of p-tau202 levels was observed in High CTE, highly significant in comparison to both Low CTE and control groups. (C) In comparison to the control, p-tau231 levels were slightly but significantly increased in participants with High CTE. (D) There was a significant fold increase in p-tau396 compared to both the control group and the Low CTE group. For (A-D) *p<0.05, **p<0.01, ***p<0.001, ANCOVA adjusting for age at death.
The High CTE group demonstrated a significant decrease in p-tau181 in comparison to the control (0.70-fold change from control, SEM=0.07, p<0.001). Participants in the High CTE group displayed a large, highly significant increase in p-tau202 levels compared to the Low CTE and control groups (3.2-fold change from control, SEM=0.29, p<0.001). This increase of p-tau202 was the largest fold increase from control observed among all epitopes studied in CTE. For p-tau231, the High CTE group demonstrated a significant increase compared to the control (1.6-fold change from control, SEM=0.09, p<0.001). Lastly, p-tau396 displayed a fold increase in High CTE, which was statistically significant from both the Low CTE and control groups (2.0-fold change from control, SEM=0.15, p<0.001).
3.3.2. Changes in AD (Figure 4A, 4B, 4C, 4D)
Fig 4: Phosphorylated tau levels in frontal cortex of low and high stage AD.
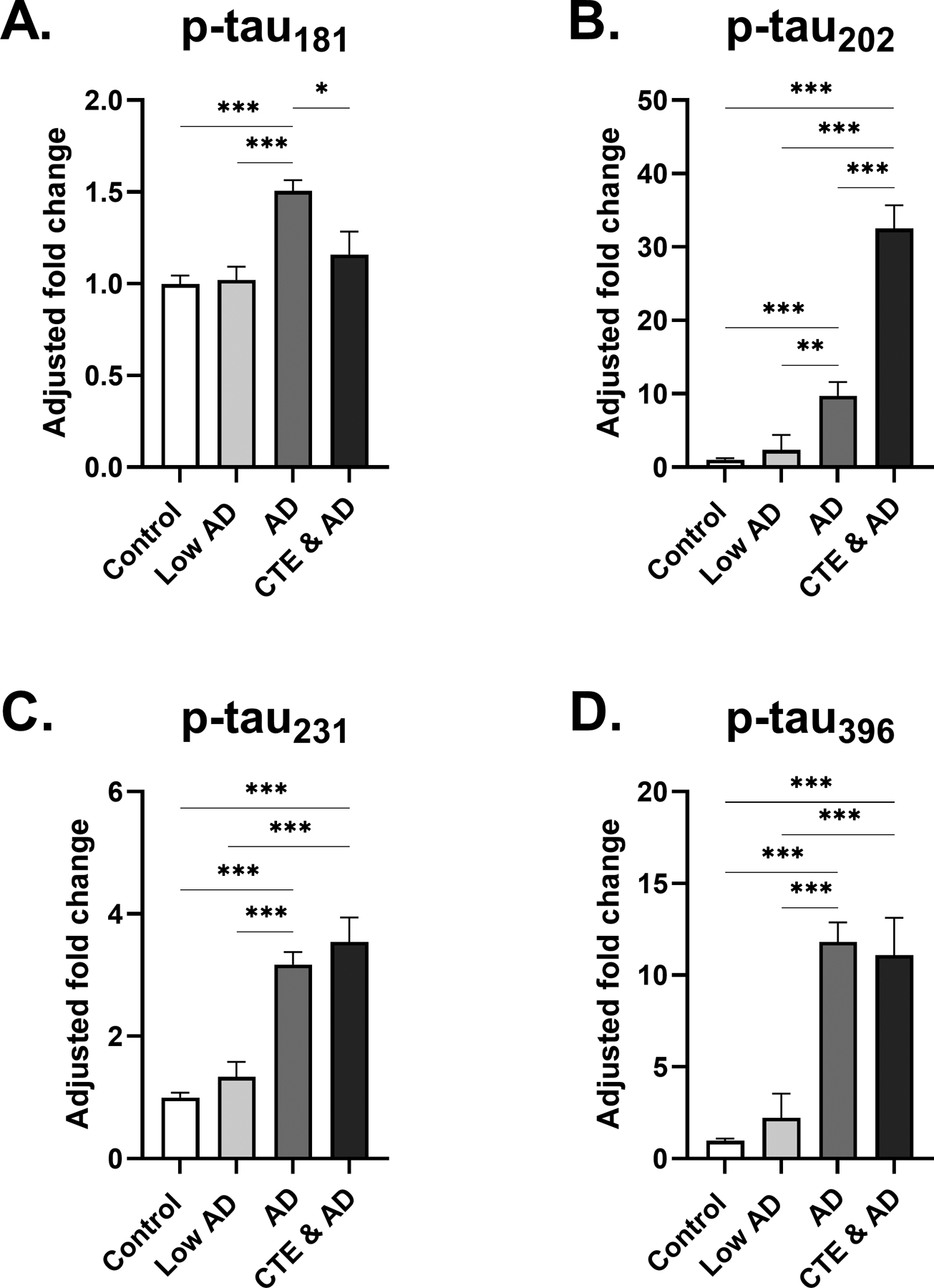
(A-D) Estimated marginal mean and SEM fold change from control is displayed. (A) A small increase in p-tau181 fold change to control was observed in the AD group, highly significant compared to both the control and Low AD group. (B) Participants with AD showed increased fold change to control of p-tau202, with less significance distinguishing from Low AD. Additionally, participants in CTE & AD group showed a highly significant, 33-fold increase in p-tau202 fold change to control. (C) Levels of p-tau231 were significantly increased in AD and CTE & AD groups in comparison to the Low AD and control groups. (D) Similar to trends observed in p-tau202 and p-tau231, participants displayed significant increases in p-tau396 levels from Low AD to AD, and from Low AD to CTE & AD. Additionally, a large 11-fold increase of p-tau396 was observed in the AD group. For (A-D) *p<0.05, **p<0.01, ***p<0.001, ANCOVA adjusting for age at death.
p-tau181, p-tau202, p-tau231, and p-tau396 all were significantly higher in AD when compared to either the control or Low AD group. The greatest increase among the epitopes was shown in p-tau396, where an 11-fold increase to control was observed in AD (SEM=2.0, p<0.001). Of the four epitopes, p-tau202 levels were increased the most in the CTE & AD group (33-fold change to control, SEM=3.2, p<0.0001). p-tau231 and p-tau396 levels were also significantly increased in the CTE & AD group compared with the control, Low AD, and AD groups. Total tau levels did not significantly change within either CTE or AD (data not shown).
3.4. Multiple regression analysis shows total years of play is a significant predictor of p-tau202 levels in CTE (Table 2a)
Table 2:
Multiple linear regression analyses of associations with p-tau epitopes in CTE and AD
| (a) Associations with tau epitopes in participants with and without CTE (excluding AD) | ||||||||
|---|---|---|---|---|---|---|---|---|
| pTau181 | pTau202 | pTau231 | pTau396 | |||||
| Predictor | β | p-value | β | p-value | β | p-value | β | p-value |
| Contact sports play (yrs) | −0.106 | 0.275 | 0.293 | 0.001 | 0.157 | 0.067 | 0.092 | 0.318 |
| Aβ1–42 (pg/mg) | −0.060 | 0.532 | 0.019 | 0.836 | 0.213 | 0.012 | 0.195 | 0.034 |
| (b) Associations with tau epitopes in participants with and without AD (excluding CTE)* | ||||||||
| pTau181 | pTau202 | pTau231 | pTau396 | |||||
| Predictor | β | p-value | β | p-value | β | p-value | β | p-value |
| Aβ1–42 (pg/mg) | 0.129 | 0.071 | 0.205 | 0.006 | 0.249 | <0.001 | 0.312 | <0.001 |
Two different models were run for each of the p-tau epitopes in CTE (a) and AD (b) using multiple linear regression analysis with total years of playing contact sports and Aβ1–42. All variables displayed underwent rank-based normalization, and both models adjusted for age at death and sex. β indicates standardized beta value. Significant associations are shown in bold.
For (a) participants with and without CTE: for pTau181 n=168, for pTau202 n=180, for pTau231 n=202, and for pTau396 n=180.
For (b) Participants with and without AD: for pTau181 n=253, for pTau202 n=244, for pTau231 n=289, and for pTau396 n=259.
Total years of playing contact sports not available for most participants in AD cohort
In order to test the hypothesis that total years of playing contact sports is associated with any of the four p-tau epitopes in CTE, we performed multiple linear regression analyses (Table 2). Controls and both Low and High CTE participants were grouped together and associations were tested between each p-tau epitope and total years of contact sports play and Aβ1–42 levels, adjusting for sex and age at death. Total years of playing contact sports was a highly significant predictor of p-tau202 (β=0.293, p=0.001). There was no significant association of total years of contact sports play with p-tau181, p-tau231, or p-tau396. Additionally, Aβ1–42 was a significant predictor of p-tau231 (β=0.213, p=0.012) and p-tau396 (β=0.195, p=0.034). Secondary linear regression models using Aβ1–40 as a covariate instead of Aβ1–42 showed similar associations between p-tau202 and total years of play (β=0.300, p=0.001). Aβ1–40 was also a significant predictor of both p-tau231 (β=0.292, p<0.001) and p-tau396 levels (β=0.345, p<0.001). A sensitivity analysis using total years of playing football rather than total years of playing contact sports showed that years of football participation was even more significantly associated with p-tau202 (β=0.383, p<0.001) as well as p-tau231 (β=0.273, p=0.001).
3.5. Multiple regression analysis shows Aβ1–42 is most closely related to p-tau396 levels in AD (Table 2B)
Multiple linear regressions were performed in order to evaluate the contributions of Aβ1–42 on the four p-tau epitopes in AD (Table 2). Participants in control, Low AD, and AD were grouped together. Data for total years of playing contact sports was not available for most participants of the AD cohort, and thus only Aβ1–42 was used as a variable, again adjusting for age at death and sex. Increasing Aβ1–42 levels in AD were significantly associated with p-tau202, p-tau231, and p-tau396, however the strongest relationship of the four epitopes was observed at p-tau396 (β=0.312, p<0.001). Subsequent linear regression models using Aβ1–40 instead of Aβ1–42 showed similar significant associations with p-tau202, p-tau231, and p-tau396, and a significant association with p-tau181 also (β=0.189, p=0.007). Again, the strongest relationship of the four epitopes with Aβ1–40 was observed at the p-tau396 epitope (β=0.530, p<0.001).
3.6. Ratio of p-tau202 to p-tau396 is significantly increased in CTE compared to AD at both early and late stages of disease (Figure 5)
Fig 5: Ratio of p-tau202 to p-tau396 is increased in low and high stage CTE compared to AD.
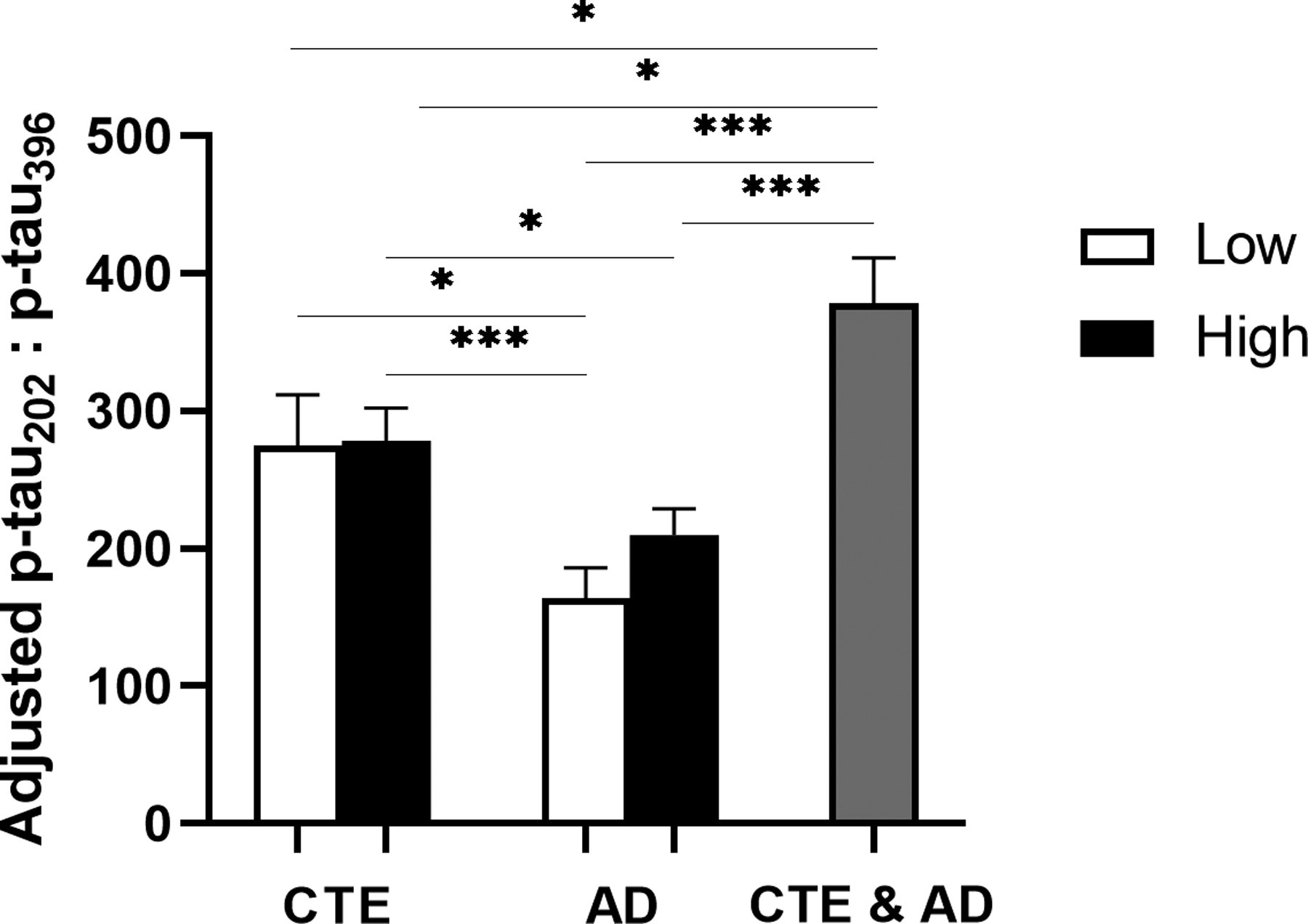
Estimated marginal mean and SEM of ratio of p-tau202 to p-tau396 is displayed. The ratio of p-tau202:p-tau396 was significantly higher in Low CTE as compared to Low AD. p-tau202:p-tau396 was also significantly increased in high stage CTE with respect to both the Low AD and AD groups. No significant differences in p-tau202:p-tau396 were observed between stages of the same disease. CTE & AD group demonstrated the greatest increase of p-tau202:p-tau396 ratio, significant from low and High CTE and Low AD and AD groups. *p<0.05, **p<0.01, ***p<0.001, ANCOVA adjusting for age at death.
Because p-tau202 demonstrated the greatest fold-increase in CTE (Fig 3) and was the only epitope significantly associated with RHI (Table 2a) and p-tau396 showed the greatest fold-increase in AD (Fig 4) and the strongest association with Aβ (Table 2b), we computed the ratio of p-tau202 to p-tau396 at both low and high stages of disease. ANCOVA adjusted for age demonstrated that the ratio of p-tau202 to p-tau396 was significantly increased in Low CTE compared to Low AD (p=0.018). High CTE p-tau202:p-tau396 ratio was also significantly higher than both early and late stages of AD (compared to Low AD: p<0.001; compared to AD: p=0.030). There were no significant differences between p-tau202:p-tau396 ratios of Low CTE and High CTE, or Low AD and AD. Lastly, the CTE & AD group had the highest levels of p-tau202 relative to p-tau396, a statistically significant increase in comparison to every other group.
In this study we exclusively used guanidine hydrochloride for protein extraction. A comparison study between guanidine hydrochloride and 2% SDS extraction solutions showed equivalent amounts of p-tau202, but less p-tau396 with 2% SDS. The two extraction techniques showed similar trends between controls, CTE, and AD participants. In addition, we compared the collapsed McKee CTE stages used here with the newly recommended consensus CTE staging system that determines low and high CTE stage based on a summation of regions involved [40] and found excellent agreement (89%) between the two staging systems in 71 cases that had complete data for the new staging determination. In addition, analysis using the consensus stages in these participants found the same p-tau levels regardless of the classification system.
4. DISCUSSION
This study examined various tau phosphorylation sites in the dorsolateral frontal cortex of participants with early and late stages of CTE and AD, in order to determine disease specific differences and similarities. Of the four epitopes studied, p-tau202 showed the greatest increase compared to control in high-stage CTE, suggesting this site is most involved in CTE. In comparison, in AD p-tau396 was increased the most, while participants with both CTE and AD had the greatest increase in p-tau202. Multiple linear regression analyses demonstrated that total years of contact sports play, a proxy for RHI, was a significant predictor of p-tau202 levels in the frontal cortex. In contrast, Aβ1–42 was most strongly associated with the p-tau396 epitope in both AD and CTE. The ratio of p-tau202 to p-tau396 was significantly higher at different stages of CTE and in CTE & AD compared to AD. Overall, in the dorsolateral frontal cortex, serine202 is the most involved tau phosphorylation site in CTE while serine396 is associated with Aβ and AD.
We combined years of play in multiple sports to look at the effect of total years of contact sport participation. Multiple linear regression analysis showed that years of play was a significant predictor of p-tau202. However, duration of play is likely not equivalent across sports types. The majority of the study participants played American football, and after limiting the sample to include only total years of football play, our results were strengthened. In addition, the association between years of play and ptau231 went from being almost significant to highly significant when only football years were included in the model, suggesting that ptau231 is also affected by RHI. Overall, in our groups football years is a main driver of the findings relating total years of contact sport participation and p-tau.
Much of what is known about tau phosphorylation has been derived from the study of AD [41] [42] [43] [19]. In line with our own findings, p-tau202 has previously been shown to be increased in AD compared to Low AD [44]. Additionally, hyperphosphorylation at p-tau202 and p-tau231 has been identified as a critical step in prompting microtubule instability [45] [46], a key component in the biological mechanism of degeneration that is likely common to AD and CTE. Here we show that in the frontal cortex in AD, p-tau396 is increased the most. Prior studies support the involvement of p-tau396 in AD and suggest that phosphorylation at this site is a vital step in disrupting normal microtubule assembly and developing neurofibrillary pathology [47] [48]. On the other hand, Neddens and colleagues found that p-tau231 showed the most evidence of progression with AD pathology using immunofluorescent techniques [44]. We also found p-tau231 to have significantly increased from Low AD to AD and to be significantly associated with Aβ in AD.
The variability of tau phosphorylation across different brain regions may in part explain why levels of p-tau181 in the frontal cortex showed no significant increase in CTE compared to controls, and only a slight increase from Low AD to AD. CSF p-tau181 is an established biomarker for AD, and blood p-tau181 is emerging as biomarker and predictor of Aβ and tau pathology [49] [50] [51]. This likely reflects tau phosphorylation changes in the temporal lobe, which is preferentially affected early in AD and later in CTE. Future studies should also focus on quantifying levels of p-tau202 and p-tau396 in CSF and blood. In a preliminary study, plasma exosomal tau levels were observed to be increased in a group of professional football athletes in comparison to controls [52]. If p-tau levels in these compartments reflect the changes observed here in the dorsolateral prefrontal cortex, then p-tau202 could potentially serve as a clinical biomarker for high stage CTE and p-tau396 as a biomarker for AD. The p-tau202:p-tau396 ratio may also be relevant for distinguishing between early stages of both diseases.
Few studies have investigated the role of p-tau202 or other phosphorylation sites in the low and high stages of CTE. A previous study using antibody CP13 to detect p-tau202 and PHF1 for p-tau396 described similar staining between CTE and AD [23]. However, this finding is constrained by both the small sample size and the design of the study. Immunohistochemistry techniques can only detect the presence, and not the relative levels of an epitope. Additionally, measurements were taken from a variety of brain regions depending on each individual case. Drawing from a much larger sample size and using biochemical measurements, our study was able to discriminate quantitative levels of tau epitopes that differed in CTE and AD. Furthermore, we demonstrated an association between years of contact sports play and levels of p-tau202.
Examining what is known about specific kinases and phosphatases that act upon these specific epitopes may provide additional insight into mechanisms of tau phosphorylation in CTE and AD. One study examining tau phosphorylation by glycogen synthase kinase 3β (GSK3β) demonstrated that phosphorylation at p-tau202 was a direct process, while p-tau396 was a two-step mechanism and required prephosphorylation at two other sites by a priming kinase and subsequently GSK3β [53]. This difference in the mechanism of phosphorylation by GSK3β at p-tau202 versus p-tau396 may in part be due to the differing pathological mechanisms of CTE and AD. In cases of CTE and traumatic brain injury (TBI) in particular, the process of tau phosphorylation and protein accumulation may be much more rapid than in AD [54]. The two-step phosphorylation at p-tau396 might correspond with a slower progression of AD pathology. Indeed, phosphorylation at p-tau396 by GSK3β has been identified as an important step in the progression of AD [55] [56] [57]. On the other hand, GSK3β may play a role in tau phosphorylation at Thr175 in CTE [58] and inhibition of GSK3 may have therapeutic effects in reducing TBI- related symptoms [59].
This study has several limitations. Although tau is phosphorylated at over 30 different sites [12] [13] we limited our evaluation to the four epitopes commonly implicated in AD and tauopathies and with available and reliable quantitative immunoassays. It is likely that there are other p-tau epitopes similarly implicated at different stages of CTE and AD that were simply not included in our study, such as p-tau217 [60]. Future studies should consider evaluating a wider variety of p-tau epitopes to more thoroughly delineate the different phosphorylation sites implicated in CTE and AD. In addition, tau phosphorylation may be altered by tau isoforms, which have been shown to change in high stage CTE [24].
CTE can be a patchy disease that is sometimes limited to the sulcal depths. However, due to the difficulty of getting cortex from the sulcus in frozen coronal slabs, we performed our measurements within the gyral crest. Given that the majority of CTE tau pathology is found at the sulcal depths [61], measuring tau from the gyri likely under-estimated the levels of p-tau in CTE in comparison to the p-tau levels in AD, especially in low stage CTE. Additionally, because pathological assessment and biochemical measurements were performed on opposite hemispheres, it is possible that the frozen section biochemistry did not always capture a tau CTE lesion. These factors may explain why we observed no differences in p-tau between controls and Low CTE. Looking at verified lesions and changes at the sulcal depths may be more helpful in identifying Low CTE changes in future studies. Although the dorsolateral prefrontal cortex is affected in early CTE, it is also affected in early stages of amyloid deposition. Future studies should aim to examine additional brain regions to clarify regionality of disease processes. It is also worth noting that a significant number of participants in the High CTE group displayed neuritic plaques, which may have obscured CTE-related changes. Lastly, although this is the largest study to date, variables such as age, sex, and presence of comorbid pathologies necessarily differed between groups. Although we statistically adjusted for some of these factors in our regression analysis, future studies with larger numbers will be necessary to confirm these findings.
In this study we investigated four of the more commonly recognized phosphorylation sites and their roles in the progressive tauopathies of CTE and AD. We found that in the dorsolateral frontal cortex, p-tau202 was consistently the most upregulated site in CTE, and was significantly predicted by total years of playing contact sports, a proxy for RHI. In AD, p-tau396 was the most increased and strongly associated with Aβ1–42. The ratio of p-tau202 to p-tau396 was significantly increased in CTE compared to AD across all stages of disease. These findings may aid in the neuropathological diagnosis and in the development of biomarkers to discriminate between CTE and AD.
ACKNOWLEDGMENTS
Funding:
This work was supported by the United States (U.S.) Department of Veterans Affairs, Veterans Health Administration, Clinical Sciences Research and Development Merit Award (I01-CX001038); Alzheimer’s Association (NIRG-305779, NIRG-362697); National Institute of Aging (RF1AG054156, R56AG057768, RF1AG057768, K23AG046377, U19AG068753, AG08122, AG054076); National Institute of Neurological Disorders and Stroke (U54NS115266, U01NS086659, K23NS102399); National Institute of Aging Boston University AD Center (P30AG13846; supplement 0572063345-5); National Heart, Lung and Blood Institute (75N92019D00031 and HHSN2682015000011); Department of Defense Peer Reviewed Alzheimer’s Research Program (PRARP #13267017); and the Concussion Legacy Foundation. This work was also supported by unrestricted gifts from the Andlinger Foundation and WWE. We gratefully acknowledge the use of resources and facilities at the Edith Nourse Rogers Memorial Veterans Hospital (Bedford, MA) as well as all the individuals whose participation and contributions made this work possible.
Declarations of interest:
Outside of the submitted work, WX received research support from the National Institutes of Health, Veterans Health Administration, Biomedical Laboratory Research and Development, and Veterans Health Administration, Clinical Sciences Research and Development Merit Awards. He reports being principal investigator and co-investigator on clinical trials, and has a patent pending regarding the diagnosis of AD using machine learning. JDC received grant support from the Department of Veterans Affairs Career Development Award and National Institute of Aging Boston University AD Center. YT reports receiving grant support to the Boston University School of Public Health. MLA has received a grant from the National Institute of Neurological Disorders and Stroke (NINDS), royalties from Oxford University, consulting fees from Corino Therapeutics, Inc., and honoraria from the International Neuropsychological Society, American Academy of Neurology, and Neuroscience Grand Rounds at Allegheny General Hospital. JM received support for the submitted work from the NIH and DOD and additional grants from the National Institute of Aging (NIA), Department of Defense, and NINDS. ACM reports grant support for other works from the NINDS/NIA, NIA, and VA, and honoraria from the University of Massachusetts, Montefiore Medical Center, Korean Dementia Society, and Texas Neurological Society. The remaining authors report no relevant conflict of interest.
REFERENCES
- [1].Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med 1973;3:270–303. [DOI] [PubMed] [Google Scholar]
- [2].Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 2005;57:128–34–discussion128–34. [DOI] [PubMed] [Google Scholar]
- [3].McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bieniek KF, Ross OA, Cormier KA, Walton RL, Soto-Ortolaza A, Johnston AE, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol 2015;130:877–89. doi: 10.1007/s00401-015-1502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. Jama 2017;318:360–70. doi: 10.1001/jama.2017.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology 2013;81:1122–9. doi: 10.1212/WNL.0b013e3182a55f7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Montenigro PH, Baugh CM, Daneshvar DH, Mez J, Budson AE, Au R, et al. Clinical subtypes of chronic traumatic encephalopathy: literature review and proposed research diagnostic criteria for traumatic encephalopathy syndrome. Alzheimers Res Ther 2014;6:1–17. doi: 10.1186/s13195-014-0068-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 2015;131:75–86. doi: 10.1007/s00401-015-1515-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stein TD, Alvarez VE, McKee AC. Concussion in Chronic Traumatic Encephalopathy. Curr Pain Headache Rep 2015;19:522. doi: 10.1007/s11916-015-0522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Armstrong RA, McKee AC, Stein TD, Alvarez VE, Cairns NJ. Cortical degeneration in chronic traumatic encephalopathy and Alzheimer’s disease neuropathologic change. Neurol Sci 2019;40:529–33. doi: 10.1007/s10072-018-3686-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Graham DI, Gentleman SM, Lynch A, Roberts GW. Distribution of beta-amyloid protein in the brain following severe head injury. Neuropathol Appl Neurobiol 1995;21:27–34. doi: 10.1111/j.1365-2990.1995.tb01025.x. [DOI] [PubMed] [Google Scholar]
- [12].Gong C-X, Liu F, Grundke-Iqbal I, Iqbal K. Post-translational modifications of tau protein in Alzheimer’s disease. J Neural Transm 2005;112:813–38. doi: 10.1007/s00702-004-0221-0. [DOI] [PubMed] [Google Scholar]
- [13].Liu F, Liang Z, Gong C-X. Hyperphosphorylation of tau and protein phosphatases in Alzheimer disease. Panminerva Med 2006;48:97–108. [PubMed] [Google Scholar]
- [14].Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med 2009;15:112–9. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- [15].Liu F, Li B, Tung E-J, Grundke-Iqbal I, Iqbal K, Gong C-X. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci 2007;26:3429–36. doi: 10.1111/j.1460-9568.2007.05955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wesseling H, Mair W, Kumar M, Schlaffner CN, Tang S, Beerepoot P, et al. Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 2020;183:1699–1713.e13. doi: 10.1016/j.cell.2020.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Luna-Muñoz J, García-Sierra F, Falcón V, Menéndez I, Chávez-Macías L, Mena R. Regional conformational change involving phosphorylation of tau protein at the Thr231, precedes the structural change detected by Alz-50 antibody in Alzheimer’s disease. J Alzheimers Dis 2005;8:29–41. doi: 10.3233/jad-2005-8104. [DOI] [PubMed] [Google Scholar]
- [18].Su JH, Cummings BJ, Cotman CW. Early phosphorylation of tau in Alzheimer’s disease occurs at Ser-202 and is preferentially located within neurites. Neuroreport 1994;5:2358–62. doi: 10.1097/00001756-199411000-00037. [DOI] [PubMed] [Google Scholar]
- [19].Zhou X-W, Li X, Bjorkdahl C, Sjogren MJ, Alafuzoff I, Soininen H, et al. Assessments of the accumulation severities of amyloid beta-protein and hyperphosphorylated tau in the medial temporal cortex of control and Alzheimer’s brains. Neurobiol Dis 2006;22:657–68. doi: 10.1016/j.nbd.2006.01.006. [DOI] [PubMed] [Google Scholar]
- [20].Mondragón-Rodríguez S, Perry G, Zhu X, Moreira PI, Acevedo-Aquino MC, Williams S. Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: implications for Alzheimer’s disease. Oxid Med Cell Longev 2013;2013:940603. doi: 10.1155/2013/940603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Šimić G, Babić Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milošević N, et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016;6:6. doi: 10.3390/biom6010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kanaan NM, Cox K, Alvarez VE, Stein TD, Poncil S, McKee AC. Characterization of Early Pathological Tau Conformations and Phosphorylation in Chronic Traumatic Encephalopathy. J Neuropathol Exp Neurol 2016;75:19–34. doi: 10.1093/jnen/nlv001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Arena JD, Smith DH, Lee EB, Gibbons GS, Irwin DJ, Robinson JL, et al. Tau immunophenotypes in chronic traumatic encephalopathy recapitulate those of ageing and Alzheimer’s disease. Brain 2020;143:1572–87. doi: 10.1093/brain/awaa071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cherry JD, Kim SH, Stein TD, Pothast MJ, Nicks R, Meng G, et al. Evolution of neuronal and glial tau isoforms in chronic traumatic encephalopathy. Brain Pathol 2020. doi: 10.1111/bpa.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Alosco ML, Mez J, Tripodis Y, Kiernan PT, Abdolmohammadi B, Murphy L, et al. Age of First Exposure to Tackle Football and Chronic Traumatic Encephalopathy. Ann Neurol 2018. doi: 10.1002/ana.25245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cherry JD, Mez J, Crary JF, Tripodis Y, Alvarez VE, Mahar I, et al. Variation in TMEM106B in chronic traumatic encephalopathy. Acta Neuropathol Commun 2018;6:115. doi: 10.1186/s40478-018-0619-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mez J, Daneshvar DH, Abdolmohammadi B, Chua AS, Alosco ML, Kiernan PT, et al. Duration of American football play and chronic traumatic encephalopathy. Ann Neurol 2019:ana.25611. doi: 10.1002/ana.25611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Montenigro PH, Alosco ML, Martin B, Daneshvar DH, Mez J, Chaisson C, et al. Cumulative Head Impact Exposure Predicts Later-Life Depression, Apathy, Executive Dysfunction, and Cognitive Impairment in Former High School and College Football Players. J Neurotrauma 2016. doi: 10.1089/neu.2016.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mez J, Solomon TM, Daneshvar DH, Murphy L, Kiernan PT, Montenigro PH, et al. Assessing clinicopathological correlation in chronic traumatic encephalopathy: rationale and methods for the UNITE study. Alzheimers Res Ther 2015;7:62. doi: 10.1186/s13195-015-0148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Au R, Seshadri S, Knox K, Beiser A, Himali JJ, Cabral HJ, et al. The Framingham Brain Donation Program: neuropathology along the cognitive continuum. Curr Alzheimer Res 2012;9:673–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ashendorf L, Alosco ML, Bing-Canar H, Chapman KR, Martin B, Chaisson CE, et al. Clinical Utility of Select Neuropsychological Assessment Battery Tests in Predicting Functional Abilities in Dementia. Arch Clin Neuropsychol 2018;33:530–40. doi: 10.1093/arclin/acx100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Galetta KM, Chapman KR, Essis MD, Alosco ML, Gillard D, Steinberg E, et al. Screening Utility of the King-Devick Test in Mild Cognitive Impairment and Alzheimer Disease Dementia. Alzheimer Dis Assoc Disord 2017;31:152–8. doi: 10.1097/WAD.0000000000000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Vonsattel J-PG, Amaya MDP, Cortes EP, Mancevska K, Keller CE. Twenty-first century brain banking: practical prerequisites and lessons from the past: the experience of New York Brain Bank, Taub Institute, Columbia University. Cell Tissue Bank 2008;9:247–58. doi: 10.1007/s10561-008-9079-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Alosco ML, Cherry JD, Huber BR, Tripodis Y, Baucom Z, Kowall NW, et al. Characterizing tau deposition in chronic traumatic encephalopathy (CTE): utility of the McKee CTE staging scheme. Acta Neuropathol 2020;140:495–512. doi: 10.1007/s00401-020-02197-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer“s Association guidelines for the neuropathologic assessment of Alzheimer”s disease: a practical approach. Acta Neuropathol 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Serrano-Pozo A, Qian J, Muzikansky A, Monsell SE, Montine TJ, Frosch MP, et al. Thal Amyloid Stages Do Not Significantly Impact the Correlation Between Neuropathological Change and Cognition in the Alzheimer Disease Continuum. J Neuropathol Exp Neurol 2016;75:516–26. doi: 10.1093/jnen/nlw026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stein TD, Montenigro PH, Alvarez VE, Xia W, Crary JF, Tripodis Y, et al. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol 2015;130:21–34. doi: 10.1007/s00401-015-1435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Motulsky HJ, Brown RE. Detecting outliers when fitting data with nonlinear regression - a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 2006;7:123–20. doi: 10.1186/1471-2105-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Templeton GF. A Two-Step Approach for Transforming Continuous Variables to Normal: Implications and Recommendations for IS Research. Cais 2011;28:1–20. doi: 10.17705/1CAIS.02804. [DOI] [Google Scholar]
- [40].Bieniek KF, Cairns NJ, Crary JF, Dickson DW, Folkerth RD, Keene CD, et al. The Second NINDS/NIBIB Consensus Meeting to Define Neuropathological Criteria for the Diagnosis of Chronic Traumatic Encephalopathy. J Neuropathol Exp Neurol. 2021. Feb 22;80(3):210–219. doi: 10.1093/jnen/nlab001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Alafuzoff I, Arzberger T, Al-Sarraj S, Bodi I, Bogdanovic N, Braak H, et al. Staging of neurofibrillary pathology in Alzheimer’s disease: a study of the BrainNet Europe Consortium. Brain Pathol 2008;18:484–96. doi: 10.1111/j.1750-3639.2008.00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–59. [DOI] [PubMed] [Google Scholar]
- [43].Koss DJ, Jones G, Cranston A, Gardner H, Kanaan NM, Platt B. Soluble pre-fibrillar tau and β-amyloid species emerge in early human Alzheimer’s disease and track disease progression and cognitive decline. Acta Neuropathol 2016;132:875–95. doi: 10.1007/s00401-016-1632-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Neddens J, Temmel M, Flunkert S, Kerschbaumer B, Hoeller C, Loeffler T, et al. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol Commun 2018;6:52–15. doi: 10.1186/s40478-018-0557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, et al. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. Journal of Neuroscience 2012;32:2430–41. doi: 10.1523/JNEUROSCI.5927-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Katsumoto A, Takeuchi H, Tanaka F. Tau Pathology in Chronic Traumatic Encephalopathy and Alzheimer’s Disease: Similarities and Differences. Front Neurol 2019;10:980. doi: 10.3389/fneur.2019.00980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Evans DB, Rank KB, Bhattacharya K, Thomsen DR, Gurney ME, Sharma SK. Tau phosphorylation at serine 396 and serine 404 by human recombinant tau protein kinase II inhibits tau’s ability to promote microtubule assembly. J Biol Chem 2000;275:24977–83. doi: 10.1074/jbc.M000808200. [DOI] [PubMed] [Google Scholar]
- [48].Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 1993;10:1089–99. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- [49].Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol 2020;19:422–33. doi: 10.1016/S1474-4422(20)30071-5. [DOI] [PubMed] [Google Scholar]
- [50].Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, et al. Plasma P-tau181 in Alzheimer“s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer”s dementia. Nat Med 2020;26:379–86. doi: 10.1038/s41591-020-0755-1. [DOI] [PubMed] [Google Scholar]
- [51].Tatebe H, Kasai T, Ohmichi T, Kishi Y, Kakeya T, Waragai M, et al. Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: pilot case-control studies including patients with Alzheimer’s disease and down syndrome. Mol Neurodegener 2017;12:63–11. doi: 10.1186/s13024-017-0206-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Stern RA, Tripodis Y, Baugh CM, Fritts NG, Martin BM, Chaisson C, et al. Preliminary Study of Plasma Exosomal Tau as a Potential Biomarker for Chronic Traumatic Encephalopathy. J Alzheimers Dis. 2016;51(4):1099–109. doi: 10.3233/JAD-151028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li T, Paudel HK. Glycogen synthase kinase 3beta phosphorylates Alzheimer’s disease-specific Ser396 of microtubule-associated protein tau by a sequential mechanism. Biochemistry 2006;45:3125–33. doi: 10.1021/bi051634r. [DOI] [PubMed] [Google Scholar]
- [54].Johnson VE, Stewart W, Smith DH. Widespread τ and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol 2012;22:142–9. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Llorens-Martín M, Jurado J, Hernández F, Avila J. GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci 2014;7:46. doi: 10.3389/fnmol.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ochalek A, Mihalik B, Avci HX, Chandrasekaran A, Téglási A, Bock I, et al. Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res Ther 2017;9:90–19. doi: 10.1186/s13195-017-0317-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kolarova M, García-Sierra F, Bartos A, Ricny J, Ripova D. Structure and pathology of tau protein in Alzheimer disease. Int J Alzheimers Dis 2012;2012:731526. doi: 10.1155/2012/731526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Moszczynski AJ, Strong W, Xu K, McKee A, Brown A, Strong MJ. Pathologic Thr175 tau phosphorylation in CTE and CTE with ALS. Neurology 2018;90:e380–7. doi: 10.1212/WNL.0000000000004899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Shim SS, Stutzmann GE. Inhibition of Glycogen Synthase Kinase-3: An Emerging Target in the Treatment of Traumatic Brain Injury. J Neurotrauma 2016;33:2065–76. doi: 10.1089/neu.2015.4177. [DOI] [PubMed] [Google Scholar]
- [60].Janelidze S, Stomrud E, Smith R, Palmqvist S, Mattsson N, Airey DC, et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nature Communications 2020;11:1683. doi: 10.1038/s41467-020-15436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].McKee AC, Stein TD, Kiernan PT, Alvarez VE. The neuropathology of chronic traumatic encephalopathy. Brain Pathol 2015;25:350–64. doi: 10.1111/bpa.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]