

Summary
Alström syndrome (AS) is a rare autosomal recessive monogenic disorder caused by mutations of the Alström syndrome 1 (ALMS1) gene, located on chromosome 2p13. It is a progressive multisystemic disease characterized mostly by obesity, sensorineural hearing loss, visual impairments, cardiomyopathy, insulin resistance and/or type 2 diabetes mellitus (T2DM), metabolic dysfunctions, non-alcoholic fatty liver disease, and chronic progressive kidney disease. Generally, the first clinical symptoms of the disease appear in the first years of life with a major variation of onset age. In this study, we aimed to examine the molecular diagnosis of a 6-year-old patient with suspected AS clinical symptoms. After applying clinical exome sequencing (CES) in the patient we found a homozygous deletion in exon 8 at the ALMS1 gene (c.2311_2312del). We identified a homozygous frameshift mutation. The reported variant was pathogenic according to the criteria of the American College of Medical Genetics and Genomics (ACMG). Thus, the patient was diagnosed with AS as a result of the combined clinical phenotype and genetic tests results. We hope the variant we found can expand the spectrum of ALMS1 variants in AS.
Keywords: ALMS1 gene, biallelic mutations, obesity, rare diseases
Alström syndrome (AS) is a rare genetic disorder caused by biallelic mutations of the Alström syndrome 1 (ALMS1) gene located on chromosome 2p13 (1). ALMS1 gene was first identified in 2002, it has a 220 kb size and contains 23 exons (2). It encodes a 41,1-kDa protein which is made up of 4,169 amino acids (2).
AS is an ultra-rare disease with a prevalence of one in 1,000,000. It is mostly characterized by ophthalmological abnormalities (photophobia, nystagmus, cone-rod dystrophy, loss of light perception) sensorineural hearing loss, obesity, cardiomyopathy, insulin resistance, metabolic dysfunctions, non-alcoholic fatty liver disease, and progressive kidney disease (2). Different ages of onset of the disease and high variability of clinical symptoms reduce the possibility of making the diagnosis in the early stages. That's why genetic analysis plays an important role in early diagnosis.
A 6-year-old female patient was consulted to our department from the pediatric endocrinology clinic due to dysmorphic features and obesity. She was born on time to healthy Turkish parents who were 5th-degree relatives. No extraordinary situation was observed in the prenatal follow-up and delivery of the patient.
The weight and height of the patient were 44,4 kg (>P97, 4.20 SDS), 131,5 cm (P97<, 2,77 SDS) respectively. Her physical examination revealed truncal obesity, wide forehead, high arched palate, acanthosis nigricans on the neck, and buffalo hump, tapered finger, and simian crease on the right hand (Online Figure, http://www.irdrjournal.com/action/getSupplementalData.php?ID=92). At 6 months of age bilateral nystagmus and photophobia were observed in her eye evaluation. Later, ophthalmoscopy showed diffuse dystrophy of the retinal pigmented epithelium at 5 years old. In the follow-up of the patient from the age of 3, a considerable weight gain was observed in anthropometric measurements according to age and height of the patient (Table 1). In the pubertal evaluation of the 5-year-old patient, thelarche was found consistent with Tanner stage 3 without axillary and pubic hair growth. Later in the laboratory tests of the patient, fasting glucose: 87 mg/dL, HbA1c: 5.4% (normal range: 4.8-5.9), insulin: 61.69 (normal: 2.6-24.9) μIU/mL, FSH: 0.92 mIU/mL (22.02.2019), LH < 0.3 mIU/mL, Estradiol: 13 pg/mL (6-27) were detected. The skeletal survey result was compatible with 8 years of age. Due to the abnormality in gonadotropin values, the patient underwent an LH-RH stimulation test. Accordingly, the patient was followed up in terms of precocious puberty after LH value was 1.34 mIU/mL.
Table 1. Patient's height and weight follow-up over the years.
| Date of Examination | Age (years) | Weight (kg) | SDa | Height (cm) | SDa | BMI (kg/m2) | SDa |
|---|---|---|---|---|---|---|---|
| 2017 | 3 | 29.0 | +4.77 | 101.0 | +0.47 | 28.4 | +5.04 |
| 2018 | 4 | 34.1 | +5.12 | 111.3 | +1.58 | 27.5 | +4.61 |
| 2019 | 5 | 38.6 | +4.78 | 118.0 | +1.94 | 27.7 | +3.96 |
| 2020 | 6 | 42.7 | +3.99 | 129.4 | +2.33 | 25.5 | +3.07 |
| 2021 | 7 | 44.8 | +3.65 | 132.8 | +2.36 | 25.4 | +2.80 |
SDa: Standard Deviation Analysis, BMI: Body Mass Index
In addition, an oral glucose tolerance test (OGTT) was applied to the patient whose clinical and laboratory results showed signs of insulin resistance. As a result of the test, 0th-minute glucose was 73 mg/dL, at 1st hour 128, and at 2nd-hour glucose value was 108 mg/dL, and total insulin was 1,144 μIU/mL. Oral metformin treatment was started in the patient who was found to have insulin resistance. The patient whose postnatal hearing screening was normal, hearing impairment was first noticed at the age of 5 years. At that point she was diagnosed with bilateral sensorineural hearing loss (SNHL) by audiometry test and was given a pair of hearing aids. An echocardiogram and abdominopelvic USG were performed on the patient, Echocardiogram was normal, however, USG showed minimal hepatosplenomegaly with grade 1 hepatic steatosis. At five years of age due to learning difficulties the Denver developmental screening test was performed on the patient. The result showed a neuromotor developmental delay in the patient and was found to be compatible with three years of age. Therefore, a brain magnetic resonance imaging (MRI) and electroencephalogram (EEG) were obtained from the patient. MRI showed evident central and peripheral cerebrospinal fluid distances, indicating a cortical atrophy, and EEG showed the presence of a background rhythm irregularity in cerebral bioelectric activity.
The proband was followed for years with a pre-diagnosis of Bardet-Biedl syndrome, however after the patient's clinical examination, we applied the clinical exome sequencing (CES) panel with the preliminary diagnosis of diseases that cause syndromic obesity such as Bardet-Biedl syndrome and Alström syndrome. As a result of the analysis, we detected a homozygous deletion in two nucleotides in the ALMS1 gene (c.2311_2312del). The mentioned deletion causes a frameshift mutation resulting in a premature termination codon (p.Ile771PhefsTer13) that causes a structural defect in the ALMS1 protein which leads to a malfuncting protein (Figure 1). We then applied Sanger sequencing both to confirm the result and to determine the carrier status of the parents. It was confirmed by Sanger sequencing that both parents were heterozygous for the same variant (Figure 1).
Figure 1.
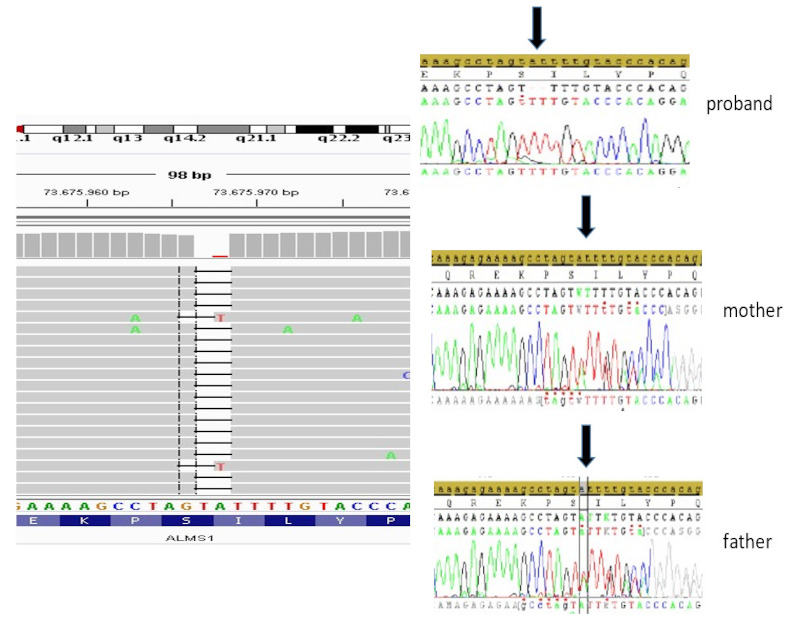
Next-generation sequencing (NGS) analysis result of the proband, proband's mother and proband's father.
In this study, we identified a homozygous mutation of the ALMS1 gene, and to the best of our knowledge, this variant has not previously been reported as homozygous in the literature (The Genome Aggregation Database (gnomAD), Leiden Open Variation Database (LOVD), and Pubmed). We hope by reporting this mutation we can expand our knowledge and spectrum of ALMS1 variants in AS.
Acknowledgements
We thank the family for allowing us to share this case. Written informed consent was obtained from the parents.
Funding:
None.
Conflict of Interest
The authors have no conflicts of interest to disclose.
References
- 1. Marshall JD, Beck S, Maffei P, Naggert JK. Alström syndrome. Eur J Hum Genet. 2007; 15:1193-1202. [DOI] [PubMed] [Google Scholar]
- 2. Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, Beck S, Boerkoel CF, Sicolo N, Martin M, Nishina PM, Naggert JK. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002; 31:74-78. [DOI] [PubMed] [Google Scholar]