

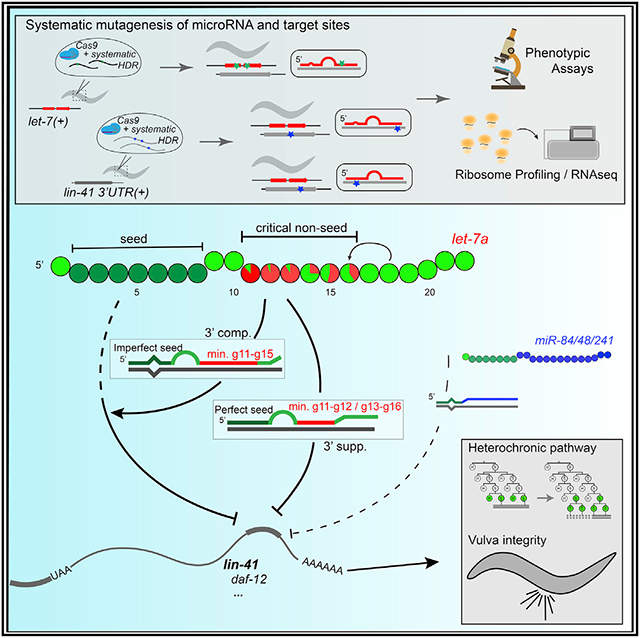
SUMMARY
Base pairing of the seed region (g2–g8) is essential for microRNA targeting; however, the in vivo function of the 3′ non-seed region (g9–g22) is less well understood. Here, we report a systematic investigation of the in vivo roles of 3′ non-seed nucleotides in microRNA let-7a, whose entire g9–g22 region is conserved among bilaterians. We find that the 3′ non-seed sequence functionally distinguishes let-7a from its family paralogs. The complete pairing of g11–g16 is essential for let-7a to fully repress multiple key targets, including evolutionarily conserved lin-41, daf-12, and hbl-1. Nucleotides at g17–g22 are less critical but may compensate for mismatches in the g11–g16 region. Interestingly, a certain minimal complementarity to let-7a 3′ non-seed sequence can be required even for sites with perfect seed pairing. These results provide evidence that the specific configurations of both seed and 3′ non-seed base pairing can critically influence microRNA-mediated gene regulation in vivo.
Graphical Abstract
In brief
Duan et al. find that microRNA-target pairing at g11–g16 is critical for the function of evolutionarily conserved microRNA let-7a; 3′ pairing is required for both perfect and imperfect seed in regulating multiple targets. These findings provide evidence that base pairing of specific microRNA non-seed nucleotides can critically contribute to target regulation.
INTRODUCTION
MicroRNAs (miRNAs) are short non-coding RNAs found in all metazoans (Friedman et al., 2009; Lee et al., 1993; Nelson and Ambros, 2021). miRNAs are transcribed from specific genomic loci and processed into precursor miRNA (pre-miRNA) consisting of the miRNA and the passenger strand in a hairpin structure (Bartel, 2018). The pre-miRNA is further processed, and the mature miRNA binds Argonaute proteins (AGO) to form the miRNA-induced silencing complex (miRISC) and base pairs with complementary sites in the 3′ untranslated region (UTR) of target RNAs. miRISC binding leads to the repression of target gene expression through translational inhibition and/or target mRNA destabilization (Bartel, 2018). miRNAs are critical for the regulation of diverse physiological processes across species (Ambros, 2004; Bartel, 2018). miRNAs regulate an estimated 60% of human genes, and the dysfunction of miRNAs is implicated in multiple diseases (Friedman et al., 2009; Paul et al., 2018).
The miRNA seed corresponds to the 6–7 contiguous nucleotides beginning at the second nucleotide (g2) from the 5′ end. Structural and biochemical studies indicate that the miRNA seed sequence is critical for target recognition and determines miRNA targeting efficacy and specificity (Salomon et al., 2015; Schirle et al., 2014). Complementarity to the miRNA seed characterizes evolutionarily conserved miRNA target sites, and genetically disrupting seed complementarity results in target de-repression (Lai, 2002; Lewis et al., 2005; Lim et al., 2005).
Organisms commonly contain multiple genes encoding miRNAs with identical seed sequences, which are grouped into seed families. miRNAs with identical seeds can in principle recognize shared targets and function additively (Abbott et al., 2005; Alvarez-Saavedra and Horvitz, 2010; Brenner et al., 2012), while miRNAs with identical seed but divergent 3′ non-seed nucleotides (g9-g22) can select distinct target sites, driven by 3′ non-seed complementarity (Broughton et al., 2016; Wahlquist et al., 2014).
let-7 (lethal-7) family miRNAs are distributed widely across animal genomes, consistent with the origin of let-7 in a bilaterian ancestor (Hertel et al., 2012; Wolter et al., 2017). The evolutionarily conserved developmental expression of let-7 family miRNAs, and the results from genetic analyses of let-7 family mutants, suggest the conservation of let-7a family functionality (Roush and Slack, 2008; Tennessen and Thummel, 2008). In nematodes, let-7 family miRNAs act in the heterochronic pathway to promote developmental cell fate transitions (Abbott et al., 2005; Reinhart et al., 2000). Similarly, let-7 family miRNAs control cellular transitions from pluripotency to differentiation in mammals and the timing of adult fates during metamorphosis in insects (Balzeau et al., 2017; Lee et al., 2016; Sokol et al., 2008).
Almost all bilaterian genomes encode at least one let-7 family isoform (let-7a), in which all nucleotides in the 3′ non-seed region are highly conserved (Figure S1A) (Hertel et al., 2012; Pasquinelli et al., 2003), suggesting that nucleotides g9-g22 of let-7a engage in critical interactions, which evolutionarily constrained the sequence. Indeed, data from genome-scale analyses of in vivo miRNA-target interactions support widespread miRNA 3′ non-seed pairing (Grosswendt et al., 2014; Helwak et al., 2013). Pairing to the 3′ non-seed region, especially g13-g16, is thought to enhance target repression in certain contexts (Brennecke et al., 2005; Grimson et al., 2007), and structural and biochemical studies of human AGO2 miRISC confirm that g13-g16 can form an A-form helix with the target to increase miRISC-target affinity and specificity (Sheu-Gruttadauria et al., 2019; Xiao and MacRae, 2020). Some miRNA target sites have imperfect seed complementarity accompanied by compensatory 3′ non-seed pairing, including Caenorhabditis elegans lin-41, for which 3′ non-seed pairing confers specificity for regulation by let-7a as opposed to other let-7 family miRNAs (Chi et al., 2012; Doench and Sharp, 2004; Grimson et al., 2007; Vella et al., 2004). However, the relative contributions of specific 3′ non-seed nucleotides to the in vivo function of miRNAs remain unclear.
Here, we report a systematic investigation of how the 3′ non-seed nucleotides contribute to the in vivo function of let-7a. We used CRISPR-Cas9 genome editing of C. elegans to introduce defined mutations into let-7a or its conserved targets. Our results indicate that base pairing of g11–g16 to targets is essential for let-7a in vivo function and that g17–g22 pairing is less critical than g11–g16 pairing but can partially compensate for mismatches in g11–g16. We confirm that the 3′ non-seed sequence of let-7a confers target specificity relative to its family paralogs. Furthermore, we show that lin-41, as well as heterochronic genes daf-12 and hbl-1, are let-7a targets for which 3′ non-seed pairing is required. Interestingly, we find that the 3′ non-seed pairing to let-7a is functionally required in certain cases of perfect seed pairing, including the natural daf-12 3′ UTR and the reconfigured endogenous lin-41 3′ UTR, suggesting that the g11–g16 nucleotides of let-7a engage in essential interactions that, in parallel with seed pairing, critically influence miRISC activity.
RESULTS
The 3′ non-seed sequence is essential for the in vivo functional specificity of let-7a compared to family paralogs
The phylogenetic distribution of let-7 family miRNA genes among genomes with miRbase 22.1 annotations includes the vast majority of bilaterians (Figures 1A and S1B) (Kozomara et al., 2019). A total of 91.5% (n = 117) of bilaterian species have at least one let-7 family isoform with no more than 3 nucleotide differences from hsa-let-7a (Figure S1C). Alignment of the isoforms closest to let-7a from each of the 117 bilaterian species indicates that the g9–g22 nucleotides exhibit conservation ranging from 86% to 99%; 75.2% of these species contain a let-7 family miRNA completely identical to let-7a (Figures 1B and S1F). This degree of conservation is not observed for other conserved miRNAs (Figures S1D-S1H). The deep conservation of let-7a suggests that let-7a is associated with essential functions that depend on the identity of the 3′ non-seed nucleotides.
Figure 1. let-7a sequence is conserved across bilaterian species.

(A) Summary of let-7 family miRNAs across bilaterian phylogeny. Bar length, number of let-7 family isoforms; bar color, sequence distance relative to hsa-let-7a-5p.
(B) Nucleotide frequency of the let-7 family isoforms most similar to hsa-let-7a-5p across bilaterians.
See also Figure S1.
To test whether let-7a 3′ non-seed nucleotides are critical for in vivo function, we used CRISPR-Cas9 to mutate the let-7a locus in C. elegans by swapping the let-7a sequence with that of its closest paralog miR-84 (Figures 2A and S1A). The temporal expression profile of miR-84 miRNA in let-7(ma341) is consistent with expression from the endogenous mir-84 locus (starting in the larvalL2 stage) combined with expression from the edited let-7 locus (peaking in the L4 stage) (Figures 2B and 2C). Meanwhile in the mir-84(n4037, null) background, miR-84 expression from the let-7(ma341) locus exhibits a temporal profile like that of normal let-7a (Figures 2D-2E), suggesting that miR-84 miRNA is expressed from the endogenous let-7 locus.
Figure 2. The 3′ non-seed sequence determines the functional specificity of let-7a among paralogs.

(A) Strategy for generation of let-7(ma341) by CRISPR-Cas9-mediated swap of the pre-let-7 sequence for the pre-mir-84 sequence. Dot-bracket notations show the pre-miRNA structures predicted by RNAfold (Denman, 1993).
(B–E) Developmental profiles of miR-84 and let-7a miRNAs by Fireplex assay. Expression is calibrated with synthetic miR-84 and let-7a oligos. Time, hours after feeding starvation-arrested L1 larvae.
(F) Representative lineages and COL-19::GFP expression patterns of V1–V4 seam cells.
(G) Seam cell numbers of young adults. n, numbers of animals tested.
(H and I) Representative expression patterns of COL-19::GFP in adults. Scale bars, 100 μm.
(J and K) Differential interference contrast (DIC) images of the vulva region of adults. Scale bars, 25 μm.
(L) Adult lethality for wild type (WT), let-7(ma341), let-7(n2853, G5C), and let-7(ma393, null). Lethal phenotypes are categorized as severe (via bursting of young adults through the vulva) or mild (via matricide of egg-laying-defective adults).
The data in (B)–(E) represent 3 biological replicas. Error bars indicate mean ± SD.
See also Figure S5. Details of the phenotypes are available in Table S1.
In wild-type (WT) larvae, seam cells (hypodermal stem cells) undergo asymmetric divisions at each larval stage: one daughter cell differentiates to Hyp7 cells (hypodermis), while the other remains a stem cell (Sulston et al., 1983). At the L4 molt, the seam cells exit the cell cycle, fuse, and produce an adult-specific cuticle structure, the lateral alae. let-7(ma341) animals exhibit heterochronic and vulva integrity defects like let-7 loss-of-function (lf), including an extra round of seam cell division after the L4 molt, incomplete adult lateral alae (Figures 2F and 2G; Table S1), reduced expression of the adult-specific hypodermal reporter COL-19::GFP (Figures 2H and 2I), and adult lethality caused by bursting at the vulva (Figures 2J-2L). These phenotypes are consistent with the let-7a lf phenotypes observed in the null (ma393) and seed (n2853) mutants, indicating that the in vivo function of let-7a cannot be substituted by its closest paralog miR-84, even when miR-84 is expressed in a developmental profile essentially identical to let-7a (Liu et al., 1995; Nelson and Ambros, 2019; Reinhart et al., 2000). Since let-7a and miR-84 share the same seed sequence, these findings indicate that the 3′ non-seed sequence is a critical determinant of the in vivo function and specificity of let-7a.
Nucleotides 11–16 are each functionally essential for let-7a in vivo
To characterize how each 3′ non-seed nucleotide contributes to the in vivo function of let-7a, we performed a single-nucleotide mutational screen using the “jump-board” strategy (Duan et al., 2020a). For each g9–g22 nucleotide, we mutated both the miRNA and passenger strands to preserve the pre-miRNA structure (Figures 3A and S2A). To confirm the expression of the mutant miRNAs, we performed small RNA sequencing of all of the mutants and found that the abundance of the mutated let-7a miRNA in all cases was greater than 50% of that of the native let-7a in WT (Figure 3B). Since let-7 is recessive in C. elegans (indicating that the 2-fold reduction of let-7a dosage is not phenocritical), we reason that any phenotypes exhibited by these mutants can be attributed to the sequence of the miRNA, not reduced expression. Note that the strains with mutations at g11–g13 also contained a WT let-7 allele on a genetic balancer umnIs25(mnDp1); hence, both WT and mutant let-7a were expressed.
Figure 3. Contribution of single 3′ non-seed nucleotides to the in vivo function of let-7a.

(A) Alignment of pre-miRNA sequences. Boxed 3′ non-seed regions.
(B) Small RNA sequencing reads from the L4 larvae that mapped to WT or mutant let-7a sequences for each single mutant. The reads mapping to WT let-7a for strains carrying mutations at g11–g13 include WT let-7a miRNA from balancer umnIs25(mnDp1). The WT reads from U21A/U22A mutants likely reflect let-7a(mutant) miRNAs whose 3′ ends had been trimmed and subsequently uridylated in vivo, resulting in artificial let-7a(+) reads. Dashed lines, 100% (top) and 50% (bottom) of RPM of let-7a(+) in WT.
(C) Quantitation of let-7a lf phenotypes: percentage of animals with abnormal COL-19:GFP expression (top), numbers of progeny per animal (center), and percentage of adult lethality (bottom). Lethal phenotypes are categorized as in Figure 2L. Abnormal COL-19:GFP patterns are classified as no Hyp7 expression (severe) or faint Hyp7 expression (mild).
(D–G) DIC images of the vulva region in adults at 25°C. Scale bars, 25 μm.
(H) Functional synergy between g18 and the critical non-seed region based on vulva integrity defect. Labels are identical to (C) (bottom).
(I) Summary of the functional merits of let-7a 3′ non-seed nucleotides. Colored circle, 3′ non-seed nucleotide; red proportion, average adult lethality caused by vulva bursting at all temperatures.
Error bars indicate mean ± SD. See also Figures S2 and S3. Details of the phenotypes are available in Table S1.
Single-nucleotide mutations at g11, g12, or g13 resulted in severe vulva integrity defects, leading to lethality and dramatically reduced fecundity similar to let-7(null) (Figures 3C and 3D). Mutation of g11, g12, or g13 also caused defects in adult alae morphogenesis and COL-19::GFP expression, which are indicative of retarded heterochronic phenotypes (Figures 3C and S2B; Table S1). Single-nucleotide mutations at g14–g16 also resulted in distinct let-7a lf phenotypes, although they were milder than g11–g13 mutations (Figures 3C-3E). By contrast, single-nucleotide mutations at g9–g10 or g17–g22 did not cause visible phenotypes (Figures 3C-3F; Table S1). These results reveal g11–g16 to be a critical non-seed region of let-7a, with g11–g13 relatively more essential than g14–g16 (Figure 3I).
Nucleotides beyond g16 can compensate for mismatches in the critical non-seed region
Mutation of g18 to any of the other 3 nucleotides did not result in let-7a lf phenotypes, nor did simultaneous mutation within the g17–g22 region of 3 (g17–g19 or g20–g22) or 6 (g17–g22) consecutive nucleotides (Figure S2C). These results stand out in contrast to the deep conservation of the let-7a g17–g22 sequence (Figure 1B). We hypothesized that g17–g22 may contribute to target repression in cases in which mismatches occur within the g11–g16 region. To test this possibility, we generated a compound mutant let-7(ma449ma435) with both U16G and U18A mutations. We observed that let-7(ma449ma435) exhibited vulva bursting and retarded adult alae phenotypes (Figures 3E-3H, S2C, and S2D) that were significantly more penetrant than for each single mutant alone. We conclude that g18, and by implication other g17–g22 nucleotides, may contribute to interactions that involve mismatches in the g14–g16 subcritical region.
It is also possible that g17–g22 pairing could be critical for target repression associated with phenotypes that we have not measured. We accordingly assessed molecular phenotypes of let-7(ma435) by ribosome profiling and RNA sequencing (RNA-seq) (Figure S3; Table S2). We found that the expression levels of multiple genes are significantly changed by the U18A mutation, including 3 putative let-7a targets with predicted pairing to g18, supporting the hypothesis that there are in vivo circumstances in which g17–g22 nucleotides are critical for proper target regulation.
De-repression of lin-41 is a major contributor to let-7a critical non-seed mutant phenotypes
The evolutionarily conserved gene lin-41/Trim71 is a direct target of let-7a across diverse phyla, and repression of lin-41/Trim71 by let-7a is essential for normal development in invertebrates and vertebrates (Ecsedi and Grosshans, 2013; Worringer et al., 2014). In C. elegans, robust repression of lin-41 by let-7a is critical for the L4-to-adult cell fate progression, and mutations disrupting the seed pairing between let-7a and its 2 complementary sites (LCSs) in the lin-41 3′ UTR result in the reiteration of L4 cell fates and severe vulva defects (Aeschimann et al., 2019). Notably, both lin-41 LCSs have imperfect seed complementarity to let-7a with a GU pair and a target A-bulge, respectively, and both LCSs contain complementarity to let-7a g11-g19, suggesting that 3′ pairing is essential to compensate for weak seed pairing to lin-41 mRNA (Figure 4A).
Figure 4. The let-7a critical non-seed nucleotides confer in vivo function by repressing lin-41 and additional 3′ targets.

(A) Pairing configurations between let-7a and lin-41 LCS1/2.
(B–E) DIC images of adult vulva regions representative of the WT lin-41(tn1541) (B), lin-41(tn1541);let-7(ma432) (C), lin-41(tn1541ma480);let-7(ma432) (D) and lin-41(tn1541ma480) (E) at 25°C. Scale bars, 25 μm. All of the genotypes tested in this figure and in Figure 5 and Figure 7 include a GFP tag on LIN-41, denoted as tn1541 (Spike et al., 2014).
(F) Vulva integrity defects reflected by adult lethality (bottom) and the number of progeny (top). Lethal phenotypes are categorized identically to Figure 2L.
(G) Representative COL-19::GFP pattern in young adults at 25°C. Scale bars, 100 μm.
(H) Models proposing that lin-41 and additional 3′ targets are de-repressed in let-7a non-seed mutants.
See also Figure S4. Details of the phenotypes are available in Table S1.
We sought to verify whether the lf phenotypes of let-7a critical non-seed mutants result from de-repression of lin-41. We used the ma432(U13A) mutant to represent the critical non-seed nucleotides due to its severe phenotypes and the relatively high conservation of g13. At the late L4 stage of WT, endogenously tagged GFP::LIN-41 is undetectable in hypodermal cells due to let-7a repression (Figure S4A) (Spike et al., 2014). In contrast, in let-7(ma432,g13), we observed elevated peri-nuclear expression of GFP::LIN-41 in seam and Hyp7 cells, suggesting the de-repression of lin-41 (Figure S4B). To confirm that the lin-41 de-repression in let-7(ma432,g13) results specifically from the disruption of g13 pairing, we restored the native pairing configurations between let-7(ma432,g13) and lin-41 by introducing compensatory mutations at both LCSs in lin-41 3′ UTR (ma480,t13) (Figure 4A). GFP::LIN-41 expression was not detectably elevated in lin-41(ma480,t13);let-7(ma432,g13), indicating that restoring g13:t13 pairing restored lin-41 repression (Figure S4C). Using qRT-PCR and ribosome profiling, we confirmed that the levels of lin-41 mRNA abundance and translation were elevated in let-7(ma432,g13) compared to the WT, and restored to normal levels in lin-41(ma480,t13);let-7(ma432,g13) (Figure S4E and S6A). In lin-41(ma480,t13);let-7(ma432,g13) animals, the let-7(lf) phenotypes caused by the ma432 mutation were substantially rescued, based on the reduced lethality, increased number of progeny, and normal adult alae (Figures 4B-4F; Table S1). The rescue of let-7(ma432,g13) phenotypes by restoring non-seed pairing indicates that de-repression of lin-41 is the major contributor to the lf phenotypes of let-7a critical non-seed mutants.
In parallel, we found that when present with WT let-7a, lin-41(ma480,t13) exhibited an elevated peri-nuclear expression of GFP::LIN-41, suggesting that disrupting the critical non-seed pairing by mutations in the target also causes lin-41 de-repression (Figure S4D). As expected, we also observed vulva integrity defects and retarded heterochronic phenotypes in lin-41(ma480) (Figures 4E and 4F; Table S1), supporting the conclusion that 3′ non-seed pairing between let-7a and lin-41 is essential for the developmental down-regulation of lin-41.
Disruption of let-7a 3′ non-seed pairing results in de-repression of additional targets, including daf-12 and hbl-1
Although lin-41(ma480,t13);let-7(ma432,g13) showed reduced lf phenotypes compared to let-7(ma432,g13), the double mutant exhibits residual defects in vulva morphogenesis and egg-laying capacity (Figures 4D-4F) and reduced COL-19::GFP expression in Hyp7 cells of adults at 25°C, indicating residual retarded heterochronic phenotypes (Figure 4G). These residual phenotypes suggest that although lin-41(ma480,t13);let-7(ma432,g13) restores lin-41 repression, the let-7(ma432) mutation may cause de-repression of additional targets that require critical non-seed pairing, referred to here as “3′ targets” (Figure 4H). Consistent with the reasoning that the phenotypes of let-7(ma432,g13) result from the combined overexpression of lin-41 and other 3′ targets, the lin-41(ma480,t13) single mutant, in which lin-41 is the only let-7a 3′ target expected to be overexpressed, exhibits weaker phenotypes than let-7(ma432,g13) (Figure 4F; Table S1).
To identify let-7a 3′ targets in addition to lin-41, we screened for genes that are de-repressed by the let-7a critical non-seed mutation using ribosomal profiling (Ribo-seq), which assesses gene expression on the translational level (Ingolia, 2016). We compared Ribo-seq profiles between WT and lin-41(ma480,t13);let-7(ma432,g13), in which lin-41 repression is restored, yet other let-7a 3′ targets are expected to be de-repressed. At the L4 stage, 351 genes were significantly overexpressed >1.5-fold in lin-41(ma480,t13);let-7(ma432,g13). To apply a stringent test for which of these differentially expressed genes could be perturbed as a result of the let-7a mutation specifically versus genes whose observed perturbation could be caused by asynchrony in staging between samples, we applied a filter to remove genes that are known to be highly dynamic over the developmental interval encompassing our sample collections (Aeschimann et al., 2017) (STAR Methods). A total of 203 genes passed this filter and were therefore judged to be likely overexpressed due to the let-7a mutation specifically (Figure 5A; Table S3). The translational levels of lin-41 and its direct downstream genes were not significantly changed compared to WT, indicating that the de-repressed genes in lin-41(ma480,t13);let-7(ma432,g13) are independent of lin-41 pathway (Figure S6A) (Aeschimann et al., 2019). To identify potential direct let-7a 3′ targets among the overexpressed genes, we used a computational approach to predict let-7a sites using seed pairing and g11–g16 pairing constraints (STAR Methods) (Veksler-Lublinsky et al., 2010). We identified 624 C. elegans genes whose 3′ UTRs contain let-7a 3′ sites, among which 8 genes are overexpressed in the g13 mutant, including the heterochronic genes daf-12 and hbl-1 (Figure 5A; Table S4).
Figure 5. let-7a represses multiple targets, including daf-12 and hbl-1, through 3′ non-seed pairing.

(A) Differential expression analysis of translatomes of lin-41(tn1541ma480);let-7(ma432) and lin-41(tn1541). Hollow points, developmentally dynamic genes for which the observed perturbation could be caused by asynchrony in staging between samples, independently of genotype; solid points, genes for which the observed perturbation is judged to likely reflect an effect of the let-7a mutation specifically (see STAR Methods; Table S3). Circular points with text labels, genes with predicted let-7a 3′ sites; triangle points, genes containing only let-7a seed-only sites.
(B) Retarded COL-19::GFP patterns characteristic of lin-41(tn1541ma480);let-7(ma432) young adults at 25°C under empty vector, daf-12(RNAi), or hbl-1(RNAi). Scale bars, 100 μm.
(C) Fold changes of mRNA abundance and translational efficiency (TE). Red points, genes with significantly increased mRNA abundance. Orange points, genes with significantly increased TE. Purple points, genes with both significantly increased TE and mRNA abundance. Circled points, genes with significantly increased RPFs in (A). Predicted let-7a 3′ targets are text labeled.
(D and E) DIC (left) and fluorescent (right) microscopy images showing expression of DAF-12::mSCARLET at L3/L4 stages at 25°C representative of lin-41(tn1541);daf-12(ma498) (D) and lin-41(tn1541);daf-12(ma498ma568) (E). Scale bars, 5 μm. All of the fluorescent images were generated with identical exposure and processing conditions.
(F) Quantification of relative fluorescent intensity of DAF-12::mSCARLET in seam cell nuclei relative to the adjacent Hyp7 nuclei. The numbers of seam cells quantified are shown under each violin plot. A total of 22 and 28 animals were scored for daf-12(ma498) at L3 and L4, respectively; 23 and 31 animals were scored for daf-12(ma498ma568) at L3 and L4, respectively.
(G) Models for daf-12 repression at L3/L4. Purple region, trinucleotide mutations that disrupt duplexing with g11–g13 of let-7a.
(H) Representative expression patterns of COL-19:GFP in adult animals. Scale bars, 100 μm.
Data in (A) and (C) represent 3 biological replicas. See also Figures S6 and S7.
The 3′ UTRs of daf-12 and hbl-1 contain multiple let-7a sites with complementarity to the critical non-seed region of let-7a (Figures S6B-S6D). We reasoned that the de-repression of daf-12 and/or hbl-1 could contribute to the residual heterochronic phenotypes of lin-41(ma480,t13); let-7(ma432,g13). Therefore, we used RNAi to knock down daf-12 or hbl-1 during the larval development of lin-41(ma480,t13);let-7(ma432,g13) and assayed for suppression of the residual lf phenotypes. Knocking down either daf-12 or hbl-1 by RNAi rescued the abnormal COL-19::GFP pattern in lin-41(ma480,t13);let-7(ma432,g13) (Figure 5B), suggesting that de-repression of daf-12 and hbl-1 contributes to the residual retarded phenotypes of lin-41(ma480,t13);let-7(ma432,g13), and that the critical non-seed pairing of let-7a is required for the repression of not only lin-41 but also additional targets, including daf-12 and hbl-1, for the L4-to-adult transition.
Previous transgenic reporter analyses suggested that daf-12 can be repressed by let-7 family miRNAs during larval development (Grosshans et al., 2005; Hammell et al., 2009). To gather additional evidence in support of a role for let-7a in the downregulation of DAF-12, we quantified the level of expression of an endogenously tagged fluorescent reporter daf-12(ma498,-mScarlet) (Ilbay and Ambros, 2019) in the L3 and L4 stages, which span the interval when let-7a is dramatically upregulated. The data confirm that endogenous DAF-12::mSCARLET expression is significantly downregulated in seam cells between the L3 and L4 stages, consistent with the repression of daf-12 by let-7a in the L4 (Figures 5D, 5F, 5G, S7B, and S7C).
To further test whether 3′ pairing to let-7a is required for the developmental downregulation of DAF-12, we used CRISPR-Cas9 editing to mutate the LCS sequences in the endogenous daf-12(ma498,mScarlet) 3′ UTR (Figure S7A) to disrupt let-7a g11-g13 complementarity. We found that compared to WT, the L4-specific reduction of DAF-12::mSCARLET in seam cells was abrogated in daf-12(ma498ma568) larvae (Figures 5E-5G). Thus, we conclude that 3′ non-seed pairing of let-7a contributes to the repression of daf-12 at the L4 stage.
daf-12(ma498ma568) exhibits a retarded COL-19::GFP expression phenotype (Figure 5H), supporting the idea that de-repression of daf-12 contributes to the residual phenotypes of lin-41(ma480);let-7(ma432). Meanwhile, daf-12(ma498ma568) exhibits relatively less penetrated phenotypes (4.6%, Figure 5H) compared to lin-41(ma480);let-7(ma432) (76.6%; Figure 5B), suggesting that the L4-to-adult cell fate transition requires the downregulation of multiple 3′ targets in addition to lin-41 and daf-12, such as hbl-1 (Figure S7E).
Repression of let-7a 3′ targets can involve translational suppression and/or mRNA decay
miRNA-mediated gene regulation can occur via translational repression or mRNA destabilization, indicated by decreased translational efficiency (TE) or reduced mRNA abundance, respectively (Bazzini et al., 2012; Giraldez et al., 2006). let-7a appears to regulate lin-41 through a combination of both modes (Bagga et al., 2005; Nottrott et al., 2006). To assess the repression mechanisms for 3′ targets other than lin-41, we performed Ribo-seq and RNA-seq analyses of lin-41(m480,t13);let-7(ma432,g13) at L4 larvae (Figures 5C and S6; Table S3). We found that the set of de-repressed let-7a 3′ targets in lin-41(ma480,t13);let-7(ma432,g13) included examples of increased mRNA abundance (e.g., grd-10) or increased TE (e.g., hbl-1), or a combination of both (e.g., daf-12) (Figure 5C). The verified let-7a targets daf-12 and hbl-1 were de-repressed in the same fashion as reported previously for seed mutants (Aeschimann et al., 2017) (Figure S5H). Thus, target recognition involving the let-7a 3′ non-seed region can elicit inhibition of TE or mRNA stability or both.
Continuous pairing at g11–g15 is required to compensate for the imperfect seed pairing between let-7a and lin-41
The involvement of 3′ pairing to compensate for imperfect seed pairing can be essential for in vivo targeting efficacy for certain miRNA-target interactions, but how the base pairing at different positions contributes to this compensatory effect remains unclear (Bartel, 2018). Moreover, detailed analyses of how 3′ pairing contributes to endogenous target repression in the context of perfect seed pairing are lacking. To investigate how 3′ pairing coordinates with seed pairing for endogenous target repression, we systematically introduced mutations designed to produce various configurations of seed and non-seed pairing between WT let-7a and the lin-41 3′ UTR (Figure 6). In each case, mutations were introduced into both lin-41 LCSs and were tested in the WT let-7a background, so the observed phenotypes should reflect the de-repression of lin-41 without confounding effects from other let-7a targets.
Figure 6. Critical 3′ non-seed pairing and seed pairing together contribute to let-7a target repression.

(A–F) Pairing configurations of let-7a to the lin-41 LCSs and associated phenotypes for lin-41(ma501) (A), lin-41(ma501ma480) (B), lin-41(ma501ma545) (C), lin-41(+) (D), lin-41(ma480) (E), and lin-41(ex11) (F), arranged in a matrix based on mismatches in seed (vertical axis) and critical non-seed region (horizontal axis). Each panel includes pairing pattern (top left), vulva defect (young adult lethality and number of progeny, top right), and heterochronic phenotype (bottom). Scale bars, 100 μm.
(G and H) Vulva integrity of lin-41 LCSs mutants with 3′ pairing mismatches in the context of imperfect (G) and perfect seed pairing (H).
(I) Models depicting the minimal pairing requirement for 3′ compensatory and 3′ supplemental effects. Statistical significance indicates relative to WT. Phenotypes were tested at 25°C. All of the lin-41 alleles tested in this figure do not contain a fluorescent protein tag.
Details of the phenotypes available in Table S1.
A previous study using mutant lin-41 LCS constructs with let-7a 3′ complementarity restricted to g13–g16 found that g13–g16 pairing alone was insufficient to compensate for imperfect seed pairing (Brancati and Grosshans, 2018). Our finding that certain single mismatches at g13 can cause strong lin-41 gain-of-function (gf) phenotypes suggests that the extent of 3′ pairing is less important than the precise pattern of pairing (Figure 6E). Through further analysis of a series of single-nucleotide mutations in the lin-41 3′ LCSs (Figure 6G), we found that disrupting base pairing at any single position in the g11–g15 complementary region results in lin-41 gf phenotypes, indicating that continuous pairing from g11 to g15 is required to compensate for the imperfect seed pairing between let-7a and lin-41 (Figure 6I).
To determine whether the requirement for g11–g15 pairing is to compensate for the imperfect seed match characteristic of the WT lin-41:let-7a interaction, we mutated both LCSs of the t11, t12, and t13 lin-41 mutants to permit consecutive g2–g8 Watson-Crick pairing (PS) to let-7a, while maintaining the single 3′ mismatch in each case. We found that the resulting compound mutants lin-41(ma501ma555,PS + t11), lin-41(ma501ma556,PS + t12), and lin-41(ma501ma480,PS + t13) exhibited substantial rescue of the gf phenotypes of lin-41(ma555,t11), lin-41(ma556,t12), and lin-41(ma480,t13), respectively (Figures 6B and 6H). This finding suggests that the requirement for pairing across g11–g15 in large part reflects compensation for imperfect seed pairing of let-7a to the lin-41 WT LCSs.
Both seed and 3′ non-seed pairing of let-7a are required for full repression lin-41
The above results suggest that the requirements for 3′ pairing may be less stringent with a perfect seed pairing to let-7a compared to the imperfect seed. To test whether 3′ pairing in the g11-g13 region is fully dispensable in the context of perfect seed pairing, we combined ma501 with a compound mutation (ma545) which simultaneously mismatches g11–g13 of let-7a (Figure 6C). Interestingly, lin-41(ma501ma545,PS + t11–13) exhibited strong vulva integrity defects and retarded COL-19::GFP expression, indicating that lin-41 was de-repressed despite the perfect seed pairing to let-7a (Figure 6C). This indicates that even with perfect seed complementarity, pairing within g11–g13 still critically contributes to the functional targeting of lin-41. We also combined ma501 with mismatch mutations at t11–t12 and t13–t16. The resulting mutants lin-41(ma501ma571,PS + t11–t12) and lin-41(ma501ma572,PS + t13–t16) did not exhibit observable lin-41 gf phenotypes (Figure 6H). We suggest that in the context of perfect seed pairing, full lin-41 repression by let-7a can occur provided that there is 3′ pairing at g11–g12 (lin-41(ma501ma572)) or g13–g16 (lin-41(ma501ma571)) (Figure 6I).
These results suggest that let-7a family paralogs miR-48, miR-84, and miR-241, which differ from let-7a substantially in their 3′ sequences, should not fully repress lin-41 even with engineered perfect seed pairing, even though their combined expression may exceed the level of let-7a at the L4 stage in C. elegans (Nelson and Ambros, 2021) (Figures 7A and 7C). Accordingly, lin-41(ma501,PS);let-7a(ma393,null) exhibited vulva integrity defects nearly as severe as let-7a(ma393,null), indicating that let-7a family paralogs do not contribute substantially to lin-41 repression, even with engineered perfect seed matches (Figure 7B).
Figure 7. Perfect seed pairing alone is insufficient for let-7a family paralogs to confer full repression of lin-41.

(A) Predicted seed-only pairing (RNAhybrid (Rehmsmeier et al., 2004) of let-7a and paralogs miR-48/84/241 to LCS1 (top) and LCS2 (bottom) of lin-41(ma501). Requirements for minimal supplemental pairing (t11–t12 pairing or t13–t16 pairing; Figure 6I) are indicated by brown or purple.
(B) Vulva integrity defects based on adult lethality (top) and number of progeny (bottom) in lin-41(tn1541), lin-41(tn1541);let-7(ma393,null) and lin-41(tn1541ma501);let-7(ma393).
(C) Predicted targeting configurations.
Details of the phenotypes available in Table S1.
The evident importance of 3′ pairing for lin-41 repression by let-7a prompted us to test whether 3′ non-seed pairing could compensate for severely compromised seed pairing. We used a previously reported allele lin-41(xe11), which creates 2 consecutive seed mismatches to let-7a in each LCS (Ecsedi et al., 2015). We confirmed that lin-41(xe11) animals exhibit strong vulva integrity defects and retarded heterochronic phenotypes (Figure 6F), indicating that extensive 3′ pairing does not render seed pairing dispensable.
DISCUSSION
A 6-nt critical non-seed pairing region of let-7a, involving g11–g16
Base paring of the 3′ non-seed region of miRNAs to targets has been implicated in compensating for weak seed pairing, for seed family isoform specificity, and target-dependent miRNA degradation (TDMD) (Bartel, 2018; Pawlica et al., 2020). However, there has been no systematic study of the contribution of individual 3′ non-seed nucleotides to the functional efficacy of miRNA-target interactions in intact animals. The functional contributions of individual 3′ nucleotides is particularly pertinent for miRNAs with evolutionarily conserved 3′ non-seed sequences (e.g., let-7a) and for their evolutionarily conserved targets (e.g., lin-41, in the cae of let-7a). Here, we used CRISPR-Cas9 genome editing for a systematic investigation of the in vivo function of individual 3′ non-seed nucleotides of let-7a in the regulation of lin-41 and other targets in C. elegans.
A key finding from our studies is that each nucleotide at g11–g16 critically contributes to the in vivo function of let-7a. This is consistent with findings from human miRISC in vitro RNA bind-n-seq (RBNS) showing that pairing to g11–g16 of let-7a is particularly crucial for enhancing the affinity of miRISC for oligonucleotide targets (McGeary et al., 2022). We further found that within g11–g16, the g11–g13 subregion is particularly important to let-7a in vivo function. This finding also agrees with the RBNS results, which show that among 4-nt blocks of target complementarity scanned along the let-7a 3′ region, pairing at g11–g14 was the most potent contributor to enhanced target affinity (McGeary et al., 2022). Therefore, both our genetic analysis in C. elegans and the RBNS analysis of human AGO2 agree on a 6-nt critical 3′ pairing region (g11–g16) of let-7a, expanded by 2 nt compared to the g13–g16 region previously identified by biochemical analysis and the co-crystallized structure of the miRISC-target complex (Grimson et al., 2007; Sheu-Gruttadauria et al., 2019).
Our results stand in contrast to a previous genetic study that indicated the functional insignificance of the 3′ non-seed region of let-7a in C. elegans, in which a compound mutation at g10, g12, and g13 of let-7a exhibited relatively mild developmental defects (Zhang et al., 2015). It is likely that since the results from Zhang et al. (2015) were obtained using exogeneous high-copy transgenes to express mutant let-7a miRNAs, the overexpression of the mutant miRNA compensated for its poor intrinsic efficacy. This highlights the importance of using genome engineering of endogenous miRNA loci so that repression efficacy can be assessed in the context of native gene dosage.
Structural analyses of human miRISC reveal that duplexing of a miRNA with perfect seed complementarity to target results in 3′ pairing at g13–g16, while g11 and g12 were structurally hindered from duplexing with the target (Sheu-Gruttadauria et al., 2019). Our results suggest that for certain target interactions, let-7a-miRISC may adopt a previously unanticipated conformation that enables g11–g16 pairing to target. For the evolutionarily conserved let-7a target Trim71/lin-41, LCS sequences conserve the combination of imperfect seed pairing accompanied by 3′ pairing involving g11 and g12. In some instances, even the specific configuration of seed mismatches is conserved (Nelson and Ambros, 2021)). We suggest that these mismatched seed configurations may promote an AGO conformation that favors the pairing of g11–g12, in addition to the previously identified g13–g16 pairing.
Although our results confirm that certain imperfect seed match configurations can be compensated for by extensive 3′ pairing, we also confirm that seed pairing is not dispensable, as lin-41 LCSs with 2 nt seed mismatches displayed strong lin-41 gf phenotypes despite extensive 3′ pairing (Figure 6F). Similarly, disrupting either the seed or 3′ complementarity results in de-repression of daf-12 in C. elegans (Figure S7D) (Aeschimann et al., 2017).
Contribution of the 3′ non-seed pairing to the target specificity of let-7 family paralogs
Broughton et al. (2016) reported that the C. elegans let-7a family paralog miR-48, which does not regulate lin-41 in WT, can substitute for let-7a in repressing lin-41 if the lin-41 LCSs were replaced by sites with perfect seed match plus 3′ pairing to miR-48. It was proposed that the weak seed pairing of let-7a to lin-41 necessitates the 3′ pairing, which differs remarkably between let-7a and miR-48, and consequently determines the targeting specificity whereby lin-41 is regulated by let-7a, not miR-48 (Brancati and Grosshans, 2018). Thus, 3′ non-seed pairing combined with imperfect seed pairing is a powerful determinant of specificity for miRNAs of the same seed family. This model is supported by our finding that miR-84, the closest family paralog of let-7a in C. elegans, does not functionally substitute for the absence of let-7a (Figure 2). We suggest that our result provides a stringent test of the model, because in our experiments, miR-84 was expressed at levels and with a temporal profile identical to WT let-7a; and unlike miR-48 in the previous reports, which differs from let-7a at every non-seed position, miR-84 has only 5 nt different from let-7a (Figure S1A). We also confirm that the in vivo functional distinction between let-7a and miR-84 primarily reflects the specificity of let-7a for targeting lin-41, as well as other targets that require 3′ non-seed pairing (Figure S5).
By mediating miRNA family targeting specificity, 3′ non-seed pairing may enable spatiotemporal specificity of target regulation. In C. elegans, hbl-1 is repressed by let-7a paralogs at the L2-to-L3 transition, and by let-7a at the L4-to-adult transition (Abrahante et al., 2003; Ilbay and Ambros, 2019). The hbl-1 3′ UTR has sites with perfect seed match that can be recognized by any paralog in the let-7 family (perfect seed only), and hence may mediate the repression of hbl-1 by let-7 family paralogs at earlier larval stages. Meanwhile, the hbl-1 3′ UTR also contains sites expected to be specifically recognized by let-7a (imperfect seed +3′ pairing), which enables the let-7a-specific repression of hbl-1 in cells that express let-7a at late larval stages (Figure S6D).
The necessity of 3′ non-seed pairing for target repression in the context of perfect seed pairing
It has long been recognized that perfect seed complementarity can be sufficient, in certain contexts, for miRISC to bind and repress a target (Brennecke et al., 2005; Doench and Sharp, 2004; Wee et al., 2012), implying that 3′ pairing may be required only to compensate for weak seed pairing. However, there is also evidence that 3′ pairing can contribute critically to target regulation for instances of perfect seed matches. For example, evolutionarily conserved target sites with perfect seed plus 3′ pairing (3′ supplemental sites) are prevalent in metazoan clades and appear to be more frequent than the 3′ compensatory sites, which have imperfect seed pairing (Friedman et al., 2009). Also, competitive binding experiments reveal that 3′ pairing can enhance miRISC affinity to perfect seed sites and enable miRNA isoforms with identical seeds to compete for target recognition (Xiao and MacRae, 2020).
Broughton et al. (2016) reported findings indicating that in the context of perfect g2–g7 seed pairing, 3′ pairing is required for the specificity of C. elegans let-7 family isoforms in the developmental repression of lin-41. Brancati and Grosshans (2018) showed that for lin-41 LCSs modified to contain a modified miR-48 site from the dot-1.1 gene with perfect g2–g8 let-7a seed pairing, let-7a was unable to fully rescue the mir-48(null) phenotypes. Our results reinforce those findings by showing that in the absence of let-7a, the let-7 family paralogs can only confer a limited degree of repression of lin-41 via perfect seed pairing alone (Figure 7). Moreover, we investigated the minimal requirements for 3′ pairing for the full repression of lin-41 containing LCSs with perfect g2–g8 seed pairing and confirmed that specific let-7a 3′ pairing is necessary to supplement perfect g2–g8 pairing, but the functional constraints on 3′ pairing are less stringent for the perfect seed pairing (Figure 6). Importantly, we show that the WT 3′ UTRs of additional let-7a targets, including the heterochronic gene daf-12, contain sites with perfect seed matches combined with extensive 3′ complementarity to let-7a, and the 3′ supplemental pairing is essential for the downregulation of DAF-12 at later developmental stages (Figure 5). These findings run counter to the assumption that perfect seed pairing should be sufficient for miRISC to confer repression and emphasize that in the native context of an intact developing animal, the miRNA 3′ non-seed pairing can be critical for the efficacy of evolutionarily conserved miRNA-target interactions.
It is noteworthy that the LCSs with perfect seed plus 3′ complementarity to let-7a in the daf-12 3′ UTR should also allow binding of the let-7 family paralogs by seed pairing. Indeed, it has been reported that daf-12 is repressed by let-7 family paralogs (mir-48, mir-84, mir-241) at the L2-to-L3 stage transition, and by let-7a at the L4 stage (Grosshans et al., 2005; Hammell et al., 2009) (Figures 5D-5F). Since 3′ pairing can render higher target binding affinity to the miRISC, we suggest that the involvement of 3′ non-seed pairing to let-7a may enable more efficacious repression of daf-12 at the L4 stage than that at earlier stages (Figure 5G).
Currently, we can only speculate about the mechanistic basis for this requirement for 3′ pairing in the context of perfect seed match. Perhaps certain structural peculiarities of a 3′ UTR (e.g., daf-12, lin-41) may render let-7a “seed-only” pairing unfavorable compared to “seed plus 3′ supplemental” pairing, or even “imperfect seed plus 3′ compensatory” pairing. It is also possible that repression by let-7a requires the recruitment of cofactors that recognize miRISC conformational features induced by 3′ pairing.
The multiplicity of C. elegans let-7a targets with 3′ non-seed pairing
Previous reports showed that lin-41 is the major let-7a target whose de-repression underlies the lf phenotypes of let-7a seed mutants (Ecsedi et al., 2015). In our study, we further confirmed that lin-41 is the major let-7a 3′ target by showing the de-repression of LIN-41 by a g13 mutation let-7(ma432), and the rescue of LIN-41 de-repression and the associated lf phenotypes by a compensatory mutation lin-41(ma480). These results were assessed using 3 approaches: (1) an endogenously tagged fluorescent LIN-41 reporter for the visualization of cell-specific expression (Figures S4A-S4D); (2) qRT-PCR to measure the global level of lin-41 transcript (Figure S4E); and (3) ribosome profiling to measure the global level of translation of lin-41 mRNA (Figure S6A). Note that we were not able to analyze the let-7(ma432) single mutant because let-7(ma432) animals exhibit strong adult lethality, which precludes propagating them in numbers that are adequate for ribosome profiling. However, a previous report showed that the translational level of lin-41 was upregulated approximately 8 times in the let-7(n2853) seed mutant (Aeschimann et al., 2017). Since let-7(ma432) exhibits stronger phenotypes than does let-7(n2853), we anticipate that lin-41 should be overexpressed to a greater degree in let-7(ma432) than in let-7(n2853). Thus, the non-significant changes in ribosome-protected footprint for lin-41 in lin-41(ma480);let-7(ma432) animals (Figure S6A) can be interpreted to indicate the restoration of normal lin-41 repression.
Our computational analysis identified 624 genes in C. elegans that contain 3′ UTR sequences predicted to bind let-7a with 3′ non-seed pairing. This finding is consistent with previous high-throughput analyses of miRNA-target chimeric ligation products, which also identified multiple let-7a targets with 3′ non-seed complementarity (Broughton et al., 2016; Grosswendt et al., 2014). By ribosome profiling, we confirmed that 3′ non-seed pairing is critical for the let-7a repression of at least 8 genes and that the overexpression of daf-12 and hbl-1 contribute to the developmental phenotypes of the let-7a g13 mutant (Figure 5). This finding indicates that in addition to lin-41, at least daf-12 and hbl-1 are also phenocritical let-7a targets in C. elegans (Ecsedi et al., 2015). We did not test the phenotypic consequences of the other overexpressed let-7a 3′ targets that are not known to be involved in the heterochronic pathway.
It is curious that the mutation of g18, which is one of two 3′ nucleotides of let-7a that shows the greatest evolutionary conservation, did not cause visible phenotypes. By ribosome profiling, we found 3 genes that were upregulated in the let-7a g18 mutant and that contain 3′ UTR sites with 3′ complementarity, including g18, suggesting that they could be let-7a targets whose proper repression depends on g18 pairing. However, caution is called for in interpreting these genes as g18 targets because their Wormbase annotations indicate that in the WT, these transcripts undergo developmental upregulation at the L4 (Harris et al., 2020), which is the stage at which our samples were collected. Thus, based on the Wormbase annotations, the increased RPF observed for these candidate g18 targets in the g18 mutant could be an artifact of slight differences in staging between samples. However, published data from translational profiling of C. elegans larvae (Aeschimann et al., 2017) argue against the above caveat, as they show that the translational levels of our candidate g18 targets were not significantly increased from L3 to L4 (Figure S3E). Moreover, in the g18 mutant, 2 of the putative target genes (chs-1 and try-1) were translationally de-repressed without significant difference in mRNA levels (Figures S3B and S3C), suggesting that in these cases, staging differences between samples are unlikely to explain the data. We thus propose that 3′ pairing to let-7a at g18 may contribute a dampening of the increase in the translational output of these developmentally upregulated mRNAs. Interestingly, in the Aeschimann et al. (2017) dataset, these putative g18 targets were not translationally de-repressed in let-7a(n2853, G5C) (Figure S3F). Although this latter difference with our results could reflect the distinct experimental setups, it is also possible that the U18A mutation may be more detrimental to the proper repression of these putative targets than the G5C seed mutation.
The evolutionary conservation of the let-7a sequence
The entire let-7a sequence is almost perfectly conserved across bilaterian phyla (Figure 1) (Wolter et al., 2017), suggesting pervasive involvement of let-7a in interactions that constrain the g9–g22 sequence. The conservation of g11–g16 of let-7a could be driven by phenocritical roles for these nucleotides in the repression of conserved targets and reinforced by a multiplicity of targets with g11–g16 pairing (John et al., 2004), supported by our identifying multiple C. elegans let-7a targets with critical non-seed pairing.
Our data do not appreciably illuminate potential mechanisms for the conservation of let-7a 3′ distal nucleotides (g17–g22) or the bridge nucleotides (g9–g10). However, although single-nucleotide or compound mutations were phenotypically tolerated in g17–g22, we observed the phenotypic effects of a g18 mutation in the context of a g16 mutation, suggesting the involvement of 3′ distal nucleotides in repressing targets with obligate mismatches to g14–g16 to let-7a. It is also possible that let-7a 3′ distal nucleotides could interact with miRISC-associated RNA-binding proteins, which recognize specific sequences. Our finding that mutations at g9–g10 were also phenotypically tolerated is consistent with the observation that let-7a is not predicted to engage in target recognition involving g9–g10 paring in C. elegans, and that fully complementary target sites that involve the bridge pairing are rare in vertebrates (Bartel, 2018). Interestingly, miRNA bridge nucleotides (g9-g10 for let-7a) can be exposed on the miRISC surface even when their sequences are complementary to the target (Sheu-Gruttadauria et al., 2019), suggesting that these nucleotides could engage in interactions external to AGO. We hypothesize that g9–g10 of let-7a may be constrained by association with conserved RNA-binding proteins that recognize these nucleotides, possibly in the context of let-7a biogenesis or target binding.
It is also possible that the identity of g9–g10 or g17–g22 nucleotides of let-7a could be critical for target repression under conditions not assayed in this study, such as various conditions of stress, and/or could be required for target interactions unrelated to the phenotypes that we monitored. The latter possibility is supported by our finding that the g18 mutant displays molecular phenotypes by Ribo-seq and RNA-seq (Figure S3). Such molecular phenotyping of other distal 3′ nucleotides of let-7a, and other “silent” nucleotides of other miRNAs, could reveal otherwise experimentally inaccessible functionalities.
Limitations of the study
There are limitations to the use of ribosome profiling of whole animals for the confirmation of in vivo miRNA targeting. In particular, the data could contain numerous false negatives, especially for genes that are broadly expressed but are only repressed by miRNAs in specific tissues or cells. For example, although we identified 8 target genes with 3′ non-seed complementarity that were upregulated in the let-7a g13 mutant, genes with such 3′ complementarity as a class were not statistically enriched (Figure S6E). Similarly, potential let-7a target genes with g18 pairing were also not statistically enriched among the de-repressed genes in the let-7a g18 mutant (data not shown; Figure S3).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact Victor Ambros (victor.ambros@umassmed.edu).
Materials availability
All C. elegans strains and reagents will be shared upon request. Data and Code Availability The raw and processed data of ribosome profiling, RNA-seq, and small RNA seq in this study have been deposited at NCBI Gene Expression Omnibus (GEO) with accession number GSE171748 (Edgar et al., 2002) and are publicly available. All original code has been deposited at GitHub (https://github.com/IsanaVekslerLublinsky/Let7_Proj_code.git.) with Zenodo DOI 6465945, and is publicly available. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Data and code availability
The raw and processed data of ribosome profiling, RNA-seq, and small RNA seq in this study have been deposited at NCBI Gene Expression Omnibus (GEO) with accession number GSE171748 (Edgar et al., 2002) and are publicly available.
All original code has been deposited at GitHub (https://github.com/IsanaVekslerLublinsky/Let7_Proj_code.git.) with Zenodo DOI 6465945, and is publicly available.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental models: organisms/strains
The genotypes, resources and identifiers (strain names) of the C. elegans strains used and the genetic alleles generated in this study are described in Table S5.
METHOD DETAILS
Phylogeny and conservation analysis
Precursor and mature miRNA sequences, and a phylogenetic tree of metazoan species were downloaded from miRbase (v22.1) (Kozomara et al., 2019). An in-house code was developed to identify miRNAs that belong to the let-7 family based on the presence of the seed sequence “GAGGUA” at g2-g7 on the mature miRNA. For each species in the collection, the number of total miRNA loci and the number of let-7 miRNA loci were counted. To calculate the distance of each member to the conserved hsa-let-7a, its sequence was aligned to the hsa-let-7a sequence and the number of mismatches in the alignment was counted. The phylogenetic trees were visualized with the program evolView v2 (He et al., 2016).
117 species encode let-7a family isoforms. From each species, the sequence of one, the most similar to let-7a, was extracted and used to construct a conservation profile. A similar analysis was done with the mir-1 and mir-34 families to obtain their conservation profiles.
Targeted mutagenesis at the let-7a genomic locus
CRISPR/Cas9 genome editing of the let-7a locus was performed using the “jump board” strategy previously described (Duan et al., 2020a) on strain VT3742, which carries the let-7(ma393) insertion of the “jump board” sequence in place of the pre-let-7 sequence, as well as the genetic balancer umnIs25(mnDp1) and a transgene oxSi1091 expressing Cas9. Templates for dsDNA HR donors were prepared by cloning the WT pre-let-7a and 500 bp of flanking sequence into pCR2.1-TOPO vector. The Q5 mutagenesis kit (NEB, Cat: E0554) was used to generate the mutant plasmids. Double-strand DNA donors were generated from the mutant plasmids by PCR with 73/106 base-pairs flanking the pre-let-7a, and the PCR product was purified by ethanol precipitation. Injection mixtures containing final concentrations of 30 ng/μL AltR_Cas-9_crRNA_INPP4A_1/2 each, 10 ng/μL AltR_Cas-9 _crRNA_dpy-10_cn64 as co-CRISPR marker (Arribere et al., 2014), 75 ng/μL Alt-R tracrRNA (IDT, Cat:1072532), 10 ng/μL each dsDNA donor in 1X duplex buffer (IDT, Cat: 11010301) were injected into the gonad of VT3742 at young adult stage. F1 dumpy animals were isolated and genotyped by PCR with let-7_SEQ_F5/R5 primers followed by an analysis of the PCR product using restriction digestion using EcoRV (NEB, Cat: N3195) and Sanger sequencing as described in Duan et al., (2020a). Mutants were backcrossed with N2 (for strains without maIs105) or VT1367(for strains with maIs105) for at least two generations.
Targeted mutagenesis at the lin-41 genomic locus
Ultramer single-strand DNA donors (lengths ranging from 115 nt to 117 nt) with 35 nt flanking homology were obtained from IDT. The injection mixture containing final concentrations of 25 ng/μL AltR_Cas-9_crRNA_lin-41_1/2 each, 15 ng/μL AltR_Cas-9_crRNA_dpy-10_cn64, 95 ng/μL Alt-R tracrRNA (IDT, Cat:1072532), 325 ng/μL each ssDNA donor in 1X duplex buffer (IDT, Cat: 11010301) were incubated at room temperature for 10 min for pre-annealing and injected into the gonad of EG9615, VT3867 or VT3873 which contain transgene oxSi1091 expressing Cas9. F1 dumpy animals were isolated and genotyped by PCR with lin-41_SEQ_F2/R2 primers, followed by an analysis of the PCR product using Sanger sequencing. Mutants were backcrossed with N2 for at least two generations.
Targeted mutagenesis at the daf-12 genomic locus
Double-stranded DNA donor (length 641 bp) containing the desired mutations plus 65/78 bp flanking homology was obtained from GENEWIZ. The injection mixture, containing final concentrations of 80 ng/μL AltR_Cas-9_crRNA_daf-12_4, 40 ng/μL AltR_Cas-9_crRNA_dpy-10_cn64, 223.3 ng/μL Alt-R tracrRNA (IDT, Cat:1072532), and 73.3 ng/μL dsDNA donor in 1X duplex buffer (IDT, Cat: 11010301), was incubated at room temperature for 10 min for pre-annealing and injected into the gonad of VT4126 which contain daf-12(ma498ma567, mScarlet with 3′ UTR InDel) as a daf-12 “jump board” and transgene oxSi1091 expressing Cas9 (Ilbay and Ambros, 2019). Genotyping and backcrossing were performed the same as the lin-41 locus.
Worm culturing and synchronization
C.elegans were cultured on nematode growth medium (NGM) and fed with E. coli HB101 unless specified. To obtain populations of synchronized developing worms, gravid adults were collected and washed twice with water. Pellets of centrifuged worms were treated with 5 mL 1 M NaOH and 1% (v/v) sodium hypochlorite for 5 min with shaking to obtain embryos, and the embryos were rinsed with M9 buffer three times. The embryos were hatched in 10 mL M9 buffer at 20°C for 16-18 h with mild shaking. Hatched L1 larvae were transferred to plates at 30-50 worms per plate and replicate plates were cultured at 15°C, 20°C, or 25°C for defined periods; samples of the population were examined by microscopy to confirm the developmental stage at the time of harvest.
Phenotypic assays for vulva defects
The adult lethality characteristic of let-7(lf), which results from rupture of the young adult animal at the vulva, was scored approximately 36 h (15°C), 24 h (20°C) or 16 h (25°C) after the animals reached developmental maturation (when at least 90% of the population had reached the adult stage). The young adult (pre-gravid) with vulva bursting phenotypes was distinguished from those without vulva bursting (matricide due to egg-laying defects) based on the visibility of breached intestinal tissues from the ruptured vulva region. To score viable progeny per adult, young adults were transferred to a fresh plate every 12 h until those capable of laying eggs had completed egg-laying. Only hatched eggs were counted.
Microscopy and heterochronic phenotypes
Differential interference contrast and fluorescent images were obtained by Zeiss.Z1 equipped with ZEISS Axiocam 503 camera. COL-19::GFP pattern images of the whole animal were obtained using a 10X objective. Adult alae and GFP::LIN-41 images were obtained using a 100X objective. DAF-12::mScarlet images were obtained using a 63X objective. Lateral hypodermal heterochronic cell lineage defects were scored by counting the number of seam cells per side of each animal with the aid of the wIs51 transgenic seam cell reporter. Patterns of expression of the adult-specific COL-19::GFP reporter were scored using the maIs105 transgenic reporter. Fluorescent images were processed by ImageJ FIJI (Schindelin et al., 2012). To quantify the DAF-12::mScarlet expression, total fluorescence signal and area of individual seam cell and Hyp7 nuclei were quantified by ImageJ FIJI, and the fluorescence signal density of one posterior and one anterior region adjacent to each nucleus was measured and used to calculate the background signal for that nucleus. The choice of conventional epifluorescence microscopy was motivated by the limitation that DIC optics was required to locate each nucleus before fluorescence illumination and that a confocal microscope with DIC capability was not available. The underlying constraint in the cases of DAF-12::mSCARLET and LIN-41::GFP is that the signals exhibit strong photobleaching, so it was necessary to initiate fluorescence image acquisition simultaneously with turning on the UV illumination, and only after having used DIC white light microscopy to identify the seam and Hyp7 nuclei at the appropriate focal plane. The photobleaching issue also prompted us to choose the cautious limit of one exposure per animal.
RNAi
Overnight cultures of HT115 bacteria expressing dsRNA (Timmons et al., 2001) were transferred to LB broth and shaken at 37°C until OD600 was between 0.4 – 0.8. The bacteria cultures were spread on NGM medium containing 100 μg/mL ampicillin and 1 mM IPTG and induced at room temperature for 24-48 h. HT115 bacteria strains for RNAi were obtained from the Ahringer library (Kamath et al., 2003).
Total RNA preparation
Harvested worms were washed with M9 medium, centrifuged, and the worm pellets were flash-frozen in liquid nitrogen. The worm pellets were thawed and lysed by adding 4X volumes of QIAzol (Qiagen, Cat: 79306) and shaking vigorously at room temperature for 15 min. The total RNA was extracted by the addition of 0.85X volume chloroform, centrifugation, and recovery of the aqueous phase, which was then re-extracted with 1 volume phenol:chloroform:isoamyl alcohol (25:24:1, pH = 5.5). Total RNA was then precipitated by adding 1 volume of isopropanol and 0.5 μL GlycoBlue (Invitrogen, Cat: AM9516), followed by incubation at −80°C for at least 30 min, and recovery by centrifugation at 25,000 rcf for 10 min at 4°C. The supernatants were then removed, and the RNA pellets were subsequently washed twice by 70% (v/v) ethanol, dried in air for 5 min, dissolved in water, and stored at −80°C.
Fireplex miRNA assay
Synchronized populations of developing worms were cultured at 20°C and harvested at 8 h (L1), 22 h (L2), 36 h (L3), 46 h (L4), and 58 h (adult) after feeding. Total RNA was extracted and miRNA levels of miR-84 and let-7a were quantified by FirePlex miRNA assay (Abcam) with customized C.elegans miRNA panel following the manufacturer’s instructions. Guava easyCyte 8HT (Millipore) was used for readout. To normalize the quantification, synthetic RNA oligonucleotides with sequences of miR-84 and let-7a (IDT) were serially diluted and subjected to FirePlex miRNA assay. Equal amounts of total RNA were used for all the samples and replicates. The amounts of miRNA in experimental samples were calculated from the standard curve generated from the serial dilution of respective synthetic RNA oligonucleotides. Note that this detection method stringently distinguishes let-7a from miR-84, as let-7a was not detected in let-7(ma341) and miR-84 was not detected in mir-84(n4037) (Figures 2C-2D).
qRT-PCR analysis
20–25 synchronized worms were cultured at 25°C and harvested at the mid-L4 stage (30 h after feeding). Total RNA was extracted as described above. Reverse transcription was performed using HiFiScript gDNA Removal RT MasterMix (CWBio, Cat: CW2020M) with 100 ng total RNA input. qPCR was performed using UltraSYBR Mixture (CWBio, Cat: CW2601) on ViiA 7 platform.
Small RNA sequencing
Synchronized populations of developing worms were cultured at 20°C and harvested at the mid-late L4 stage (45 h after feeding). Total RNA was extracted as described above. The small RNA sequencing libraries were constructed using NEBNext multiplex small RNA library prep set (NEB, Cat: E7300), and sequenced by Illumina NextSeq 500 system. The adaptor sequences were trimmed from the 3′ end of the raw reads by Cutadapt/1.9 using default parameters (Martin, 2011). To quantify the wild type or mutant let-7a miRNAs, the trimmed reads were mapped with Bowtie2/2.3.4.3 to either wild type or mutant let-7a sequences indexed with -c using parameters -end-to-end -N 0 -no-1mm-upfront -L 22 (Langmead and Salzberg, 2012). To quantify the total small RNA reads, trimmed reads were size filtered by Cutadapt/1.9, and reads with a length between 18-25 bp were kept. The filtered reads were mapped to C. elegans genome WBCel235 by star/2.7.6a with default parameters (Dobin et al., 2013). The numbers of reads that mapped uniquely to the genome were used to calculate the RPM of wild type and mutant let-7a. Gene counting was done by subread1.6.2/featureCounts (Liao et al., 2014). The total number of miRNA reads that mapped uniquely to the genome were used to calculate the RPMs of wild type and mutant let-7a.
Ribosome profiling
Worm harvesting
Synchronized populations of developing worms were cultured at 20°C for 45 h after feeding. Harvested worms were harvested by M9, washed with water three times, and incubated at room temperature for 10 min to allow digestion of intestinal bacteria. Worms were then pelleted by centrifuge at 4,500 rcf for 2 min at room temperature and residual water was removed until the total volumes were twice as the worm pellets. The samples were then flashed frozen by liquid nitrogen and stored at −80°C.
Monosome preparation
Concentrated lysis buffer was added to each frozen sample to a final concentration of 20 mM Tris-HCl (pH = 7.4), 150 mM NaCl, 5mM MgCl2, 0.5X Protease Inhibitor (Sigma, Cat:P2714), 1 mM DTT, 0.1 mg/mL cycloheximide (Millipore, Cat:C4859), 1% (v/v) Triton X-100 and 5 U/mL Turbo DNase (Invitrogen, Cat:AM2238) (Ingolia et al., 2012), and worm pellets were kept on ice until fully thawed. Suspended worms were transferred to 400 μm silica beads tube (OPS Diagnostic, Cat: PFAW-400-100-04) and lysed in bead beater homogenizer for 4 min at 4°C. Lysates were then centrifuged at 25,000 rcf for 10 min at 4°C, and supernatants were collected. To generate monosomes, RNase I (Invitrogen, Cat: AM2294) was added to a final concentration of 0.2 U per μl of harvested worm pellet. The digestion was incubated at room temperature for 40 min with gentle rotation and then quenched by adding SUPERase RNase inhibitor (Invitrogen, Cat: AM2694) at 4 U per RNase I unit. The lysates were then loaded onto 5-40% (m/v) sucrose gradients prepared with lysis buffer without Triton X-100 and centrifuged at 32,000 rpm for 3 h at 4°C in an SW41Ti rotor (Beckman Coulter, Cat:331362). The sucrose gradients were fractionated using BR-188 Density Gradient Fractionation System with 60% (m/v) sucrose as chase solution, and monosome fractions were collected according to OD254 profiles.
RPF cloning
3.5 X volumes of QIAzol reagent were added to the gradient fractions containing monosomes, and RNA was extracted and separated by 17.5% denaturing PAGE. A synthetic RNA oligonucleotide with a length of 30 nt was used as a size marker. The gel was stained by Sybr Gold (Invitrogen, Cat: S11494) at room temperature for 10 min, and a gel slice containing RNA of approximately 30 nt was excised and ground by an RNase-free pellet pestle (Fisher Scientific, Cat: 12-141-364). RNA was extracted from the gel slice by adding 500 μL of 300 mM NaAc (pH = 5.5), 1 mM EDTA, and 0.25% (m/v) SDS and mildly shaking overnight at room temperature (Ingolia et al., 2012). The gel granules were excluded using Spin-X tube filter (Millipore, Cat: CLS8160) and the RNA was then concentrated by ethanol precipitation, dissolved in water, and stored at −80°C. 5′ phosphorylation and 3′ dephosphorylation were performed with T4 PNK (NEB, Cat: M0201S) following the manufacturer’s instructions, and the products were subjected to phenol/chloroform extraction and ethanol precipitation. cDNA libraries were constructed using QIAseq miRNA Library Kit (Qiagen, Cat:331505 & 331595) following the manufacturer’s instructions, except that the amplifying PCR was conducted with 9-12 cycles, and sequencing was performed using Illumina NextSeq 500 system.
RPF data analysis
The adaptor sequences were trimmed from the 3′ end of the raw reads by the Cutadapt/1.9 using default parameters (Martin, 2011). Reads were size filtered to keep only reads with a length between 26-34 bp (a range that fits ribosome-protected fragments) (Aeschimann et al., 2015). The rRNA and tRNA reads were removed by initially mapping with Bowtie2/2.3.4.3 to C. elegans rRNA and tRNA sequences from WBCel235 with default parameters (Chan and Lowe, 2009, 2016; Langmead and Salzberg, 2012), and the remaining reads were mapped to the C. elegans genome WBcel235 by Star/2.5.3 with default parameters. The p-offset of the 5′ end of the mapped reads and the monosome periodicity were determined by plastid/0.4.8 as quality control, and gene counting on exons was generated by plastid_cs/0.4.8 with p-offset adjusted (Dunn and Weissman, 2016; Santos et al., 2019). Differential expression analysis was performed by DESeq2 with default parameters (Kucukural et al., 2019; Love et al., 2014). Genes with a max raw count smaller than 10 were excluded from the analysis. Volcano plots were generated using ggplot2 (Hadley, 2016).
To identify genes whose observed expression perturbation between mutant and wild-type samples could be due to slight differences in staging between sample populations, the gene expression changes between the mutants and wild-type were compared with the expression changes between different developmental time points published previously (Aeschimann et al., 2019). Of particular interest were genes reported to change in N2 between 28 and 30 h, and/or between 30 and 32 h at 25°C (Aeschimann et al., 2017), which corresponds to the L4 harvest time point at 20°C used in this study. A gene was flagged as potentially non-specifically perturbed owing to imperfect synchrony if the normalized RPF fold change that we observed between mutant and wild-type was smaller than 1.5 times of the reported RPF fold change between two developmental time points in the wild-type (Aeschimann et al., 2017).
RNAseq of mRNA after ribosomal RNA depletion
To ensure mRNA representation in our samples regardless of poly(A) status we enriched for mRNA in our samples using ribosomal RNA depletion with rRNA anti-sense oligos and RNaseH (Duan et al., 2020b). Worm samples for RNA-seq were aliquoted from the ribosome profiling harvests before the lysis step and frozen separately. The total RNA was extracted as described above. To enrich for mRNA, rRNA was depleted as described in Duan et al., (2020b). rRNA-depleted mRNA samples were then purified by RNA Clean & Concentrator-5 Kit (ZYMO, Cat: R1015) (Zhang et al., 2012). The RNA-seq libraries were constructed by NEBNext Ultra II RNA Library Prep kit (NEB, Cat: E7775, E7335, E7500) and sequenced by Illumina NextSeq 500 system (Duan et al., 2020b).
RNA-seq and translational efficiency data analysis
The adaptor sequences were trimmed from RNA-seq data and reads shorter than 15 nt were filtered out from the analysis by Cutadapt/1.9. tRNA, signal recognition particle RNA (srpR), and residual cytoplasmic rRNA reads were removed by initial mapping with Bowtie2/2.3.4.3, and the remaining reads were mapped to C. elegans genome WBcel235 by Star/2.5.3 with default parameters. Gene counting was done by featureCounts(Subread/1.6.2) (Liao et al., 2014). Differential expression analysis was performed by DESeq2 with default settings (Kucukural et al., 2019; Love et al., 2014). To calculate the translational efficiency (TE), the genes with a max raw count smaller than 10 for either RNA-seq or ribosome profiling were excluded from the analysis. The TE was calculated by dividing normalized ribosome profiling counts by normalized RNA-seq counts for each replica. Significance was calculated by the Student t-test. Volcano plots were generated using ggplot2.
Target prediction
3′ UTR sequences of C. elegans genes were downloaded from the WormBase Parasite website (Howe et al., 2016, 2017) for the genomic version WBcel235. The UTRs were sorted and filtered to remove redundant sequences. The final dataset contained 15,058 3′UTR sequences for 13975 genes (some genes have more than one 3′UTR isoform). The target sites of let-7a sequence that obey one of the following criteria were predicted using the algorithm developed in Veksler-Lublinsky et al., (2010): (1) perfect Watson-Crick (W/C) complementarity (perfect match) to positions 2-7; (2) W/C match to positions 2-7, but allowing 1 target bulge in positions 5-7; (3) W/C match to positions 2-8, but allowing 1 GU/Mismatch (MM) in positions 5-8; (4) W/C match to positions 2-7, but allowing 1 mRNA bulge in positions 2-4; (5) W/C match to positions 2-8, but allowing 1 GU/MM in positions 2-4.
For sites that obey one of the above criteria, a flanking region of an additional 20 nt after the seed was extracted. The interaction duplex between the full site and the miRNA was then calculated using RNAduplex (Lorenz et al., 2011). The duplex was parsed to identify both the seed type and the non-seed type for each reported interaction as follows: (A) Seed type: “1” 2-7 full match; “2” 2-7 + 1 target bulge on 5-7 or 2-8 with 1GU/MM 5-8; “3” 2-7 + 1 target bulge on 2-4 or 2-8 with 1 GU/MM 2-4. (B) NonSeed type: “1” perfect match to positions 11-13 or 12-14 or 13-16; “2” match allowing GUs to positions 11-13 or 12-14 or 13-16; “−1” none of the above. Note that our unpublished data suggest that a single GU in 3′ non-seed region may not significantly affect miRNA repressing efficacy. For genes that have multiple UTR sequences in the analysis, UTRs with redundant duplexes were filtered out from the final report.
In the output results (Table S4 and Data S1), site configurations are categorized in (SeedType, NonSeedType) format. For SeedType, ‘1’ indicates sites with g2-g7 perfect seed pairing; ‘2’ indicates sites with mildly imperfect seed pairing (1 GU/mismatch at g5-g8 or 1 target nucleotide bulge at t5-t7); ‘3′ indicates sites with severely imperfect seed pairing (1 GU/mismatch at g2-g4 or 1 target nucleotide bulge at t2-t4). For NonSeedType, ‘1’ indicates sites with at least 3 consecutive Watson-Crick pairing to g11-g16 of let-7a (g13 must be pairing); ‘2’ indicates sites with at least 3 consecutive pairing to g11-g16 of let-7a with GU wobble (g13 must be pairing); ‘−1’ indicates other sites which are considered as no critical non-seed pairing.
QUANTIFICATION AND STATISTICAL ANALYSIS
In Figure 5A, DEseq2 was used for differential expression analysis of ribosome occupancy, significantly perturbed genes were defined by FC > 1.5 and p.adj <0.1 by DEseq2. In Figure 5C, DEseq2 was used for differential expression analysis of mRNA abundance, genes with significantly increased mRNA abundance were defined by FC > 1.5, p.adj <0.1 by DESeq2; Student’s t test was used for differential expression analysis of TE, genes with significantly increased TE were defined by FC > 1.5, p < 0.1 by Student’s t test. In Figures 2G, 3C, 4F, 5F, 6A-6H, and 7B, the p-values representation is as follow: 0.05–0.01(*); 0.01–0.001(**); 0.001–0.0001(***); <0.0001(****) by Student’s t test (two-tailed, unpaired). Error bars indicate mean ± SD. Significance tests were conducted with Prism 8. Statistical details can be found in corresponding figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E.coli HB101 | N/A | N/A |
| E.coli HT115 RNAi clones | Kamath et al., 2003 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 1X duplex buffer | IDT | Cat#11010301 |
| QIAzol Lysis Reagent | Qiagen | Cat#79306 |
| GlycoBlue | Invitrogen | Cat#AM9516 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#P2714 |
| Turbo DNase | Invitrogen | Cat#AM2238 |
| 400 μm silica beads | OPS Diagnostic | Cat#PFAW-400-100-04 |
| RNase I | Invitrogen | Cat#AM2294 |
| SUPERase.In RNase Inhibitor | Invitrogen | Cat#AM2694 |
| SYBR Gold Nucleic Acid Gel Stain | Invitrogen | Cat#S11494 |
| T4 PNK | NEB | Cat#M0201S |
| RNase Inhibitor, Murine | NEB | Cat#M0314S |
| Thermostable RNase H | MCLAB | Cat#HTRH-100 |
| Critical commercial cssays | ||
| Q5 site-directed mutagenesis kit | NEB | Cat#E0552S |
| QIAseq miRNA Library kit | Qiagen | Cat#331505 |
| QIAseq miRNA NGS 96 Index IL | Qiagen | Cat#331565 |
| KAPA Library Quantification Kit | KAPA Biosystems | Cat#KK4824 |
| RNA Clean & Concentrator-5 Kit | Zymo | Cat#R1015 |
| NEBNext Multiplex Small RNA Library Prep Set | NEB | Cat#E7300 |
| NEBNext Ultra II RNA Library Prep kit | NEB | Cat#E7775 |
| NEBNext Multiplex oligo for Illumina | NEB | Cat#E7500 E7335 E7710 E7730 |
| HiFiScript gDNA Removal cDNA Synthesis Kit | CWBIO | CW2020M |
| UltraSYBR Mixture (Low ROX) | CWBIO | CW2601L |
| Deposited data | ||
| Raw and processed data of ribosome profiling | This paper | GEO: GSE171726 |
| Raw and processed data of RNA-seq | This paper | GEO: GSE171733 |
| Raw and processed data of small RNA seq | This paper | GEO: GSE171747 |
| Code of target prediction algorithm | This paper | GitHub: https://github.com/IsanaVekslerLublinsky/Let7_Proj_code.git; Zenodo DOI: 6465945 |
| C.elegans Reference Genome WBCel235 | Ensemble/iGenome (Illumina) | https://support.illumina.com/sequencing/sequencing_software/igenome.html |
| miRBase release 22.1 | miRBase | http://www.mirbase.org/ftp.shtml |
| Experimental models: Organisms/strains | ||
| C.elegans: Strain N2 | Caenorhabditis Genetics Center | WormBase Strain: N2 |
| C.elegans: Strain VT1313 (maIs105 V; mir-84(n4037) X) | This paper | VT1313 |
| C.elegans: Strain VT1367 (maIs105 V) | This paper | VT1367 |
| C.elegans: Strain VT2692 (maIs105 V; let-7(n2853) X) | This paper | VT2692 |
| C.elegans: Strain VT3439 (maIs105 V; let-7(ma341) X) | This paper | VT3439 |
| C.elegans: Strain VT3455 (wIs51 V) | This paper | VT3455 |
| C.elegans: Strain VT3460 (wIs51 V; let-7a(ma341) X) | This paper | VT3460 |
| C.elegans: Strain VT3479 (maIs105 V; let-7(ma341); mir-84(n4037) X) | This paper | VT3479 |
| C.elegans: Strain VT3585 (lin-41(ma378) I; maIs105 V; let-7(ma341) X) | This paper | VT3585 |
| C.elegans: Strain VT3639 (lin-41(ma378) I; maIs105 V) | This paper | VT3639 |
| C.elegans: Strain VT3645 (mnDp1(umnIs25) (X;V),+/+, maIs105 V; let-7(ma393) X) | Duan et al., 2020a; 2020b | VT3645 |
| C.elegans: Strain VT3648 (mnDp1(umnIs25) (X;V),+/+, maIs105 V; let-7(ma341) X) | This paper | VT3648 |
| C.elegans: Strain VT3729 (mnDp1(umnIs25) (X;V),+/+, maIs105 V; let-7(ma432ma435) X) | This paper | VT3729 ma432ma435 is also labeled as ma428 |
| C.elegans: Strain VT3742 (oxSi1091(Pmex-5::Cas-9(smu-2 introns) unc-119+) II; mnDp1(umnIs25) (X;V)/+ V; let-7(ma393) X) | This paper | VT3742 |
| C.elegans: Strain VT3793 (mnDp1(umnIs25) (X;V),+/+,maIs105 V; let-7(ma431) X) | This paper | VT3793 |
| C.elegans: Strain VT3794 (mnDp1(umnIs25) (X;V),+/+,maIs105 V; let-7(ma432) X) | This paper | VT3794 |
| C.elegans: Strain VT3795 (mnDp1(umnIs25) (X;V),+/+,maIs105 V; let-7(ma433) X) | This paper | VT3795 |
| C.elegans: Strain VT3796 (mnDp1(umnIs25) (X;V),+/+,maIs105 V; let-7(ma434) X) | This paper | VT3796 |
| C.elegans: Strain VT3797 (maIs105 V; let-7(ma435) X) | This paper | VT3797 |
| C.elegans: Strain VT3798 (maIs105 V; let-7(ma436) X) | This paper | VT3798 |
| C.elegans: Strain VT3799 (maIs105 V; let-7(ma437) X) | This paper | VT3799 |
| C.elegans: Strain VT3825 (mnDp1(umnIs25) (X;V),+/+,maIs105 V; let-7(ma448) X) | This paper | VT3825 |
| C.elegans: Strain VT3826 (mnDp1(umnIs25) (X;V),+/+,maIs105 V; let-7(ma449) X) | This paper | VT3826 |
| C.elegans: Strain VT3827 (maIs105 V; let-7(ma450) X) | This paper | VT3827 |
| C.elegans: Strain VT3828 (maIs105 V; let-7(ma451) X) | This paper | VT3828 |
| C.elegans: Strain VT3829 (maIs105 V; let-7(ma452) X) | This paper | VT3829 |
| C.elegans: Strain VT3835 (maIs105 V; let-7(ma453) X) | This paper | VT3835 |
| C.elegans: Strain VT3836 (maIs105 V; let-7(ma454) X) | This paper | VT3836 |
| C.elegans: Strain VT3843 (maIs105 V; let-7(ma455) X) | This paper | VT3843 |
| C.elegans: Strain VT3844 (maIs105 V; let-7(ma456) X) | This paper | VT3844 |
| C.elegans: Strain VT3867 (lin-41(tn1541) I; oxSi1091 II; mnDp1(umnIs25) (X;V)/+ V; let-7(ma432) X) | This paper | VT3867 |
| C.elegans: Strain VT3868 (lin-41(tn1541) I; mnDp1(umnIs25) (X;V)/+ V; let-7(ma432) X) | This paper | VT3868 |
| C.elegans: Strain VT3873 (oxSi1091 II; mnDp1(umnIs25) (X;V)/+ V; let-7(ma432) X) | This paper | VT3873 |
| C.elegans: Strain VT3874 (maIs105 V; let-7(ma476) X) | This paper | VT3874 |
| C.elegans: Strain VT3875 (maIs105 V; let-7(ma477) X) | This paper | VT3875 |
| C.elegans: Strain VT3876 (maIs105 V; let-7a(ma478) X) | This paper | VT3876 |
| C.elegans: Strain VT3877 (mnDp1(umnIs25) (X;V),+/+, maIs105 V; let-7(ma449ma435) X) | This paper | VT3877 ma449ma435 is also labeled as ma479 |
| C.elegans: Strain VT3878 (lin-41(tn1541ma480) I; let-7(ma432) X) | This paper | VT3878 |
| C.elegans: Strain VT3879 (lin-41(tn1541ma480) I; maIs105 V; let-7(ma432) X) | This paper | VT3879 |
| C.elegans: Strain VT3945 (lin-41(tn1541ma501) I; maIs105 V) | This paper | VT3945 |
| C.elegans: Strain VT3949 (lin-41(tn1541ma480) I) | This paper | VT3949 |
| C.elegans: Strain VT3950 (lin-41(tn1541ma480) I; maIs105 V) | This paper | VT3950 |
| C.elegans: Strain VT3954 (lin-41(tn1541ma501) I; mnDp1(umnIs25) (X;V),+/+,maIs105 V; let-7(ma393) X) | This paper | VT3954 |
| C.elegans: Strain VT3959 (lin-41(tn1541) I; mnDp1(umnIs25) (X;V),+/+,maIs105 V; let-7(ma393) X) | This paper | VT3959 |
| C.elegans: Strain VT3974 (lin-41(ma501ma545) I; maIs105 V) | This paper | VT3974 ma501ma545 is also labeled as ma511 |
| C.elegans: Strain VT3975 (lin-41(xe11) I; maIs105 V) | This paper | VT3975 |
| C.elegans: Strain VT4009 (lin-41(ma501ma572) I; maIs105 V) | This paper | VT4009 ma501ma572 is also labeled as ma554 |
| C.elegans: Strain VT4058 (lin-41(ma501ma480) I; maIs105 V) | This paper | VT4058 ma501ma480 is also labeled as ma504 |
| C.elegans: Strain VT4060 (lin-41(ma480) I; maIs105 V) | This paper | VT4060 |
| C.elegans: Strain VT4091 (lin-41(ma501ma555) I; maIs105 V) | This paper | VT4091 ma501ma555 is also labeled as ma552 |
| C.elegans: Strain VT4092 (lin-41(ma501ma556) I; maIs105 V) | This paper | VT4092 ma501ma556 is also labeled as ma553 |
| C.elegans: Strain VT4093 (lin-41(ma501ma571) I; maIs105 V) | This paper | VT4093 ma501ma571 is also labeled as ma554 |
| C.elegans: Strain VT4094 (lin-41(ma555) I; maIs105 V) | This paper | VT4094 |
| C.elegans: Strain VT4095 (lin-41(ma556) I; maIs105 V) | This paper | VT4095 |
| C.elegans: Strain VT4123 (lin-41(ma564) I; maIs105 V) | This paper | VT4123 |
| C.elegans: Strain VT4124 (lin-41(ma565) I; maIs105 V) | This paper | VT4124 |
| C.elegans: Strain VT4125 (lin-41(ma566) I; maIs105V) | This paper | VT4125 |
| C.elegans: Strain VT4126 (oxSi1091 II; maIs105 V; daf-12(ma498ma567) X) | This paper | VT4126 |
| C.elegans: Strain VT4128 (lin-41(tn1541) I; maIs105 V; daf-12(ma498ma568) X) | This paper | VT4128 |
| C.elegans: Strain VT4129 (lin-41(tn1541) I; maIs105 V; daf-12(ma498) X) | This paper | VT4129 |
| C.elegans: Strain DG3913 (lin-41(tn1541) I) | Caenorhabditis Genetics Center | WormBase: DG3913 |
| Oligonucleotides | ||
| see Table S6 | N/A | N/A |
| Recombinant DNA | ||
| Plasmid: pBlueScript SK(+) let-7a(mir-84 swap) | This paper | pBS-09 |
| Plasmid: pCR2.1-TOPO let-7a(U9G) | This paper | pCR-32 |
| Plasmid: pCR2.1-TOPO let-7a(A10U) | This paper | pCR-33 |
| Plasmid: pCR2.1-TOPO let-7a(G11U) | This paper | pCR-12 |
| Plasmid: pCR2.1-TOPO let-7a(G12U) | This paper | pCR-34 |
| Plasmid: pCR2.1-TOPO let-7a(U13A) | This paper | pCR-25 |
| Plasmid: pCR2.1-TOPO let-7a(U14A) | This paper | pCR-13 |
| Plasmid: pCR2.1-TOPO let-7a(G15A) | This paper | pCR-14 |
| Plasmid: pCR2.1-TOPO let-7a(U16G) | This paper | pCR-35 |
| Plasmid: pCR2.1-TOPO let-7a(A17U) | This paper | pCR-36 |
| Plasmid: pCR2.1-TOPO let-7a(U18A) | This paper | pCR-26 |
| Plasmid: pCR2.1-TOPO let-7a(A19U) | This paper | pCR-15 |
| Plasmid: pCR2.1-TOPO let-7a(G20C) | This paper | pCR-39 |
| Plasmid: pCR2.1-TOPO let-7a(U21A) | This paper | pCR-40 |
| Plasmid: pCR2.1-TOPO let-7a(U22A) | This paper | pCR-16 |
| Plasmid: pCR2.1-TOPO let-7a(U18C) | This paper | pCR-37 |
| Plasmid: pCR2.1-TOPO let-7a(U18G) | This paper | pCR-38 |
| Plasmid: pCR2.1-TOPO let-7a(U16G + U18A) | This paper | pCR-41 |
| Plasmid: pCR2.1-TOPO let-7a(17–22) | This paper | pCR-42 |
| Plasmid: pCR2.1-TOPO let-7a(A17U + U18A + A19U) | This paper | pCR-43 |
| Plasmid: pCR2.1-TOPO let-7a(G20C + U21A + U22A) | This paper | pCR-44 |
| Plasmid: pCR2.1-TOPO let-7(INPP4A, Jump Board, null) | This paper | pCR-07 |
| Software and algorithms | ||
| let-7a target prediction algorithm | This paper | https://github.com/IsanaVekslerLublinsky/Let7_Proj_code.git Zenodo DOI: 6465945 |
| ImageJ-FIJI | Schindelin et al., 2012 | https://imagej.net/Fiji |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| SnapGene Viewer | SnapGene | https://www.snapgene.com/snapgene-viewer/ |
| Jalview2 | Waterhouse et al., 2009 | https://www.jalview.org/ |
| Adobe Illustrator CS5 | Adobe Inc. | https://www.adobe.com/products/illustrator.html |
| Cutadapt/1.9 | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| Bowtie2/2.3.4.3 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR/2.7.6a | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| featureCounts (Subread/1.6.2) | Liao et al., 2014 | http://subread.sourceforge.net/ |
| plastid/0.4.8 | Dunn and Weissman, 2016 | https://plastid.readthedocs.io/en/latest/index.html |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| RStudio(2019) | RStudio | https://rstudio.com/ |
| ggplot2 | Hadley, 2016 | https://ggplot2.tidyverse.org/ |
| Other | ||
| SW 41 Ti swinging-bucket rotor | Beckman-Coulter | Cat#331362 |
| Spin-X centrifuge tube filter | Millipore | Cat#CLS8160 |
| Fisherbrand™ RNase-Free Disposable Pellet Pestles | Fisher Scientific | Cat#12-141-364 |
Highlights.
Single nucleotide at g11–g16 are critical for in vivo function of microRNA let-7a
let-7a 3′ pairing mediates both perfect and imperfect seed interactions
A multiplicity of let-7a target genes is regulated through 3′ non-seed pairing
The let-7a 3′ sequence cannot be replaced by that of a close family paralog
ACKNOWLEDGMENTS
We thank Xantha Karp, Charles Nelson, and Alejandro Vasquez-Rifo for commenting on the manuscript. This research was supported by funding from NIH grants R01GM088365, R01GM034028, and R35GM131741 (to V.A.). Some C. elegans strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110745.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
REFERENCES
- Abbott AL, Alvarez-Saavedra E, Miska EA, Lau NC, Bartel DP, Horvitz HR, and Ambros V (2005). The let-7 MicroRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev. Cell 9, 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahante JE, Daul AL, Li M, Volk ML, Tennessen JM, Miller EA, and Rougvie AE (2003). The Caenorhabditis elegans hunchback-like gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Dev. Cell 4, 625–637. [DOI] [PubMed] [Google Scholar]
- Aeschimann F, Kumari P, Bartake H, Gaidatzis D, Xu L, Ciosk R, and Grosshans H (2017). LIN41 post-transcriptionally silences mRNAs by two distinct and position-dependent mechanisms. Mol. Cell 65, 476–489.e4. [DOI] [PubMed] [Google Scholar]
- Aeschimann F, Neagu A, Rausch M, and Grosshans H (2019). let-7 coordinates the transition to adulthood through a single primary and four secondary targets. Life Sci. Alliance 2, e201900335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeschimann F, Xiong J, Arnold A, Dieterich C, and Grosshans H (2015). Transcriptome-wide measurement of ribosomal occupancy by ribosome profiling. Methods 85, 75–89. [DOI] [PubMed] [Google Scholar]
- Alvarez-Saavedra E, and Horvitz HR (2010). Many families of C. elegans microRNAs are not essential for development or viability. Curr. Biol 20, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V (2004). The functions of animal microRNAs. Nature 431, 350–355. [DOI] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, and Fire AZ (2014). Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, and Pasquinelli AE (2005). Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122, 553–563. [DOI] [PubMed] [Google Scholar]
- Balzeau J, Menezes MR, Cao S, and Hagan JP (2017). The LIN28/let-7 pathway in cancer. Front. Genet 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2018). Metazoan MicroRNAs. Cell 173, 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Lee MT, and Giraldez AJ (2012). Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 336, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancati G, and Grosshans H (2018). An interplay of miRNA abundance and target site architecture determines miRNA activity and specificity. Nucleic Acids Res. 46, 3259–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, and Cohen SM (2005). Principles of microRNA-target recognition. PLoS Biol. 3, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner JL, Kemp BJ, and Abbott AL (2012). The mir-51 family of microRNAs functions in diverse regulatory pathways in Caenorhabditis elegans. PLoS One 7, e37185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton JP, Lovci MT, Huang JL, Yeo GW, and Pasquinelli AE (2016). Pairing beyond the seed supports MicroRNA targeting specificity. Mol. Cell 64, 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, and Lowe TM (2009). GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 37, D93–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, and Lowe TM (2016). GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44, D184–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Hannon GJ, and Darnell RB (2012). An alternative mode of microRNA target recognition. Nat. Struct. Mol. Biol 19, 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denman RB (1993). Using RNAFOLD to predict the activity of small catalytic RNAs. BioTechniques 15, 1090–1095. [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, and Sharp PA (2004). Specificity of microRNA target selection in translational repression. Genes Dev. 18, 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Choi S, Nelson C, and Ambros V (2020a). Engineering essential genes with a "jump board" strategy using CRISPR/Cas9. MicroPubl Biol. 2020. 10.17912/micropub.biology.000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Sun Y, and Ambros V (2020b). RNA-seq with RNase H-based ribosomal RNA depletion specifically designed for C. elegans. MicroPubl Biol. 2020. 10.17912/micropub.biology.000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JG, and Weissman JS (2016). Plastid: nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genomics 17, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecsedi M, and Grosshans H (2013). LIN-41/TRIM71: emancipation of a miRNA target. Genes Dev. 27, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecsedi M, Rausch M, and Grosshans H (2015). The let-7 microRNA directs vulval development through a single target. Dev. Cell 32, 335–344. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, and Bartel DP (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Van Dongen S, Inoue K, Enright AJ, and Schier AF (2006). Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science 312, 75–79. [DOI] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, and Bartel DP (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans H, Johnson T, Reinert KL, Gerstein M, and Slack FJ (2005). The temporal patterning microRNA let-7 regulates several transcription factors at the larval to adult transition in C. elegans. Dev. Cell 8, 321–330. [DOI] [PubMed] [Google Scholar]
- Grosswendt S, Filipchyk A, Manzano M, Klironomos F, Schilling M, Herzog M, Gottwein E, and Rajewsky N (2014). Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol. Cell 54, 1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley W (2016). Ggplot2 (Springer Science+Business Media, LLC; ). [Google Scholar]
- Hammell CM, Karp X, and Ambros V (2009). A feedback circuit involving let-7-family miRNAs and DAF-12 integrates environmental signals and developmental timing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U S A 106, 18668–18673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TW, Arnaboldi V, Cain S, Chan J, Chen WJ, Cho J, Davis P, Gao S, Grove CA, Kishore R, et al. (2020). WormBase: a modern model organism information resource. Nucleic Acids Res. 48, D762–D767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Zhang H, Gao S, Lercher MJ, Chen WH, and Hu S (2016). Evolview v2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 44, W236–W241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwak A, Kudla G, Dudnakova T, and Tollervey D (2013). Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel J, Bartschat S, Wintsche A, Otto C, and The Students of the Bioinformatics Computer, Lab; and Stadler PF (2012). Evolution of the let-7 microRNA family. RNA Biol. 9, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe KL, Bolt BJ, Cain S, Chan J, Chen WJ, Davis P, Done J, Down T, Gao S, Grove C, et al. (2016). WormBase 2016: expanding to enable helminth genomic research. Nucleic Acids Res. 44, D774–D780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe KL, Bolt BJ, Shafie M, Kersey P, and Berriman M (2017). WormBase ParaSite - a comprehensive resource for helminth genomics. Mol. Biochem. Parasitol. 215, 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilbay O, and Ambros V (2019). Regulation of nuclear-cytoplasmic partitioning by the lin-28-lin-46 pathway reinforces microRNA repression of HBL-1 to confer robust cell-fate progression in C. elegans. Development 146, dev183111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT (2016). Ribosome footprint profiling of translation throughout the genome. Cell 165, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, and Weissman JS (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc 7, 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, and Marks DS (2004). Human MicroRNA targets. PLoS Biol. 2, e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Birgaoanu M, and Griffiths-Jones S (2019). miRBase: from microRNA sequences to function. Nucleic Acids Res. 47, D155–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukural A, Yukselen O, Ozata DM, Moore MJ, and Garber M (2019). DEBrowser: interactive differential expression analysis and visualization tool for count data. BMC Genomics 20, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC (2002). Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet 30, 363–364. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Han S, Kwon CS, and Lee D (2016). Biogenesis and regulation of the let-7 miRNAs and their functional implications. Protein Cell 7, 100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, and Ambros V (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, and Bartel DP (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, and Johnson JM (2005). Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433, 769–773. [DOI] [PubMed] [Google Scholar]
- Liu Z, Kirch S, and Ambros V (1995). The Caenorhabditis elegans heterochronic gene pathway controls stage-specific transcription of collagen genes. Development 121, 2471–2478. [DOI] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA package 2.0. Algorithms Mol. Biol 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMB J. 17, 3. [Google Scholar]
- McGeary SE, Bisaria N, Pham TM, Wang PY, and Bartel DP (2022). MicroRNA 3′-compensatory pairing occurs through two binding modes, with affinity shaped by nucleotide identity and position. Elife 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C, and Ambros V (2019). Trans-splicing of the C. elegans let-7 primary transcript developmentally regulates let-7 microRNA biogenesis and let-7 family microRNA activity. Development 146, dev172031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C, and Ambros V (2021). A cohort of Caenorhabditis species lacking the highly conserved let-7 microRNA. G3 11, jkab022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottrott S, Simard MJ, and Richter JD (2006). Human let-7a miRNA blocks protein production on actively translating polyribosomes. Nat. Struct. Mol. Biol 13, 1108–1114. [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, McCoy A, Jimenez E, Salo E, Ruvkun G, Martindale MQ, and Baguna J (2003). Expression of the 22 nucleotide let-7 heterochronic RNA throughout the Metazoa: a role in life history evolution? Evol. Dev 5, 372–378. [DOI] [PubMed] [Google Scholar]
- Paul P, Chakraborty A, Sarkar D, Langthasa M, Rahman M, Bari M, Singha RS, Malakar AK, and Chakraborty S (2018). Interplay between miRNAs and human diseases. J. Cell. Physiol 233, 2007–2018. [DOI] [PubMed] [Google Scholar]
- Pawlica P, Sheu-Gruttadauria J, MacRae IJ, and Steitz JA (2020). How complementary targets expose the microRNA 3′ end for tailing and trimming during target-directed microRNA degradation. Cold Spring Harb. Symp. Quant. Biol 84, 179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmsmeier M, Steffen P, Hochsmann M, and Giegerich R (2004). Fast and effective prediction of microRNA/target duplexes. Rna 10, 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, and Ruvkun G (2000). The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901–906. [DOI] [PubMed] [Google Scholar]
- Roush S, and Slack FJ (2008). The let-7 family of microRNAs. Trends Cell Biol. 18, 505–516. [DOI] [PubMed] [Google Scholar]
- Salomon WE, Jolly SM, Moore MJ, Zamore PD, and Serebrov V (2015). Single-molecule imaging reveals that Argonaute reshapes the binding properties of its nucleic acid guides. Cell 162, 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos DA, Shi L, Tu BP, and Weissman JS (2019). Cycloheximide can distort measurements of mRNA levels and translation efficiency. Nucleic Acids Res. 47, 4974–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle NT, Sheu-Gruttadauria J, and MacRae IJ (2014). Structural basis for microRNA targeting. Science 346, 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu-Gruttadauria J, Xiao Y, Gebert LF, and MacRae IJ (2019). Beyond the seed: structural basis for supplementary microRNA targeting by human Argonaute2. EMBO J. 38, e101153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol NS, Xu P, Jan YN, and Ambros V (2008). Drosophila let-7 microRNA is required for remodeling of the neuromusculature during metamorphosis. Genes Dev. 22, 1591–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spike CA, Coetzee D, Eichten C, Wang X, Hansen D, and Greenstein D (2014). The TRIM-NHL protein LIN-41 and the OMA RNA-binding proteins antagonistically control the prophase-to-metaphase transition and growth of Caenorhabditis elegans oocytes. Genetics 198, 1535–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, and Thomson JN (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol 100, 64–119. [DOI] [PubMed] [Google Scholar]
- Tennessen JM, and Thummel CS (2008). Developmental timing: let-7 function conserved through evolution. Curr. Biol 18, R707–R708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, and Fire A (2001). Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263, 103–112. [DOI] [PubMed] [Google Scholar]
- Veksler-Lublinsky I, Shemer-Avni Y, Kedem K, and Ziv-Ukelson M (2010). Gene bi-targeting by viral and human miRNAs. BMC Bioinformatics 11, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella MC, Choi EY, Lin SY, Reinert K, and Slack FJ (2004). The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3′UTR. Genes Dev. 18, 132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlquist C, Jeong D, Rojas-Munoz A, Kho C, Lee A, Mitsuyama S, van Mil A, Park WJ, Sluijter JP, Doevendans PA, et al. (2014). Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 508, 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, and Barton GJ (2009). Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25 (9), 1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee LM, Flores-Jasso CF, Salomon WE, and Zamore PD (2012). Argonaute divides its RNA guide into domains with distinct functions and RNA-binding properties. Cell 151, 1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolter JM, Le HH, Linse A, Godlove VA, Nguyen TD, Kotagama K, Lynch A, Rawls A, and Mangone M (2017). Evolutionary patterns of metazoan microRNAs reveal targeting principles in the let-7 and miR-10 families. Genome Res. 27, 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worringer KA, Rand TA, Hayashi Y, Sami S, Takahashi K, Tanabe K, Narita M, Srivastava D, and Yamanaka S (2014). The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell Stem Cell 14, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, and MacRae IJ (2020). Robust differential microRNA targeting driven by supplementary interactions in vitro. RNA 26, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Artiles KL, and Fire AZ (2015). Functional relevance of "seed" and "non-seed" sequences in microRNA-mediated promotion of C. elegans developmental progression. RNA 21, 1980–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Theurkauf WE, Weng Z, and Zamore PD (2012). Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly(A) selection. Silence 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw and processed data of ribosome profiling, RNA-seq, and small RNA seq in this study have been deposited at NCBI Gene Expression Omnibus (GEO) with accession number GSE171748 (Edgar et al., 2002) and are publicly available.
All original code has been deposited at GitHub (https://github.com/IsanaVekslerLublinsky/Let7_Proj_code.git.) with Zenodo DOI 6465945, and is publicly available.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.