

Summary
Gene expression signature-based inference of functional connectivity within and between genetic perturbations, chemical perturbations, and disease status can lead to the development of actionable hypotheses for gene function, chemical modes of action, and disease treatment strategies. Here, we report a FuSiOn-based genome-wide integration of hypomorphic cellular phenotypes that enables functional annotation of gene network topology, assignment of mechanistic hypotheses to genes of unknown function, and detection of cooperativity among cell regulatory systems. Dovetailing genetic perturbation data with chemical perturbation phenotypes allowed simultaneous generation of mechanism of action (MOA) hypotheses for thousands of uncharacterized natural products fractions (NPFs). The predicted MOAs span a broad spectrum of cellular mechanisms, many of which are not currently recognized as ‘druggable’. To enable use of FuSiOn as a hypothesis generation resource, all associations and analyses are available within an open-source web-based GUI (http://fusion.yuhs.ac)
eTOC:
FuSiOn employs genome-wide integration of quantitative genetic-perturbation and chemical-perturbation phenotypes to assemble a biological mechanism-driven similarity network. This network can be queried to return testable mechanism of action hypotheses for genes and chemicals of unknown function, and to establish connections between those mechanisms and other cell biological processes.
Graphical Abstract
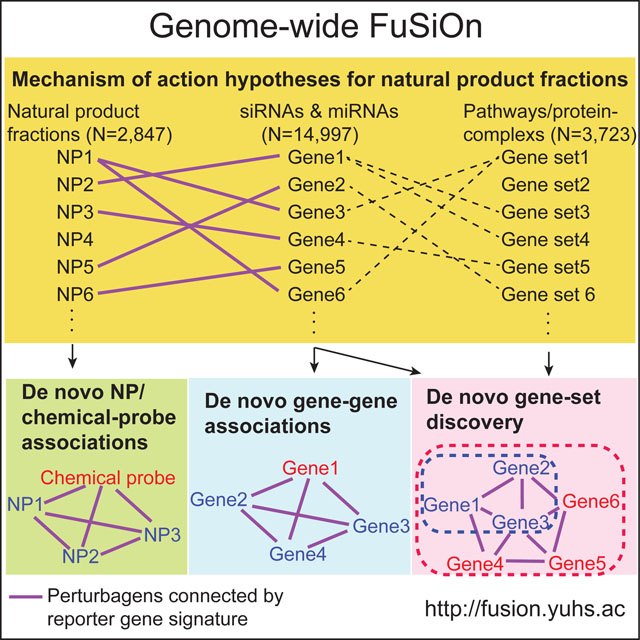
Introduction
Functional Signature Ontology (FuSiOn) was conceived as a ‘guilt-by-association’ hypothesis generator for natural product mechanism of action discovery (Potts et al., 2013). Natural products (NPs) are rich in chemical diversity with structure subject to co-evolution with biological systems, thus they may engage targets not currently recognized as chemically addressable. Specifically, NPs have remained an attractive resource for drug discovery in disease, especially in cancer. A significant barrier associated with NPs discovery is the purification of metabolites from producing organisms and discovery of biological mechanism of action. With FuSiOn, we sought to help overcome these barriers through construction of a scalable high-throughput discovery approach to deliver statistically prioritized testable mechanism of action hypotheses for thousands of complex marine microbe-derived NPFs (3–6 bioactive compounds/fraction) in parallel. We measured expression of 6 highly variable ‘reporter genes’, whose collective expression served as a quantitative indicator of cell state changes in response to perturbations from siRNA’s targeting the kinome, miRNA mimics, and NPFs. Perturbation signatures from all three libraries are clustered together to produce ‘guilt by association’ hypotheses. Evaluation of NPFs in this fashion allows for high-throughput iterative prioritization based on attractive functional consequences on the cells. In addition to stratification of samples for follow-up, this enables development of bio-assay guided purification schemes. This approach has successfully assigned function to previously uncharacterized miRNA’s and siRNA’s and linked NPs to cellular mechanism of action (Potts et al., 2013, Potts et al., 2015, Vaden et al., 2017).
Here, we report a genome-wide FuSiOn inclusive of 14,272 distinct human gene perturbations, 725 human miRNA perturbations, and 2,847 chemicals consisting of 2776 marine-derived NPFs and 71 purified NPs and chemicals. This comprehensive feature matrix was used to generate a map of functional associations between all genes in the genome and assess the overall topology of the functional network in a biological setting. We employed this map, together with experimental evaluation, to expand gene membership within known mechanistic processes and to discover previously unrecognized mechanistic relationships among gene networks. Finally, we integrated chemical perturbations with the genome-wide genetic functional network to assign biological mechanisms of action to a large number of NPs; a subset of which underwent experimental validation. Notably, we find our NPs are predicted to engage a diverse array of biological functions in human cells, many of which are not currently recognized as druggable. These associations and predictions have been made available as a searchable web-based GUI to enable community-based hypothesis testing (http://fusion.yuhs.ac).
Results
Phenotypic distributions of genetic and chemical perturbations
To produce a genome-wide FuSiOn map and to accommodate newly acquired NPFs, we employed a previously optimized bead-based multiplex-high throughput assay platform to measure mRNA expression of eight pre-selected endogenous reporter genes (ACSL5, ALDOC, BNIP3, BNIP3L, LOXL2, NDRG1, and two stable genes (PPIB and HPRT) as internal normalization controls after exposure of HCT116 colon cancer cells to 14,272 siRNA pools, 725 miRNA mimics, and 2,847 chemical perturbations consisting of mostly NPFs. Normalized reporter gene expression values for genetic and chemical perturbations showed near normal distribution (Figure S1A, Data S1). A two-way hierarchical clustering revealed large multi-directional changes in probe magnitude in response to perturbations, suggesting FuSiOn has the potential to discriminate between many distinct signature classes (Figure 1A).
Figure 1. FuSiOn retrieves genetic and chemical functionalogues.

(A) Two-way hierarchical clustering of normalized reporter expression
(B) Frequency of perturbations resulting in 0 to 6 probes in the moveable range. The 362 perturbations in which 0 probes move are defined as “silent”.
(C-D) Density distributions of probe RMS values for (C) 14,997 genetic (KS test p<2.2E-16) and (D) 2,847 chemicals (KS test p<2.2E-16)
(E-F) CDF of p-values assessing similarity (Pearson correlation) among pairwise combinations of (E) miRNAs with the same seed sequence compared to those with different seed sequences and (F) miRNA’s with the same predicted targets (top 10% of context scores) compared to similarities of those with different predicted targets (KS p < 2.2E-16)
(G) CDF of predicted seed effect p-values among pairwise combinations of siRNAs with the same seed region.
(H) Overlap of siRNA oligos with significant ‘seed effect’ versus unexpressed genes (HG p<2E-16)
* All experiments performed in triplicate, unless otherwise indicated. Values are means. Error bars plotted as ± 1 SD. *p < 0.05; **p < 0.01. red dotted lines indicate p=.05
To help formalize the reporting sensitivity, we sought to annotate “silent” perturbations with little to no consequence on reporter gene expression. For each of the 6 “dynamic” reporters, we arbitrarily defined the range in which the reporter is silent within 1 standard deviation of the mean of reporter variation among all samples. A perturbation with no functional effect was simply defined as one for which all 6 reporters were silent. Using this metric, 362 perturbations out of a total of 17,844, had no effect, corresponding to 2.0% of the total perturbation set. This suggests FuSiOn has some level of discriminatory power for 98.0% of all tested perturbations (Figure 1B).
A Euclidean distance based similarity matrix was built for all possible pairs of genetic-genetic and genetic-chemical perturbations. Statistical significance (p-values and FDR q-values) was assessed by permutation resampling in two directions. Overall p-value distribution indicates enrichment of statistically similar relationships among both genetic (Figure S1B) and chemical (Figure S1C) perturbations. We considered two genes to have a significant ‘functional signature’ or to be ‘functionalogues’ if the FDR q-value for the pairwise distance was less than 0.1. The combination of the six reporter genes has discriminative potential for classifying different perturbation classes as evidenced by significantly shifted root mean square sum values from the controls for genetic (D = 0.81, p-value < 2.2e-16; Figure 1C) and chemical perturbations (D = 0.46, p-value < 2.2e-16; Figure 1D).
Consistency with known miRNA biology
As with the original iteration of FuSiOn, miRNA behavior was evaluated to assess clustering sensitivity and specificity. The biological activities of miRNAs are primarily specified by the miRNA seed sequence (generally nucleotides 2–8), which anneal to complementary sequences on target mRNAs to suppress mRNA stability and translation. The miRNA library employed here was composed of 725 synthetic miRNAs, corresponding to 702 unique mature sequences, and 108 unique seed sequences present in 2 or more distinct miRNAs. As expected, miRNAs with the same seed sequence are more highly correlated in FuSiOn to one another than are miRNA’s with different seeds (Figure S1D). 65.8% of pairwise correlations between miRNAs with the same seed are statistically significant (Pearson p<0.05) compared to only 5.7% of miRNAs with different seeds (Figure 1E). Seed pairing to target mRNA is not always sufficient for repression, and other context-specific parameters have been shown to boost efficacy (Grimson et al., 2007). These include AU-rich nucleotide composition near the target site, target site position on the 3’ UTR, 3’ supplementary pairing outside the seed region, and target abundance in the mRNA. TargetScan miRNA context scores uses all these criteria in addition to seed sequence pairing to rank relative confidence in predicted mRNA target sites. (Garcia et al., 2011). We find miRNA’s with the same targets are more significantly correlated in FuSiOn, and this correlation is context score dependent. The set of targets with higher context scores (Figure 1F, Figure S1E) show higher correlations than when we consider all targets (Figure S1F,G)
Deciphering siRNA seed-based effects in FuSiOn
Given that siRNAs can produce miRNA-like effects driven by seed sequences (Zhong et al., 2014), we sought to employ a computational strategy to detect siRNA functional signatures dominated by seed effects. The siRNA ‘seed’ region was defined as nucleotides 2–8, corresponding to 2,896 unique seeds represented by at least 2 siRNA oligos. For each seed class, pairwise Euclidean distances of siRNA’s with the same seed were compared to distances between those siRNA’s and all others using a KS test and corrected with an FDR. Out of the 427,424 possible pairwise distances we calculated, we found that 26,237 (5.5%) were significantly (p<.05) close due to probable seed effect (Figure 1G). We noted that, of the siRNAs whose signatures are driven by seed-based effects, a significant proportion (HG p-value < 2.2E-16, Figure 1H) correspond to genes which are not expressed in our host cell line, HCT116. These siRNAs are expected to have no “on-target” functional consequence in HCT116 cells and were filtered from downstream analyses. Collectively, these results suggest FuSiOn has the ability to group together biological perturbations with similar mechanisms spanning a wide range of biological functions.
Expansion of gene membership within known biological pathways and networks
We next sought to directly evaluate consistency of the genome-wide “FuSiOn-derived” similarity matrix with gene pathways curated though independent orthogonal efforts by determining if genes assigned to the same manually curated gene sets also had significantly similar FuSiOn signatures. Pairwise Euclidean distances between all genes were derived from their associated FuSiOn measurements, and we considered two genes to have significantly close functional signatures if the FDR corrected q-value was less than 0.1. “Gene set” memberships were extracted from several distinct public resources (Figure 2A). A hypergeometric test was used to evaluate correspondence of significant FuSiOn-derived Euclidean distances among genes within versus between these “gene sets”. Significant overlap was detected within sets derived from the Molecular Signature Database Version 3.0 (C2;p<2.2E-208), GO terms (C5;p=1.5E-21) (Liberzon et al., 2011) and comprehensive resource of mammalian protein complexes (CORUM;p=2.4E-25) (Figure 2A). (Ruepp et al., 2010). In contrast, no significant association was found with the synthetic lethal genetic relationships reported in the DAISY database (p=0.12) (Jerby-Arnon et al., 2014). We note that synthetic lethal interactions tend to appear between genes participating in mechanistically distinct biological processes and would thus not be expected to be enriched for short FuSiOn-based Euclidean distances.
Figure 2: Reannotation of biological gene pathways with FuSiOn.

(A) HG p-values for gene set enrichment amongst FuSiOn edges. Sets: MSigDB V3 (C2, p<2.2E-208; and C5, p=1.5E-21), CORUM (p=2.4E-25), STRING activation edges (p=2.19E-13), and DAISY synthetic lethal database (p=0.12)
(B-C) p-values (KS test) indicating enrichment of FuSiOn similarity edges for each set in (B) CORUM and (C) C2
(D-E) (D) ‘5196_TNF-alpha/NF-kappa B signaling complex’ and (E)178_Respiratory chain complex I (holoenzyme) mitochondrial were significant in (B-C). Red=pre-annotated genes; blue = added genes. Length and line thickness of edges are proportional to Euclidean distances.
Given gene sets in the C2 and CORUM databases were enriched for genes with significant functional signatures in FuSiOn, we examined the diversity of biological function underpinning this enrichment. For each gene set annotated in the C2 and CORUM databases, we used a Kolmogorov-Smirnov statistic to determine if pairwise Euclidean distances between members of the same annotated pathways were significantly shorter than distances from those genes to all other siRNA’s in the screen. We found 13.0% of gene sets in C2 and 23.8% of gene sets in CORUM were significant (KS p<0.05), spanning multiple biological annotations (Data S2; Figure 2B,C). This suggests FuSiOn has a reasonable capacity to accurately assemble mechanistic gene networks across a wide variety of cell biological activities.
We next explored whether the scale of FuSiOn could be leveraged to expand gene membership in cell autonomous functional pathways. For every gene set in which we detected significantly close associations between members (Figure 2B,C; KS p<0.05), we developed a query-based algorithm to search for additional genes outside of the set with similar functional signatures. Distances between each siRNA and the median centroid of each gene set were calculated, and resulting p-values were corrected with an FDR adjustment. Overall, 1834 new assignments were made to 201 gene sets (Data S3). Among these, TNFSF8 and IGFBP3 significantly associated with the ‘TNF-alpha/NF-kappa B signaling complex’ from CORUM. TNFSF8 is a cytokine belonging to the TNF ligand family, and there are numerous reports on the role of IGFBP3 in regulating the TNF-alpha pathway (Lee et al., 2011, Zhang et al., 2013, Zhang and Steinle, 2014) (Figure 2D). Additionally, multiple genes known to be involved in mitochondrial maintenance were assigned to the C2 gene set ‘Respiratory chain complex I (holoenzyme mitochondrial)’ including MTND5 (a core subunit of complex I), MTATP8 (mitochondrial membrane ATP synthetase), MRPL13 (involved in mitochondrial organelle biogenesis), and ESSRA, also known as ERR-alpha, known to regulate expression of genes involved in oxidative phosphorylation and mitochondrial biogenesis (Eskiocak et al., 2014) (Figure 2E).
De Novo network construction by FuSiOn
Given strong indications of reasonable concordance between FuSiOn assigned functional relationships and known biology, we next deployed the FuSiOn similarity matrix for de novo functional gene network construction. We first centered this effort on existing physical protein-protein interaction (PPI) networks associated with activation edges in the STRING database (N=17,561) (Szklarczyk et al., 2011) We observed four-fold enrichment of the activation edges in the FuSiOn functionalogues (FDR < 10%) with near machine zero significance in the hypergeometric test (p = 2.19E-13) (Figure 2A). We further categorized STRING PPI relationships by a K-core score, which measures the degree of interconnectivity of a sub-graph in which each node has a degree of at least K. For example, a trimeric complex (3 nodes, 3 edges) has k=2 (2 connections per node). Within the FuSiOn similarity matrix, high K-core scores correlated with the significance of FuSiOn similarity between gene pairs (Figure 3A). This observation indicates that gene products within densely connected protein complexes are more likely to share a discrete biological function than those within more sparsely connected protein complexes. We employed this information to generate de novo functional hypotheses for each gene in the FuSiOn dataset. For each perturbagen in FuSiOn, we collected the 500 most similar perturbagens and subjected these sets to gene-level MCODE network-cluster analysis. This algorithm is designed to search for enrichment of gene product interactions (PPIs) among members in a gene list (Bader and Hogue, 2003). Given that K-core scores positively correlate with FuSiOn significance (Figure 3A), we filtered for PPIs with a K-core score of at least 2 (minimum of 3 interactions amongst 3 nodes). Thus, a complex will be associated with a given siRNA or miRNA query if there is a protein complex with at least 3 members detectable amongst the list of the 500 closest genes to the query (Figure 3B). Enriched PPI complexes were detected for 13,158 genetic perturbations out of the 14,997 in FuSiOn. Of those, 8,018 perturbations were associated with MCODE complexes enriched for pre-annotated biological function (HG q-value < 0.1). Collectively, these results indicate FuSiOn has reasonable capacity to discriminate between functional PPIs. Therefore, discrete queries within the FuSiOn similarity matrix are likely to return testable hypotheses for previously unanticipated functional interactions among protein subcomplexes (Data S4).
Figure 3: Network analysis of FuSiOn siRNA perturbations.

(A) CDF of the p-values for the FuSiOn edges represented in the PPI network grouped by the minimal k-core membership compared to genetic perturbations with no physical interaction (background)
(B) The top 500 closest siRNAs to a query perturbation (red) were subjected to an MCODE analysis to detect for enrichment of PPI’s. PPI’s were further filtered to select for a minimal of 3 (k>=2) proteins in each complex
(C) AP clustering of the siRNA perturbations by their functional signatures using Euclidean distance as a similarity metric. Nodes are colored according to cluster membership
(D) FuSiOn network drawn by force-directed graph(n edges= 188,802)
(E) Cluster 28 includes four COPI genes (red) and 6 proteasomal subunits (box)
(F-G) HCT116 cells were treated with siRNAs targeting (F) ARCN1 or (G) 5 proteasomal subunits for 72 hours. Depletion of target proteins and effects of reciprocal depletion were determined by immunoblot
Given that we can detect coherent PPI complexes in FuSiOn, we next sought to derive functional gene sets entirely from the FuSiOn similarity matrix. The 14,050 siRNA pools with detectable FuSiOn signatures were segmented into functional subgroups using Affinity Propagation Clustering (APC) (Frey and Dueck, 2007). This method was chosen as it is a deterministic clustering method that defines, in a data-driven fashion, both the number and membership of clusters emerging from a given similarity matrix. This method parsed at least 527 clusters (Figure 3C, Data S5), 51.2% of which are significantly enriched (HG q < 0.1) for a biological function. These clusters therefore offer significant opportunity to recognize and evaluate previously unknown functional relationships and interactions within the genome.
FuSiOn network architecture
A total of 189,086 significant genetic interactions between 5,598 unique genetic perturbations detected from the similarity matrix (FDR < 10%) were subjected to network construction using a force-directed graph drawing algorithm. The global FuSiOn network displayed a distinct bimodal structure (Figure 3D) when compared to a network drawn with random permutations of the FuSiOn similarity matrix (Figure S2A). Complex networks can be classified into random, scale-free, or hierarchical networks, depending on network topology. The FuSiOn network exhibited typical scale-free network topology as determined by evenly distributed clustering coefficients (Figure S2B) and power law degree distribution of the 5,598 nodes (Figure S2C). Network modularity is defined as the fraction of edges that fall within modules minus expected fraction from random network (Newman, 2006). The FuSiOn network exhibited highly modular network structure (modularity = 0.523) compared to a randomized network (modularity = 0.091) (Figure S2D),suggesting the presence of a small number of genetic hubs or submodules possibly involved in diverse biological functions. In comparison to other biological networks, the FuSiOn network shares network properties with the co-expression based biological network that was characterized by highly modular and scale free network properties (Carlson et al., 2006).
Leveraging the genome-scale FuSiOn network for biological discovery
Given the scale-free, modular network properties of FuSiOn, we sought to discriminate between distinct gene modules and characterize biological diversity within the resulting subnetwork to help identify new mechanistic gene modules; assign new gene function; and uncover cooperativity between known genes and pathways. A random walk-trap algorithm detected 903 modules (subnetworks) in the FuSiOn network, 28 of which included 10 or more genes (Figure S3A). Seven of the 28 clusters are associated with at least one known biological function (HG q<.1) (Data S6). For instance, cluster 1 is enriched with genes involved in amino acid metabolism and lysosome function, cluster 9 with JAK-STAT signaling, cluster 27 with calcium and chemokine signaling, and cluster 28 with the proteasome. We note that the statistical power required to detect significant functional associations with a given cluster, by this method, is limited by sample size.
To generate and test a FuSiOn-driven biological hypothesis, we examined cluster 28, as it contained a protein complex, coatomer I (COPI) (FDR q<.03), which we previously identified as a molecular linchpin that supports survival of KRASmut/LKB1mut lung adenocarcinomas through an unknown mechanism (Kim et al., 2013). Three COPI subunits, COPA, COPZ1, and ARCN1, are interconnected by FuSiOn and associated with 44 genes by two or more edges, six of which encode proteasome subunits (FDR q<.1;Figure 3E). The proteasome is critical for sustaining oncogenesis through supporting higher rates of protein synthesis and destabilizing tumor suppressors proteins such as p53 or other anti-apoptotic proteins. As a result, inhibiting its function is one of the clinically approved regimes for treating multiple myeloma (Crawford et al., 2011). To experimentally evaluate functional interactions between COPI and the proteasome, we examined the consequences of siRNA-mediated depletion of members of one complex on the abundance of members of the other complex. Notably, siRNAmediated depletion of the COPI subunit, archain1 (ARCN1) reduced the abundance of 3/5 proteasomal subunits clustering with COPI- PSMA3, PSMA5, and PSMA6 (Figure 3F; S2E). PSMA3 displayed partial but statistically meaningful depletion of cognate mRNA levels. However, reduction in mRNA levels were less evident for PSMA5 and PSMA6 suggesting more indirect effects on their protein concentrations, perhaps via destabilization due to unbalanced stoichiometry with PSMA3 (Figure S3B). Depletion of proteasome subunits had no effect on ARCN1 (Figure 3G). Together, these observations support a previously unknown functional and directional connection between these 2 protein complexes in which COPI integrity is required to maintain proteasome subunit stoichiometry.
High-throughput parsing of NP mechanism of action
In addition to the genome-scale miRNA mimic and siRNA libraries, the FuSiOn perturbagen set included 2,847 chemicals, 2,776 of which were NPFs from a total of 199 unique bacteria and marine species. The remaining chemicals were synthetic and pure NPs. For the fraction library production, metabolite extracts from clonal bacterial cultures were separated into either 9 or 20 fractions per strain by reverse-phase C18 chromatography. Each fraction is estimated to have anywhere from 3–6 active metabolites, and successively numbered fractions may contain the same or similar metabolites.
We subjected the chemical FuSiOn dataset to AP-clustering (Figure 4A) and overlaid strain of origin annotations. Overall, we found that the NP fractions separated into 164 clusters with NP fractions from each species were distributed throughout(Data S5). We considered that this result could be due to either incoherence in the dataset, or diversity in metabolites produced by individual organisms, but with multiple organisms producing metabolites with similar functional consequences on the cells. To help differentiate between these possibilities, we characterized a representative subset of the NP fractions by liquid-chromatography mass spectrometry (LC/MS) and evaluated diversity in metabolite profiles. We first focused our efforts on the metabolites produced from SN-B-022. We found the more polar (early) fractions from SN-B-022 (Figure 4A, red box; fractions 1–9) clustered independently from the less polar (later) fractions (Figure 4A, green box; fractions 11–20). A comparison of LC/MS spectra revealed a distinct peak corresponding to rhodomycin in the representative late fraction (Figure 4B). The observation that these fractions cluster separately from each other suggests that SN-B-022 produces at least two classes of compounds (rhodomycin and at least one other unknown metabolite) with different chemical profiles that are functionally distinguishable by FuSiOn. A well-recognized challenge associated with NPFs is de-convolute activity associated with chemical synergism from activity associated with a single fraction. However, successively numbered fractions with similar FuSiOn signatures that contain common shared metabolites discoverable with LC/MS provide strong evidence of a single chemical responsible for shared activity across fractions
Figure 4: Clustering of NPFs reveals common functions.

(A) AP clustering of the chemical perturbations according to their functional signatures using Euclidean distances as a similarity metric. Nodes are colored according to strain annotations with pure chemicals colored white. Highlighted clusters are zoomed in to the right
(B) LC/MS trace of SN-B-022–5 compared to SN-B-022–16. The peak corresponding to rhodomycin is highlighted in blue
(C) LC/MS trace of SN-C-004–17 compared to SN-C-002–11. The common metabolites in both fractions is highlighted in blue
(D) One-way hierarchical cluster comparing the functional signatures of SN-A-022–6 to XCT-790 (15 μM)
(E) Correlation of viability values in response to SN-A-022–6 (1.65 μg/mL) and AUC in response to oligomycin A in a panel of 10 NSCLC cancer cell lines. Pearson R and p-values are indicated
(F) Fluorescent staining of Parkin-YFP and DAPI (nuclear) for Hela-Parkin YFP cells in response to SN-A-022–6 (10 μg/mL) or compared to no treatment(scale bar=10 μm)
(G) Relative oxygen consumption rates (OCR) of Hela-Parkin YFP cells, normalized to total protein levels, in response to either no treatment (blue), 1 μg/mL (green) or 10 μg/mL (red) of SN-A-022–6 7 hours post-chemical treatment
We next considered that fractions from different organisms may cluster together due to the presence of a common metabolite. We noted a subset of sequential fractions from SN-C-004 (fractions 13–18; Figure 4A, blue box) clustered with the majority of the fractions produced by SN-C-002 (14 out of 20). The LC/MS spectra revealed an unknown metabolite selectively shared among these fractions, indicating that the functional activity is most likely driven by the same active metabolite produced by two different species (Figure 4C).
XCT-790 and SN-A-022–6 disrupt mitochondrial energy production
The above observations indicated reasonable coherence in the NPs data set, which we next evaluated by examination of a guilt-by-association hypothesis linked to pure chemicals in the perturbation file. We noted that fraction SN-A-022–6 clustered closely with XCT790 (Figure 4D). XCT790 is a known estrogen receptor related alpha (ERRα) inhibitor, however, we previously described it to have a potent activity against mitochondrial energy production, independent of its ERRα inhibitory effect (Eskiocak et al., 2014). We leveraged our recently published study in which our library was screened for viability across a panel of 26 lung cancer cell lines (Nichols et al., 2018). We compared viability in response to SN-A-022–6 (1.65 μg/mL) to that of oligomycin A, a mitochondrial ATPase synthetase inhibitor, derived in an independent study across the same panel of cell lines(Seashore-Ludlow et al., 2015), and found a high correlation (Figure 4E). Thus, we hypothesized that SN-A-022–6 is clustering with XCT790 due to common mechanism promoting mitochondrial mysregulation. To directly evaluate SN-A-022–6 for XCT790-like effects on mitochondrial function, we examined consequences on stress-induced mitochondrial turnover. The Parkin E3-ligase is recruited to damaged mitochondria to induce autophagy-mediated organelle clearance in a process known as mitophagy. Cellular Parkin redistribution from a diffuse to a punctate pattern indicates initiation of this process, and was robustly induced in HeLa Parkin-YFP cells upon exposure to SN-A-022–6 (Figure 4F). Furthermore, this was associated with reduced oxygen consumption in a dose-dependent manner (Figure 4G), indicating that, like XCT790, SN-A-022–6 is a mitochondrial poison.
These observations indicate that, using a sparse quantitative feature, FuSiOn is reasonably powered to accurately cluster complex NPFs and chemicals together according to similar mechanism-of-action. Thus, FuSiOn can help to decipher the primary functional consequence of chemicals with multiple targets and aid in the deconvolution of complex mixtures to identify distinct metabolites responsible for a phenotype of interest.
Functional landscape of NPFs
Finally, we sought to integrate the chemical and genetic datasets with AP-clustering. Representation of the output as a graph indicated extensive interleaving of the genetic and chemical perturbations within phenotypic subnetworks (Data S5; Figure 5A). To estimate the diversity of cell biological systems differentially engaged by the NP collection, pre-annotated gene sets from the public domain were subjected to enrichment analysis using the genome-scale similarity profile for each NPF. To do this, we first selected 1,280 NPF’s with RMS values > 0.6 (Figure 1D) and devised a computational pipeline to associate gene pathways to chemicals. For each NP, we computed distances from the NP to members of each gene set (‘in class’) and compared to distances from the NP to all other siRNA’s (‘out of class’) with a KS statistic. (Figure S4A). The most commonly perturbed biological processes (FDR q <.1) by a large number of NPs were those represented by proteasome components and cell cycle proteins (Figure 5B). Additional biological processes associated with distinct NPs included APC-CDC20 regulation, spliceosome, TGF-β signaling, cell-cell junction, IL signaling, DNA replication and repair, mitochondrial energy production, and translation(Data S7; Figure 5B, S5A). As an additional avenue to generate mechanism of action hypotheses for NPs, we assigned NPFs to pre-annotated gene sets (C2, CORUM) using similar methods as described in Figure 2B–C (Data S3). Of note, consistent with activity as a mitochondrial poison, XCT-790 mapped to the gene set 2914_Respiratory chain complex I beta subunit mitochondrial from the CORUM database (p=0.028; Figure 5C)
Figure 5: Functional landscape of NPFs.

(A) AP clustering of all perturbation datasets according to their functional signatures using Pearson distances as a similarity metric. Nodes are colored according to dataset of origin.
(B) A KS test was used to discover gene sets (KEGG) with similar functional signatures to each chemical perturbation. Number of NPs assigned by FDR 10% cutoff ( adjusted KS test p values) are represented in parenthesis. Boxes are colored according to gene set size and box sizes are drawn proportional to numbers of chemicals
(C) XCT-790 was re-assigned to the gene-set from the CORUM database ‘2914 respiratory chain complex I beta subunit mitochondrial’. Blue= chemical; red= gene set members. Edge lengths and widths are proportional to similarities (Euclidean distance)
(D) MCODE analysis found significant enrichment of PPI’s relating to endocytic pathways for the query NPF, SN-B-040-C. Edges are colored according to distances from the query and nodes are colored according to RMS
(E) LC/MS trace of SN-B-040-C compared to pure ikarugamycin. The peak corresponding to ikarugamycin is highlighted in red
(F) MCODE analysis found significant enrichment of PPI’s relating to the proteasome for the query NPF, SN-C-002–11. Edges are colored according to distances from the query and nodes are colored according to RMS
(G) Protein expression of the short-lived protein, REDD1 after exposure to the known proteasome inhibitor, MG132 (10 μM), and to NPs SN-C-002–11 and SN-004–11 (10μg/mL) for 30 minutes prior to cycohexamide treatments for the indicated times. Cyclohexamide (10μg/mL; CHX) treatment inhibits synthesis of new proteins
(H) Cells were pre-treated with 10 μg/mL of NPF, DMSO, or 10 μM of MG132 for 24 hours, then lysed and mixed with an AMC-tagged proteasome substrate. Fluorescence was read at the indicated time points post-substrate incubation. Proteasome activity is measured as percent reduction in fluorescence relative to DMSO
We next sought a method for facile detection of NPFs potentially engaging a given biological process of interest. To do this, we used MCODE to detect PPI clusters within the 500 top-ranked genetic functionalogues associated with each NPF. As before, we filtered for complexes with a K-core score of at least 2 (Figure 3B; minimum of 3 interactions). MCODE-associated complexes were detected for 2480/2847 chemicals, 1660 of which are significantly associated with a pre-annotated biological function (HG q<.1) (Data S4). As a test-of-concept we selected endocytosis as a starting biological process of interest to link to NP mechanism. NPFs associated with MCODE clusters were searched for those with the highest number of interconnecting PPI edges belonging to endocytic protein complexes as defined by KEGG (153 proteins with 480 PPIs among them). SN-B-040-C was identified as a top ranked candidate via FuSiOn association with the endocytic proteins AP2A1, AP2M1, and SYNJ1 (Figure 5D). The LC/MS spectra of SN-B-040-C contained a peak corresponding to ikarugamycin (Figure 5E), a NP previously described as an inhibitor of clathrin-mediated endocytosis in the context of non-small cell lung cancer (Elkin et al., 2016). We next selected SN-C002/SN-C-004 (from Figure 4A (blue box), 4C) as NP fractions of interest to link to a biological process. This search returned a proteasome PPI cluster within the SN-C-00211 functionalogues (Figure 5F). To evaluate if SN-C-002–11 may affect proteasome activity, we assessed its capacity to stabilize the short half-life protein REDD1, which is known to be regulated by proteasome degradation. REDD1 protein accumulation was induced by culturing HCT116 cells for 24 hours in hypoxic conditions (1% O2). Cells were then treated with SN-C-002–11 or MG132, a known proteasome inhibitor, for 30 minutes followed by treatment with cyclohexamide (10 μg/mL) to prevent synthesis of new proteins. Notably, SN-C-002–11 inhibited degradation of REDD1 to a similar extent as MG132 (Figure 5G). SN-C-002–11 was also able to directly inhibit proteasomal activity as measured by monitoring proteasome-dependent cleavage of a fluorescent proteasome substrate peptide in vitro (Figure 5H). Importantly, SN-C-004–11, an earlier fraction of SN-C-004 which clustered away from the SN-C-002/SN-C-004 cluster did not induce stabilization of REDD1 or inhibit proteasomal activity. This provides strong evidence that SN-C-002–11 acts to affect proteasomal function and further highlights the use of FuSiOn to deliver productively evaluable mechanistic hypotheses.
Discussion
Deciphering the molecular underpinnings of cell biological systems and decoding modes of action of bioactive chemicals are rate-limiting steps for advancing pharmaceuticals that appropriately target newly discovered disease mechanisms. Critical needs include a broad-scale functional annotation of intervention targets within the cell regulatory systems perturbed in disease, collection of features that allow these to be identified in patients, and assignment of chemicals that can intersect these targets. The objective of this study was to help address these gaps though production of an open access hypothesis generation engine that allows operators to identify chemical and genetic “functionalogues” of a gene or biological process of interest. Following on from pioneering work which demonstrated the power of perturbation-induced gene expression signatures as a mechanistic pattern-matching tool (Hughes et al., 2000) and building on our previously developed platform, we have implemented a genome-scale annotation of protein function and chemical mode-of-action in human cells that produces verifiable mode-of-action hypotheses for previously uncharacterized gene products and chemical entities.
Sparse quantitative gene expression-derived functional signatures were employed as cross-modality phenotypic discriminators to link concordant cellular responses to 14,272 distinct human gene perturbations, 725 miRNA mimics, and 2776 marine microbe-derived NPs. We leveraged this comprehensive feature matrix to generate a map of functional associations between all genes in the genome, assess the overall topology of the functional network in a biological setting, and simultaneously predict biological response mechanisms for thousands of uncharacterized chemicals.
De novo construction of a genome-wide functional signature association map was a key goal for establishing a comprehensive framework from which to generate guilt-by-association hypotheses. Perturbation expression signatures associated with over 14,000 individual human genes were used to construct a genome-scale functional similarity network. This inclusive network was credentialed for concordance with goldstandard functional subnetworks; was shown to accurately expand gene membership within subnetworks supporting key cell biological processes; was shown to identify previously unrecognized functional modules; and was shown to predict verifiable mechanistic interactions among protein sub-complexes.
As previously noted, NPs remain an attractive source of drug discovery, as their structure is subject to co-evolution with biological systems. Thus, they may engage targets not currently recognized as druggable. However, a significant barrier associated with NP utilization is purification of active metabolites from producing organisms and annotation of biological mechanism of action. Here, ~2700 NPFs containing uncharacterized chemical entities (3–6 bioactive metabolites per fraction) derived from marine bacteria were profiled for integration into the genome-wide functional interaction map described above. The resulting guilt-by-association hypotheses helped to predict the primary functional consequence of metabolites with multiple targets by computationally assigning hundreds of metabolites to hundreds of human cell biological processes. This will aid in eventual deconvolution of complex mixtures to identify distinct metabolites responsible for a phenotype of interest.
By nature, the ability to capture gene-gene and gene-chemical relationships with the approach developed here depends on whether the given perturbation has any measurable consequences within the chosen reference cell that are detectable by the chosen probe set. As such, many bona fide mechanistic associations have not been detected in this study. In addition, the scale of the data matrix and the technologies deployed will unavoidably return false associations at some frequency. However, the orthogonal credentialing described here indicates numerous informative relationships have been returned that can productively accelerate focused discovery campaigns. Therefore, we anticipate FuSiOn to serve as an iterative hypothesis generator that accelerates discovery of novel interactions among cellular pathways, annotation of previously unrecognized functions for microRNAs and genes, and identification of new chemicals that can intercept biological activities not currently druggable. To enable community-based discovery and hypothesis testing, we have made all associations, annotations, and pre-computed relationships described within the manuscript available as a query-able web-based GUI (http://fusion.yuhs.ac).
STAR Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael A. White (Michael.a.white@pfizer.com)
Experimental Model and Subject Details
Cell Lines:
Cell lines used in this study are described in the key resources table. HCT-116 cell line used in this study was purchased from ATCC (The American Type Culture Collection). HCT-116 cells and HeLa-Parkin cells were maintained in DMEM (Gibco) supplemented with either 5% FBS (HCT-116) (Gibco) or 10% FBS (Hela-Parkin) with 1% antibiotics (Gibco) at 37°C in a humidified atmosphere containing 5% CO2.
Method Details
Natural products library
The natural products fraction library used for this study is composed of extracts from 600 marine-derived bacterial strains and 20 marine invertebrates (19 sponges, 1 tunicate). The library of microbial NP fractions was derived from marine-derived Actinomycetes (400), Firmicutes (150) and alpha-proteobacteria (50). These bacteria were cultivated from marine sediment samples collected in Tonga, the Gulf of Mexico (Texas, Louisiana), estuaries in South Carolina, and the Bahamas. A variety of techniques were utilized to isolate strains, including the use of nutrient-limited isolation media, such as those composed of only humic or fulvic acid, the use of small-molecule signaling compounds (N-acylhomoserine lactones, siderophores) that mimic the natural environment of the bacteria of interest, and isolation of spores using density gradient ultracentrifugation. Selection of bacterial isolates was carried out based on morphological appearance and followed up by phylogenetic characterization using 16S rRNA analysis using the Universal 16S rRNA primers FC27 and RC 1492 for the majority of the phylogentic analysis. 16S rRNA sequences were compared to sequences in available databases using the Basic Local Alignment Search Tool.
To generate the fraction library used in this study, bacterial strains were fermented in 5 × 2.8 L Fernbach flasks each containing 1 L of a seawater based medium (10 g starch, 4 g yeast extract, 2 g peptone, 1 g CaCO3, 40 mg Fe2(SO4)3·4H2O, 100 mg KBr) and shaken at 200 rpm for seven days at 27 ºC. After seven days of cultivation, XAD7-HP resin (20 g/L) was added to adsorb the organic products, and the culture and resin were shaken at 200 rpm for 2 h. The resin was filtered through cheesecloth, washed with deionized water, and eluted with acetone to give a crude extract, with an average of 2.0 g of crude extract/strain. Further fractionation of the bacterial crude extracts (~500 mg) was accomplished using an Isco medium pressure automatic purification system (equipped with UV and ELSD detectors) using reversed phase C18 chromatography (gradient from 90:10 H2O:CH3CN to 0:100 H2O:CH3CN over 25 minutes, RediSep Rf Gold High Performance column with 600 mg capacity). Fermentation of each bacterial strain gives rise to a total of either 10 or 20 natural product fractions/stran. All natural product fractions in the library are standardized to a concentration of 10 mg/mL in DMSO. All fractions have been analyzed by low resolution LC/MS using an Agilent Model 6130 single quadrupole instrument. About 1/3 of the fractions have been analyzed for high resolution MS using a Thermo Velos Orbitrap.
Due to the nature of the fractionation approach, there is by design, the potential for sequential fractions to contain the same compounds. For example, if the peak for staurosporine is split between fraction 6 and 7, one could expect to have a similar biological signature between the sequential fractions.
Proteasome Activity
Proteasome activity was measured with a fluorometric kit from Abcam (ab107921), which utilizes a AMC tagged peptide substrate. In the presence of proteasome activity, AMC will be released and proteasome activity can be measured using fluorescent units as a surrogate. HCT116 cells were seeded at a density of 300,000 cells/well in 6 well format and allowed to adhere for 24 hours. Cells were then pre-treated with either 10 μg/mL of NPF (SN-C-002–11 or SN-C-004–11), with DMSO, or with 10 μM MG-132 for 24 hours in biological triplicates. Following a 24 hour incubation, cells were trypsinized, pelleted and resuspended in 250 μL of lysis buffer (1% NP-40 buffer). 20μL of lysate was plated in 96 well format, with volume equalized to 100 uL per well with assay buffer. 1 μL of AMC-tagged proteasome substrate was added to each well, and the plate was incubated at 37 degrees, protected from light. Fluorometric readings in a microplate reader were made at 2 inutes, 20 minutes, and 50 minutes post-substrate incubation. All fluorometric values were normalized to DMSO at the same time point
Cell culture, transfection and Immunoblot analysis
For siRNA transfection, 200,000 cells in 2 ml of growth medium were added to a 0.5 ml mixture of 100 pmole siRNA and 4 μl of Lipofectamine RNAiMAX reagent (Invitrogen, #13778) per well of 6-well plate following manufacturer’s protocol. After 72 hours of transfection, cell lysates were prepared using either RIPA buffer or in 50nM Tris (pH 6.8), 2% SDS and 10% glycerol. 10 μg of each sample was separated in 8–16% TGX gel (Biorad, #456–1105), transferred onto 0.2 μm PVDF membrane, incubated with primary antibodies dissolved in PBST buffer with 5% BSA at 4°C overnight, washed twice with TBST buffer, incubated with proper secondary antibody conjugated with HRP in TBST buffer with 5% skim milk for two hours at room temperature, washed and detected using ECL reagents (Amersham) following manufacturer’s protocol. Primary antibodies for immunoblot analyses were purchased from Cell Signaling Technology (PSMA3; 12446S, PSMA5; 2457S, PSMA6; 2459S, PSMB5; 12919S, PSMB7; 13207S, REDD1;2516, β-tubulin;2128,and Abcam (Archain; ab96725, β-actin; ab8227).
REDD1 degradation assay
Cells were plated in 6 well dishes at a density of 300K cells/well and allowed to adhere for 24 hours. Cells were then transferred to a hypoxic incubator (2% oxygen) and incubated for 24 hours. Cells were treated with either 10 μM of MG132, 10 μg/mL SN-002–11, or 10 μg/mL SN-004–11 and allowed to incubate for 30 minutes. Following incubation, cells were treated with 10 μg/mL of cycloheximide and individual wells were harvested at 0,10,30,60, and 120 minutes post-cyclohexamide treatment and analyzed for REDD1 protein levels via Western blot.
qPCR
For qRT-PCR, siRNA transfected cells were subjected to RNA isolation (Qiagen #74134), cDNA synthesis (Enzynomics #EZ005S), and qRT-PCR (Enzynomics #RT501) using primer sequences listed in the Key Resources table.
Seahorse Assay
An XF-24 Extracellular Flux Analyzer (Seahorse Bioscience) was used for measurement of oxygen consumption and extracellular acidification rates. Hela cells stably expressing Parkin fused to YPF were seeded at 40,000 cells per well in a 24-well Seahorse-specific plate (Seahorse Bioscience) in 500 microliters standard culture media (10% FBS and DMEM supplemented with penicillin and streptomycin). The cells were allowed to attach overnight. At the start of treatment, the cells were treated with the appropriate compound in 200 microliters of standard culture media then cultured for seven hours with treatment. Following the completion of treatment, the media was aspirated and the cells were equilibrated in XF Base Medium Minimum DMEM (supplemented with 25 millimolar glucose, 2 millimolar glutamine, and 1 millimolar sodium pyruvate). Oligomycin (1 micromolar final), FCCP (1 micromolar final), and rotenone (100 nanomolar final) were used to assess the function of the electron transport chain after treatment. Oxygen consumption and extracellular acidification rates were normalized to cell number.
Imaging of Fluorescent protein
HeLa cells stably expressing Parkin fused to YFP were seeded on glass coverslips in standard culture media (10% FBS and DMEM supplemented with penicillin and streptomycin) and allowed to adhere overnight. Cells were treated for four hours with the appropriate compound-treatment condition prepared in warmed, standard culture media. Following the completion of treatment, the media was aspirated and the cells fixed with a 4% PFA solution for fifteen minutes. The solution was aspirated and the cells washed one time with 50 mM ammonium chloride. Cells were permeabilized with 0.1% Triton-X-100 for 10 minutes, washed two times with 1×PBS, then mounted with DAPI-containing ProLong Gold.
Cell based high throughput screens and library reagents
Quantification of reporter gene expression and library screening was performed as previously described (Potts et al., 2013). The natural products library was collected as described above and screened at a final concentration of 60mg/mL for a total treatment time of 21 hours. We used the miRIDIAN microRNA library (Dharmacon Catalog #CS-00005, lot # 01823). The siRNA library was purchased from Dharmacon (siGenome lot # 050915) and screened as pools of 4 oligos. Both the miRNA and the siRNA libraries were screened at a final concentration of 50nM for a total treatment time of 72 hours.
Liquid Chromatography/Mass Spectrometry
LC-MS data was acquired on an Agilent 1100 Series HPLC with an Agilent Model 6130 Single Quadruple Mass Spectrometer and a photodiode array detector. The system was equipped with a reversed-phase C18 column (Phenomenex Luna, 150 mm × 4.6 mm, 5 μm) and operated at a flow rate of 0.7 mL/min. All samples were analyzed using a gradient solvent system from 10% to 99% CH3CN (0.1% formic acid) over 15 min to afford compounds The gradient used for all samples was 90:10 H20 (0.1% formic acid):CH3CN (0.1% formic acid) to 1:99 H20 (0.1% formic acid):CH3CN (0.1% formic acid) over 17 minutes, then 1:99 H20 (0.1% formic acid):CH3CN (0.1% formic acid) for 10 minutes. Detection was carried out at four UV wavelengths (210, 254, 280, 330 nm) and in dual mode MS (positive and negative ion).
Quantification and Statistical Analyses
Data Normalization
Eight cell-based reporter gene expression profiles collected for 14,272 siRNA pools, 725 miRNA mimics and 3,144 natural product fractions were normalized as follows. First, to normalize different cell numbers across wells, the six background-corrected reporter gene expression values per well were divided by the geometric mean of the two internal control probes, HPRT and PPIB. Second, this version includes genome-scale siRNA perturbations and significantly expanded natural product-perturbations assayed in multiple batches, thus, it inevitably accompanies batch-to-batch signal variations. To account for them, the six in-well normalized reporter values were further divided by the medians of the in-plate control wells (up to 10 non-targeting siRNAs or vehicles per plate) and log2-transformed. Duplicated perturbations were averaged, and mean of the triplicate normalized values for each reporter per perturbation was used for further analysis.
Similarity matrix construction
In this study, Euclidean distance was used to quantify the similarity between expression profiles of different perturbations since it takes into account the magnitude of variation, unlike other correlation-based metrics. To assess statistical significance of a similarity between perturbation A and B, background distance distributions for perturbation A and B were generated, respectively. For this, perturbation labels for each of the six reporter genes were permuted 100K times and the background distance distributions were obtained by estimating Euclidean distance from perturbation A to the 100K permuted data points, then, repeated for perturbation B. These two-directional background density distributions were used to estimate two empirical p-values for each pair of perturbations, which are usually similar to each other, and a more conservative (greater) p-value was chosen to represent its statistical significance for the pair. For the genetic and chemical perturbation pairs, only genetic perturbations were permuted to provide a single p-value. False discovery rates (FDRs) were estimated by fitting a beta-uniform mixture (BUM) model to the estimated P values using the dnet package for R (Fang and Gough, 2014). Alternative FDRs by Benjamini-Hotchberg (BH) method were also provided, which were useful especially when BUM model fails to fit to estimated p-values. All data processing, permutation, and p-value estimation were carried out using R. To investigate the correlation between genetic associations by FuSiOn (FDR < 10%, N = 177,744) and preconceived gene sets, we conducted hypergeometric test using various public gene sets; i.e. activation relationships of STRING PPI database (N = 17,561), C2 (N = 960,121) and C5 (N = 9,394,552) gene sets of MSIGDB v4.0, synthetic lethal relationships (N = 2,365) detected by DAISY algorithm, and miRNA-siRNA target relationships (N = 4,145) reported in TargetScan.
Gene set analysis
To achieve systems-level functional annotation of a perturbation, gene set analysis was performed for each of 14,997 genetic and 3,144 chemical perturbations against 3,723 unique pre-annotated gene sets obtained from CORUM (Ruepp et al., 2008), C2 MSigDB (Liberzon et al., 2011) and PCDq protein complex (Kikugawa et al., 2012) after removing redundancy. If a query gene is included in a target gene set, it was removed from the gene set before an analysis. Additionally, as the off-target effect of siRNA and miRNA is mostly driven by the seed sequence, genes in a gene set whose siRNA pools have at least one seed matching oligo to a query siRNA or miRNA were also censored from the gene set. After applying these filters, only gene sets containing between 3 and 200 members were subjected for an analysis. On average, 3,300 gene sets were used for an analysis for each perturbation. To identify overrepresented gene sets by functionalogues, an array of distance values for a perturbation from the similarity matrix was subjected to Kolmogorov-Smirnov (K-S) test iteratively for the qualifying gene sets.
FuSiOn network analysis
Significant genetic interactions (N = 189,086) by FuSiOn under FDR 10% were subjected to network construction and visualization using a force-directed graph drawing algorithm implemented in the R package “igraph”. For comparisons, a randomized network was prepared by sampling the same number of interactions between random pairs of genetic perturbations (N = 14,997). After removing nodes and edges with cluster size less than 10 disconnected from the main network, FuSiOn network consisting of 5,598 nodes and 188,802 edges were subjected to further analysis. The walk-trap algorithm (‘walktrap.community’ function, step = 4) implemented in the R package “igraph” was used for the detection of clusters in the FuSiOn network. Out of the 903 detected clusters, twenty-eight network clusters with ten or more nodes were selected for subsequent functional analysis for the detection of representative gene sets (N = 3,723) based on hypergeometric tests. Modularity Q value was estimated with the R package ‘igraph’ with the parameters as follows: maximized modularity without weight.
MCODE and PPI analysis
Collections of manually curated protein-protein interactions (PPI) were retrieved from the mentha databse (https://mentha.uniroma2.it/). Cytoscape plugin Molecular Complex Detection (MCODE) detects highly interconnected regions in a network(Bader and Hogue, 2003). We implemented the MCODE algorithm with R using ‘sna’ and ‘igraph’ packages for the batch-mode running of the entire perturbations with the parameters as follows: minimum K-core = 2, maximum depth = 20, node score cutoff = 0.2, degree cutoff = 2, haircut = T, fluff = F, include loop = F, and duplicated edge = F. The MCODE parameter k-core measures degree of interconnectivity of a sub-graph in which each vertex has degree at least k. For example, a triangle (3 nodes, 3 edges) is a 2-core (2 connections per node). We identified PPI subnetworks formed by 500 top ranked functionalogues by Euclidean distance for each of the genetic and chemical perturbations. Among the activation interactions between human proteins, those with the highest confidence score (> 0.9, N = 17,561) were extracted from the STRING database v10.0.
Detection of siRNA seed effect
The seed sequence of an 19mer siRNA oligo was determined to be from positions 2 to 8. We selected seed sequences for further analysis in which there were at least 5 siRNA oligos containing the seed. For each seed sequence, we calculated pairwise Euclidean distances between all siRNAs containing the seed. A NULL distribution of the distances between the siRNAs containing that seed compared to the rest of the siRNAs in the screen was calculated and a p-value was determined based on this distribution. P-values were corrected with an FDR correction. Gene expression data for HCT116 was downloaded from the cancer cell line encyclopedia (Barretina et al., 2012) and unexpressed genes were annotated to be those in which RNAseq based FPKM <1 and Affymetrix quantile normalized expression values were <5.
Affinity propagation clustering
Affinity propagation clustering was performed as previously described (Kim et al., 2016, Witkiewicz et al., 2015, McMillan et al., 2018). Clustering analysis was performed with the affinity propagation clustering (APC) algorithm using the ‘apcluster’ package in R. APC is a deterministic clustering method that identifies the number of clusters and cluster ‘exemplars’ (that is, the cluster centroid or the data point that is the best representative of all the other data points within that cluster) entirely from the data6, giving it an advantage over non-deterministic methods subject to a biased randomized initialization step, such as hierarichial clustering or methods in which the number of clusters has to be pre-specified, such as k-means clustering.
APC performs clustering by passing messages between the data points. It takes as input a square matrix representing pairwise similarity measures between all data points. The algorithm views each data point as a node in a network and is initialized by connecting all the nodes together, where edges between nodes are proportional to Euclidean distance. The algorithm then iteratively transmits messages along the edges, pruning edges with each iteration until a set of clusters and exemplars emerges.
Two real-valued messages are passed between nodes. The ‘responsibility’ message computes how well-suited point i is to choose point k as an exemplar, given all the other candidate exemplars, k′, and is updated by:
The availability message, a(i,k), computes how appropriate it is for point i to select point k as an exemplar, taking into account all the other points for which k is an exemplar, i′, and is given by:
In the above equation, a(i, k) is set to the self-responsibility, r(k, k), plus the sum of the positive responsibilities candidate k receives from other points. The entire sum is thresholded at 0, with a negative availability indicating that it is inappropriate for point i to choose point k as an exemplar so the tie is severed. The self-availability, a(k, k), reflects the accumulated evidence that point k is an exemplar and is updated with the following rule, which reflects the evidence that k is an exemplar based on the positive responsibilities sent to k from all points, and is updated by:
In the first iteration, all points are considered equally likely to be candidate exemplars, and a(i, k) is set to 0 and s(i, k) is set to the input similarity measure between points i and k. The above rules are then iteratively updated until a clear, stable set of clusters and exemplars emerges.
In our implementation, we first used the algorithm to identify an initial set of exemplars and clusters from the data matrix. The exemplars were then clustered together and this procedure was repeated until no more clusters emerged, identifying a hierarchical structure of clusters. Networks were drawn with cytoscape31 in the following manner. All members of the primary clusters are interconnected, and edge lengths are drawn to be proportional to Euclidean distances. Edge lengths between exemplars that cluster together are also drawn to be proportional to Euclidean distances. The entire map was rendered in a two-dimensional display using a cytoscape built-in spring-embedded algorithm.
Enrichment of functional signatures in annotated gene sets
A modification of a Kolmogorov-Smirnov statistic was made to determine if we can detect a functional enrichment between siRNAs annotated as being in the same manually curated functional classes or gene sets. Gene sets were downloaded from the CORUM database (Ruepp et al., 2008) and MSigDB version 3 (Liberzon et al., 2011), and were filtered to be inclusive of gene sets with between 5 and 250 members. The C2 database was further filtered for gene sets annotated as belonging to either KEGG, Reactome, PID, or BioCarta. For a given gene set, we calculated pairwise distances between genes included in the set to every other gene included in the whole genome siRNA perturbation dataset. If we can detect a significant overall functional enrichment for a given gene set, then we would expect pairwise distances between siRNAs annotated as being in the same gene set to be significantly shorter than distances between those same siRNAs and the remaining siRNAs in the genome library. For a pathway, to determine the degree to which distances in a set are located towards the bottom of a ranked list of distances, and thus lower relative to background, the following equation was used:
where v(j) is the position of each pairwise distance between genes in the same gene set in the ordered list of distances, t is binomial coefficient , where k is the number of genes included in the gene set and m is the total number of siRNA’s assayed not included in the gene set.
To determine a p-value, 5,000 permutations of randomized sorting of genes of the same set size was performed, and urandom was calculated. The resulting p-value was determined to be:
Reannotation of gene sets
Of the gene sets in which a significant functional signature is detectable (Data S2), pairwise Euclidean distances between members in the gene set are significantly shorter than when compared to a background distribution. Therefore, we can generate a vector that describes an overall gene set functional signature by collapsing the 6 probe values to the median for members of the set. We then calculated Euclidean distances from query chemicals and siRNAs not included in the gene set to the gene set vector. In order to generate a p-value, we randomly permuted genes into groups of the same gene set size and calculated distance from the query to each random permutation. The p-value was calculated to be
The p-value was corrected with a Bonferroni correction.
Other statistical analyses
All other statistical analyses were performed using that stats package in R. Statistical tests used and all corresponding p-values and numbers are indicated in the results section and corresponding figure legends.
Data and code availability
Datasets generated in this study and all corresponding analyses are included as supplementary datasets and via a queryable website (http://fusion.yuhs.ac). Source code will be available upon request by the lead contact.
Additional Resources
All normalized data has been made available as a queryable website (http://fusion.yuhs.ac)
Supplementary Material
Highlights:
Generation of genome-wide gene/gene and gene/chemical similarity maps
Annotation of novel gene function and cooperatively among biological pathways
Parallel MOA predictions for thousands of NPFs with diverse cell biological effects
Significance:
Chemistry-first new target nomination campaigns leverage large-scale uncharacterized chemical diversity as a de novo discovery tool unconstrained by preconceived notions of mechanistic relationships. While often advantaged by rich chemical equity and a predisposition to return pharmacologically addressable targets, a major bottleneck is identification of molecular mechanisms of action for compounds with attractive phenotypic properties. This is especially true for natural products, a historically productive source of effective drugs. To help mitigate this bottleneck, we have developed a resource that facilitates massively parallel generation of testable molecular mechanism of action hypotheses for uncharacterized or poorly characterized cellular perturbagens; including purified chemicals, biological metabolites and natural product mixtures.
Acknowledgements
This research was supported by a NIH grant U41AT008718 to MAW and JBM from the NIH/NCCIH, grant I-1689 from the Welch Foundation to JBM, grant to HSK from Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2014R1A1A2057232, 2017R1A2B2006777) and grant 2018H1D3A2000524 to SBK from the NRF of Korea through the Brain Pool program. EAM was supported by NIH training grant 5T32GM8203-27. R.M.V was supported by CPRIT training grant RP140110 and NIH training grant 5T32CA124334-09.
Footnotes
Declarations of Interest
The authors have no conflicts of interest to report. Michael White, Rachel Vaden, and Elizabeth McMillan are current employees of Pfizer, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- BADER GD & HOGUE CW 2003. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics, 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARRETINA J, CAPONIGRO G, STRANSKY N, VENKATESAN K, MARGOLIN AA, KIM S, WILSON CJ, LEHAR J, KRYUKOV GV, SONKIN D, REDDY A, LIU M, MURRAY L, BERGER MF, MONAHAN JE, MORAIS P, MELTZER J, KOREJWA A, JANE-VALBUENA J, MAPA FA, THIBAULT J, BRIC-FURLONG E, RAMAN P, SHIPWAY A, ENGELS IH, CHENG J, YU GK, YU J, ASPESI P JR., DE SILVA M, JAGTAP K, JONES MD, WANG L, HATTON C, PALESCANDOLO E, GUPTA S, MAHAN S, SOUGNEZ C, ONOFRIO RC, LIEFELD T, MACCONAILL L, WINCKLER W, REICH M, LI N, MESIROV JP, GABRIEL SB, GETZ G, ARDLIE K, CHAN V, MYER VE, WEBER BL, PORTER J, WARMUTH M, FINAN P, HARRIS JL, MEYERSON M, GOLUB TR, MORRISSEY MP, SELLERS WR, SCHLEGEL R. & GARRAWAY LA 2012. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483, 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARLSON MR, ZHANG B, FANG Z, MISCHEL PS, HORVATH S. & NELSON SF 2006. Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC Genomics, 7, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRAWFORD LJ, WALKER B. & IRVINE AE 2011. Proteasome inhibitors in cancer therapy. J Cell Commun Signal, 5, 101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELKIN SR, OSWALD NW, REED DK, METTLEN M, MACMILLAN JB & SCHMID SL 2016. Ikarugamycin: A Natural Product Inhibitor of ClathrinMediated Endocytosis. Traffic, 17, 1139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESKIOCAK B, ALI A. & WHITE MA 2014. The estrogen-related receptor alpha inverse agonist XCT 790 is a nanomolar mitochondrial uncoupler. Biochemistry, 53, 4839–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FANG H. & GOUGH J. 2014. The ‘dnet’ approach promotes emerging research on cancer patient survival. Genome Med, 6, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FREY BJ & DUECK D. 2007. Clustering by passing messages between data points. Science, 315, 972–6. [DOI] [PubMed] [Google Scholar]
- GARCIA DM, BAEK D, SHIN C, BELL GW, GRIMSON A. & BARTEL DP 2011. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol, 18, 1139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIMSON A, FARH KK, JOHNSTON WK, GARRETT-ENGELE P, LIM LP & BARTEL DP 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell, 27, 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUGHES TR, MARTON MJ, JONES AR, ROBERTS CJ, STOUGHTON R, ARMOUR CD, BENNETT HA, COFFEY E, DAI H, HE YD, KIDD MJKING, A. M., MEYER, M. R., SLADE, D., LUM, P. Y., STEPANIANTS, S. B.SHOEMAKER, D. D., GACHOTTE, D., CHAKRABURTTY, K., SIMON, J., BARD, M. & FRIEND, S. H. 2000. Functional discovery via a compendium of expression profiles. Cell, 102, 109–26. [DOI] [PubMed] [Google Scholar]
- JERBY-ARNON L, PFETZER N, WALDMAN YY, MCGARRY L, JAMES D, [Google Scholar]
- SHANKS E, SEASHORE-LUDLOW B, WEINSTOCK A, GEIGER T, CLEMONS PA, GOTTLIEB E. & RUPPIN E. 2014. Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell, 158, 1199–209. [DOI] [PubMed] [Google Scholar]
- KIKUGAWA S, NISHIKATA K, MURAKAMI K, SATO Y, SUZUKI M, ALTAF-UL-AMIN M, KANAYA S. & IMANISHI T. 2012. PCDq: human protein complex database with quality index which summarizes different levels of evidences of protein complexes predicted from h-invitational protein-protein interactions integrative dataset. BMC Syst Biol, 6 Suppl 2, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM HS, MENDIRATTA S, KIM J, PECOT CV, LARSEN JE, ZUBOVYCH I, SEO BY, KIM J, ESKIOCAK B, CHUNG H, MCMILLAN E, WU S, DE BRABANDER J, KOMUROV K, TOOMBS JE, WEI S, PEYTON M, WILLIAMS N, GAZDAR AF, POSNER BA, BREKKEN RA, SOOD AK, DEBERARDINIS RJ, ROTH MG, MINNA JD & WHITE MA 2013. Systematic identification of molecular subtype-selective vulnerabilities in nonsmall-cell lung cancer. Cell, 155, 552–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM J, MCMILLAN E, KIM HS, VENKATESWARAN N, MAKKAR G, RODRIGUEZ-CANALES J, VILLALOBOS P, NEGGERS JE, MENDIRATTA S, WEI S, LANDESMAN Y, SENAPEDIS W, BALOGLU E, CHOW CB, FRINK RE, GAO B, ROTH M, MINNA JD, DAELEMANS D, WISTUBA II, POSNER BA, SCAGLIONI PP & WHITE MA 2016. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature, 538, 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE YC, JOGIE-BRAHIM S, LEE DY, HAN J, HARADA A, MURPHY LJ & OH Y. 2011. Insulin-like growth factor-binding protein-3 (IGFBP-3) blocks the effects of asthma by negatively regulating NF-kappaB signaling through IGFBP-3Rmediated activation of caspases. J Biol Chem, 286, 17898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIBERZON A, SUBRAMANIAN A, PINCHBACK R, THORVALDSDOTTIR H, TAMAYO P. & MESIROV JP 2011. Molecular signatures database (MSigDB) 3.0. Bioinformatics, 27, 1739–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCMILLAN EA, RYU MJ, DIEP CH, MENDIRATTA S, CLEMENCEAU JR, VADEN RM, KIM JH, MOTOYAJI T, COVINGTON KR, PEYTON M, HUFFMAN K, WU X, GIRARD L, SUNG Y, CHEN PH, MALLIPEDDI PL, LEE JY, HANSON J, VORUGANTI S, YU Y, PARK S, SUDDERTH J, DESEVO C, MUZNY DM, DODDAPANENI H, GAZDAR A, GIBBS RA, HWANG TH, HEYMACH JV, WISTUBA I, COOMBES KR, WILLIAMS NS, WHEELER DA, MACMILLAN JB, DEBERARDINIS RJ, ROTH MG, POSNER BA, MINNA JD, KIM HS & WHITE MA 2018. Chemistry-First Approach for Nomination of Personalized Treatment in Lung Cancer. Cell, 173, 864–878 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEWMAN ME 2006. Modularity and community structure in networks. Proc Natl Acad Sci U S A, 103, 8577–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLS BA, OSWALD NW, MCMILLAN EA, MCGLYNN K, YAN J, KIM MS, SAHA J, MALLIPEDDI PL, LADUKE SA, VILLALOBOS PA, RODRIGUEZ-CANALES J, WISTUBA II, POSNER BA, DAVIS AJ, MINNA JD, MACMILLAN JB & WHITEHURST AW 2018. HORMAD1 Is a Negative Prognostic Indicator in Lung Adenocarcinoma and Specifies Resistance to Oxidative and Genotoxic Stress. Cancer Res, 78, 6196–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POTTS MB, KIM HS, FISHER KW, HU Y, CARRASCO YP, BULUT GB, OU YH, HERRERA-HERRERA ML, CUBILLOS F, MENDIRATTA S, XIAO G, HOFREE M, IDEKER T, XIE Y, HUANG LJ, LEWIS RE, MACMILLAN JB & WHITE MA 2013. Using functional signature ontology (FUSION) to identify mechanisms of action for natural products. Sci Signal, 6, ra90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POTTS MB, MCMILLAN EA, ROSALES TI, KIM HS, OU YH, TOOMBS JE, BREKKEN RA, MINDEN MD, MACMILLAN JB & WHITE MA 2015. Mode of action and pharmacogenomic biomarkers for exceptional responders to didemnin B. Nat Chem Biol, 11, 401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUEPP A, BRAUNER B, DUNGER-KALTENBACH I, FRISHMAN G, MONTRONE C, STRANSKY M, WAEGELE B, SCHMIDT T, DOUDIEU ON, STUMPFLEN V. & MEWES HW 2008. CORUM: the comprehensive resource of mammalian protein complexes. Nucleic Acids Res, 36, D646–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUEPP A, WAEGELE B, LECHNER M, BRAUNER B, DUNGER-KALTENBACH I, FOBO G, FRISHMAN G, MONTRONE C. & MEWES HW 2010. CORUM: the comprehensive resource of mammalian protein complexes−−2009. Nucleic Acids Res, 38, D497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEASHORE-LUDLOW B, REES MG, CHEAH JH, COKOL M, PRICE EV, COLETTI ME, JONES V, BODYCOMBE NE, SOULE CK, GOULD J, ALEXANDER B, LI A, MONTGOMERY P, WAWER MJ, KURU N, KOTZ JD, HON CS, MUNOZ B, LIEFELD T, DANCIK V, BITTKER JA, PALMER, [Google Scholar]
- M., BRADNER, J. E., SHAMJI, A. F., CLEMONS, P. A. & SCHREIBER, S. L. 2015. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov, 5, 1210–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZKLARCZYK D, FRANCESCHINI A, KUHN M, SIMONOVIC M, ROTH A, MINGUEZ P, DOERKS T, STARK M, MULLER J, BORK P, JENSEN LJ & VON MERING C. 2011. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res, 39, D561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VADEN RM, OSWALD NW, POTTS MB, MACMILLAN JB & WHITE MA 2017. FUSION-Guided Hypothesis Development Leads to the Identification of N(6),N(6)-Dimethyladenosine, a Marine-Derived AKT Pathway Inhibitor. Mar Drugs, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WITKIEWICZ AK, MCMILLAN EA, BALAJI U, BAEK G, LIN WC, MANSOUR J, MOLLAEE M, WAGNER KU, KODURU P, YOPP A, CHOTI MA, YEO CJ, MCCUE P, WHITE MA & KNUDSEN ES 2015. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun, 6, 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Q, JIANG Y, MILLER MJ, PENG B, LIU L, SODERLAND C, TANG J, KERN TS, PINTAR J. & STEINLE JJ 2013. IGFBP-3 and TNF-alpha regulate retinal endothelial cell apoptosis. Invest Ophthalmol Vis Sci, 54, 5376–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Q. & STEINLE JJ 2014. IGFBP-3 inhibits TNF-alpha production and TNFR-2 signaling to protect against retinal endothelial cell apoptosis. Microvasc Res, 95, 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHONG R, KIM J, KIM HS, KIM M, LUM L, LEVINE B, XIAO G, WHITE MA & XIE Y. 2014. Computational detection and suppression of sequence-specific off-target phenotypes from whole genome RNAi screens. Nucleic Acids Res, 42, 8214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets generated in this study and all corresponding analyses are included as supplementary datasets and via a queryable website (http://fusion.yuhs.ac). Source code will be available upon request by the lead contact.
Additional Resources
All normalized data has been made available as a queryable website (http://fusion.yuhs.ac)