

Abstract
Introduction:
We examined the ability of plasma hyperphosphorylated tau (p-tau)181 to detect cognitive impairment due to Alzheimer’s disease (AD) independently and in combination with plasma total tau (t-tau) and neurofilament light (NfL).
Methods:
Plasma samples were analyzed using the Simoa platform for 235 participants with normal cognition (NC), 181 with mild cognitive impairment due to AD (MCI), and 153 with AD dementia. Statistical approaches included multinomial regression and Gaussian graphical models (GGMs) to assess a network of plasma biomarkers, neuropsychological tests, and demographic variables.
Results:
Plasma p-tau181 discriminated AD dementia from NC, but not MCI, and correlated with dementia severity and worse neuropsychological test performance. Plasma NfL similarly discriminated diagnostic groups. Unlike plasma NfL or t-tau, p-tau181 had a direct association with cognitive diagnosis in a bootstrapped GGM.
Discussion:
These results support plasma p-tau181 for the detection of AD dementia and the use of blood-based biomarkers for optimal disease detection.
Keywords: Alzheimer’s Disease, P-tau, Plasma biomarkers, Network analysis, Neuropsychology
1 |. INTRODUCTION
The National Institute on Aging and Alzheimer’s Association (NIA-AA) Research Framework defines Alzheimer’s disease (AD) by its underlying pathophysiological processes.1 In vivo biomarkers to measure AD neuropathological changes are critical for the early detection and treatment of AD.2–8 There are three classifications of biomarkers: brain amyloidosis (A), neurodegeneration (N), and tau pathology (T). Cerebrospinal fluid (CSF) and positron emission tomography (PET) imaging are gold standards for the detection of AD pathophysiology and provide a direct window into the central nervous system (CNS) to identify amyloid beta (Aβ) and hyperphosphorylated tau (p-tau).1 Magnetic resonance imaging (MRI) is routine for the assessment of neuronal loss (e.g., atrophy),9 but CSF analysis for proteins such as total tau (t-tau) and neurofilament light (NfL) also provides insight into the severity of neurodegeneration.10 These approaches are viewed as invasive and/or expensive, calling on the need for scalable biomarker measurements with similar accuracy.1,11 The development of more practical biomarkers will permit large-scale implementation, both in research and clinical settings.
Through advancements in ultra-sensitive immunoassay and mass spectrometry technology, low abundance proteins can be detected in the blood, offering an exciting avenue for biomarker development.3 Plasma biomarkers of neurodegeneration have been of interest in the field of AD and AD-related dementias, especially NfL and t-tau, which reflect neuronal damage and cell death or degeneration.12,13 Sugarman et al.14 found that higher levels of plasma NfL discriminated AD dementia from those with normal cognition (NC) and mild cognitive impairment (MCI); in addition, plasma NfL was associated with disease severity and worse neuropsychological test performance across multiple domains, both at baseline and longitudinally. The results were less promising for plasma t-tau, which discriminated AD dementia from NC only, weakly correlated with neuropsychological function, and did not predict longitudinal outcomes. These results are consistent with previous studies15–17 that support plasma NfL as a more accurate biomarker of AD neurodegeneration compared to t-tau.
Recent efforts have targeted immunoassay developments to detect p-tau in the blood.18 Hyperphosphorylated tau is a hallmark AD pathology and a precipitant of neurodegeneration and cognitive and functional decline in AD,3,9,19 making it a clear target for biomarker investigation to facilitate disease detection, diagnosis, and assess therapeutic response. With new developments in p-tau immunoassay technologies, plasma p-tau shows potential as a feasible biomarker of AD pathology.20,21 While multiple tau phospho-forms measured in CSF have support in detecting AD-type tau pathology, such as threonine 217 (p-tau217) and threonine 231 (p-tau231),22,23 threonine 181 (p-tau181) has been widely characterized in early plasma biomarker analysis.3,24 Plasma p-tau181 concentration is associated with worse neuropsychological test performance,25,26 correlates with longitudinal gray matter atrophy and increased odds of conversion to AD dementia,17,25,27–29 and distinguishes AD dementia from other neurodegenerative disorders (e.g., frontotemporal lobar degeneration).3,11,28,30,31 Furthermore, plasma p-tau181 was highly predictive of AD pathology in a recent autopsy study; despite being taken 8 years prior to death, the biomarker was sensitive to post mortem AD tauopathy.25
Research on the ability of plasma p-tau181 to accurately detect the clinical manifestation of AD is still in its infancy. Existing studies have focused on p-tau181 in isolation; the diagnostic utility of plasma p-tau181 compared to and in conjunction with other plasma biomarkers remains unclear. This is an important limitation as a panel of plasma biomarkers will likely have optimal diagnostic accuracy32,33 as opposed to any one plasma protein in isolation.
The objective of this study was to examine the association between plasma p-tau181 and cognitive diagnostic status (i.e., NC, MCI due to AD, and AD dementia), in isolation and in conjunction with plasma t-tau and NfL, as well as to test the association between plasma p-tau181 and neuropsychological test performance. Our sample included participants from the Boston University (BU) Alzheimer’s Disease Research Center (ADRC) Clinical Core. Sugarman et al.14 examined the usefulness of t-tau and NfL in the BU ADRC sample. We therefore leveraged both the sample and the previously quantified plasma biomarkers to examine the predictive validity of p-tau181. We hypothesized that p-tau181, t-tau, and NfL would independently discriminate participants with MCI and AD dementia from NC and that a panel of combined biomarkers would outperform each on its own. Our hypotheses were tested using traditional regression-based approaches (e.g., multinomial regression), in addition to a network modeling approach that assessed the conditional association of biomarkers, diagnostic and daily function variables, neuropsychological test performance, and relevant demographic variables in a single model.
Network theory suggests that entities such as neurological diseases are best understood from an inclusive approach, with various predictors and corollaries examined in concert. Network models represent a joint conditional probability distribution in which nodes (i.e., variables) are connected with edges (i.e., directed or undirected associations between variables).34 These models are data-driven attempts to understand complex systems.35 While under-utilized, network models have been used in research on biomarkers in AD,36–39 and most recently in one study of blood-based biomarkers.16 We contend that blood-based biomarkers will have optimal clinical utility as part of a panel of biological, cognitive, and behavioral data to assist with clinical detection and diagnosis of AD (i.e., to provide a full clinical profile of an individual patient).
2 |. METHODS
2.1 |. Participants and design
This study included participants from the BU ADRC Clinical Core Registry. The BU ADRC is one of ≈30 centers funded by the NIA and provides data to the National Alzheimer’s Coordinating Center (NACC) to promote collaborative research on AD. A detailed description of the BU ADRC is provided elsewhere.14,40–43 The present sample and study design are similar to the Sugarman et al.14 study that examined plasma NfL and t-tau in the BU ADRC. The BU ADRC follows older adults with and without cognitive impairment. All participants are English-speaking older adults with adequate visual acuity and hearing. Participants are excluded for a history of a serious mental illness (e.g., bipolar disorder, schizophrenia), non-AD/ADRD neurological disorders (e.g., brain tumor, multiple sclerosis), or medical conditions that preclude study participation. The BU ADRC protocol involves annual neurological examination, a clinical and medical interview, neuropsychological testing, measures of daily function, and other procedures.
Beginning in 2008, voluntary blood draws were initiated. All participants included in the current study provided a plasma sample, were evaluated, and received a diagnosis of NC, MCI due to AD, or AD dementia made during the same visit as samples were obtained. Neuropsychological and diagnostic data were used that were closest in time to the participants’ initial blood draw, which did not necessarily correspond to their first ADRC visit. No follow-up data were included in the current study. Procedures were approved by the BU Medical Center Institutional Review Board. Participants (or their legally authorized representatives) provided written informed consent prior to participation in the BU ADRC protocol.
2.2 |. Plasma biomarker collection and analysis
Non-fasting blood samples were collected for all participants. Blood was collected into plastic dipotassium ethylene diaminetetraacetic acid tubes, and processed according to standard procedures, with plasma aliquoted and frozen at −80°C. Frozen plasma aliquots were shipped on dry ice to the University of Gothenburg (Sweden) for batch analysis. Plasma p-tau181 concentration was measured using an in-house single molecule array method on an HD-X analyzer (Quanterix), as previously described in detail.11 The lower limit of quantification (LLoQ) was 1.0 pg/mL, with a dynamic range of 1.0 to 128.0 pg/mL. An in-house Simoa method was used to measure plasma NfL concentration (Quanterix), as described by Gisslén et al.44 The LLoQ was 1.9 pg/mL, with a dynamic range of 1.9 to 1800 pg/mL. Plasma T-tau concentration was measured using Tau 2.0 kit and the HD-1 analyzer (Quanterix) with an LLoQ of 0.061 pg/mL and a dynamic range of 0.061 to 360 pg/mL. The measurements were performed in one round of experiments, using one batch of reagents. Intra-assay coefficients of variation were below 10% for all the biomarkers.
2.3 |. Diagnostic procedures
All participants included in this study had a research cognitive diagnosis of NC, MCI due to AD, or AD dementia. Diagnoses were made by a BU ADRC multidisciplinary diagnostic consensus panel, after presentation and discussion of all examination and test findings (including review of structural MRI, if available); neuropsychological test scores; functional measures; as well as social, family, and medical history. Plasma biomarker data were not used in the adjudication of cognitive syndromes or suspected etiologies. Established criteria were used for AD dementia45,46 and MCI due to AD47,48 diagnoses. As an ADRC, we followed the NACC Uniform Data Set (UDS) diagnostic criteria for cognitive syndromes and suspected etiologies. There have been three versions of the UDS over time. For versions 1 and 2 of the UDS, AD dementia diagnoses were based on the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the Alzheimer’s Disease and Related Disorders Association (ADRDA) diagnostic criteria.45 For version 3, MCI and dementia diagnoses were based on the 2011 NIA-AA criteria.46,48 Regardless of the UDS version for MCI and dementia due to AD, MCI diagnoses were made based on evidence of impairment on neuropsychological test scores (i.e., 1.5 standard deviations below the normative mean) in the absence of functional impairment. A dementia diagnosis required the presence of functional impairment in the context of objective impairment on neuropsychological testing. Reported complaints and/or progressive worsening were required for both MCI and dementia diagnoses.
Dementia severity was rated using the Clinical Dementia Rating (CDR) Dementia Staging Instrument.49,51 CDR ratings encompass several domains including orientation, memory, judgment/problem solving, home and hobbies, community affairs, and personal care. CDR Sum of Boxes (CDR-SB) was used as the primary index from this measure following research suggesting comparable or improved diagnostic utility and desirable psychometric properties compared to the CDR global score.51,52 The Functional Activities Questionnaire (FAQ), a measure of instrumental activities of daily living, was also collected to monitor functional change over time.53 CDR scores were informed by the report of a study partner and FAQ scores were based on the informant version of the measure.
2.4 |. Neuropsychological tests
Consistent with the NACC-UDS,54,55 participants underwent a standardized neuropsychological examination to assess cognition across several domains. Tests administered and examined in this study included the Mini-Mental State Examination (MMSE), the Wechsler Adult Intelligence Scale-IV Digit Span (Forward and Backward; WAIS-IV DSF and DSB), Trail Making Test Parts A and B (TMT-A and TMT-B, respectively), Semantic Fluency (Animal and Vegetable Fluency), the short form Boston Naming Test (BNT), and the Wechsler Memory Scale, Revised Logical Memory Delayed Recall (LM-II). Participants were also administered the Neuropsychological Assessment Battery (NAB) List Learning Test (Trials 1–3, Short Delay [SD], and Long Delay [LD]).56 Neuropsychological outcomes included the primary indices of the tests that are routinely used and interpreted in both clinical and research settings. Scores reflect total correct for all measures except for TMT-A and B, for which total time was used (in seconds).
2.5 |. Demographic variables and apolipoprotein E ε4 allele status
Demographic information was collected on all participants, including age (years), sex (male or female), self-reported race (re-coded as non-Hispanic White versus other), and years of education. Blood was drawn and used for apolipoprotein E (APOE) genotyping (ε4 carriers vs. non-carriers).
2.6 |. Statistical analytic plan
Analyses were performed using the statistical programming language R (version 4.0.2; R Development Core Team).57 We examined the performance of p-tau181 in discriminating diagnostic groups relative to and in conjunction with plasma NfL and t-tau (NC, MCI due to AD, AD dementia). We assessed the relative contribution of p-tau181 in a multinomial model with plasma NfL and t-tau, demographic variables (i.e., age, sex, self-reported race, and years of education), and APOE ε4 allele genotype status as additional predictors. The Hosmer and Lemeshow test58 suggested no evidence of a poorly fitting model, χ2(16, N = 569) = 12.18, P = 0.732. Standardized effects are reported. Sensitivity models examined different combinations of the biomarkers, demographic variables, and APOE ε4 allele status to qualitatively compare receiver operating characteristic (ROC) analyses using the multiclass area under the curve (mAUC) statistic.59 Discrimination accuracy was categorized according to guidelines suggested in Hosmer and Lemeshow (AUC = 0.50: no discrimination; AUC = 0.70–0.80: acceptable discrimination; AUC = 0.80–0.90: excellent discrimination; AUC ≥ 0.90: outstanding discrimination).60 We assessed an additional model with all predictors and the CDR-SB as the criterion. Given the distribution of CDR-SB, we initially used Poisson regression.61 However, there was evidence for violation of residual deviance, χ2(559, N = 569) = 1845.45, P < 0.001. A negative binomial generalized linear model was thus used, which improved the model, χ2(559, N = 569) = 516.24, P = 0.902. Robust standard errors were interpreted and incidence rate ratios were computed using the delta method.62 Nonparametric bootstrapping of all models was conducted with 10,000 samples. Because missing data were rare (≤ 1%), pairwise deletion was used for regression models. Effect sizes were interpreted according to Cohen.63
Partial correlation analyses using a Holm correction were conducted to examine the relationship between p-tau181 and neuropsychological test performance.64 These analyses controlled for demographic variables (age, sex, self-reported race, and years of education) and APOE ε4 allele status. These analyses were conducted to maintain consistency with our previous study involving this sample, and to compare and clarify our effects in the network models (see below).14 Pearson, polychoric, and polyserial correlations were used depending on whether the variables were continuous, ordinal, or mixed, respectively. We also conducted these analyses using Spearman’s rank correlation for comparison. While there were a larger number of missing values for these variables (> 5% for 7/12 neuropsychological variables and > 10% for 1/12 variables), pairwise deletion was implemented to maintain consistency with our regression models. The association between neuropsychological measures and NfL and t-tau in this sample were previously reported by Sugarman et al.14
We conducted a network analysis to determine the variables with the greatest number and magnitude of associations in the network, and whether patterns or groupings of variables could be discerned. We also examined which variables would be most important and influential in determining or enabling the associations between other variables in the network (i.e., variables that would foster connections by serving as “central junctions” for other associations). We used undirected graphs due to (1) the cross-sectional nature of our data, (2) to avoid stringent assumptions regarding feedback loops, and (3) for clear interpretation of edge-weight parameters as the strength of unique associations.65
Gaussian graphical models (GGMs) were estimated using the least absolute shrinkage and selection operator (LASSO)66 regularization and a stepwise unregularized model search algorithm (ggmModSelect),65 as well as, for comparison, a model retaining edges based on traditional statistical significance (α = 0.05). The input matrix was estimated with Pearson, polychoric, and polyserial correlations depending on whether the variables were continuous, ordinal, or mixed, respectively. Variables in the model included plasma p-tau181, NfL, and t-tau; diagnostic and functional measures (cognitive diagnostic status, CDR-SB, FAQ); demographic variables (age, sex, self-reported race, and years of education); APOE ε4 allele status; and neuropsychological measures.
Unlike the previously discussed analyses, there was concern for circularity in our network models as the CDR, FAQ, and neuropsychological measures were available during the multidisciplinary diagnostic consensus panels at which diagnoses were made. Causal graph models were not estimated due to these concerns for circularity. In addition, concerns were limited to retained edges between certain variables (i.e., the CDR, FAQ, and neuropsychological measures) and diagnosis. This relationship would not impact the retention of edges/effects between these variables and each other (e.g., the association between FAQ and CDR), as well as other variables included in the model (including biomarkers, which were our primary interest). As such, circularity would only impact 14 out of 529 possible direct effects. These effects were not interpreted. Given that the purpose of network theory is to include all relevant variables, we judged the value of inclusion to outweigh any limitation on the findings.
Network metrics of interest included node strength (i.e., the absolute sum of edge weights), closeness and between centrality (i.e., the average distance and shortest path length between nodes), and expected influence (the sum of edges extending from a given node).67 These measures assess the importance and influence of variables for the overall network, rather than simply examining individual connections or edges. Network stability was assessed with a non-parametric bootstrap of standardized edge weights and a person-dropping bootstrap of centrality measures with 10,000 samples. Strength, closeness, and expected influence met established thresholds of stability (see supporting information for details).65 The regularized network was interpreted due to the wide intervals of bootstrapped weights using the unregularized model search algorithm and the lack of sparsity in these results (see supporting information). Edge weights reflect a standardized partial correlation that has been regularized and bootstrapped.
Network fit was evaluated with the extended Bayesian information criterion (EBIC68), the root mean square error of approximation (RMSEA69) and the Tucker-Lewis index (TLI70). Networks were visualized with eigenmodels, a latent modeling approach designed to create meaningful graphical space.71 Latent dimensions were estimated for the x- and y-axes (model-based decomposition and regression) with parameters derived from Markov chain Monte Carlo (MCMC) simulation. These dimensions can be interpreted in a similar manner to a traditional factor analysis.
3 |. RESULTS
3.1 |. Demographic and plasma biomarker results
A total of 569 individuals were included in this study, including 235 diagnosed as NC at the time of their plasma sample (41.3%), 181 diagnosed as MCI (31.8%), and 153 diagnosed as AD dementia (26.9%). The median age of the sample was 75 years (range 53–94) and 56.4% were female. Demographic and clinical characteristics are presented in Table 1. The range of values for blood-based biomarkers fell within the validated dynamic ranges for the assays (p-tau181: 1.3–124.1 pg/mL; NfL: 3.0–121.5 pg/mL; t-tau: 0.6–37.7 pg/mL).
TABLE 1.
Demographic and clinical characteristics by diagnostic group
| NC (n = 235) | MCI (n = 181) | AD dementia (n = 153) | ||
|---|---|---|---|---|
| Demographic variables | ||||
| Age | 72.38 (7.69) | 74.96 (7.25) | 76.82 (8.13) | |
| Sex (male: female) | 37:63 | 42:58 | 56:44 | |
| Race (non-Hispanic White: non-White) | 90:10 | 75:25 | 92:8 | |
| Years of education | 16.56 (2.54) | 15.52 (2.77) | 14.95 (2.95) | |
| Genetic | ||||
| APOE ε4 allele status (yes: no) | 33:67 | 33:67 | 58:42 | |
| Neuropsychological measures | ||||
| Animal Fluency | 22.08 (5.27) | 16.98 (5.13) | 10.45 (5.37) | |
| BNT | 28.23 (1.65) | 25.31 (4.49) | 20.27 (7.23) | |
| DSF | 6.99 (0.93) | 6.40 (1.03) | 6.13 (1.21) | |
| DSB | 5.30 (1.23) | 4.64 (1.25) | 3.61 (1.42) | |
| LM-II | 14.70 (3.74) | 11.02 (4.66) | 2.34 (3.44) | |
| MMSE | 29.39 (0.91) | 28.20 (1.68) | 21.12 (6.21) | |
| NAB Trials 1–3 | 23.69 (4.53) | 18.21 (5.01) | 9.94 (4.91) | |
| NAB SD | 8.39 (2.36) | 5.32 (2.59) | 1.24 (1.75) | |
| NAB LD | 8.41 (2.41) | 4.91 (2.79) | 0.72 (1.46) | |
| TMT-A | 28.78 (8.80) | 36.97 (18.03) | 63.08 (38.88) | |
| TMT-B | 69.50 (28.47) | 127.76 (78.14) | 223.30 (88.42) | |
| Vegetable Fluency | 15.71 (3.97) | 12.93 (3.89) | 6.64 (3.98) | |
| Functional | ||||
| CDR Sum of Boxes | 0.07 (0.26) | 0.37 (0.64) | 6.53 (4.27) | |
| FAQ | 0.23 (1.99) | 0.65 (1.89) | 14.27 (9.74) | |
| Biomarkers | ||||
| NfL | 15.43 (10.51) | 17.61 (9.89) | 26.57 (17.45) | |
| P-tau181 | 16.05 (11.07) | 18.10 (10.01) | 25.92 (15.62) | |
| T-tau | 3.20 (2.73) | 3.29 (2.39) | 3.70 (2.99) |
Note. Values reflect mean or proportion, with standard deviation in parentheses.
Abbreviations: AD, Alzheimer’s disease; BNT, Boston Naming Test; CDR, Clinical Dementia Rating; DSF & DSB, Digit Span Forward and Backward; FAQ, Functional Activities Questionnaire; LM-II, Logical Memory Delayed Recall; MCI, mild cognitive impairment; MMSE, Mini-Mental State Examination; NAB SD & LD, Neuropsychological Assessment Battery List Learning Test, Short and Long Delay; NC, normal control; NfL, neurofilament light; P-tau181, hyperphosphorylated tau; TMT-A & B, Trail Making Test; Parts A and B; T-tau, total tau.
3.2 |. Plasma biomarkers and diagnostic status
The initial multinomial model showed that higher p-tau181 levels were associated with significantly higher conditional odds of an AD dementia diagnosis (conditional odds ratio [COR] = 1.47, 95% confidence interval [CI: 1.06, 2.08], P = .008), but not an MCI diagnosis (COR = 1.13, 95% CI [0.89, 1.44], P = .310; see Table 2 and Figure S1 in supporting information). Higher NfL levels were also associated with significantly higher conditional odds of an AD dementia diagnosis (COR = 2.53, 95% CI [1.71, 3.65], P < .001), but not an MCI diagnosis (COR = 1.25, 95% CI [0.93, 1.67], P = .115). Higher t-tau levels were not associated with higher conditional odds of an AD dementia or MCI diagnosis.
TABLE 2.
Multinomial model contrasting the normal control group versus each of the other groups (N = 569)
| NC vs. | β z | COR | 95% CI | Wald Z | P | |
|---|---|---|---|---|---|---|
| (Intercept) | MCI | 0.067 | 1.07 | (0.06, 17.27) | 0.047 | 0.962 |
| AD dementia | 1.811 | 6.12 | (0.16, 205.35) | 1.115 | 0.265 | |
| P-tau181 | MCI | 0.126 | 1.13 | (0.89, 1.44) | 1.015 | 0.310 |
| AD dementia | 0.385 | 1.47 | (1.06, 2.08) | 2.650 | 0.008** | |
| T-tau | MCI | 0.056 | 1.06 | (0.83, 1.35) | 0.491 | 0.624 |
| AD dementia | 0.184 | 1.20 | (0.91, 1.58) | 1.402 | 0.161 | |
| NfL | MCI | 0.220 | 1.25 | (0.93, 1.67) | 1.578 | 0.115 |
| AD dementia | 0.929 | 2.53 | (1.71, 3.65) | 5.573 | <0.001*** | |
| Age | MCI | 0.024 | 1.02 | (0.99, 1.06) | 1.491 | 0.136 |
| AD dementia | 0.018 | 1.02 | (0.98, 1.06) | 0.979 | 0.327 | |
| Race | MCI | 1.067 | 2.91 | (1.51, 5.52) | 3.578 | <0.001*** |
| AD dementia | −0.300 | 0.74 | (0.27, 1.83) | −0.696 | 0.487 | |
| Sex | MCI | −0.394 | 0.67 | (0.43, 1.05) | −1.809 | 0.070 |
| AD dementia | −1.072 | 0.34 | (0.21, 0.59) | −4.171 | <0.001*** | |
| Years of education | MCI | −0.124 | 0.88 | (0.82, 0.96) | −3.060 | 0.002** |
| AD dementia | −0.232 | 0.79 | (0.72, 0.88) | −4.897 | <0.001*** | |
| APOE ε4 allele status | MCI | 0.037 | 1.04 | (0.67, 1.64) | 0.162 | 0.871 |
| AD dementia | 1.125 | 3.08 | (1.80, 5.12) | 4.394 | <0.001*** |
Abbreviations: AD, Alzheimer’s disease; APOE, apolipoprotein E; CI, confidence interval; COR, conditional odds ratio; MCI, mild cognitive impairment; NC, normal control; NfL, neurofilament light; P-tau181, hyperphosphorylated tau; T-tau, Total tau.
In the full model (i.e., demographics, APOE ε4 allele status, and all plasma biomarkers), the accuracy for discriminating diagnostic groups fell in the “acceptable” range (mAUC = 0.749). In a model with demographic variables and APOE ε4 allele status (i.e., without plasma biomarkers), the discrimination accuracy was at the bottom of the “acceptable” range (mAUC = 0.706). Models with plasma biomarkers, and without demographic variables and APOE ε4 allele status, all fell below the range of “acceptable” discrimination (Table 3). Models with individual plasma biomarkers, along with demographic variables and APOE ε4 allele status, fell in the range of “acceptable” discrimination: NfL, mAUC = 0.743; p-tau181, mAUC = 0.730; and t-tau, mAUC = 0.711. In models with and without p-tau181, the independent contribution of t-tau was negligible (i.e., adding it to the model with demographic variables and APOE ε4 allele status resulted in a ΔmAUC of 0.005, whereas removing it from models resulted in a ΔmAUC between 0.000 and 0.002).
TABLE 3.
Multiclass area under the curve statistics for multinomial models including blood-based biomarkers, demographic variables, and APOE ε4 allele status
| Model | mAUC | Descriptor |
|---|---|---|
| All predictors | 0.749 | Acceptable |
| Demographic variables and APOE status | 0.706 | Acceptable |
| P-tau181 only | 0.634 | Below |
| T-tau only | 0.543 | Below |
| NfL only | 0.658 | Below |
| P-tau181, NfL, demographic variables, and APOE ε4 allele status | 0.748 | Acceptable |
| P-tau181, t-tau, demographic variables, and APOE ε4 allele status | 0.730 | Acceptable |
| NfL, t-tau, demographic variables, and APOE ε4 allele status | 0.745 | Acceptable |
| P-tau181, demographic variables, and APOE ε4 allele status | 0.730 | Acceptable |
| T-tau, demographic variables, and APOE ε4 allele status | 0.711 | Acceptable |
| NfL, demographic variables, and APOE ε4 allele status | 0.743 | Acceptable |
Note. Models compare AD dementia and MCI due to AD groups to normal controls. Descriptors based on Hosmer and Lemeshow.60
Abbreviations: APOE, apolipoprotein E; mAUC, multiclass area under the curve statistic; NfL, neurofilament light; P-tau181, hyperphosphorylated tau; T-tau, total tau. Demographic predictors included age, race, sex, years of education, and APOE ε4 allele status (carriers vs. non-carriers).
3.3 |. Plasma biomarkers and CDR-SB
The negative binomial generalized linear model showed a statistically significant effect of p-tau181 on CDR-SB (incident rate ratio [RR] = 1.29, 95% CI [1.01, 1.39], P = .003; see Table 4). Plasma NfL was also associated with CDR-SB (RR = 1.72, 95% CI [1.70, 2.59], P < .001). In contrast, there was no statistically significant effect of t-tau on CDR-SB (RR = 1.07, 95% CI [1.00, 1.30], P = .345).
TABLE 4.
Negative binomial generalized linear model predicting CDR Sum of Boxes (N = 569)
| β z | SE | RR | 95% CI | P | |
|---|---|---|---|---|---|
| (Intercept) | 1.77 | 1.16 | 5.85 | (0.51, 48.31) | 0.128 |
| P-tau181 | 0.25 | 0.09 | 1.29 | (1.01, 1.39) | 0.003** |
| T-tau | 0.06 | 0.07 | 1.07 | (1.00, 1.30) | 0.345 |
| NfL | 0.54 | 0.11 | 1.72 | (1.70, 2.59) | <0.001*** |
| Age | 0.00 | 0.01 | 1.00 | (0.98, 1.03) | 0.951 |
| Race | −0.61 | 0.25 | 0.54 | (0.37, 0.99) | 0.015* |
| Sex | −0.37 | 0.16 | 0.69 | (0.38, 0.70) | 0.018* |
| Years of education | −0.10 | 0.03 | 0.91 | (0.85, 0.95) | 0.001** |
| APOE ε4 allele status | 0.58 | 0.15 | 1.78 | (1.42, 2.60) | <0.001*** |
Abbreviations: APOE, apolipoprotein E; CDR, Clinical Dementia Rating; CI, confidence interval; NfL, neurofilament light; P-tau181, hyperphosphorylated tau; RR, incident rate ratio; SE, robust standard error; T-tau, total tau.
3.4 |. P-tau181 and neuropsychological test performance
Partial correlation models that controlled for demographic variables and APOE ε4 allele status showed that p-tau181 was associated with all neuropsychological measures except TMT-A. Higher p-tau181 was associated with lower MMSE scores (r = –0.20, 95% CI [–0.28, –0.12], P < .001), DSF (r = −0.11, 95% CI [–0.19, –0.02], P = .01), DSB (r = –0.09, 95% CI [–0.17, –0.01], P = .03), animal fluency (r = –0.18, 95% CI [–0.26, –0.10], P < .001), vegetable fluency (r = –0.15, 95% CI [–0.23, –0.07], P < .001), BNT (r =–0.17, 95% CI [–0.25, –0.09], P < .001), LM-II (r = –0.16, 95% CI [–0.24, –0.08], P < .001), NAB Trial 1–3 (r = –0.14, 95% CI [–0.22, –0.06], P < .001), NAB SD (r = –0.14, 95% CI [–0.22, –0.06], P < .001), and NAB LD (r = –0.13, 95% CI [–0.21, –0.05], P < .001), as well as slower performance on TMT-B, r = 0.13, 95% CI [0.05, 0.21], P < .001. These findings were consistent with models using Spearman’s rank correlation (see supporting information).
3.5 |. Network analysis
Neuropsychological measures loaded highly on a latent dimension in the regularized GGM, especially MMSE, NAB Trials 1–3, NAB SD and LD, and LM-II (Figure 1). The second latent dimension was comprised primarily of CDR-SB and FAQ scores (i.e., functional status and disease progression). The biomarker variables loaded similarly on the two latent dimensions and were clustered near TMT-A and TMT-B; age; sex; APOE ε4 allele status; and, to a lesser extent, cognitive diagnostic status. These groupings are suggestive of “subnetworks” that could be loosely interpretable as factors (i.e., “microsystems” of the network). Out of 529 possible edges, 138 (26%) were retained after regularization and 45 (8%) were retained after bootstrapping, suggesting an adequately sparse model. Thus, we judged that the network adequately controlled for false positive associations. There was also adequate model fit, χ2(184, N = 569) = 620.46, EBIC = 27,444.53, RMSEA = 0.06, TLI = 0.95.
FIGURE 1.
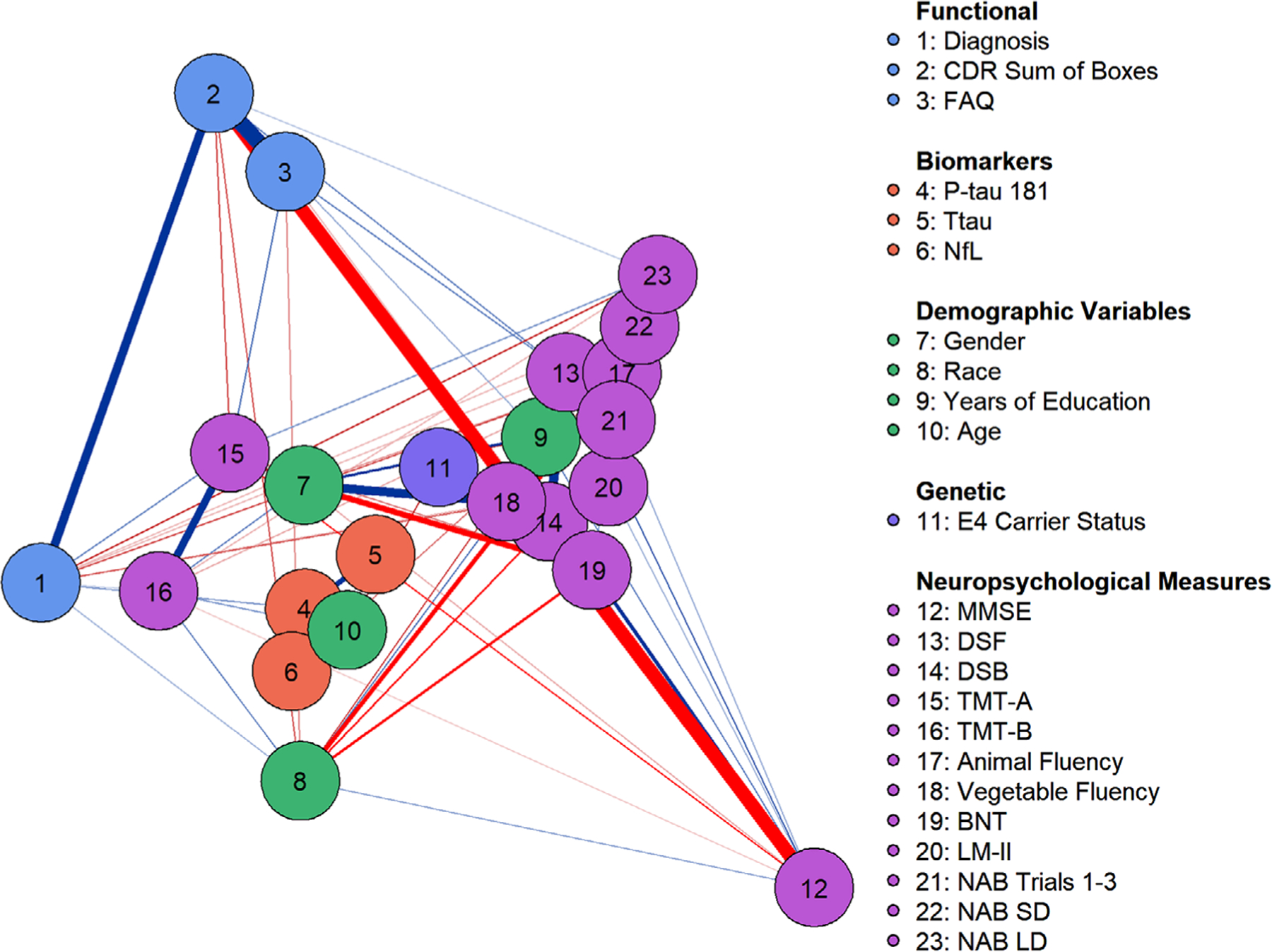
Gaussian graphical model with least absolute shrinkage and selection operator regularization. Latent dimensions were estimated with Markov chain Monte Carlo simulation. Nodes on the right side of the visualization load highly on dimension 1 and nodes toward the top load highly on dimension 2. Node location reflects variable loading on the two latent dimensions. Positive associations are depicted as blue. Edge width reflects the magnitude of association and edge saturation (i.e., darkness) reflects the likelihood of the association. Node colors reflect artificial groupings from the legend. BNT, Boston Naming Test; CDR, Clinical Dementia Rating; DSF & DSB, Digit Span Forward and Backward; FAQ, Functional Activities Questionnaire; LM-II, Logical Memory Delayed Recall; MMSE, Mini-Mental State Examination; NAB SD & LD, Neuropsychological Assessment Battery List Learning Test, Short and Long Delay; NfL, neurofilament light; P-tau181, hyperphosphorylated tau; TMT-A & B, Trail Making Test, Parts A and B; Ttau, total tau
Cognitive diagnostic status was connected to several edges (see supporting information), including TMT-B (βz = 0.18, 95% CI [0.11, 0.25], animal fluency (βz = –0.08, 95% CI [–0.14, –0.01]), vegetable fluency (βz = –0.09, 95% CI [–0.16, –0.03]), LM-II (βz = –0.21, 95% CI [–0.29, –0.13]), NAB SD (βz = –0.07, 95% CI [–0.14, –0.01]), and NAB LD (βz = –0.17, 95% CI [–0.25, –0.10]). This was expected given that neuropsychological test performance is used to assist with adjudication of consensus diagnostic status. More importantly, cognitive diagnosis was only connected to p-tau181 (βz = 0.07, 95% CI [0.01, 0.12]) and not plasma NfL or t-tau. None of the neuropsychological measures were connected to p-tau181. NfL was connected to FAQ scores (βz = 0.08, 95% CI [0.02, 0.13]) and animal fluency (βz = –0.09, 95% CI [–0.16, – 0.03]). The discrepancy between these findings and results from partial correlation models reflects regularization (i.e., the removal of connections that weaken the overall predictive accuracy of the network model).
Among centrality measures, CDR-SB and cognitive diagnostic status ranked highly for closeness and betweenness centrality (Figure 2). Vegetable fluency and NAB Trials 1–3 ranked highly for expected influence. None of the biomarkers ranked highly for closeness centrality. However, p-tau181 ranked relatively highly for betweenness centrality and expected influence and NfL ranked highly for betweenness centrality. These measures, therefore, can be considered important and/or influential as “central junctions” for other connections in the overall network. In contrast, t-tau was not highly ranked on any centrality measure. There were relatively modest differences in centrality measures between the models derived from regularization, stepwise model selection, and statistical significance.
FIGURE 2.
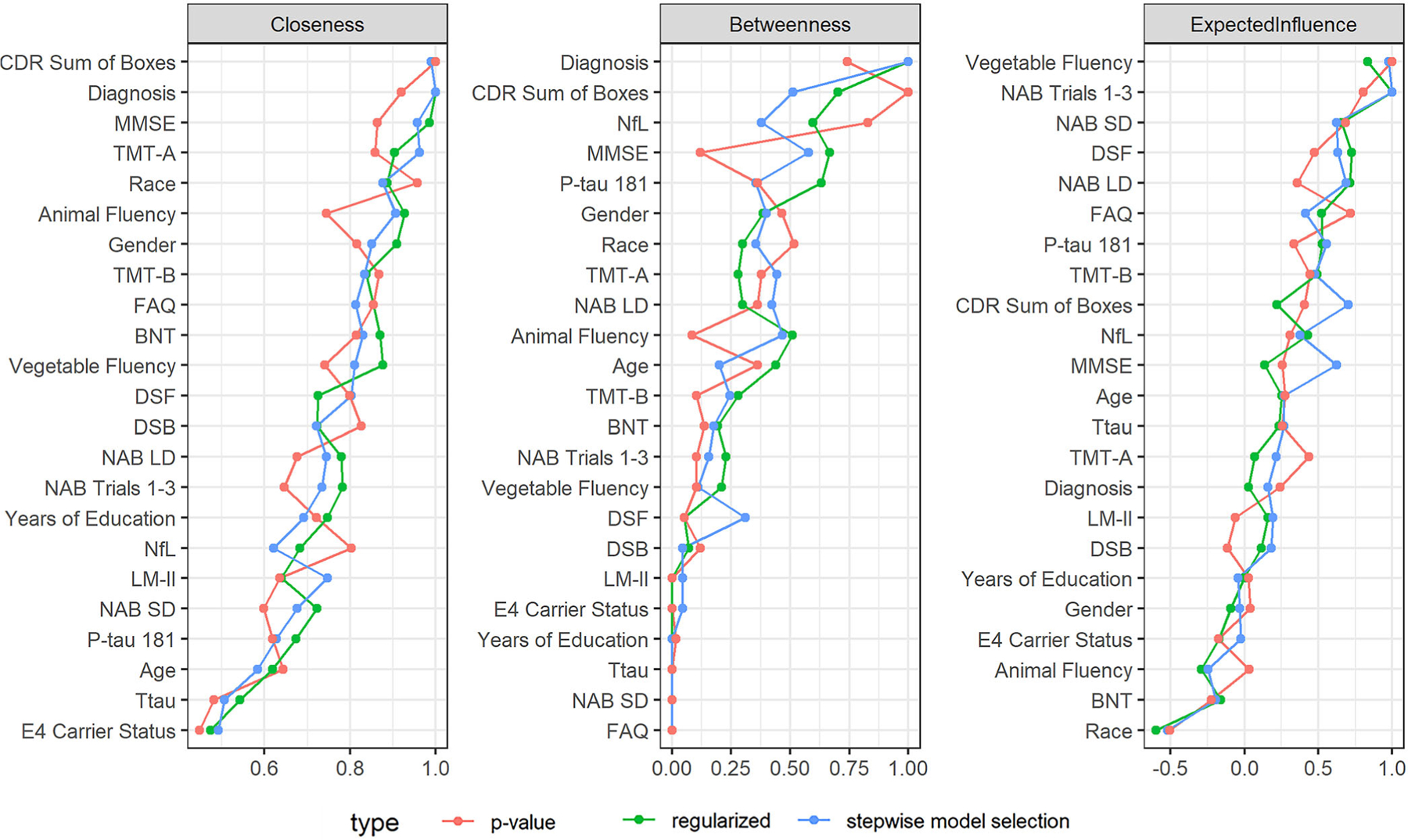
Network centrality measures with variables ranked from greatest to least. Centrality measures reflect the direct and indirect connections between nodes in a network and each node’s influence in fostering connections. Closeness centrality reflects the average number of intervening nodes between a node and all other nodes. Betweenness centrality reflects how often a node fosters connections between nodes (i.e., lies on the shortest path length). Expected influence reflects the magnitude and direction of all edges connected to a node. BNT, Boston Naming Test; CDR, Clinical Dementia Rating; DSF & DSB, Digit Span Forward and Backward; FAQ, Functional Activities Questionnaire; LM-II, Logical Memory Delayed Recall; MMSE, Mini-Mental State Examination; NAB SD & LD, Neuropsychological Assessment Battery List Learning Test, Short and Long Delay; NfL, neurofilament light; P-tau181, hyperphosphorylated tau; TMT-A & B, Trail Making Test, Parts A and B; Ttau, total tau
4 |. DISCUSSION
The present study evaluated the ability of plasma p-tau181 to detect cognitive impairment due to AD among 569 community-dwelling older adults from the BU ADRC Clinical Core. This study expanded on our previous work on plasma NfL and t-tau14 by examining these plasma biomarkers in conjunction with plasma p-tau181; we examined their association with clinical diagnosis and additional characteristics of the sample including dementia severity, functioning, and neuropsychological test performance. Higher levels of plasma p-tau181 accurately discriminated participants with NC from AD dementia but not MCI due to AD. Models that included plasma biomarkers in combination with demographic variables and APOE ε4 allele status provided the greatest predictive accuracy of diagnostic groups, supporting arguments for a focus on combined biomarkers in AD research.72 Higher plasma p-tau181 concentrations were associated with greater dementia severity and worse neuropsychological test performance. We did not find adequate discrimination for diagnosis of MCI due to suspected AD, which is inconsistent with previous studies examining p-tau181.27,11,73
In addition to traditional regression models, we examined a network model of associations among plasma biomarkers, diagnostic and functional measures, demographic variables, APOE ε4 allele status, and neuropsychological measures. Network models provide a data-driven method to investigate complex systems and to examine numerous associations simultaneously. Unlike plasma NfL or t-tau, plasma p-tau181 showed a direct relationship with cognitive diagnostic status in the model. Plasma p-tau181 was also connected to both NfL and t-tau, which were indirectly associated with cognitive diagnostic status. Such connections provide tentative support for complex biological interactions among p-tau181, myelin and axonal loss, and neuronal dysfunction in the clinical manifestation of AD (as reviewed in Nasrabady et al.19). However, neurodegeneration would perhaps best be characterized by a longitudinal graph model, such as a hidden Markov model,74,75 in which causal inferences could be explicit. Future longitudinal studies should confirm these findings and pathways.
Unlike t-tau, p-tau181 and NfL ranked highly on network centrality measures, suggesting that they explained important variance in direct and indirect associations within the network. That is, these plasma biomarkers were highly influential in fostering connections and impacting other nodes. In addition, analysis of latent dimensions in the network suggested groupings based on (1) objective cognitive performance and (2) functional status/disease progression. Plasma biomarkers were associated with these groupings in a pattern similar to the node reflecting research diagnosis (i.e., AD dementia, MCI due to AD, and NC), suggesting the biomarkers provide important and unique information on both sets of clinical characteristics. Plasma p-tau181 was central to the network more broadly and with more stability than NfL. In contrast, t-tau had little impact or relevance to direct and indirect associations in the network. In sum, these findings suggest that plasma p-tau181 and NfL captured important aspects of the clinical profile in our sample, not otherwise captured by demographic variables and APOE ε4 allele status, or traditional measures of cognitive and daily function.
Growing research has examined the association between plasma p-tau181 and the clinical presentation of AD.11,25,27,28,29,74 Plasma p-tau181 concentrations have been shown to correspond to AD progression27 and to predict AD neuropathology irrespective of diagnosis.28 Karikari et al.11 found that p-tau181 levels differentiated among NC, MCI, and AD groups in their discovery (n = 37), validation (n = 989), and primary care (n = 105) cohorts. The lowest concentrations were found among young adults and older individuals (> 60 years) diagnosed as NC, higher concentrations were associated with Aβ-positive NC and Aβ-negative MCI groups, and the highest concentrations were found among Aβ-positive MCI and AD groups. Higher plasma p-tau181 levels have also been observed among individuals with MCI who converted to AD dementia compared to non-converters.25 In a recent study, Moscoso et al.74 found that longitudinal increases in p-tau181 and NfL were independently associated with cognitive decline across the AD spectrum, as well as hypometabolism and atrophy. Further, through analysis of biomarkers of neurodegeneration, as measured by MRI and fluorodeoxyglucose PET scans, they were able to differentiate the associated pattern of neurodegeneration for each plasma biomarker. Unlike plasma NfL, plasma p-tau181 was associated with an AD-typical neurodegenerative pattern specific to Aβ-positive individuals.
In the present study, p-tau181 levels were able to discriminate between individuals with AD dementia compared to NC, and were associated with neuropsychological test performance and a staging instrument of dementia severity. Although our results conflict with the aforementioned reports of p-tau181 levels distinguishing an MCI diagnosis from NC and AD dementia,27,11,73 these findings (i.e., association with neuropsychological test performance, CDR score) provide some support for the use of p-tau181 as an early prognostic biomarker for AD. Previous studies assessing p-tau181 levels varied in their blood draw protocols, in which assays were collected from both fasting and non-fasting individuals. Björkqvist et al.32 suggested that replication of plasma collection protocols was important for reproduction of biomarker results. Thus, our lack of findings in MCI individuals may derive from our use ofa non-fasting blood draw and additional proteins interfering with the assay analysis.20,29
Lack of biomarker or pathological confirmation of AD may also help to explain our lack of association with MCI. Lantero Rodriguez et al.28 found that plasma p-tau181 predicted AD neuropathology post mortem but did not distinguish MCI by clinical diagnosis across three time-points. Compared to dementia, MCI diagnosis is particularly heterogeneous and unstable (e.g., associated with mixed pathology and even psychiatric factors76). Two recent studies found that plasma p-tau181 better distinguished MCI from NC when stratified by Aβ status and that Aβ-positive individuals with MCI have similar elevations to individuals with AD dementia.17,77 While our MCI group had suspected AD etiology, it was not restricted to amnestic MCI and there might have been heterogeneous pathology. Finally, we focused on p-tau181; other p-tau isotopes may be more sensitive to the early stages of AD pathology, with greater association to neocortical neurofibrillary pathology and better predictive power for conversion.78–85
In our mAUC analyses, models included plasma biomarkers (individually and in combination), analyzed with demographic variables and APOE ε4 allele status. We found discrimination accuracy within the acceptable range for predicting MCI and AD dementia. The model including all three biomarkers together was the superior predictive model, which is unsurprising when attempting to measure a disease of mixed pathologies such as AD.22,86–88 Using plasma measures of p-tau181, NfL, and t-tau, we were able to evaluate AD tauopathy, and non-specific neuronal injury and neurodegeneration.12,13,19 Plasma p-tau181 and NfL independently strengthened the effectiveness of diagnostic prediction. However, in models with and without p-tau181, the independent contribution of t-tau was negligible. This suggests that the cost of acquiring both t-tau and p-tau181 might outweigh the diagnostic benefit, favoring p-tau181 alone (i.e., adding or subtracting t-tau from various models resulted in a negligible change in overall predictive diagnostic accuracy). Our network analyses also suggested the importance of both p-tau181 and NfL in capturing various aspects of our sample’s clinical presentation (via centrality measures). Multiple plasma biomarkers will be needed to accommodate the heterogeneity and interconnectedness of AD pathology, and further research will be needed to confirm which provides optimal clinical utility.86,87
Scalable and non-invasive biomarkers of AD neuropathological changes will be critical for detection and implementation of treatment efforts,2–8 and to meet the demands of clinical trials.72 Knopman et al.72 suggest that validated blood-based measurements would overcome economic and operational barriers for precision hypothesis testing (targeted interventions for specific disease mechanisms at optimal times in the disease course) and could improve classification and staging (a major source of trial failure). These measurements could also increase the diversity and generalizability of samples. There are concerns for how plasma biomarkers could translate to personal medicine. Largent et al.89 proposed that a validated direct-to-consumer plasma p-tau test could benefit personal health care. However, the authors also noted that such a test could be misused if broadly available to the general population.
There are several limitations to our findings. While there is increasing support for the accuracy and reliability of plasma biomarkers for detecting AD pathology,3,20,27,14,77 we did not have a gold standard biomarker of AD or neurodegeneration, neither were the participants separated according to Aβ status. Nonetheless, our study provides a highly relevant example for the complementary use of plasma biomarkers in a primary or secondary care clinical setting in which gold standard diagnostic methods may not be feasible. The study was cross-sectional, limiting our ability to make causal inferences. Some of our findings differed from our previous analyses with this sample (e.g., the ability of t-tau to discriminate individuals with AD vs. NC at baseline; see Sugarman et al.14). These discrepancies likely resulted from the different statistical models used (i.e., multinomial models versus analysis of covariance).
Our sample and all ADRC samples are most representative of a clinic-based population (i.e., individuals who are concerned with memory problems). Thus, our results have limited generalizability to the general public. Similarly, the sample only included participants with cognitive impairment due to suspected AD. While there were few other suspected etiologies in the BU ADRC at the time of this study and we intentionally focused on AD, AD is commonly comorbid with other neurodegenerative diseases. Limiting the sample to suspected AD may reduce the generalizability of the findings. Finally, we followed NACC UDS diagnostic criteria, which included a revision in version 3. This change in diagnostic criteria may have had some impact on our findings, albeit minimal.
5 |. CONCLUSIONS
The present results suggest that plasma p-tau181 provides unique diagnostic and clinical information and support its implementation in routine biomarker assessment related to the detection of AD dementia. The results further emphasize the importance of multiple biomarkers to capture different aspects of AD for optimal disease detection. A validated plasma biomarker panel could provide a less invasive, scalable means to transform clinical and research trials related to AD,73 and perhaps personal health care.89
Supplementary Material
RESEARCH IN CONTEXT.
1. Systematic review:
We reviewed the literature with traditional sources (e.g., PubMed). While several recent studies have examined plasma hyperphosphorylated tau (p-tau)181, the clinical usefulness of blood-based biomarkers remains poorly understood. Network studies are also needed to examine associations between various predictors of Alzheimer’s disease (AD) given its complex and mixed pathology.
2. Interpretation:
The present results provide further evidence that plasma p-tau181 provides unique diagnostic and clinical information and support its implementation in routine biomarker assessment related to the detection of AD dementia. The results also emphasize the importance of multiple biomarkers to capture different aspects of AD for optimal disease detection.
3. Future directions:
Longitudinal studies are needed to clarify the clinical usefulness of plasma p-tau181, particularly in terms of its role in the early detection of AD. Plasma biomarker–pathological correlation studies will also be essential for validation. In particular, a validated plasma biomarker panel could provide a less invasive, scalable means to transform clinical and research trials related to AD, and perhaps personal health care.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (P30AG013846; U01NS093334, U01NS086659–01; RF1AG05416; K23NS102399) and the Department of Veterans’ Affairs (I01-CX001038). This publication was also supported by a Pilot Grant from the Boston University Alzheimer’s Disease Center (AG013846), as well as the National Center for Advancing Translational Sciences, National Institutes of Health, through BU-CTSI Grant Number 1UL1TR001430. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018–02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG-720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809–2016862), the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21–831376-C, #ADSF-21–831381-C, and #ADSF-21–831377-C), the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2019–0228), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), and the UK Dementia Research Institute at UCL. KB is supported by the Swedish Research Council (#2017–00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809–2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017–0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), the European Union Joint Program for Neurodegenerative Disorders (JPND2019–466-236), and the National Institute of Health (NIH), USA, (grant #1R01AG068398–01). TKK was funded by the Brightfocus Foundation (#A2020812F), the Swedish Alzheimer Foundation (Alzheimerfonden; #AF-930627), the Swedish Brain Foundation (Hjärnfonden; #FO2020–0240), the Swedish Parkinson Foundation (Parkinsonfonden), the Swedish Dementia Foundation (Demensförbundet), the Agneta Prytz-Folkes & Gösta Folkes Foundation (#2020–00124), the Aina (Ann) Wallströms and Mary-Ann Sjöbloms Foundation, the Anna Lisa and Brother Björnsson’s Foundation, Gamla Tjänarinnor, and the Gun and Bertil Stohnes Foundation. KWT is supported by the US Department of Veterans Affairs (# IK2 CX002065). YT is supported by grants from the National Institutes of Health (K23 NS102399, R01 HL141774–02, R01 OH011511–01, R01 HD090191, R01AG062348, U01 NS093334, RF1 AG054156). MOC is supported by grants from the National Institutes of Health (R01–1AG070883–01, NIH R01 PAR-19–070, P30AG13846, PAR-19–070) RA is supported by the NIH/NIA (AG033040, AG049899, AG049810, AG054156, AG055337, AG016495, AG062109, AG059011, AG061340, AG063635, AG068753), the Alzheimer’s Drug Discovery Foundation (VMF-14–318524). GJ is supported by the National Institutes of Health (U01-AG068057, R56-AG069130, and R01-AG069453). TS is supported by the Department of Veterans Affairs, Veterans Health Administration, Clinical Sciences Research and Development Merit Award (I01-CX001038); National Institute of Aging (RF1AG054156, RF1AG057768, U19AG068753); National Institute of Neurological Disorders and Stroke (U54NS115266). AM is supported by the National Institutes of Health (U19AG068753, R01 AG058822, R01 AG062348, U01NS093334, P30 AG13846), the Department of Veterans Affairs (1I01BX004613, BX002466), the National Institute of Aging (U54NS115266, RF1 AG057902), and the Department of Defense (AZ170038). RK is supported by the Department of Defense (CDMRP/GWIRP GW180103). RS is supported by the NINDS/NIA (R01NS11965, R01AG062624, U01NS093334, P30-AG13846). JM is supported by the National Institute of Health (P30AG13846, R01AG029672, U01AG032984, R01AG048927, U01NS093334, RF1AG057902, R01AG062348, R01AG061028, U54NS115266, R01AG062602, U19AG068753) and by the Department of Defense (W81XWH1810580). MA is supported by the National Institutes of Health (P30AG013846; U01NS093334, U01NS086659–01; RF1AG05416; K23NS102399) and the Department of Veterans’ Affairs (I01-CX001038).
Footnotes
CONFLICTS OF INTEREST
AB receives royalties and consulting fees from Oxford University Press, Elsevier, and Sage Therapeutics. He has also received consulting fees from Artis Senior Living Benchmark. AM received honoraria for speaking from the Korean Society of Neuroscience, the University of Maryland, and the Texas Neurological Society. HZ has served on scientific advisory boards for Eisai, Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, Nervgen, AZTherapies, and CogRx; has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, and Biogen; and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). KB has served as a consultant, on advisory boards, or on data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu, Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics, and Siemens Healthineers, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. KT receives payments from the US Department of Veterans Affairs and the Alzheimer’s Association. LG serves as a consultant for Rebion and Cognoptix, and has received travel expenses for HIH grant reviews. MA serves as a consultant for Corino Therapeutics, Inc. He has received honoraria from the International Neuropsychological Society, the Allegheny General Hospital, the American Academy of Neurology, and the University of California, San Francisco. RA serves as a consultant or on scientific advisory board for Signant Health, Biogen, and GSK. RS is a member of the Mackey-White Committee of the NFL Players Association. He is a paid consultant to Biogen (Cambridge, MA, USA) and Eli Lilly (Indianapolis, IN, USA). He receives royalties for published neuropsychological tests from Psychological Assessment Resources, Inc. (Lutz, FL, USA) and is a member of the Board of Directors of King-Devick Technologies (Chicago, IL, USA). The remaining authors have nothing to disclose. The content is solely the responsibility of the authors. There is no sponsor.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
REFERENCES
- 1.Jack CR, Bennett DA, Blennow K, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14(4):535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berti V, Osorio RS, Mosconi L, Li Y, De Santi S, de Leon MJ. Early Detection of Alzheimer’s Disease with PET Imaging. Neurodegener Dis 2010;7(1–3):131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blennow K, Zetterberg H. Biomarkers for Alzheimer’s disease: current status and prospects for the future. J Intern Med 2018;284(6):643–663. [DOI] [PubMed] [Google Scholar]
- 4.Blennow K, Zetterberg H, Minthon L, et al. Longitudinal stability of CSF biomarkers in Alzheimer’s disease. Neurosci Lett 2007;419(1):18–22. [DOI] [PubMed] [Google Scholar]
- 5.Blennow K, Shaw LM, Stomrud E, et al. Predicting clinical decline and conversion to Alzheimer’s disease or dementia using novel Elecsys Aβ(1–42), pTau and tTau CSF immunoassays. Sci Rep 2019;9(1):19024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement 2016;12(3):292–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jack CR, Wiste HJ, Schwarz CG, et al. Longitudinal tau PET in ageing and Alzheimer’s disease. Brain 2018;141(5):1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol 2016;15(7):673–684. [DOI] [PubMed] [Google Scholar]
- 9.Jack CR, Holtzman DM. Biomarker Modeling of Alzheimer’s Disease. Neuron 2013;80(6):1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou W, Zhang J, Ye F, et al. Plasma neurofilament light chain levels in Alzheimer’s disease. Neurosci Lett 2017;650:60–64. [DOI] [PubMed] [Google Scholar]
- 11.Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol 2020;19(5):422–433. [DOI] [PubMed] [Google Scholar]
- 12.Zetterberg H Neurofilament Light: a Dynamic Cross-Disease Fluid Biomarker for Neurodegeneration. Neuron 2016;91(1):1–3. [DOI] [PubMed] [Google Scholar]
- 13.Wirth M, Madison CM, Rabinovici GD, Oh H, Landau SM, Jagust WJ. Alzheimer’s Disease Neurodegenerative Biomarkers Are Associated with Decreased Cognitive Function but Not β-Amyloid in Cognitively Normal Older Individuals. J Neurosci 2013;33(13):5553–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugarman MA, Zetterberg H, Blennow K, et al. A longitudinal examination of plasma neurofilament light and total tau for the clinical detection and monitoring of Alzheimer’s disease. Neurobiol Aging 2020;94:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Illán-Gala I, Lleo A, Karydas A, et al. Plasma tau and neurofilament light in frontotemporal lobar degeneration and Alzheimer’s disease. Neurology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raket LL, Kühnel L, Schmidt E, Blennow K, Zetterberg H, Mattsson-Carlgren N. Utility of plasma neurofilament light and total tau for clinical trials in Alzheimer’s disease. Alzheimers Dement Diagn Assess Dis Monit 2020;12(1):e12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simrén J, Leuzy A, Karikari TK, et al. The diagnostic and prognostic capabilities of plasma biomarkers in Alzheimer’s disease. Alzheimers Dement 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashton NJ, Leuzy A, Karikari TK, et al. The validation status of blood biomarkers of amyloid and phospho-tau assessed with the 5-phase development framework for AD biomarkers. Eur J Nucl Med Mol Imaging 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nasrabady SE, Rizvi B, Goldman JE, Brickman AM. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun 2018;6(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zetterberg H Blood-based biomarkers for Alzheimer’s disease—An update. J Neurosci Methods 2019;319:2–6. [DOI] [PubMed] [Google Scholar]
- 21.Zou K, Abdullah M, Michikawa M. Current Biomarkers for Alzheimer’s Disease: from CSF to Blood. J Pers Med 2020;10(3):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashton NJ, Pascoal TA, Karikari TK, et al. Plasma p-tau231: a new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol 2021;141(5):709–724. 10.1007/s00401-021-02275-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janelidze S, Berron D, Smith R, et al. Associations of Plasma Phospho-Tau217 levels with tau positron emission tomography in early Alzheimer Disease. JAMA Neurol 2021;78(2):149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barthélemy NR, Horie K, Sato C, Bateman RJ. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J Exp Med 2020;217(11):e20200861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Connor A, Karikari TK, Poole T, et al. Plasma phospho-tau181 in presymptomatic and symptomatic familial Alzheimer’s disease: a longitudinal cohort study. Mol Psychiatry 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai C-L, Liang C-S, Lee J-T, et al. Associations between plasma biomarkers and cognition in patients with Alzheimer’s Disease and amnestic mild cognitive impairment: a cross-sectional and longitudinal study. J Clin Med 2019;8(11):1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med 2020;26(3):379–386. [DOI] [PubMed] [Google Scholar]
- 28.Lantero Rodriguez J, Karikari TK, Suárez-Calvet M, et al. Plasma p-tau181 accurately predicts Alzheimer’s disease pathology at least 8 years prior to post-mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol (Berl) 2020;140(3):267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang C-C, Chiu M-J, Chen T-F, Chang H-L, Liu B-H, Yang S-Y. Assay of Plasma Phosphorylated Tau Protein (Threonine 181) and Total Tau Protein in Early-Stage Alzheimer’s Disease. J Alzheimers Dis 2018;61(4):1323–1332. [DOI] [PubMed] [Google Scholar]
- 30.Mielke MM, Hagen CE, Xu J, et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement 2018;14(8):989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med 2020;26(3):387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Björkqvist M, Ohlsson M, Minthon L, Hansson O. Evaluation of a Previously Suggested Plasma Biomarker Panel to Identify Alzheimer’s Disease. PLoS ONE 2012;7(1):e29868. Block ML. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhiman K, Blennow K, Zetterberg H, Martins RN, Gupta VB. Cerebrospinal fluid biomarkers for understanding multiple aspects of Alzheimer’s disease pathogenesis. Cell Mol Life Sci 2019;76(10):1833–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buntine WL. Operations for Learning with Graphical Models. J Artif Intell Res 1994;2:159–225. 10.1613/jair.62 [DOI] [Google Scholar]
- 35.Barabási A-L. The network takeover. Nat Phys. 2012;8(1):14–16. [Google Scholar]
- 36.Wang T, Ren Z, Ding Y, et al. FastGGM: an efficient algorithm for the inference of Gaussian graphical model in biological networks. PLOS Comput Biol 2016;12(2):e1004755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ortiz A, Munilla J, Álvarez-Illán I, Górriz JM, Ramírez J. Exploratory graphical models of functional and structural connectivity patterns for Alzheimer’s Disease diagnosis. Front Comput Neurosci 2015;9. 10.3389/fncom.2015.00132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dyrba M, Mohammadi R, Grothe MJ, Kirste T, Teipel SJ. Gaussian Graphical Models Reveal Inter-Modal and Inter-Regional Conditional Dependencies of Brain Alterations in Alzheimer’s Disease. Front Aging Neurosc 2020;12. 10.3389/fnagi.2020.00099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peterson CB, Osborne N, Stingo FC, Bourgeat P, Doecke JD, Vannucci M. Bayesian modeling of multiple structural connectivity networks during the progression of Alzheimer’s disease. Biometrics 2020;76(4):1120–1132. 10.1111/biom.13235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ashendorf L, Jefferson AL, Green RC, Stern RA. Test–retest stability on the WRAT-3 reading subtest in geriatric cognitive evaluations. J Clin Exp Neuropsychol 2009;31(5):605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gavett BE, Lou KR, Daneshvar DH, Green RC, Jefferson AL, Stern RA. Diagnostic Accuracy Statistics for Seven Neuropsychological Assessment Battery (NAB) Test Variables in the Diagnosis of Alzheimer’s Disease. Appl Neuropsychol Adult 2012;19(2):108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jefferson AL, Wong S, Gracer TS, Ozonoff A, Green RC, Stern RA. Geriatric performance on an abbreviated version of the Boston Naming Test. Appl Neuropsychol 2007;14(3):215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cousins CC, Alosco ML, Cousins HC, et al. Nailfold capillary morphology in Alzheimer’s disease dementia. J Alzheimers Dis 2018;66(2):601–611. [DOI] [PubMed] [Google Scholar]
- 44.Gisslén M, Price RW, Andreasson U, et al. Plasma concentration of the neurofilament light protein (NFL) is a biomarker of CNS injury in HIV infection: a cross-sectional study. EBioMedicine 2016;3:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34(7):939–939. [DOI] [PubMed] [Google Scholar]
- 46.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7(3):263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winblad B, Palmer K, Kivipelto M, et al. Mild cognitive impairment – beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med 2004;256(3):240–246. [DOI] [PubMed] [Google Scholar]
- 48.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7(3):270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hughes CP, Berg L, Danziger W, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140(6):566–572. [DOI] [PubMed] [Google Scholar]
- 50.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43(11):2412–2414. [DOI] [PubMed] [Google Scholar]
- 51.Cedarbaum JM, Jaros M, Hernandez C, et al. Rationale for use of the Clinical Dementia Rating Sum of Boxes as a primary outcome measure for Alzheimer’s disease clinical trials. Alzheimers Dement 2013;9(1,Supplement):S45–S55. [DOI] [PubMed] [Google Scholar]
- 52.O’Bryant SE, Waring SC, Cullum CM, et al. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer’s Research Consortium study. Arch Neurol 2008;65(8):1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pfeffer RI, Kurosaki TT, Harrah CH, Chance JM, Filos S. Measurement of functional activities in older adults in the community. J Gerontol 1982;37(3):323–329. [DOI] [PubMed] [Google Scholar]
- 54.Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer’s Coordinating Center (NACC) Database: the Uniform Data Set. Alzheimer Dis Assoc Disord 2007;21(3):249–258. [DOI] [PubMed] [Google Scholar]
- 55.Weintraub S, Salmon D, Mercaldo N, et al. The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord 2009;23(2):91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iverson GL, Brooks BL, White T, Stern RA. Neuropsychological Assessment Battery: introduction and advanced interpretation. In The Neuropsychology Handbook 3rd Ed. New York, NY, US: Springer Publishing Company; 2008:279–343. [Google Scholar]
- 57.Core Team R. R: A Language and Environment for Statistical Computing R Foundation for Statistical Computing. Vienna, Austria. 2020. https://www.R-project.org/. [Google Scholar]
- 58.Fagerland MW, Hosmer DW. A generalized Hosmer–Lemeshow goodness-of-fit test for multinomial logistic regression models. Stata J 2012;12(3):447–453. [Google Scholar]
- 59.Hand DJ, Till RJ. A simple generalisation of the area under the ROC curve for multiple class classification. Mach Learn 2001;45(2):171–186. [Google Scholar]
- 60.Hosmer DW, Lemeshow S. Applied Logistic Regression New York, NY: John Wiley & Sons; 2000. [Google Scholar]
- 61.Long S Regression Models for Categorical and Limited Dependent Variables Thousand Oaks, CA: Sage Publications; 1997. [Google Scholar]
- 62.Long JS, Freese J. Regression Models for Categorical Dependent Variables Using Stata 2nd ed.. College Station, TX: Stata Press; 2006. [Google Scholar]
- 63.Cohen J Statistical Power Analysis for the Behavioral Sciences L. Erlbaum Associates. 2nd ed.. Hillsdale, NJ: Hillsdale, NJ. 1988. [Google Scholar]
- 64.Holm S A simple sequentially rejective multiple test procedure. Scand J Stat 1979;6(2):65–70. [Google Scholar]
- 65.Epskamp S, Borsboom D, Fried EI. Estimating psychological networks and their accuracy: a tutorial paper. Behav Res Methods 2018;50(1):195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Epskamp S, Cramer AO, Waldorp LJ, Schmittmann VD, Borsboom D. qgraph: network visualizations of relationships in psychometric data. J Stat Softw 2012;48(4):1–18. [Google Scholar]
- 67.Robinaugh DJ, Millner AJ, McNally RJ. Identifying highly influential nodes in the complicated grief network. J Abnorm Psychol 2016;125(6):747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen J, Chen Z. Extended Bayesian information criteria for model selection with large model spaces. Biometrika 2008;95(3):759–771. 10.1093/biomet/asn034 [DOI] [Google Scholar]
- 69.MacCallum RC, Browne MW, Sugawara HM. Power analysis and determination of sample size for covariance structure modeling. Psychol Methods 1996;1(2):130–149. 10.1037/1082-989x.1.2.130 [DOI] [Google Scholar]
- 70.Bollen KA. Sample size and Bentler and Bonett’s nonnormed fit index. Psychometrika 1987;52(1):161–161. 10.1007/bf02293963 [DOI] [Google Scholar]
- 71.Jones PJ, Mair P, McNally RJ. Visualizing psychological networks: a tutorial in R. Front Psychol 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knopman DS, Haeberlein SB, Carrillo MC, et al. The National Institute on Aging and the Alzheimer’s Association Research Framework for Alzheimer’s disease: perspectives from the Research Roundtable. Alzheimers Dement 2018;14(4):563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moscoso A, Grothe MJ, Ashton NJ, et al. Longitudinal associations of blood phosphorylated tau181 and neurofilament light chain with neurodegeneration in Alzheimer disease. JAMA Neurol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eddy SR. Profile hidden Markov models. Bioinformatics 1998;14(9):755–763. [DOI] [PubMed] [Google Scholar]
- 75.Rabiner L, Juang B. An introduction to hidden Markov models. IEEE ASSP Mag 1986;3(1):4–16. 10.1109/massp.1986.1165342 [DOI] [Google Scholar]
- 76.Sugarman MA, Alosco ML, Tripodis Y, Steinberg EG, Stern RA. Neuropsychiatric symptoms and the diagnostic stability of mild cognitive impairment. J Alzheimers Dis 2018;62(4):1841–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Karikari TK, Benedet AL, Ashton NJ, et al. Diagnostic performance and prediction of clinical progression of plasma phospho-tau181 in the Alzheimer’s Disease Neuroimaging Initiative. Mol Psychiatry 2021;26(2):429–442. [DOI] [PubMed] [Google Scholar]
- 78.Ewers M, Buerger K, Teipel SJ, et al. Multicenter assessment of CSF-phosphorylated tau for the prediction of conversion of MCI. Neurology 2007;69(24):2205–2212. [DOI] [PubMed] [Google Scholar]
- 79.Hampel H, Bürger K, Pruessner JC, et al. Correlation of cerebrospinal fluid levels of tau protein phosphorylated at threonine 231 with rates of hippocampal atrophy in Alzheimer Disease. Arch Neurol 2005;62(5):770–773. [DOI] [PubMed] [Google Scholar]
- 80.Buerger K, Teipel SJ, Zinkowski R, et al. CSF tau protein phosphorylated at threonine 231 correlates with cognitive decline in MCI subjects. Neurology 2002;59(4):627–629. [DOI] [PubMed] [Google Scholar]
- 81.Hampel H, Buerger K, Zinkowski R, et al. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer Disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry 2004;61(1):95–102. [DOI] [PubMed] [Google Scholar]
- 82.Suárez-Calvet M, Karikari TK, Ashton NJ, et al. Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer’s continuum when only subtle changes in Aβ pathology are detected. EMBO Mol Med 2020;12(12): e12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brickman AM, Manly JJ, Honig LS, et al. Plasma p-tau181, p-tau217, and other blood-based Alzheimer’s disease biomarkers in a multi-ethnic, community study. Alzheimer’s Dement 2021;17(8):1353–1364. 10.1002/alz.12301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Buerger K, Ewers M, Pirttilä T, et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain 2006;129(11):3035–3041. [DOI] [PubMed] [Google Scholar]
- 85.Janelidze S, Stomrud E, Smith R, et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat Commun 2020;11(1):1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age: neuropathologies and Cognition. Ann Neurol 2018;83(1):74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain 2016;139(11):2983–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007;69(24):2197–2204. 10.1212/01.wnl.0000271090.28148.24 [DOI] [PubMed] [Google Scholar]
- 89.Largent EA, Wexler A, Karlawish J. The Future Is P-Tau—Anticipating Direct-to-Consumer Alzheimer Disease Blood Tests. JAMA Neurol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.