

Summary
Candida albicans is a leading cause of death due to systemic fungal infections. Poor patient outcomes are attributable to the limited number of antifungal classes and the increasing prevalence of drug resistance. Protein kinases have emerged as rewarding targets in the development of drugs for diverse diseases, yet kinases remain untapped in the quest for new antifungals. Here, we performed a comprehensive analysis of the C. albicans kinome to identify genes for which loss-of-function confers hypersensitivity to the two most widely deployed antifungals, echinocandins and azoles. Through this analysis, we found a role for the casein kinase 1 (CK1) homologue Hrr25 in regulating tolerance to both antifungals as well as target-mediated echinocandin resistance. Follow-up investigations established that Hrr25 regulates these responses through its interaction with the SBF transcription factor. Thus, we provide insights into the circuitry governing cellular responses to antifungals and implicate Hrr25 as a key mediator of drug resistance.
Subject areas: Biological sciences, Molecular biology, Microbiology
Graphical abstract
Highlights
-
•
Screening Candida albicans kinase mutants reveals 47 regulators of antifungal tolerance
-
•
Hrr25 is important for growth and cell wall/membrane stress tolerance
-
•
Hrr25 enables target-mediated echinocandin resistance
-
•
Hrr25 interacts with the SBF transcription factor complex
Biological sciences; Molecular biology; Microbiology
Introduction
Invasive fungal infections exert a devastating impact on human health worldwide, with attributable mortality rates estimated at 1.5 million deaths per year (Brown et al., 2012; Fisher et al., 2018). In the last two decades, the prevalence of systemic fungal disease has significantly risen because of the increasing number of individuals with compromised immune function (Enoch et al., 2017; Pfaller and Diekema, 2010). Candida species, and in particular C. albicans, are the leading cause of hospital-acquired bloodstream infections in North America, with devastating mortality rates of ∼40% (Brown et al., 2012; Pfaller and Diekema, 2007; Pfaller et al., 2019; Wisplinghoff et al., 2004). Despite the detrimental health risks associated with pathogenic fungi, only three major classes of antifungal drugs exist to treat systemic fungal infections. The echinocandins inhibit (1,3)-β-D-glucan biosynthesis, a major structural component of the fungal cell wall; the azoles inhibit ergosterol biosynthesis, the major sterol of fungal cell membranes; and the polyenes directly bind to and extract ergosterol from the plasma membrane (Lee et al., 2021). Alarmingly, the increase in the prevalence of pathogenic fungi that are resistant to these antifungal classes has contributed to therapy failures in the clinic (Lee et al., 2021), highlighting the urgent need to develop antifungals to combat invasive fungal diseases.
Protein kinases (PKs) are key cell signaling enzymes that serve as richly rewarding drug targets to treat diverse diseases ranging from cancer to metabolic disorders (Zhang et al., 2009). However, kinases remain completely untapped as a target class in the quest for new, mechanistically distinct antifungals, even though select studies have demonstrated they are important for cellular processes that influence C. albicans growth, virulence, and drug resistance. For instance, the target of rapamycin (TOR) kinase plays a major role in governing cell growth and proliferation in response to nutrient availability, and pharmacological inhibition of TOR kinase signaling potentiates azole activity in vitro and in mammalian models of fungal pathogenesis (Shekhar-Guturja et al., 2016a; 2016b). Another notable example is protein kinase C (Pkc1), a key regulator of cellular stress responses. Genetic or pharmacological inhibition of C. albicans Pkc1 enhances azole and echinocandin efficacy and attenuates virulence in a mouse model of disseminated candidiasis (LaFayette et al., 2010; Munro et al., 2007; Sussman et al., 2004). Finally, pharmacological inhibition of the casein kinase Yck2 restores echinocandin efficacy against a drug-resistant C. albicans strain (Caplan et al., 2020). Thus, PKs are attractive targets for antifungal drug development.
Large-scale functional genomic analyses are a valuable strategy to investigate the diverse roles of PKs in the model yeast Saccharomyces cerevisiae, as well as pathogenic fungi (Bimbó et al., 2005; Lee et al., 2016b; de Souza et al., 2013; Wang et al., 2011; Youn et al., 2017). In C. albicans, systematic approaches have been used to evaluate the contributions of PKs in growth and stress tolerance (Bar-Yosef et al., 2018; Blankenship et al., 2010; Ramírez-Zavala et al., 2013). Screening a homozygous transposon mutant library of non-essential C. albicans protein kinases for sensitivity to different stressors revealed an expanded role for kinase signaling in cell wall integrity relative to S. cerevisiae (Blankenship et al., 2010). In addition, a collection of protein kinases and their regulators under the control of a tetracycline-inducible promoter was used to define the impact of kinase overexpression on filamentous growth of C. albicans (Bar-Yosef et al., 2018; Ramírez-Zavala et al., 2013). Finally, the gene replacement and conditional expression (GRACE) library has been employed to study genes important for fitness, drug susceptibility, and virulence in C. albicans (Caplan et al., 2018; Chen et al., 2018; Costa et al., 2019; Fu et al., 2021; Lee et al., 2016a; O’Meara et al., 2015; Roemer et al., 2003). Yet, given that this functional genomics resource remains limited in genome coverage, a comprehensive phenotypic analysis of the C. albicans kinome in antifungal susceptibility has yet to be performed.
Here, we leveraged an expanded GRACE collection that covers all C. albicans PKs and select PK regulators. Using this comprehensive resource, we conducted systematic phenotypic screens to identify genes required for antifungal drug tolerance and identified PKs with previously undescribed roles in this trait. We focused on the essential casein kinase 1 (CK1) homologue Hrr25, as this was identified as a previously unestablished regulator of both echinocandin and azole susceptibility. Further analyses suggested that Hrr25 regulates antifungal tolerance and resistance through physical interactions with the SBF transcription factor in C. albicans. Overall, our work provides insights into the cellular signaling governing antifungal tolerance and resistance and uncovers a role for Hrr25 in these important traits.
Results
Generation of the GRACE PK library and characterization of PKs important for C. albicans growth
The original GRACE library encompassed an extensive collection of 2,356 conditional expression mutants, which covered ∼40% of the C. albicans genome (Roemer et al., 2003). In this system, one allele of a given gene is deleted and the second allele in the diploid pathogen is controlled by a tetracycline-repressible promoter (tetO) (Roemer et al., 2003). Thus, conditional repression of a gene of interest can be achieved by the addition of tetracycline or the analogue doxycycline (DOX), enabling the analysis of both essential and non-essential gene function (Figure 1A). Recently, we expanded the GRACE library by 866 additional GRACE mutants (termed GRACEv2) (Fu et al., 2021). All 108 PKs present in the C. albicans genome and 55 select PK regulators were included in this set (Figure 1B), which were compiled by searching the Candida Genome Database (CGD) for all genes annotated with this function and cross-referenced to a previous study that generated a tetracycline-inducible PK expression library (Ramírez-Zavala et al., 2013; Skrzypek et al., 2017).
Figure 1.

Systematic screening of the GRACE PK collection to identify kinases important for fungal growth
(A) Schematic of GRACE strains. In this system, one allele of a given gene is deleted and the second allele in the diploid genome is placed under the control of a tetracycline-repressible promoter (tetO).
(B) Leveraging the GRACE (blue) and GRACEv2 (purple) libraries provides comprehensive coverage of C. albicans PKs and select PK regulators.
(C) Colony images of hits from the essentiality screen. The GRACE PK library was grown overnight in YPD with and without DOX (100 μg/mL). Cells were then transferred onto YNB agar with or without DOX (100 μg/mL) and following 48 h of incubation at 30°C, images were taken using a ChemiDoc XRS + imager. Sixteen of the 163 mutant strains exhibited severe growth defects following DOX-mediated transcriptional repression.
(D) Growth curve validation of screening hits. All 16 GRACE strains identified as hits were subjected to an overnight growth in YPD at 30°C and a second overnight growth in YPD with 100 μg/mL of DOX before cells were pinned with a microplate replicator into 96-well plates containing liquid YNB supplemented with 100 μg/mL of DOX . Cells were incubated for 24 h and the optical density (OD600) of each well was measured every hour. Area under the curve (AUC) was calculated by the trapezoidal method using GraphPad Prism. Error bars represent SEM of technical quadruplicates. Testing was repeated as an independent biological replicate to confirm results.
Given that essential genes are compelling targets for antifungal drug development, we first set out to define those PKs and PK regulators that are required for growth in C. albicans. To this end, the PK library was grown overnight in YPD followed by a subculture into fresh YPD in the presence or absence of a high concentration of DOX (100 μg/mL). Following growth under these conditions for 24 h, cells were stamped onto minimal medium in the presence or absence of DOX (100 μg/mL) and colony images were acquired following a 48-h incubation at 30°C. Of the 163 genes assessed, transcriptional repression of 16 resulted in little to no growth in both technical replicates (Figure 1C). These signaling regulators governed diverse biological processes including cell cycle, organelle organization, cytoskeletal organization, and response to stress. The DOX-dependent growth defects of the 16 strains identified as hits were confirmed by performing growth curves in liquid medium (Figure 1D). Fourteen of the 16 genes identified are annotated as being essential in C. albicans, with PKC1 and CBK1 homozygous deletion mutants reported as viable albeit displaying significant growth defects (Fu et al., 2021). Thus, we describe a conditional gene repression library of all PKs and select PK regulators in C. albicans and determine which kinases are important for growth in this fungal pathogen.
Discovery of C. albicans PKs important for tolerance to echinocandins and azoles
We leveraged the GRACE PK collection to identify genes important for antifungal tolerance, defined as the ability of drug-susceptible strains to grow and survive in the presence of an antifungal at concentrations above the minimum inhibitory concentration (MIC), a trait that can contribute to therapeutic failure (Berman and Krysan, 2020). We monitored growth in the presence of the two most widely deployed classes of antifungals, the echinocandins and azoles. The 163 arrayed mutants were grown overnight in the presence of low (0.05 μg/mL) or high (20 μg/mL) concentrations of DOX to permit the assessment of essential genes or maximize transcriptional repression of nonessential genes, respectively. Cells were subsequently inoculated into fresh medium with the corresponding DOX concentration in addition to the echinocandin caspofungin or the azole fluconazole at concentrations that impaired growth of the parental strain by ∼15%. Growth of each strain was assessed by measuring optical density (OD600) after 24 and 48 h for caspofungin and fluconazole, respectively (Table S3). All screens were performed in technical duplicate and growth was averaged between replicates, given that we observed a positive correlation in all conditions tested (Figure S1). Strains with a significant growth defect in the presence of the antifungal and DOX without exhibiting a significant growth defect in DOX alone were deemed hits.
This analysis identified 47 genes important for antifungal tolerance. Specifically, transcriptional repression of 31 genes conferred hypersensitivity to caspofungin; 14 hits were obtained under the low DOX condition, 8 hits were identified under the high DOX condition, and 9 hits were identified in both screens. Furthermore, transcriptional repression of 16 genes conferred hypersensitivity to fluconazole, with 5 hits identified upon incubation in low DOX, 8 hits obtained upon incubation in high DOX, and 3 hits identified using either DOX concentration (Figure 2A). We identified known regulators of antifungal tolerance, reassuring the validity of our screening approach (Figure 2B). For the caspofungin susceptibility screen, these included PKs involved in the cell wall stress response (BCK1, MKK2, MKC1, PRK1, and SWE1), genes associated with septin function or organization (CDC11, CDC12, and GIN4), and casein kinase 2 regulatory subunits (CKB1 and CKB2) (Blankenship et al., 2010). For the fluconazole susceptibility screen, these included the stress-activated MAPK HOG1 and the RAM signaling regulator CBK1 (Blankenship et al., 2010; Chauhan et al., 2007; Song et al., 2008). Our screens also identified many genes yet to be implicated in antifungal tolerance, including several cell cycle-related genes (CDC7, DBF4, PHO85, CLB4, CAK1, PCL1, KIN4, RAD53, and CSK1), and those involved in RNA metabolism (BUD32, CTK1, and RIO1) for caspofungin and fluconazole, respectively. We also identified Hrr25 as the only kinase important for tolerance to both antifungals that was yet to be implicated in this trait (Figure 2B). Thus, we provide a comprehensive functional genomic analysis of C. albicans PKs important for regulating echinocandin and azole tolerance unveiling several previously unestablished regulators.
Figure 2.

Functional genomic screening of the GRACE PK library identifies regulators of antifungal drug tolerance
(A) Scatter (XY) plots depicting the growth of each strain in the presence and absence of caspofungin or fluconazole upon DOX-mediated transcriptional repression. The GRACE PK library was grown overnight with low DOX (0.05 μg/mL) or high DOX (20 μg/mL). Cells were subsequently inoculated into YPD with the corresponding DOX, in the presence and absence of caspofungin (15 ng/mL) or fluconazole (0.8 μg/mL). Growth (OD600) was measured after 24 and 48 h of incubation at 30°C for the caspofungin and fluconazole screens, respectively. Data points are averages of technical duplicates. Dotted lines indicate the median absolute deviation (MAD) cut off values for each condition. Hits are labeled and displayed in orange. See also Figure S1.
(B) Hits identified from the antifungal drug susceptibility screens. Previously established and unestablished regulators of antifungal tolerance are distinguished by gray and red lines, respectively. Functional groups are indicated by colored boxes (see legend). PKs are specified in bold text. PK regulators are unbolded.
Hrr25 is a key modulator of cell wall and cell membrane stress tolerance
To prioritize kinases for further analysis, we focused on uncharacterized hits that overlapped from the echinocandin and azole screens as they represent attractive targets for the development of potentiators for both antifungal classes (Figure 2B). This narrowed our focus to the casein kinase 1 (CK1) Hrr25. Recently, the CK1 family has attracted attention in the field of antifungal drug discovery through the validation of Yck2 as a promising therapeutic target to combat candidiasis (Caplan et al., 2020). Notably and in contrast to Yck2, Hrr25 is essential for C. albicans growth offering an additional advantage as a potential single-agent antifungal target (Figures 1C and 1D). To verify that Hrr25 was important at mediating azole and echinocandin tolerance, we performed dose-response assays using the HRR25 conditional-expression strain. The susceptibility of each strain to the antifungal was determined by measuring the MIC80. In comparison to the wild-type strain in the absence of DOX, DOX-mediated transcriptional repression of HRR25 conferred >8-fold hypersensitivity to caspofungin and 4-fold hypersensitivity to fluconazole (Figure 3A and S2), confirming the role of this kinase in regulating antifungal tolerance.
Figure 3.

Hrr25 is required for cell wall and membrane stress tolerance and enables target-mediated echinocandin resistance
(A) Transcriptional repression of HRR25 reduces caspofungin and fluconazole tolerance of C. albicans. Wild type (CaSS1) and tetO-HRR25/Δ strains were grown overnight in YPD and then subjected to an additional overnight culture in the absence and presence of 0.05 μg/mL DOX. Dose-response assays were performed in YPD with 2-fold dilution gradients of the antifungal, in the presence or absence of DOX (0.5 μg/mL) as indicated. Growth (OD600) was measured after 72 h of incubation at 30°C, averaged between technical duplicates, and normalized to the wild-type drug-free control (see color bar). All assays were performed in biological duplicate. See also Figure S2.
(B) Transcriptional repression of HRR25 reduces echinocandin and azole susceptibility in C. albicans clinical isolates. DPL15 represents an echinocandin-resistant clinical isolate harboring a mutation in the drug target gene FKS1. CaCi-2 and CaCi-17 represent azole-tolerant and azole-resistant clinical isolates obtained from a patient undergoing azole therapy. Parental and HRR25 conditional-expression strains were grown overnight in YPD and then subject to an additional overnight culture in the absence and presence of DOX (0.5 μg/mL for DPL15 and CaCi-2 backgrounds; 5 μg/mL for CaCi-17 background to achieve sufficient transcriptional repression). Dose-response assays were performed as described in (a), in the presence or absence of DOX (0.5 μg/mL for DLP15 and CaCi-2 backgrounds; 5 μg/mL for CaCi-17 background) as indicated. Growth (OD600) was measured after 72 h. All assays were performed in biological duplicate. See also Figure S2.
(C) Transcriptional repression of HRR25 enhances sensitivity to the cell wall-perturbing agent calcofluor white and the membrane-perturbing agent sodium dodecyl sulfate. Strains were cultured as described in (b) and subject to dose-response assays as described in (a). Growth was measured after 48 h for calcofluor white and 72 h for sodium dodecyl sulfate. All assays were performed in biological duplicate. See also Figure S2.
(D) Deletion of SWI4 and SWI6 confers hypersensitivity to cell wall and membrane stress. Wild type (CB420) and swi4Δ/Δ swi6Δ/Δ strains were grown overnight in YPD and dose-response assays were performed as described in (a). Growth was measured after 72 h for caspofungin and fluconazole and 48 h for calcofluor white and sodium dodecyl sulfate. All assays were performed in biological duplicate.
Next, we investigated whether Hrr25 was important for enabling bona fide resistance to both the echinocandins and azoles. To do so, we placed both alleles of HRR25 under the control of the tetO promoter in an echinocandin-resistant (DPL15) clinical isolate (Park et al., 2005), as well as azole-tolerant (CaCi-2) and azole-resistant (CaCi-17) clinical isolates (White, 1997a). In the absence of DOX, HRR25 was significantly overexpressed relative to wild type, whereas addition of DOX significantly reduced expression in all strain backgrounds (Figure S2). Interestingly, repression of HRR25 expression enhanced caspofungin efficacy by at least 8-fold against an echinocandin-resistant clinical isolate (DPL15), which harbors a mutation in the drug target, FKS1 (Figure 3B). Transcriptional repression of HRR25 increased fluconazole sensitivity by at least 8-fold against the azole-tolerant clinical isolate (CaCi-2), whereas DOX-dependent hypersensitivity to azole was not observed for the azole-resistant isolate (CaCi-17) that both overexpresses drug efflux pumps and harbors a mutant form of the azole target gene ERG11 that impedes azole binding (Figure 3B) (Hill et al., 2015; White, 1997a, 1997b).
Finally, to investigate whether genetic impairment of HRR25 conferred hypersensitivity to other cell wall and cell membrane stressors, we performed dose-response assays using the cell wall-perturbing agent calcofluor white and the membrane-perturbing agent sodium dodecyl sulfate. DOX-mediated transcriptional repression of HRR25 conferred 4-fold hypersensitivity to calcofluor white for the echinocandin-resistant clinical isolate (DPL15), relative to the parental control in the absence of DOX, as determined by the MIC80 (Figure 3C). Genetic depletion of HRR25 increased sodium dodecyl sulfate sensitivity by at least 2- and 4-fold for the azole-tolerant (CaCi-2) and azole-resistant (CaCi-17) clinical isolates, respectively. Taken together, we conclude Hrr25 regulates tolerance to both cell wall and cell membrane stresses and enables target-mediated echinocandin resistance.
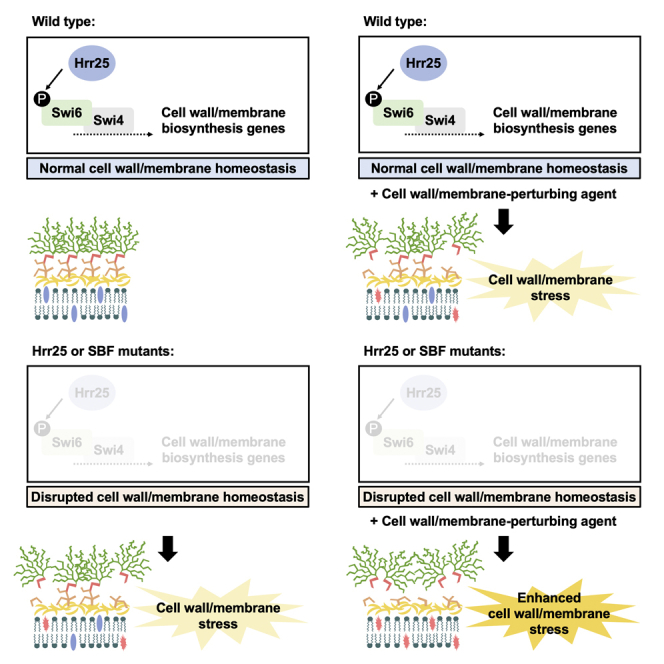
Hrr25 regulates cell wall and membrane stress tolerance through its interaction with the SBF complex
In budding yeast, Hrr25 mediates the DNA damage response through its interaction with the SBF complex, a dimeric complex comprising the cell-cycle regulated transcription factor Swi6 and the DNA binding subunit Swi4 (Ho et al., 1997). Aside from its essential role in cell cycle and DNA repair, SBF participates in the cell wall integrity program as a target of Slt2, the terminal MAPK of the Pkc1 signaling cascade in S. cerevisiae (Levin, 2011). In C. albicans, SBF mediates cell wall and cell membrane stresses through interaction with the Slt2 ortholog Mkc1, and the transcriptional regulator Cas5 (LaFayette et al., 2010; Xie et al., 2017). Indeed, we confirmed homozygous deletion of SWI4 and SWI6 in C. albicans resulted in 8-fold and 4-fold hypersensitivity to the cell wall-perturbing agents, caspofungin and calcofluor white, respectively, as well as a 16- and 4-fold hypersensitivity to the cell membrane-perturbing agents, fluconazole and sodium dodecyl sulfate (Figure 3D), similar to that observed upon transcriptional repression of HRR25.
To test the hypothesis that Hrr25 orchestrates tolerance to cell wall and cell membrane stresses through the SBF complex, we tested whether Hrr25 physically interacts with Swi6 by co-immunoprecipitation. Hrr25 was His6-FLAG3-tagged on both alleles in a strain harboring tandem affinity purification (TAP)-tagged Swi6 on one allele. The His6-FLAG3-tag did not alter the function of Hrr25 given that Hrr25 is essential and this strain was viable under basal conditions. We verified functionality of TAP-tagged Swi6 protein by confirming it did not alter tolerance to caspofungin (Figure S3). We detected an enrichment of Hrr25 following purification of TAP-Swi6, in the strain harboring both TAP-tagged Swi6 and His6-FLAG3-tagged Hrr25, relative to the control strain which lacked TAP-tagged Swi6 (Figure 4A). These results support a model in which Hrr25 regulates cell wall and cell membrane stress tolerance in C. albicans through its interaction with the SBF transcriptional factor complex.
Figure 4.

Hrr25 regulates cell wall andmembrane stress tolerance and homeostasis through its interaction with Swi6
(A) Hrr25 physically associates with Swi6 in C. albicans. C-terminally TAP-tagged Swi6 was affinity purified with immunoglobulin G (IgG) beads followed by tobacco etch virus (TEV)-protease cleavage and a second round of purification using calmodulin beads. The His6-FLAG3-tagged Hrr25 was monitored by western blot analysis and detected using an α-FLAG antibody. Swi6 pull down was confirmed using an α-TAP antibody. Input samples confirm the expression of tagged proteins. Testing was repeated as an independent biological replicate to confirm results. See also Figure S3.
(B and C) Hrr25 and SBF mutants exhibit a cell wall damage response in the absence of exogenous stress. Wild type (CaSS1), tetO-HRR25/Δ, and tetO-CCT8/Δ strains were grown overnight in YPD and then subjected to an additional overnight culture in presence or absence of 0.05 μg/mL DOX. Wild type (CB420) and the swi4Δ/Δ swi6Δ/Δ strains were subject to a single overnight culture in YPD. Strains were then diluted to an OD600 of 0.1 in YPD and grown at 30°C for 4 h in shaking conditions in either the presence or absence of DOX as indicated (0.05 μg/mL for tetO-CCT8/Δ and 5 μg/mL for tetO-HRR25/Δ and wild type). Cells were stained with calcofluor white (CFW) (10 μg/mL) to visualize chitin and imaged by DIC and fluorescence microscopy. Assays were performed in biological duplicate. Scale bar indicates 10 μm and applies to all images. The Corrected Total Cell Fluorescence (CTCF) was calculated for each image using ImageJ. Means are graphed with the error bars representing SD. Signal was compared to the wild-type control and significance was determined by two-way ANOVA (Tukey’s test) or unpaired t-test. Asterisks indicate level of significance (∗∗∗∗p-value < 0.0001).
Finally, we further explored how Hrr25 regulated responses to cell wall stress, given its profound impact on echinocandin tolerance and resistance. We reasoned that either Hrr25 was important for activating cell wall stress response pathways upon detection of diverse cell wall perturbations or depletion of the kinase itself impaired cell wall homeostasis such that the addition of a cell wall stressor exacerbated the effect leading to impaired growth. To address these possibilities, we questioned whether Hrr25 and SBF mutants exhibited a cell wall damage response in the absence of exogenous stress, which would be consistent with the second model. We measured abundance of chitin, an essential component of the fungal cell wall, which accumulates as a well-characterized compensatory response to cell wall stress (Lesage and Bussey, 2006). Strains were grown overnight in YPD, followed by subculture in fresh medium. At mid-log phase cells were stained with calcofluor white , a fluorescent dye that binds primarily to chitin (Roncero and Durán, 1985), and imaged by microscopy. We observed increased calcofluor white staining in cells lacking Hrr25 or SBF, relative to their respective parental controls (Figures 4B and 4C). These observations were consistent with that observed for a control tetO-CCT8/cct8Δ strain, a known cell wall integrity regulator that plays a role in the folding and assembly of cytoskeletal proteins (Caplan et al., 2018). Thus, our observations implicate a role for Hrr25 in maintaining cell wall homeostasis through interactions with the SBF complex.
Discussion
In this study, we leveraged a comprehensive collection of GRACE mutants that encompass all PKs and select PK regulators in the C. albicans genome to identify key modulators of azole and echinocandin susceptibility. Within this collection of 163 mutants, we identified 31 genes important for tolerance to caspofungin and 16 genes important fluconazole susceptibility. Although some of these regulators were previously reported as important modulators of antifungal-induced stress, many had yet to be implicated in these processes. For instance, we uncovered several cell cycle-associated PKs in tolerance to caspofungin, expanding the repertoire of regulators that couple cell cycle dynamics to cell wall integrity (Kono et al., 2016; Xie et al., 2017). Moreover, we identified several genes encoding factors involved in RNA metabolism as being important for withstanding azole-induced stress, providing a new connection between RNA metabolism and antifungal tolerance. Notably, genetic interaction data from the model yeast S. cerevisiae has uncovered a negative genetic interaction between ERG11 and a gene involved in RNA catabolic processes, the translation termination factor SUP35, further suggesting a functional connection between these cellular processes (Costanzo et al., 2016; van Leeuwen et al., 2016; Usaj et al., 2017). Our results highlight that expansion of functional genomic resources to provide comprehensive genome coverage is critical to further define vulnerabilities in fungal pathogens.
Through our chemical-genetic screening efforts, we identified and characterized the CK1 homologue Hrr25 as a previously unestablished modulator of tolerance to both cell wall and membrane stress in C. albicans. Previous studies have implicated this kinase as a core modulator of DNA repair pathways, spindle assembly, and meiosis (Brockman et al., 1992; Fish et al., 1995; Vancura et al., 1994). Hence, the mechanism by which Hrr25 enabled tolerance to antifungal-induced stress in C. albicans remained elusive. Through co-immunoprecipitation, we detected a physical interaction between Hrr25 and the regulatory subunit of the SBF complex, Swi6. In S. cerevisiae, exposure to cell wall damage causes SBF to regulate the transcription of cell wall-related genes including FKS2, which encodes a stress-induced alternative subunit of 1,3-β-D-glucan synthase that is the target of echinocandins (Levin, 2011). In C. albicans, the transcription factors Swi4 and Swi6 have been reported to enable tolerance to cell wall and ergosterol biosynthesis inhibitors, and have also been reported to function downstream of another core signaling kinase, Pkc1 (Caplan et al., 2018; LaFayette et al., 2010). The physical interaction we observed between Swi6 and Hrr25 motivates future exploration of potential crosstalk between Hrr25 and Pkc1 in regulating drug-induced stress responses. Under basal conditions, SBF drives the cell cycle specific transcription of genes involved in membrane and cell wall biosynthesis (Iyer et al., 2001). Consistent with this, Hrr25 and SBF mutant cells exhibited a cell wall damage response even in the absence of exogenous stress, suggesting compromise of this signaling cascade imposes stress on the cell. Overall, our results implicate SBF as an effector of Hrr25 in regulating both cell wall and membrane homeostasis.
PKs are one of the most intensively pursued protein classes in pharmaceutical drug development, particularly in the field of oncology (Zhang et al., 2009). In recent years, kinase inhibitors have shown therapeutic promise as antimicrobials (Carette et al., 2018; Derbyshire et al., 2014). Considering the importance of Hrr25 for C. albicans growth under basal and drug-induced stress conditions, it may represent an attractive target for both mono- and combination therapies. Importantly, we identified Hrr25 as a key regulator of echinocandin resistance as depletion of this kinase enhanced caspofungin efficacy against a drug-resistant C. albicans strain that harbors mutations in the drug target gene FKS1. Given the emergence of Fks1-mediated resistance in C. albicans in clinical settings, identifying regulators of echinocandin resistance that could be targeted by pharmacological inhibitors provides a much-needed strategy to combat drug-resistant infections. Indeed, Hrr25 was identified as a putative target of a kinase inhibitor (GW461484) that possessed both single-agent and echinocandin-potentiating activity against a C. albicans clinical isolate (Caplan et al., 2020). Although targeting a fungal kinase with known human homologs raises concerns for host toxicity, species-selectivity can be improved through chemo-structural approaches (Caplan et al., 2020; Juvvadi et al., 2019; Whitesell et al., 2019). Overall, our functional analysis of the C. albicans kinome advances our understanding of the circuitry governing antifungal drug tolerance and expands the repertoire of fungal targets for therapeutic intervention.
Limitations of study
One notable caveat to the interpretation of this study is that there may be instances where the DOX repression system results in incomplete repression of the target gene. In particular, work has shown that for genes whose function can be fulfilled with very low expression, it is difficult to fully repress gene expression and therefore loss-of-function phenotypes may not be observed. Based on previous observations in S. cerevisiae and E. coli, one would expect a higher rate of these cases among genes with a low baseline level of expression (Arita et al., 2021). Another limitation of this work is that characterization of the C. albicans kinome was restricted to essentiality and antifungal tolerance and other phenotypes such as tolerance to host-relevant stress conditions and morphogenesis would be of interest to pursue in future work. Finally, we chose to follow-up on Hrr25, as it was a previously unestablished hit in both our echinocandin and azole susceptibility screens; however, many of the other hits in our screens were previously unreported regulators of antifungal-induced stress and require further experimentation to elucidate the mechanisms by which they govern antifungal tolerance.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse IgG (Magnetic Bead Conjugate) | Cell Signaling | Cat# 5873; RRID: AB_10835692 |
| Anti-TAP Polyclonal Antibody | Invitrogen | Cat# CAB1001; RRID: AB_10709700 |
| Monoclonal ANTI-FLAG® M2-Peroxidase (HRP) antibody | Sigma-Aldrich | Cat# A8592; RRID: AB_439702 |
| Chemicals, peptides, and recombinant proteins | ||
| Doxycycline Hydrochloride (DOX) | BioBasic | Cat# DB0889 |
| Caspofungin | Merck | N/A |
| Fluconazole | Sequoia Research Products Ltd | N/A |
| Benzonase® Nuclease | Sigma-Aldrich | Cat# 70746 |
| Tobacco etch virus (TEV) | Sigma-Aldrich | Cat# T4455 |
| Calmodulin Sepharose beads | Sigma-Aldrich | Cat# GE17-0529-01 |
| Experimental models: Organisms/strains | ||
| See Table S1 for fungal strains used | This paper | N/A |
| C. albicans GRACE library | Merck/Genome Canada | N/A |
| Oligonucleotides | ||
| See Table S2 for primers used | This paper | N/A |
| Recombinant DNA | ||
| See Table S1 for plasmids used | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com |
| TreeView3 | Keil et al. (2016) | https://bitbucket.org/TreeView3Dev/treeview3/src/master/ |
| ImageJ | Schneider et al. (2012) | https://imagej.nih.gov/ij/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by lead contact Leah E. Cowen (leah.cowen@utoronto.ca).
Materials availability
The use of the GRACE library requires the execution of a fully executed MTA with Merck Sharp & Dohme Corp. Consequently, interested laboratories should contact Scott Walker at scott.walker@merck.com and provide full contact information of the requesting PI, full contact information of the PI’s institution that handles MTAs, and a brief statement of the proposed use of the strain collection(s) requested by the PI. Subsequently, a representative from Merck will contact Leah E. Cowen at leah.cowen@utoronto.ca to arrange distribution. All other unique/stable reagents generated in this study will be provided without restriction as long as stocks remain available and reasonable compensation is provided by requestor to cover processing and shipment.
Experimental model and subject details
Strains and culture conditions
Archives of strains were maintained at −80°C in 25% glycerol. All strains were cultured in YPD (1% yeast extract, 2% bactopeptone, 2% glucose) at 30°C, unless otherwise specified. Active cultures were maintained on solid YPD (2% agar) at 4°C for no longer than 1 month. Strains and plasmids used in this study are listed Table S1. Oligonucleotides used in this study are listed in Table S2.
Assembling the GRACE PK library
To obtain a list of all known and putative PKs and select PK regulators, the Candida Genome Database (CGD) was searched for all genes annotated with this function and cross-referenced our list to a previous study that generated a comprehensive tetracycline-inducible protein kinase expression library (Ramírez-Zavala et al., 2013). To assemble the GRACE PK library, we leveraged 82 strains from the original GRACE library (Roemer et al., 2003) and 81 strains from the GRACEv2 collection (Fu et al., 2021). List of all strains in the GRACE PK library are listed in Table S3.
Method details
Essentiality testing
Strains were pinned into 96-well flat-bottom microtiter plates (Sarstedt) containing liquid YPD and grown overnight at 30°C in static conditions. Cells were transferred via 96-well replicator (∼0.5 μL) into liquid YPD medium using in the absence and presence of 100 μg/mL DOX (Doxycycline Hydrochloride, BioBasic, DB0889) and grown at 30°C overnight. DMSO was the solvent for DOX. Using a microplate metal replicator, ∼2 μL of cells were stamped onto YNB plates (0.17% yeast nitrogen base without amino acids and without ammonium sulfate, with 2% glucose, 0.1% glutamic acid (MSG), and 2% agar), supplemented with histidine, and with or without DOX (100 μg/mL) in technical duplicate. Following 48 h of incubation at 30°C, plates were imaged using the ChemiDoc Imaging System. Mutants that exhibited severe growth defects or no growth in the presence of DOX but grew well in the absence of DOX in both replicates were deemed as hits.
Growth curves
Overnight cultures for strains were prepared as described for essentiality testing on solid medium. Cells were transferred via 96-well pinner (∼0.5 μL) into liquid YNB, supplemented with histidine, with or without DOX (100 μg/mL) in 96-well flat-bottom microtiter plates (Sarstedt). Each strain was tested in technical quadruplicate. Growth (OD600) at 30°C was assessed using a CG-12 Cell-Grower Robot, third generation (S&P Robotics) every 30 minutes for a total of 24 hours. Area under the curve (AUC) was calculated by the trapezoidal method using GraphPad Prism.
Antifungal drug susceptibility screen
DOX, caspofungin (provided by Merck) and fluconazole (Sequoia Research Products) were dissolved in DMSO. GRACE PK mutants were pinned into 96-well flat-bottom microtiter plates (Sarstedt) containing liquid YPD and grown overnight at 30°C in static conditions. The next day, cells were transferred via 96-well pinner (∼0.5 μL) into liquid YPD with 0.05 μg/mL or 20 μg/mL DOX and grown for another 24 hours. To perform the screen, cells were transferred via 96-well pinner (∼0.5 μL) into liquid YPD containing DOX (0.05 μg/mL or 20 μg/mL), and YPD containing DOX and 15 ng/μL caspofungin or 0.8 μg/mL fluconazole. Following 24 and 48 hours of incubation at 30°C for the caspofungin and fluconazole screen, respectively, absorbance was measured at 600 nm using a spectrophotometer (Molecular Devices) to assess growth. Screens were performed in technical duplicate and growth of replicates were averaged. Strains with growth defects in the antifungal drug (OD600 < 1.5 MADs below the median for caspofungin; OD600 < 3 MADs below the median for fluconazole) without exhibiting significant growth defects in the absence of antifungal (OD600 > 7 or > 4 MADs below the median for YPD at 24 and 48 hours, respectively) were deemed as hits.
Dose response assays
DOX, caspofungin and calcofluor white (Sigma-Aldrich) were dissolved in DMSO. Fluconazole was dissolved in DMSO or ddH2O. Sodium dodecyl sulfate was dissolved in ddH2O. Cultures were grown overnight in YPD at 30°C while shaking (200 rpm). All conditional expression strains and the parental control were subcultured to an OD600 of 0.1 in YPD with and without the indicated DOX concentration and grown at 30°C while shaking (200 rpm) overnight. Dose response assays were performed in 96-well flat-bottom microtiter plates (Sarstedt) in a final volume of 0.1 mL/well with 2-fold dilutions of each compound in YPD medium. For conditional expression mutants, dose response assays were set up in the presence and absence of the indicated DOX concentration. Approximately 2×103 cells from an overnight culture were inoculated into each well of the 96-well plate and plates were incubated at 30°C in static conditions for the indicated time frame. Absorbance was measured at 600 nm using a spectrophotometer (Molecular Devices) to assess growth. Growth was normalized to the wild-type no-drug controls. Each strain was tested in technical duplicate in two biological replicates. Dose response data was quantitatively displayed with color using the program Java TreeView3.
Cell wall staining and fluorescent microscopy
Strains were grown overnight in YPD at 30°C while shaking (200 rpm). GRACE strains and the parental control were subcultured to an OD600 of 0.1 in YPD with and without the indicated DOX concentration and grown at 30°C while shaking (200 rpm) overnight. Strains were then subcultured to an OD600 of 0.1 into YPD with and without the indicated DOX concentration at 30°C while shaking for 4 hours. Cells were washed in PBS, and resuspended in PBS to an OD600 of 3, and stained with calcofluor white (10 μg/mL) before imaging using a Zeiss Axio Imager.MI (Carl Zeiss) and AxioCam Mrm with AxioVision 4.7 software at 100x magnification. Calcofluor white was viewed under the 4′,6-diamidino-2-phenylindole (DAPI) hybrid filter.
Immunoprecipitation of Hrr25
Strains were grown overnight in YPD at 30°C while shaking (200 rpm) and used to inoculate 50 mL of YPD at an OD600 of 0.2 in a 500 mL Erlenmeyer flask. Cultures were grown for 5 hours at 30°C while shaking (200 rpm), harvested by centrifugation (3000 rpm at 4°C for 5 minutes), washed once with ice-cold ddH2O, snap frozen in liquid nitrogen, and stored at −80°C. Pellets were resuspended in 1 mL of ice-cold lysis buffer (100 mM Tris pH 8.0, 100 mM NaCl, 10% glycerol, 1 mM DTT, 0.05% Tween 20, 1 mM PMSF, Roche™ PhosStop™, cOmplete™ Protease Inhibitor Cocktail) and vortexed with acid-wash glass beads for 2 × 4 minutes at 4°C, with 7 minutes on ice between beatings. Lysates were recovered by a stacked transfer (3000 rpm at 4°C for 5 minutes) and cell debris was removed by centrifugation for 10 minutes at max speed at 4°C and an additional spin for 5 minutes at max speed. Following treatment with Benzonase® Nuclease (Sigma-Aldrich) (20 minutes at 4°C with end-over-end rocking), supernatants were recovered by ultracentrifugation using Beckman TL-100 (60,000 rpm at 4°C for 1.5 hours). Protein concentrations were determined using Bradford assays and normalized to 2 mg/mL. To serve as input controls, a portion of each sample was diluted (1:1) with 2× Laemmli buffer (Laemmli, 1970).
Anti-TAP immunoprecipitations were performed with Mouse IgG (Magnetic Bead Conjugate) (Cell Signaling Technology). Beads were washed 2× with ddH2O, 2× with lysis buffer (described previously), and 50 μL was added to 1 mL lysate (1 mg/mL). Samples were incubated for 2 hours at 4°C with end-over-end rocking, followed by 5× washes with lysis buffer, and 3× washes with Tobacco etch virus (TEV) protease buffer (10 mM Hepes-KOH pH 8.0, 150 mM NaCl, 0.1% NP-40, 0.5 mM EDTA, 1 mM DTT, 1 mM PMSF, Roche™ PhosStop™). Beads were resuspended in 300 μL TEV buffer containing ∼100 units TEV protease (Sigma-Aldrich) and incubated overnight at 4°C with end-over-end rocking.
Calmodulin sepharose beads (Sigma-Aldrich; 60 μL per pull-down) were washed 3× with calmodulin binding buffer (10 mM β-mercaptoethanol, 10 mM Hepes-KOH pH 8.0, 150 mM NaCl, 1 mM MgOAc, 1 mM imidazole, 0.1% NP-40, 2 mM CaCl2, 1 mM PMSF, Roche™ PhosStop™, cOmplete™ Protease Inhibitor Cocktail) and distributed in tubes. Supernatant was transferred from the IgG beads to a fresh tube, 300 μL of calmodulin binding buffer was added, and the mixture was incubated for 5 minutes end-over-end at room temperature. Supernatant was combined with the first wash, and this was repeated twice to obtain a combined TEV eluate of 1200 μL. To the eluate, 5 μL of 1 M CaCl2 was added and mixed by inversion. Supernatant was transferred to the tube containing the calmodulin-sepharose beads and incubated for 2 hours at 4°C with end-over-end rocking. The slurry was transferred to a Bio-spin column and the flow through recovered in an Eppendorf tube. Beads were washed 1× with calmodulin-binding buffer followed by 2× washes with calmodulin rinsing buffer (50 mM NH4HCO3 pH 8.0, 75 mM NaCl, 1 mM MgOAc, 1 mM imidazole, 2 mM CaCl2, 1 mM PMSF, Roche™ PhosStop™, cOmplete™ Protease Inhibitor Cocktail). Beads were resuspended in 100 μl of 1x Laemmli buffer and boiled for 5 minutes to elute the proteins.
Western blot analysis
Protein samples were analyzed by western immunoblotting on 10% SDS-PAGE gels. Proteins were wet-transferred onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories) for 1 hour at room temperature with an ice pack. Blots were blocked with 5% skim milk in tris-buffered saline with 0.1% Tween 20 (TBS-T) for 1 hour at room temperature. Molecular weights were determined by electrophoresis of marker proteins from the PageRuler pre-stained protein ladder (Invitrogen).
TAP epitopes were detected using a 1:3000 dilution of anti-TAP Polyclonal Antibody (Invitrogen, #CAB1001) and anti-Rabbit FITC secondary antibody diluted 1:3000 in the block solution. FLAG epitopes were detected using Monoclonal ANTI-FLAG® M2-Peroxidase (HRP) antibody (Sigma-Aldrich, #A8592) diluted 1:3000 in TBS-T. Blots were washed with TBS-T before detecting the signals using the Clarity™ Western ECL Substrate (Bio-Rad). All blots were performed in at least biological duplicate.
Reverse transcriptase quantitative PCR (RT-qPCR)
Strains were grown overnight in YPD at 30°C while shaking (200 rpm). GRACE strains and the parental control were subcultured to an OD600 of 0.1 in YPD with and without DOX (0.5 μg/ml or 5 μg/ml) and grown at 30°C while shaking (200 rpm) overnight. The next day, strains were subcultured into YPD (with and without the corresponding DOX) to an OD600 of 0.1 for all wild-type controls and an OD600 of 0.4 for HRR25-depleted strains while shaking for 5 hours at 30°C. Cultures were pelleted and samples were flash-frozen with liquid nitrogen and stored at −80°C. Cell lysis was performed with the MiniBeadBeater-16 (BioSpec Products) using acid-washed glass beads (Sigma G8772-500 g) (4× for 45 seconds, with 1 minute on ice in between). RNA extraction and subsequent DNase treatment was performed with QIAGEN RNeasy kit and Invitrogen DNA-free DNA Removal Kit, followed by cDNA synthesis using iScript cDNA Synthesis Kit (BioRad). The BioRad CFX-384 Real-Time System and Applied Biosystems Fast SYBR Green Master Mix were used to perform RT-qPCR (10 μl reaction volume in a 384-well plate). The following PCR cycling conditions were used for amplification: 95°C for 3 min, then 95 °C for 10 s and 60 °C for 30 s, for 40 cycles. The melt curve was generated using the following: 95 °C for 10 s and 65°C for 10 s with an increase of 0.5°C per cycle up to 95°C. All reactions were performed in biological duplicates and in technical triplicates using the primer pairs oLC6141/oLC6142 (HRR25), oLC2285/oLC2286 (ACT1), and oLC1988/oLC1989 (TEF1).
Strain construction
CaLC7462
To place HRR25 under the control of a doxycycline-repressible promoter in a clinical isolate background (CaCi-2, CaLC79), a repair cassette containing the TAR-tetO promoter was amplified from genomic DNA from CaLC3786 using oLC5850 and oLC5851, which contain homology immediately upstream and downstream of the HRR25 start codon. The plasmid pLC1041, which contains CRISPR machinery and guide directed to HRR25, was digested with Kpn1-HF and Sac1-HF. The plasmid and repair were transformed into CaCi-2. NAT resistant-transformants were PCR tested for correct integration with primers oLC4714/oLC5853 and for lack of the wild-type promoter using primers oLC5852/oLC5853.
CaLC8121
To place HRR25 under the control of a doxycycline-repressible promoter in an azole-resistant clinical isolate (CaCi-17, CaLC91), a repair cassette containing the TAR-tetO promoter was amplified from genomic DNA from CaLC3786 using primer pair oLC5850/oLC5851, which contain homology immediately upstream and downstream of the HRR25 start codon. The plasmid pLC1041, which contains CRISPR machinery and guide directed to HRR25, was digested with Kpn1-HF and Sac1-HF. The plasmid and repair were transformed into DPL15. NAT resistant-transformants were PCR tested for correct integration with primers oLC4714/oLC5853 and for lack of the wild-type promoter using primers oLC5852/oLC5853. The NAT marker was excised and NAT sensitive colonies were additionally tested with oLC4714/oLC5853 to confirm integration of tetO and oLC5852/oLC5853 to ensure no wild type copies of the promoter were present.
CaLC8125
To place HRR25 under the control of a doxycycline-repressible promoter in an echinocandin-resistant FKS1T1922C homozygous mutant (DPL15, CaLC990), a repair cassette containing the TAR-tetO promoter was amplified from genomic DNA from CaLC3786 using primer pair oLC5850/oLC5851, which contain homology immediately upstream and downstream of the HRR25 start codon. The plasmid pLC1041, which contains CRISPR machinery and guide directed to HRR25, was digested with Kpn1-HF and Sac1-HF. The plasmid and repair were transformed into DPL15. NAT resistant-transformants were PCR tested for correct integration with primers oLC4714/oLC5853 and for lack of the wild-type promoter using primers oLC5852/oLC5853. The NAT marker was excised and NAT sensitive colonies were additionally tested with oLC4714/oLC5853 to confirm integration of tetO and oLC5852/oLC5853 to ensure no wild type copies of the promoter were present.
CaLC7471
C-terminal 6xHis3-Flag tagging of HRR25 was achieved by a transient CRISPR in CaLC3393 (SWI6-TAP/SWI6). The sgRNA was generated by gene specific primers oLC9126/oLC9127 as well as the universal primers oLC6926/oLC6927/oLC6928/oLC6929 from pLC1081. The HRR25 tagging cassette was amplified from pLC1084 by oLC9125/oLC8270. NAT-resistant colonies were PCR tested for correct integration with primers oLC9129/oLC6942 and for absence of untagged allele using primers oLC9129/oLC9130.
CaLC8115
C-terminal 6xHis3-Flag tagging of HRR25 was achieved by a transient CRISPR in a wild type SN95 background (CaLC239). The sgRNA was generated by gene specific primers oLC9126/oLC9127 as well as the universal primers oLC6926/oLC6927/oLC6928/oLC6929 from pLC1081. The HRR25 tagging cassette was amplified from pLC1084 by oLC9125/oLC8270. NAT-resistant colonies were PCR tested for correct integration with primers oLC9129/oLC6942 and for absence of untagged allele using primers oLC9129/oLC9130.
Quantification and statistical analysis
GraphPad Prism was used to generate graphical displays and perform tests of significance. Statistical details and replicate numbers for each experiment are provided in the corresponding figure legends. Values of p < 0.05 were considered significant.
Acknowledgments
We thank all current and past Cowen lab members for helpful discussions, and in particular, Luke Whitesell and Zhongle Liu for their input on the biochemical experiments. We thank Merck and Genome Canada for making the GRACE and HET C. albicans mutant collections available; and André Nantel and the National Research Council of Canada for their distribution. L.E.C. is supported by the Canadian Institutes of Health Research (CIHR) Foundation grant (FDN-154288) and National Institutes of Health (NIH) R01 grants (R01AI127375 and R01AI162789); L.E.C. is a Canada Research Chair (Tier 1) in Microbial Genomics & Infectious Disease and co-Director of the CIFAR Fungal Kingdom: Threats & Opportunities program. Y.L. is supported by a CIHR Frederick Banting and Charles Best Canada Graduate Scholarship – Doctoral Award.
Author contributions
Y.L., N.R., and L.E.C. designed the experiments and interpreted the data. Y.L. performed the experiments. Y.L. and D.L. constructed GRACE strains. S.D.L. provided guidance on the biochemical experiments. Y.L. and N.R. wrote the original draft of the manuscript, and all other authors reviewed and edited the final manuscript.
Declaration of interests
L.E.C. is a co-founder and shareholder in Bright Angel Therapeutics, a platform company for development of antifungal therapeutics.
Published: June 17, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104432.
Supporting Citations
The following reference appears in the Supplemental Information: Hussein et al. (2011); Liu and Myers (2017); Min et al. (2016); Noble and Johnson (2005); Veri et al. (2018).
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information requested to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Arita Y., Kim G., Li Z., Friesen H., Turco G., Wang R.Y., Climie D., Usaj M., Hotz M., Stoops E.H., et al. A genome-scale yeast library with inducible expression of individual genes. Mol. Syst. Biol. 2021;17 doi: 10.15252/msb.202110207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Yosef H., Gildor T., Ramírez-Zavala B., Schmauch C., Weissman Z., Pinsky M., Naddaf R., Morschhäuser J., Arkowitz R.A., Kornitzer D. A global analysis of kinase function in Candida albicans hyphal morphogenesis reveals a role for the endocytosis regulator akl1. Front. Cell. Infect. Microbiol. 2018;8:17. doi: 10.3389/fcimb.2018.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman J., Krysan D.J. Drug resistance and tolerance in fungi. Nat. Rev. Microbiol. 2020;18:319–331. doi: 10.1038/s41579-019-0322-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bimbó A., Jia Y., Poh S.L., Karuturi R.K.M., den Elzen N., Peng X., Zheng L., O’Connell M., Liu E.T., Balasubramanian M.K., Liu J. Systematic deletion analysis of fission yeast protein kinases. Eukaryot. Cell. 2005;4:799–813. doi: 10.1128/EC.4.4.799-813.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship J.R., Fanning S., Hamaker J.J., Mitchell A.P. An extensive circuitry for cell wall regulation in Candida albicans. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman J.L., Gross S.D., Sussman M.R., Anderson R.A. Cell cycle-dependent localization of casein kinase I to mitotic spindles. Proc. Natl. Acad. Sci. U S A. 1992;89:9454–9458. doi: 10.1073/pnas.89.20.9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G.D., Denning D.W., Gow N.A.R., Levitz S.M., Netea M.G., White T.C. Hidden killers: human fungal infections. Sci. Transl. Med. 2012;4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- Caplan T., Polvi E.J., Xie J.L., Buckhalter S., Leach M.D., Robbins N., Cowen L.E. Functional genomic screening reveals core modulators of echinocandin stress responses in Candida albicans. Cell. Rep. 2018;23:2292–2298. doi: 10.1016/j.celrep.2018.04.084. [DOI] [PubMed] [Google Scholar]
- Caplan T., Lorente-Macías Á., Stogios P.J., Evdokimova E., Hyde S., Wellington M.A., Liston S., Iyer K.R., Puumala E., Shekhar-Guturja T., et al. Overcoming fungal echinocandin resistance through inhibition of the non-essential stress kinase Yck2. Cell. Chem. Biol. 2020;27:269–282.e5. doi: 10.1016/j.chembiol.2019.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette X., Platig J., Young D.C., Helmel M., Young A.T., Wang Z., Potluri L.-P., Moody C.S., Zeng J., Prisic S., et al. Multisystem analysis of Mycobacterium tuberculosis reveals kinase-dependent remodeling of the pathogen-environment interface. mBio. 2018;9 doi: 10.1128/mBio.02333-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan N., Kruppa M., Calderone R. The Ssk1p response regulator and Chk1p histidine kinase mutants of Candida albicans are hypersensitive to fluconazole and voriconazole. Antimicrob. Agents. Chemother. 2007;51:3747–3751. doi: 10.1128/AAC.00929-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Mallick J., Maqnas A., Sun Y., Choudhury B.I., Côte P., Yan L., Ni T.j. h., Li Y., Zhang D., et al. Chemogenomic profiling of the fungal pathogen Candida albicans. Antimicrob. Agents. Chemother. 2018;62 doi: 10.1128/AAC.02365-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A.C.B.P., Omran R.P., Correia-Mesquita T.O., Dumeaux V., Whiteway M. Screening of Candida albicans GRACE library revealed a unique pattern of biofilm formation under repression of the essential gene ILS1. Sci. Rep. 2019;9:9187. doi: 10.1038/s41598-019-45624-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M., VanderSluis B., Koch E.N., Baryshnikova A., Pons C., Tan G., Wang W., Usaj M., Hanchard J., Lee S.D., et al. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353:aaf1420. doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire E.R., Zuzarte-Luís V., Magalhães A.D., Kato N., Sanschagrin P.C., Wang J., Zhou W., Miduturu C.v., Mazitschek R., Sliz P., et al. Chemical interrogation of the malaria kinome. Chembiochem. 2014;15:1920–1930. doi: 10.1002/cbic.201400025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch D.A., Yang H., Aliyu S.H., Micallef C. In: Methods in Molecular Biology. Lion T., editor. Humana Press; 2017. The changing epidemiology of invasive fungal infections; pp. 17–65. [DOI] [PubMed] [Google Scholar]
- Fish K.J., Cegielska A., Getman M.E., Landes G.M., Virshup D.M. Isolation and characterization of human casein kinase I∊ (CKI), a novel member of the CKI gene family. J. Biol. Chem. 1995;270:14875–14883. doi: 10.1074/jbc.270.25.14875. [DOI] [PubMed] [Google Scholar]
- Fisher M.C., Hawkins N.J., Sanglard D., Gurr S.J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science. 2018;360:739–742. doi: 10.1126/science.aap7999. [DOI] [PubMed] [Google Scholar]
- Fu C., Zhang X., Veri A.O., Iyer K.R., Lash E., Xue A., Yan H., Revie N.M., Wong C., Lin Z.-Y., et al. Leveraging machine learning essentiality predictions and chemogenomic interactions to identify antifungal targets. Nat. Commun. 2021;12:6497. doi: 10.1038/s41467-021-26850-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J.A., O’Meara T.R., Cowen L.E. Fitness trade-offs associated with the evolution of resistance to antifungal drug combinations. Cell. Rep. 2015;10:809–819. doi: 10.1016/j.celrep.2015.01.009. [DOI] [PubMed] [Google Scholar]
- Ho Y., Mason S., Kobayashi R., Hoekstra M., Andrews B. Role of the casein kinase I isoform, Hrr25, and the cell cycle-regulatory transcription factor, SBF, in the transcriptional response to DNA damage in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U S A. 1997;94:581–586. doi: 10.1073/pnas.94.2.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein B., Huang H., Glory A., Osmani A., Kaminskyj S., Nantel A., Bachewich C. G1/S transcription factor orthologues Swi4p and Swi6p are important but not essential for cell proliferation and influence hyphal development in the fungal pathogen Candida albicans. Eukaryot. Cell. 2011;10:384–397. doi: 10.1128/EC.00278-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V.R., Horak C.E., Scafe C.S., Botstein D., Snyder M., Brown P.O. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- Juvvadi P.R., Fox D., Bobay B.G., Hoy M.J., Gobeil S.M.C., Venters R.A., Chang Z., Lin J.J., Averette A.F., Cole D.C., et al. Harnessing calcineurin-FK506-FKBP12 crystal structures from invasive fungal pathogens to develop antifungal agents. Nat. Commun. 2019;10:4275. doi: 10.1038/s41467-019-12199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil C., Leach R.W., Faizaan S.M., Bezawada S., Parsons L., Baryshnikova A. Treeview 3.0 (beta 1)—visualization and analysis of large data matrices. Zenodo. 2016;10:5281. [Google Scholar]
- Kono K., Al-Zain A., Schroeder L., Nakanishi M., Ikui A.E. Plasma membrane/cell wall perturbation activates a novel cell cycle checkpoint during G1 in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U S A. 2016;113:6910–6915. doi: 10.1073/pnas.1523824113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- LaFayette S.L., Collins C., Zaas A.K., Schell W.A., Betancourt-Quiroz M., Gunatilaka A.A.L., Perfect J.R., Cowen L.E. PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, Calcineurin, and Hsp90. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.A., Robbins N., Xie J.L., Ketela T., Cowen L.E. Functional genomic analysis of Candida albicans adherence reveals a key role for the Arp2/3 complex in cell wall remodelling and biofilm formation. PLoS Genet. 2016;12 doi: 10.1371/journal.pgen.1006452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.-T., So Y.-S., Yang D.-H., Jung K.-W., Choi J., Lee D.-G., Kwon H., Jang J., Wang L.L., Cha S., et al. Systematic functional analysis of kinases in the fungal pathogen Cryptococcus neoformans. Nat. Commun. 2016;7:12766. doi: 10.1038/ncomms12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Puumala E., Robbins N., Cowen L.E. Antifungal drug resistance: molecular mechanisms in Candida albicans and beyond. Chem. Rev. 2021;121:3390–3411. doi: 10.1021/acs.chemrev.0c00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen J., Pons C., Mellor J.C., Yamaguchi T.N., Friesen H., Koschwanez J., Ušaj M.M., Pechlaner M., Takar M., Ušaj M., et al. Exploring genetic suppression interactions on a global scale. Science. 2016;354:aag0839. doi: 10.1126/science.aag0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage G., Bussey H. Cell wall assembly in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2006;70:317–343. doi: 10.1128/MMBR.00038-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin D.E. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: the cell wall integrity signaling pathway. Genetics. 2011;189:1145–1175. doi: 10.1534/genetics.111.128264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Myers L.C. Mediator tail module is required for Tac1-activated CDR1 expression and azole resistance in Candida albicans. Antimicrob. Agents. Chemother. 2017;61 doi: 10.1128/AAC.01342-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min K., Ichikawa Y., Woolford C.A., Mitchell A.P. Candida albicans gene deletion with a transient CRISPR-Cas9 system. mSphere. 2016;1 doi: 10.1128/mSphere.00130-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro C.A., Selvaggini S., de Bruijn I., Walker L., Lenardon M.D., Gerssen B., Milne S., Brown A.J.P., Gow N.A.R. The PKC, HOG and Ca2+ signalling pathways co-ordinately regulate chitin synthesis in Candida albicans. Mol. Microbiol. 2007;63:1399–1413. doi: 10.1111/j.1365-2958.2007.05588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble S.M., Johnson A.D. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot. Cell. 2005;4:298–309. doi: 10.1128/EC.4.2.298-309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Meara T.R., Veri A.O., Ketela T., Jiang B., Roemer T., Cowen L.E. Global analysis of fungal morphology exposes mechanisms of host cell escape. Nat. Commun. 2015;6:6741. doi: 10.1038/ncomms7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S., Kelly R., Kahn J.N., Robles J., Hsu M.-J., Register E., Li W., Vyas V., Fan H., Abruzzo G., et al. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob. Agents. Chemother. 2005;49:3264–3273. doi: 10.1128/AAC.49.8.3264-3273.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller M.A., Diekema D.J. Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 2007;20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller M.A., Diekema D.J. Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol. 2010;36:1–53. doi: 10.3109/10408410903241444. [DOI] [PubMed] [Google Scholar]
- Pfaller M.A., Diekema D.J., Turnidge J.D., Castanheira M., Jones R.N. Twenty years of the SENTRY antifungal surveillance program: results for Candida species from 1997–2016. Open. Forum. Infect. Dis. 2019;6:S79–S94. doi: 10.1093/ofid/ofy358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Zavala B., Weyler M., Gildor T., Schmauch C., Kornitzer D., Arkowitz R., Morschhäuser J. Activation of the Cph1-dependent MAP kinase signaling pathway induces white-opaque switching in Candida albicans. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer T., Jiang B., Davison J., Ketela T., Veillette K., Breton A., Tandia F., Linteau A., Sillaots S., Marta C., et al. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol. Microbiol. 2003;50:167–181. doi: 10.1046/j.1365-2958.2003.03697.x. [DOI] [PubMed] [Google Scholar]
- Roncero C., Durán A. Effect of calcofluor white and Congo red on fungal cell wall morphogenesis: in vivo activation of chitin polymerization. J. Bacteriol. 1985;163:1180–1185. doi: 10.1128/jb.163.3.1180-1185.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar-Guturja T., Tebung W.A., Mount H., Liu N., Köhler J.R., Whiteway M., Cowen L.E. Beauvericin potentiates azole activity via inhibition of multidrug efflux, blocks Candida albicans morphogenesis, and is effluxed via Yor1 and circuitry controlled by Zcf29. Antimicrob. Agents. Chemother. 2016;60:7468–7480. doi: 10.1128/AAC.01959-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar-Guturja T., Gunaherath G.M.K.B., Wijeratne E.M.K., Lambert J.-P., Averette A.F., Lee S.C., Kim T., Bahn Y.-S., Tripodi F., Ammar R., et al. Dual action antifungal small molecule modulates multidrug efflux and TOR signaling. Nat. Chem. Biol. 2016;12:867–875. doi: 10.1038/nchembio.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrzypek M.S., Binkley J., Binkley G., Miyasato S.R., Simison M., Sherlock G. The Candida Genome Database (CGD): incorporation of Assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucleic. Acids. Res. 2017;45:D592–D596. doi: 10.1093/nar/gkw924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y., Cheon S.A., Lee K.E., Lee S.-Y., Lee B.-K., Oh D.-B., Kang H.A., Kim J.-Y. Role of the RAM network in cell polarity and hyphal morphogenesis in Candida albicans. Mol. Biol. Cell. 2008;19:5456–5477. doi: 10.1091/mbc.e08-03-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza C.P., Hashmi S.B., Osmani A.H., Andrews P., Ringelberg C.S., Dunlap J.C., Osmani S.A. Functional analysis of the Aspergillus nidulans kinome. PLoS. One. 2013;8 doi: 10.1371/journal.pone.0058008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman A., Huss K., Chio L.-C., Heidler S., Shaw M., Ma D., Zhu G., Campbell R.M., Park T.-S., Kulanthaivel P., et al. Discovery of cercosporamide, a known antifungal natural product, as a selective Pkc1 kinase inhibitor through high-throughput screening. Eukaryot. Cell. 2004;3:932–943. doi: 10.1128/EC.3.4.932-943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usaj M., Tan Y., Wang W., VanderSluis B., Zou A., Myers C.L., Costanzo M., Andrews B., Boone C. TheCellMap.org: a web-accessible database for visualizing and mining the global yeast genetic interaction network. G3 (Bethesda) 2017;7:1539–1549. doi: 10.1534/g3.117.040220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vancura A., Sessler A., Leichus B., Kuret J. A prenylation motif is required for plasma membrane localization and biochemical function of casein kinase I in budding yeast. J. Biol. Chem. 1994;269:19271–19278. doi: 10.1016/s0021-9258(17)32163-4. [DOI] [PubMed] [Google Scholar]
- Veri A.O., Miao Z., Shapiro R.S., Tebbji F., O’Meara T.R., Kim S.H., Colazo J., Tan K., Vyas V.K., Whiteway M., et al. Tuning Hsf1 levels drives distinct fungal morphogenetic programs with depletion impairing Hsp90 function and overexpression expanding the target space. PLoS. Genet. 2018;14 doi: 10.1371/journal.pgen.1007270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Zhang S., Hou R., Zhao Z., Zheng Q., Xu Q., Zheng D., Wang G., Liu H., Gao X., et al. Functional analysis of the kinome of the wheat scab gungus Fusarium graminearum. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T.C. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents. Chemother. 1997;41:1482–1487. doi: 10.1128/AAC.41.7.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T.C. The presence of an R467K amino acid substitution and loss of allelic variation correlate with an azole-resistant lanosterol 14alpha demethylase in Candida albicans. Antimicrob. Agents. Chemother. 1997;41:1488–1494. doi: 10.1128/AAC.41.7.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L., Robbins N., Huang D.S., McLellan C.A., Shekhar-Guturja T., LeBlanc E.v., Nation C.S., Hui R., Hutchinson A., Collins C., et al. Structural basis for species-selective targeting of Hsp90 in a pathogenic fungus. Nat. Commun. 2019;10:402. doi: 10.1038/s41467-018-08248-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisplinghoff H., Bischoff T., Tallent S.M., Seifert H., Wenzel R.P., Edmond M.B. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- Xie J.L., Qin L., Miao Z., Grys B.T., Diaz J.D.L.C., Ting K., Krieger J.R., Tong J., Tan K., Leach M.D., et al. The Candida albicans transcription factor Cas5 couples stress responses, drug resistance and cell cycle regulation. Nat. Commun. 2017;8:499. doi: 10.1038/s41467-017-00547-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn J.-Y., Friesen H., Nguyen Ba A.N., Liang W., Messier V., Cox M.J., Moses A.M., Andrews B. Functional analysis of kinases and transcription factors in Saccharomyces cerevisiae using an integrated overexpression library. G3 (Bethesda) 2017;7:911–921. doi: 10.1534/g3.116.038471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Yang P.L., Gray N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information requested to reanalyze the data reported in this paper is available from the lead contact upon request.