

Abstract
In this study, we evaluate the impact of whole genome and transcriptome analysis (WGTA) on predictive molecular profiling and histologic diagnosis in a cohort of advanced malignancies. WGTA was used to generate reports including molecular alterations and site/tissue of origin prediction. Two reviewers analyzed genomic reports, clinical history, and tumor pathology. We used National Comprehensive Cancer Network (NCCN) consensus guidelines, Food and Drug Administration (FDA) approvals, and provincially reimbursed treatments to define genomic biomarkers associated with approved targeted therapeutic options (TTOs). Tumor tissue/site of origin was reassessed for most cases using genomic analysis, including a machine learning algorithm (Supervised Cancer Origin Prediction Using Expression [SCOPE]) trained on The Cancer Genome Atlas data. WGTA was performed on 652 cases, including a range of primary tumor types/tumor sites and 15 malignant tumors of uncertain histogenesis (MTUH). At the time WGTA was performed, alterations associated with an approved TTO were identified in 39 (6%) cases; 3 of these were not identified through routine pathology workup. In seven (1%) cases, the pathology workup either failed, was not performed, or gave a different result from the WGTA. Approved TTOs identified by WGTA increased to 103 (16%) when applying 2021 guidelines. The histopathologic diagnosis was reviewed in 389 cases and agreed with the diagnostic consensus after WGTA in 94% of non‐MTUH cases (n = 374). The remainder included situations where the morphologic diagnosis was changed based on WGTA and clinical data (0.5%), or where the WGTA was non‐contributory (5%). The 15 MTUH were all diagnosed as specific tumor types by WGTA. Tumor board reviews including WGTA agreed with almost all initial predictive molecular profile and histopathologic diagnoses. WGTA was a powerful tool to assign site/tissue of origin in MTUH. Current efforts focus on improving therapeutic predictive power and decreasing cost to enhance use of WGTA data as a routine clinical test.
Keywords: biomarker, diagnostic, WGTA, pathology, precision medicine, oncology, cancer of unknown primary, machine learning
Introduction
The development of next‐generation sequencing (NGS) followed by its integration into clinical oncology form the foundation of precision oncology. The profound role that NGS has had in expanding our knowledge of the genetics underpinning and driving oncogenesis is not disputed. Likewise, comprehensive whole genome and transcriptome analysis (WGTA) approaches remain the dominant mechanism by which new clinically relevant biomarkers and drug sensitivities can be identified in patient subgroups. Here, we investigate how this technology is used in current pathology practice, such as in the sequencing of targeted gene panels as well as whole exome, genome, and transcriptome sequencing. As the price of sequencing decreases, healthcare institutions offer a varied selection of genetic testing to help manage oncology patients, but the precise role of more comprehensive tumor somatic genetic testing remains uncertain. Panel sequencing including a collection of actionable genes is often preferred, in selected patients or all patients depending on institutions, as it is relatively rapid and inexpensive and yields data specifically focused on actionable and informative targets linked to approved therapies. Whole exome or genome sequencing is now sometimes considered, especially in malignancies with no known driver, as a relatively unbiased approach to genetic characterization. Whole transcriptome sequencing is increasingly relevant as a gold standard to assess genetic rearrangements such as translocations, while also providing information on gene expression levels. Genome and transcriptome‐wide testing presents a much broader scope of information compared to panel sequencing, and is now even being offered commercially. However, clinicians' understanding of how to use the detailed data from either approach is still evolving. These molecular advancements have the potential to have a huge impact on the practice of anatomical pathologists, whose expertise is centered on making accurate and clinically relevant diagnosis, as well as selecting and interpreting the most appropriate battery of prognostic and predictive biomarkers.
While advanced molecular techniques are being adopted for clinical trials focused on precision oncology, the interpretation of these assays is an ongoing challenge. Recently published clinical trials that utilized sequencing data show that, while these approaches can provide a hitherto unmatched level of insight into the mechanisms driving metastatic cancers, the lack of availability of novel targeted therapies and of relevant clinical trials evaluating effectiveness means that the clinical benefit of NGS is often limited [1, 2, 3]. However, some evidence also indicates the benefits of treating using off‐label drugs based on molecular profiling in hard‐to‐treat cancers [4]. Most common targetable molecular alterations that are supported by high levels of evidence, such as systematic reviews and randomized controlled trials, can be assessed using immunohistochemistry (IHC) or small gene panels in routine practice [5, 6]. The remaining targets identified in broader sequencing approaches, based on the available targeted therapies in 2021, usually involve off‐label or repurposed drug use, for which evidence of level 1 therapeutic efficacy is lacking or under investigation [7, 8]. No studies to date have compared the detection rate of biomarkers associated with institutionally approved or reimbursed therapies, comparing WGTA and conventional histopathology workup.
We reviewed a series of cases in which whole genome and transcriptome sequencing was performed as part of BC Cancer's Personalized Oncogenomic (POG) study in Canada between 2012 and 2019, along with routine molecular profiling and tumor histopathology. Our goal was to evaluate the impact of integrating WGTA with pathological analysis and how these data informed the pathologic diagnosis and could identify cases eligible for treatment with institutionally approved and/or reimbursed targeted therapeutic options (TTOs).
Materials and methods
Consent and institutional review board process
This research project was approved by the BC Cancer Research Ethics Board (protocols H14‐00681 and H12‐00137). Cancer patients with advanced disease, the majority of whom had failed conventional treatment and/or did not have known cancer drivers by conventional testing, and fulfilled the inclusion criteria, were consented, by providing a written informed consent, for tumor profiling using RNA‐Seq (tumor) as well as whole genome sequencing (tumor and blood) (Clinicaltrials.gov ID: NCT02155621).
Tissue biopsy and processing
A fresh tissue biopsy was mandatory for all patients who participated in the study. Samples were taken from metastatic or recurrent tumors primarily under imaging guidance. The samples were snap‐frozen and anchored in a small amount of optimal‐cutting‐temperature (OCT) compound for cryosectioning. Those samples were used for DNA and RNA extraction as well as frozen sections for histologic correlation. Matching normal DNA was extracted from peripheral blood leukocytes. The snap‐frozen tissue specimens, either from needle core biopsies or surgical resections, were cryosectioned at 50 μm for nucleic acid and protein extraction and 5 μm for hematoxylin–eosin staining every 200 μm. Cases were excluded if tumor content was less than 40% by pathology review. The intervening sections were placed into RNAse‐free Eppendorf tubes. Only a small amount of OCT compound was used to bind the tissue to the chuck of the cryostat as OCT is known to inhibit downstream extraction and PCR steps [9, 10].
Library construction and NGS
Paired‐end DNA and RNA sequencing libraries were generated at Canada's Michael Smith Genome Sciences Centre, and sequenced on Illumina platforms (San Diego, CA, USA): HiSeq 2500 using V3 or V4 chemistry and paired‐end 125 base reads, or HiSeqX using v2.5 chemistry and paired‐end 150 base reads of 125 bp or 150 bp paired‐end reads. Average coverage for WGS was 80–100× on frozen tumor tissue DNA and 30–40× on germline DNA from blood. A minimum of 200× coverage was required for targeted amplicon reads.
cDNA libraries were prepared from biopsy samples using strand‐specific RNA‐Seq Sample Preparation kit (stranded, polyA+) from Illumina. RNA sequencing was performed on the Illumina HiSeq 2500 platform or the NextSeq500 using v2 chemistry, targeting a minimum of 200 million paired‐end reads per sample.
RNA expression analysis
RNA‐Seq data were analyzed using JAGuar v2.0.3 [11] and subsequently processed using previously published Genome Sciences Centre pipelines to yield exon‐ and transcript‐level read counts and RPKM (Reads Per Kilobase of transcript per Million mapped reads) values based on Ensembl 69 gene models [11]. Gene‐level RPKM values were calculated using a collapsed gene model.
Fold change for each gene was calculated by dividing each gene's RPKM value against an average of the RPKM values for the gene in a compendium of adjacent normal tissue samples from the Illumina Human BodyMap 2.0 project. A percentile ranking of the RPKM of each gene against reference datasets from tumors of The Cancer Genome Atlas (TCGA, https://tcga-data.nci.nih.gov/tcga/) [12] was used to identify genes with aberrant expression and to prioritize genes of interest.
The expression correlation analysis was two‐pronged – preliminary expression correlation analysis for tumor typing was undertaken relative to the entire set of normal and tumor transcriptomes in TCGA. Two‐way analysis of variance was used to identify genes that distinguished each pair of TCGA tumor types. This resulted in a set of 3,000 genes that were the most informative in explaining patterns of variance amongst all TCGA tumor types. A spearman correlation was calculated for this set of genes from the tumor sample against each TCGA sample. These pairwise correlations were clustered by the disease status (tumor or normal) and cancer type of the TCGA samples. The cancer set with the highest median correlation was determined to be representative of the closest cancer type for the sample. In addition, the Supervised Cancer Origin Prediction Using Expression (SCOPE, version 1.0 at https://github.com/jasgrewal/cancerscope [13]) algorithm was used to obtain pan‐cancer classification scores to determine cancer type of origin.
Genome analysis
WGS normal and tumor data for each patient were aligned to the GRCh37 reference human genome using BWA v0.5.7 (v0.5.7 for up to 125 bp reads and v0.7.6a for 150 bp reads). [14] Duplicate reads were marked using Picard (v1.38, https://github.com/broadinstitute/picard). The tumor and normal WGS samples were compared to identify somatic events. Somatic single‐nucleotide variants (SNVs) were called using SAMtools (v0.1.17), Strelka v1.0.6, and MutationSeq v1.0.2 [15, 16, 17]. Strelka v0.4.62 was also used to call small insertions and deletions. The somatic variant annotation was completed with snpEff 3.2 based on Ensembl gene models (v69), COSMIC v64, and dbSNP v137. Loss of heterozygosity (LOH) events were determined with APOLLOH v0.1.1 [18, 19]. Somatic copy number variants were identified using CNAseq v0.0.6 (https://www.bcgsc.ca/platform/bioinfo/software/cnaseq). Tumor content (purity) and estimated average ploidy were determined by manual review of copy number and LOH data. Amplifications were defined as regions with copy number of more than twice the average tumor ploidy. Structural variants and gene fusions were identified with ABySS and Trans‐ABySS [20, 21, 22].
Microbial and viral integration detection analysis was done using an in‐house pipeline and BioBloomTools [23]. Microsatellite instability (MSI) detection was based on MSIsensor (v0.2) [24]. Mutation signatures were classified using a non‐negative least squares deconvolution based on COSMIC v2 mutation signatures (https://cancer.sanger.ac.uk/cosmic/signatures_v2), computed from SNVs called by Strelka using a previously described approach [25].
Determination of tumor type
Each case was first reviewed by the patient's primary oncologist who provided specific questions and relevant clinical information to the bioinformatic team. The genomic and transcriptomic findings, combined with specific clinical observations and questions, guided a literature search to identify prognostic and/or predictive evidence surrounding the patient's WGTA profile. Soon after, during weekly tumor board sessions involving a multidisciplinary group including pathologists, medical oncologists, bioinformaticians, and computational biologists as part of the WGTA project, data for each case were compiled from (1) gene expression, (2) whole genome analysis, (3) histomorphologic assessment, and (4) clinical background. The consensus emerging from all four evidence sources was defined as the final diagnosis, which was retrospectively compared to the initial diagnosis rendered by the pathologist reliant on histomorphology and clinical assessment. The individual processes for data assessment are described in detail below:
Expression correlation analysis for tumor typing was undertaken relative to the entire set of normal and tumor transcriptomes in TCGA as well as the Illumina Body Map 2.0.
The whole genome analysis identified somatic and germline variants of interest. These variants could either be indicative of treatment response or resistance or, in some cases, provide additional information regarding the cancer diagnosis. These mutation‐based evidence points for treatment management and/or diagnosis would arise from the literature review performed by computational biologists as part of the analytic pipeline for each case.
The histomorphology assessment was gathered from previous pathology reports including diagnostic biopsies and/or resection specimens. All cases were reviewed centrally by an expert specialty practice pathologist prior to being presented to the multidisciplinary group.
Finally, relevant clinical history and imaging was presented by the treating medical oncologist for each case. Of note, this information was provided to the genome analysts/bioinformaticians and pathologists at least 4 weeks prior to the discussion of the case in order to facilitate any relevant clinical interpretation.
Overall, a combination of whole genome analysis, mutational burden, machine learning driver gene expression analysis, gene expression‐based pathway analysis, morphologic report, IHC, imaging, and other clinical metrics was used to suggest a tumor type/site of origin to the oncology clinical team. This final diagnosis was used in our study for comparison against the initial pathology diagnosis as well as the machine learning‐based algorithm result. The cases were labeled as malignant tumor of uncertain histogenesis (MTUH) when a specific diagnosis, including site and tissue type, could not be rendered after the pathology workup of tumor tissue (from biopsy or resection specimen). We recognize that the clinical oncology literature refers to tumors of unknown origin as primary of unknown origin or carcinoma of unknown primary (CUP), but we use the MTUH terminology to help clarify that the types of tissues within a given organ site are varied and crucial in understanding the pathogenesis of a malignancy. SCOPE, a previously validated whole transcriptome‐based pan‐cancer method powered by a neural network algorithm, was also used to predict cancer type of origin in retrospective analysis based on gene expression [13].
Review of the biomarkers detected by WGTA and the molecular pathology workup
The WGTA and molecular pathology reports were reviewed for all cases (n = 652) looking for biomarkers associated with TTO either approved by the National Comprehensive Cancer Network (NCCN) (categories 1, 2A, and 2B), Food and Drug Administration (FDA), or Health Canada. Since the study is retrospective, we compared the first date of approval/recommendation to the date when the case was reviewed at the tumor board meeting to determine if the biomarker and associated TTO were approved at the time of WGTA. We also reviewed mutations associated with treatment resistance, based on NCCN consensus, because although not associated with TTO they were likely to have an impact on management.
Review of the histopathologic report and comparison to SCOPE prediction of site and/or tissue of origin
We then reviewed the pathology report diagnosis of cases (n = 389) based on availability and the presence of a TCGA comparator for the same tumor type. The latter was important because we required the TCGA gene expression data as a comparator for a given tumor type to formulate WGTA‐based diagnostic prediction using SCOPE. When an alternate site and/or tissue of origin was suggested at the tumor board, orthogonal testing using IHC or Sanger sequencing was used to validate the WGTA findings. We then compared the final diagnosis given at the time of the analysis of each case, during tumor boards, to the pathology diagnosis just prior to the meeting (see section ‘Determination of tumor type’).
Results
Cohort demographics, clinical metrics, and sequencing data
WGTA was performed on a cohort of 652 unique cases with advanced cancer sequenced between 2012 and 2019. Patients were 36% males and 64% females, and the 5‐year survival was 45% with a median survival of 3 years. The cohort selection process and tumor types breakdown are summarized in Figure 1.
Figure 1.
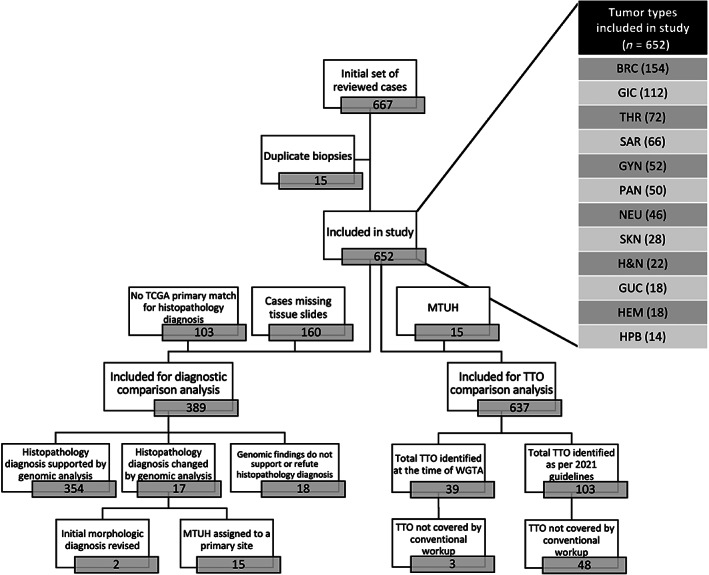
Cohort selection flowchart and tumor types breakdown. BRC, breast carcinoma; GIC, gastrointestinal carcinoma; THR, thoracic carcinoma; SAR, bone and soft tissue sarcomas; GYN, gynecologic carcinoma; PAN, pancreatic carcinoma; NEU, central neural system neoplasm; SKN, cutaneous malignancy; H&N, head and neck carcinoma; GUC, genitourinary carcinoma; HEM, hematologic malignancy; HPB, hepatobiliary carcinoma.
Conventional molecular pathology workup and WGTA showed comparable detection rates for biomarkers associated with approved TTO
Among non‐MTUH (n = 637), WGTA identified biomarkers associated with approved TTO in 39 (6%) cases (2 cases had two TTO) at the time of the analysis (2012–2019, Figure 2 and supplementary material, Table S1). These included 13 ERBB2 amplifications (11 breast carcinomas and 2 colorectal carcinomas), 9 EGFR oncogenic mutations and 8 ALK rearrangements in lung adenocarcinomas, 5 KIT oncogenic mutations in gastrointestinal stromal tumor (GIST), 3 cases with MSI (lung and pancreatic carcinomas), 2 BRAF p.V600E (c.1799T>A) mutations in melanomas, and 1 BRCA2 mutation in a breast carcinoma (c.667C>G, p.H223D). Of these alterations, three (0.4%) were not included in the routine molecular pathology workup at the time of the initial analysis (ERBB2 in two colorectal adenocarcinomas, and MSI in a squamous cell carcinoma of the lung) (Figure 3). In four (0.6%) cases, the predictive biomarker testing from the initial pathology workup was different to the WGTA results (ERBB2 amplifications), and in three (0.4%) cases the initial testing failed (ALK fusion and EGFR p.L858R [c.2573T>G] mutations).
Figure 2.
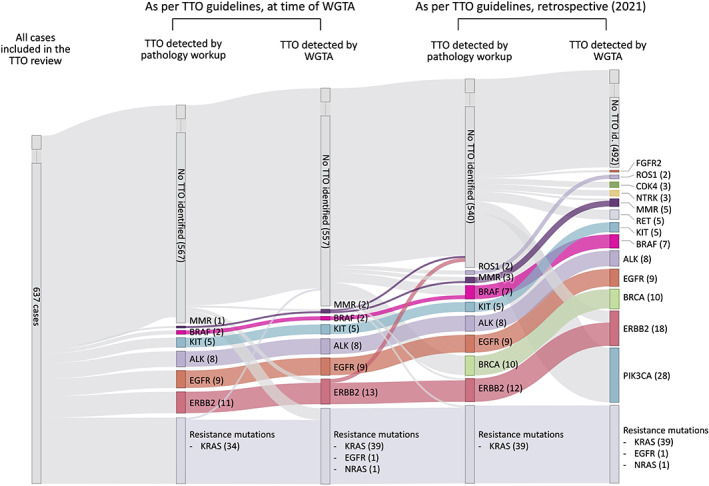
Longitudinal progression of the number of TTO as per guidelines from two different time periods and using two different testing approaches: conventional pathology workup and WGTA (N = 637).
Figure 3.
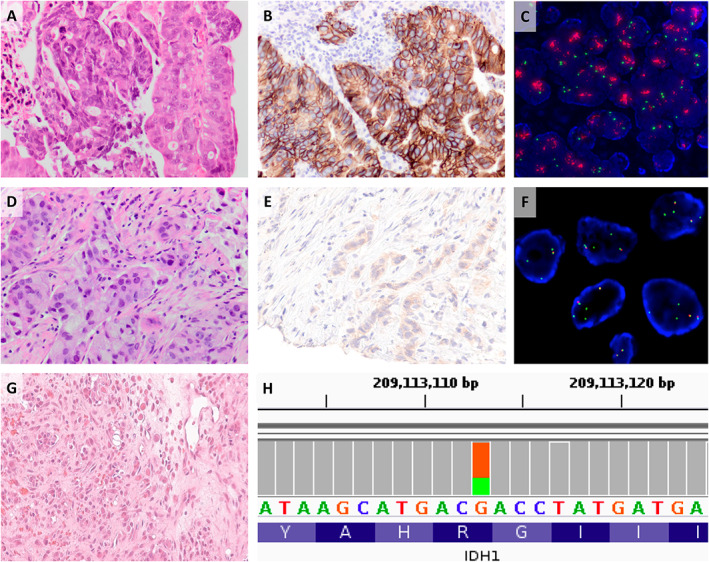
Detection of clinically significant molecular alterations by WGTA. (A–C) Detection of an incidental HER2 amplification in a CRC. (D–F) ALK fusion in NSCLC, missed on FISH analysis. (G, H) Detection of an IDH1 mutation in a MUTH supported a diagnosis of cholangiocarcinoma.
When reviewing those same cases according to the current NCCN biomarker guidelines, the number of cases with alterations associated with a TTO increased to 103 as new actionable targets are approved (PIK3CA and BRCA1/2 oncogenic mutations; ROS1, RET, NTRK1/2, and FGFR2 rearrangement; and CDK4 amplification), or previous targets are approved for use in different tumor sites (ERBB2 in colorectal and lung adenocarcinoma; BRAF in colorectal adenocarcinoma; and MSI in pancreatic, colorectal, and lung carcinomas). Of those 103 cases, 48 (47%) would not have been included in the current molecular pathology workup (1 case had two TTOs), including 29 cases of HER2‐negative and ER/PR‐positive breast carcinomas with PIK3CA oncogenic mutation (60%), 6 ERBB2 amplification lung and colorectal carcinomas (13%), 5 RET fusions in medullary thyroid carcinomas and lung adenocarcinomas (10%), 3 CDK4 amplifications in dedifferentiated liposarcomas (6%), 3 NTRK1/2 fusions (papillary thyroid carcinomas, mammary carcinoma, and glioblastoma multiforme) (6%), MSI in 2 lung carcinomas (4%), and 1 cholangiocarcinoma with a FGFR fusion (2%) (supplementary material, Table S1).
Genomic alterations associated with a resistant phenotype were identified in 41 cases; 39 oncogenic KRAS mutations and 1 oncogenic NRAS mutation in colorectal adenocarcinomas, and an exon 20 insertion in a lung carcinoma. Of these KRAS mutations, 34 (83%) were tested as part of the conventional pathology workup, with one of these testing attempts having failed. The remainder were not tested even though they would have been expected to be part of routine workup at the time of WGTA (5 KRAS mutations). In addition, two abnormalities were not identified as they were not part of testing panel (one NRAS mutation and EGFR exon 20 insertion) at the time.
Within the group of 15 MTUH, a case where the histopathologic diagnosis was revised to cholangiocarcinoma was shown to have a FGFR2 fusion, and another case, revised to colorectal adenocarcinoma, had a KRAS oncogenic mutation.
Most pathologic diagnoses were supported by the WGTA and clinical analysis
Of the 389 cases where the initial pathology diagnosis was compared to the diagnostic consensus after the WGTA directed tumor board, the integrated review of whole genome analysis, gene expression correlation to TCGA tumor types, and clinicopathologic reports agreed with the original pathologic diagnosis in 94% of cases (n = 374), excluding 15 MTUH (Figure 4).
Figure 4.
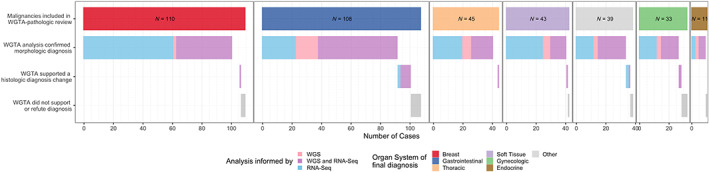
Analysis comparing the initial pathology diagnosis to the diagnostic consensus delivered after WGTA review.
In two cases (0.5% of total), the original pathology report diagnosis was found to be incorrect after molecular analysis and review. One case was initially diagnosed as a vulvar adenocarcinoma not otherwise specified (NOS), but gene expression analysis matched closely with the profile of breast ductal adenocarcinomas. This triggered a pathologic review and the diagnosis was changed to an HER2‐amplified mammary‐like adenocarcinoma of the vulva [12]. As a result, the patient was treated with an ERBB2 inhibitor. The second case was initially diagnosed as adenocarcinoma of likely ovarian origin, but comprehensive gene expression and mutational profiles analysis including high expression of HNF1β, NAPSA (Napsin A), and GPC3 (Glypican‐3); an inactivating mutation in ARID1A; and copy gains in HNF1β and ERBB2 genes supported the diagnosis of ovarian clear cell carcinoma.
Finally, in 18 cases (5%), WGTA was non‐contributory, and neither supported nor refuted the initial pathological diagnosis. These tumors were often pancreatobiliary malignancies (6/18, 33%) and uterine carcinosarcomas (3/18, 17%). Tumor content (ranging from 25 to 90%), biopsy site, and patient characteristics were not significantly different from the rest of the cohort.
Comprehensive WGTA identified a cell lineage in all MTUH
In 15 cases, the initial clinicopathologic workup could not confidently assign a tumor site or type. Nine were initially diagnosed as adenocarcinoma of unknown origin, two as squamous cell carcinoma, three as carcinoma, and one as an unclassifiable malignancy. We identified a likely cell lineage for all 15 MTUH using the WGTA and clinical history. The post‐WGTA diagnoses included cholangiocarcinoma (n = 3), esophageal squamous cell carcinoma (n = 2), colorectal adenocarcinoma (n = 2), papillary renal cell carcinoma (n = 1), pancreatic adenocarcinoma (n = 1), lung adenocarcinoma (n = 1), Ewing sarcoma (n = 1), bladder adenocarcinoma (n = 1), high‐grade serous carcinoma of the ovary (n = 1), salivary gland adenocarcinoma NOS (n = 1), and anal squamous cell carcinoma (n = 1). RNA‐Seq alone was sufficient to establish a diagnosis in 4/15 cases. In the remaining 11/15 cases, the diagnosis was jointly determined using RNA‐Seq and whole genome analysis. Pathology reports including IHC results and clinical context were also reviewed for each case. In all 15 cases, the new diagnosis was incorporated in management by the treating team to plan additional testing, treatment, and counsel the patients on their disease.
Automated machine learning‐based RNA‐Seq analysis can confirm diagnoses except in low tumor cellularity liver biopsies
In addition to the combined review of RNA‐Seq, genome mutation, and clinicopathologic data, we also explored the use of an automated machine learning approach for predicting diagnosis from RNA‐Seq data. The SCOPE algorithm was used to assess the potential of automated tools in aligning diagnoses from RNA‐Seq data in precision oncology workflows [13]. SCOPE matched the predicted final diagnosis in 273 of the 374 cases (73%). When looking at tumor types with 10 or more cases, the SCOPE algorithm had the highest rate of success with breast carcinoma (91% accuracy, n = 98/108), ovarian carcinoma (81% accuracy, n = 17/21), and lung adenocarcinoma (76% accuracy, n = 28/37) as opposed to pancreatic adenocarcinoma (25% accuracy, n = 5/20), which were missed most often by the method (see supplementary material, Table S2 for classifier statistics by cancer type). Among the low‐success tumor types (tumor type accuracy <40%), we found that the mispredictions mostly resulted from the identification of a histologically similar cancer (36% of mispredictions, n = 14/39); 20 of the 39 mispredictions originated from liver biopsies, leading us to investigate the impact of biopsy site and tumor content accurate diagnosis prediction from RNA‐Seq (supplementary material, Figure S1). We observed a significant association between biopsy site and SCOPE outcome (p = 0.008, chi‐square test, N = 282/374 only biopsy sites with a minimum of 10 samples considered). In biopsy sites with at least 10 samples, tumor content was found to be significantly associated with SCOPE prediction in liver biopsies only (N = 124/282, p adjusted = 0.017, unpaired t‐test), where predictions were biased toward hepatobiliary and gastrointestinal malignancies (Figure 5 and supplementary material, Figure S2). When considered independent of the biopsy site, SCOPE's predictions were not found to be influenced by tumor content, suggesting that it is the distinct expression profiles contributed by normal cells from the metastatic site, rather than the proportion of normal cells, which is the greatest confounding factor. Examining MTUH cases, SCOPE's predictions were accurate in matching the final revised diagnosis in 8/15 cases, and in 3/15 matched the revised diagnosis confidently when accounting for biopsy site bias.
Figure 5.
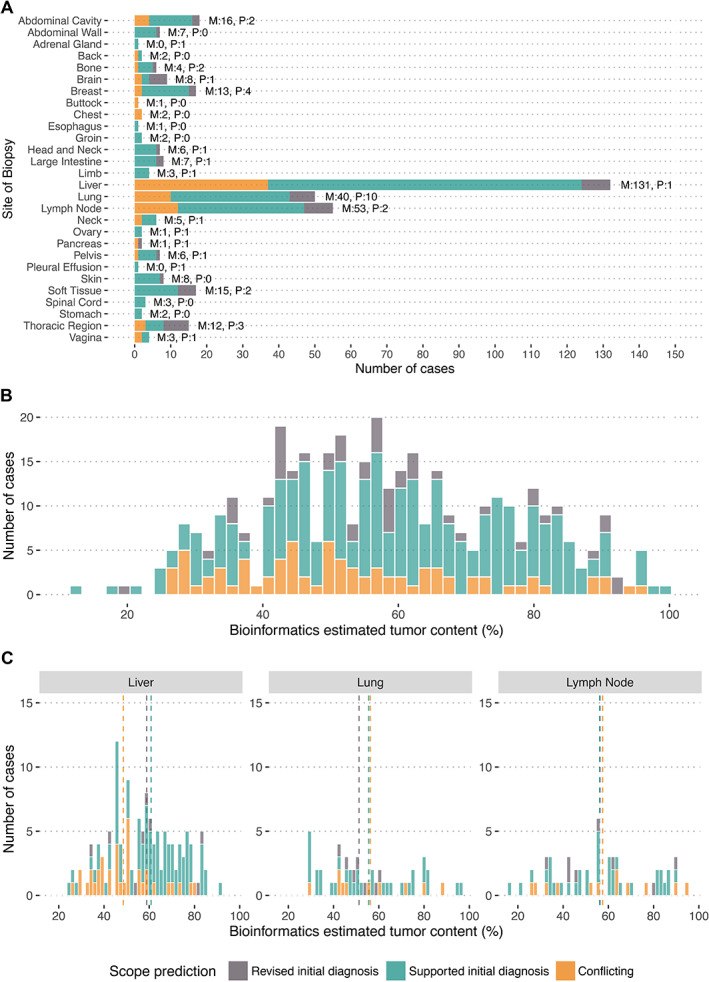
Impact of biopsy site and tumor content on the ability of an automated RNA‐Seq based classifier (SCOPE) to provide the correct putative diagnosis in the POG cohort. (A) The outcome from SCOPE is shown separated by the site of biopsy of the tumor. M and P indicate the number of metastatic and primary/relapse samples, respectively. (B) The distribution of cases across all biopsy sites is shown as a function of tumor content. (C) The majority of samples arose from three biopsy sites, lymph node, lung, and liver, indicated in each of the panels. Liver biopsies with low tumor content led to the highest number of conflicting (incorrect) assessments from SCOPE. A statistically significant association was found between SCOPE misprediction in liver biopsies and tumor content (p < 0.001, Wilcox test, not shown in figure).
Discussion
We present data from a cohort study where WGTA was performed to characterize a heterogeneous population of advanced malignancies in a Canadian academic center. In our selected cohort, WGTA identified predictive biomarkers that were associated with TTOs approved by major health institutions. Over the span of the study, the number of TTO‐associated biomarkers significantly increased secondary to rapidly evolving recommendation guidelines. The pathologic diagnosis was supported by WGTA in most cases with a previously assigned tumor type, and the analysis proved effective to assign cell lineage in MTUH cases (cases without a pathology‐assigned tissue of origin).
The number of predictive biomarkers identified from WGTA is progressing over time but is still limited by the availability of approved TTOs
Over the span of the study, the number of biomarkers associated with approved TTOs almost tripled. Many of these emerging biomarkers were not accounted for in our local panel at the time of writing this manuscript. Our results show that, so far, targeted panels could adjust quickly to the rapid progression of predictive biomarkers; however, there might come a point where WGTA becomes more efficient than the current approach. For example, predictive assays such as homologous recombinant deficiency score, mutational signatures, and precise mutational burden analysis are mostly unique to WGTA and have shown potential to help clinical management. Those were not taken into account in our study as they are not yet part of the clinical standard. In the coming years, as these metrics are tested for clinical use in clinical trials, they may emerge as clearly superior to current clinical tests. In that scenario, WGTA would enter routine clinical practice.
WGTA had the potential to improve therapeutic options over the conventional pathology workup, which included histomorphology and low‐plex molecular testing, in 0.9% of cases. In some situations, the biomarkers were absent from the routine molecular pathology workup at the time of WGTA, while in others it was missed because of technical failures in the conventional assays. By identifying potentially actionable targets, WGTA results may have facilitated enrollment into clinical trials in additional cases but, as guidance of management is currently approved for only a few predictive biomarkers, it had little influence on reimbursed or institutionally recommended therapies. Results from the SHIVA trial in 2015 showed that off‐label management guided by large gene panel results did not improve patient outcome over standard treatment [26]. A more recent precision oncology trial based on WGTA concluded that it was only beneficial in a minor subgroup of patients for whom therapeutic options were depleted [27]. Outcome analysis and assessment of off‐label treatment options were outside the scope this analysis. At this time, the lack of treatment options endorsed by major health institutions, even within a broad clinical trial context, is a key obstacle to better understand how WGTA might be useful to guide therapeutic management and understand resistance in routine oncology care.
WGTA helps in the investigation of MTUH
Gene expression profiling as a means to classify MTUH was demonstrated to be feasible by several studies in the early 2000s [28, 29, 30, 31, 32]. Although some studies suggested outcome benefits and prognostic use of using gene expression panels to guide management, the only available randomized clinical trial did not show benefit, and the ESMO clinical guideline for CUP diagnosis and treatment does not recommend gene expression profiling [33, 34, 35, 36]. The earlier studies agree that gene expression profiles were useful prognostic markers [37]. The microarrays used in these studies did not, however, have the same coverage as RNA‐Seq and lacked mutational analysis. A recent analysis of 200 CUPs using a sequencing panel including 236 cancer genes showed high rate of clinically relevant genomic alterations, some of which had treatment implications [38]. In our series, WGTA identified a tissue of origin in all our MTUH cases and identified TTO or drug‐resistant phenotype in some. Our data show that the combined expression and mutational analysis added value for most MTUH cases. It now remains to be demonstrated whether this information can lead to improved patient outcome in this treatment‐resistant group of patients. It is important to note that, due to the often poorly differentiated and advanced nature of these cases, there were no gold standard diagnoses and, although post‐WGTA testing could be done, some uncertainty about the tissue of origin could still remain after WGTA.
Machine learning approaches hold promise for assisting in the interpretation of complex genomic data. Applying machine learning to cancer diagnosis based on RNA‐Seq data showed some success in correctly identifying tumor types in our cohort but was hindered by the combined presence of normal tissue from the metastatic biopsy site. Further innovations such as single‐cell sequencing may resolve this issue by being able to separately profile each cell, and then combine data from those cells predicted to be cancerous. Currently, in practice, the effect of possible tissue contamination could be accounted for during case discussion at molecular tumor board meetings in which molecular data are reviewed along with the clinical and pathology context. In some tumor types, such as the pancreatobiliary malignancies and the uterine carcinosarcomas, the SCOPE algorithm often failed to identify a pathologic diagnosis. In the former group, the abundant liver tissue contamination may have biased the analysis, similar to low cellularity liver metastasis while, in the case of carcinosarcomas, the biphasic nature of the malignant tissue and the varying ratio of each component within each sample have likely limited the analysis. More research is needed to investigate the predictive potential of algorithms derived from machine learning, and genome and transcriptome‐wide data will be crucial in training as this field evolves.
Several limitations affect the interpretation of our results, such as the heterogeneity of our cohort. Our cohort study was highly selected based on patient eligibility and enrollment (strong preference was given to cases with no known driver alterations), and included mostly breast, gastrointestinal, thoracic, and gynecologic malignancies, while underrepresenting lymphoid, head and neck, central nervous system, endocrine, and genitourinary tumors. This study focuses on the molecular and diagnostic pathology workup, including diagnosis and molecular profiling, and as such was not designed to investigate an outcome effect. Finally, we had no control group to assess the effectiveness of changes in histologic diagnosis or molecular subtype.
Conclusion
Our experience with WGTA as part of this project highlighted its utility for identifying the site of origin in MTUH, along with the complexity of the interpretation of its data, especially in metastatic cancers where expression profiles are influenced by the site of sampling. However, the technology is limited within the scope of institutionally supported TTO‐associated biomarkers by the few validated actionable genomic and transcriptomic targets. There has been a marked increase in the number of predictive biomarkers over the course of this study, some of which need to be integrated as part of the routine molecular pathology workup in our center. The future of precision oncology testing is still unclear and may see a transition to more comprehensive sequencing approaches as the variety of predictive biomarkers progresses, but the current standard is robust and adaptable. We acknowledge that cancer remains an often fatal disease and our understanding of the precise mechanisms of oncogenesis and the response to therapeutics is far from complete. For forward‐looking, research‐centric organizations, WGTA represents a powerful modality to discover new clinically relevant biomarkers, drug sensitivities, and novel therapeutic axes.
Author contributions statement
BT‐C designed the study. BT‐C, JKG, MRJ, EP, YS, EC, THL, DDWT, LW and EZ collected the data. BT‐C, CD, LH, BH, DI, AFL, CHL, TN, DFS, BSS, BS, TS, CBG and SY carried out pathology review. BT‐C, JKG, MRJ and EP analyzed the data. BT‐C and JKG designed and generated the figures. BT‐C and JKG wrote the original draft of the manuscript. EP, DGH, ANK, DDWT, AJM, KM, JRN, BSS, DAR, JL, MAM, CBG, SJMJ and SY revised the manuscript.
Supporting information
Figure S1. Impact of cancer type on the ability of an automated RNA‐Seq based classifier (SCOPE) to provide the correct putative diagnosis in the POG cohort
Figure S2. Impact of tumor content on the ability of an automated RNA‐Seq based diagnostic (SCOPE) to provide the correct putative diagnosis in the POG cohort
Table S1. List of actionable mutations detected by WGTA
Table S2. Performance statistics for accurate diagnosis from the automated RNA‐Seq based cancer classifier (SCOPE)
Acknowledgements
This work would not be possible without the participation of our patients and families, the POG team, Canada's Michael Smith Genome Sciences Centre technical platforms, the support of the BC Cancer Foundation, and their donors. We also want to acknowledge the help of Lindsay Zibrik with the submission process.
No conflicts of interest were declared.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1. Robinson DR, Wu YM, Lonigro RJ, et al. Integrative clinical genomics of metastatic cancer. Nature 2017; 548: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017; 23: 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pleasance E, Titmuss E, Williamson L, et al. Pan‐cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nat Cancer 2020; 1: 452–468. [DOI] [PubMed] [Google Scholar]
- 4. Massard C, Michiels S, Ferté C, et al. High‐throughput genomics and clinical outcome in hard‐to‐treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov 2017; 7: 586–595. [DOI] [PubMed] [Google Scholar]
- 5. Marchevsky AM, Wick MR. Evidence levels for publications in pathology and laboratory medicine. Am J Clin Pathol 2010; 133: 366–367. [DOI] [PubMed] [Google Scholar]
- 6. Merlin T, Weston A, Tooher R. Extending an evidence hierarchy to include topics other than treatment: revising the Australian 'levels of evidence'. BMC Med Res Methodol 2009; 9: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. CEBM . Levels of Evidence. 2009. [Accessed 3 February 2022]. Available from: https://www.cebm.ox.ac.uk/resources/levels-of-evidence/oxford-centre-for-evidence-based-medicine-levels-of-evidence-march-2009
- 8. Evidence‐Based Medicine Working Group . Evidence‐based medicine. A new approach to teaching the practice of medicine. JAMA 1992; 268: 2420–2425. [DOI] [PubMed] [Google Scholar]
- 9. Jamshidi F, Pleasance E, Li Y, et al. Diagnostic value of next‐generation sequencing in an unusual sphenoid tumor. Oncologist 2014; 19: 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thibodeau ML, Reisle C, Zhao E, et al. Genomic profiling of pelvic genital type leiomyosarcoma in a woman with a germline CHEK2:c.1100delC mutation and a concomitant diagnosis of metastatic invasive ductal breast carcinoma. Cold Spring Harb Mol Case Stud 2017; 3: a001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Butterfield YS, Kreitzman M, Thiessen N, et al. JAGuaR: junction alignments to genome for RNA‐seq reads. PLoS One 2014; 9: e102398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grewal JK, Eirew P, Jones M, et al. Detection and genomic characterization of a mammary‐like adenocarcinoma. Cold Spring Harb Mol Case Stud 2017; 3: a002170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grewal JK, Tessier‐Cloutier B, Jones M, et al. Application of a neural network whole transcriptome‐based pan‐cancer method for diagnosis of primary and metastatic cancers. JAMA Netw Open 2019; 2: e192597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics 2010; 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ding J, Bashashati A, Roth A, et al. Feature‐based classifiers for somatic mutation detection in tumour‐normal paired sequencing data. Bioinformatics 2012; 28: 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saunders CT, Wong WS, Swamy S, et al. Strelka: accurate somatic small‐variant calling from sequenced tumor‐normal sample pairs. Bioinformatics 2012; 28: 1811–1817. [DOI] [PubMed] [Google Scholar]
- 18. Cingolani P, Platts A, Wang le L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin) 2012; 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ha G, Roth A, Lai D, et al. Integrative analysis of genome‐wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple‐negative breast cancer. Genome Res 2012; 22: 1995–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Birol I, Jackman SD, Nielsen CB, et al. De novo transcriptome assembly with ABySS. Bioinformatics 2009; 25: 2872–2877. [DOI] [PubMed] [Google Scholar]
- 21. Robertson G, Schein J, Chiu R, et al. De novo assembly and analysis of RNA‐seq data. Nat Methods 2010; 7: 909–912. [DOI] [PubMed] [Google Scholar]
- 22. Simpson JT, Wong K, Jackman SD, et al. ABySS: a parallel assembler for short read sequence data. Genome Res 2009; 19: 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chu J, Sadeghi S, Raymond A, et al. BioBloom tools: fast, accurate and memory‐efficient host species sequence screening using bloom filters. Bioinformatics 2014; 30: 3402–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Niu B, Ye K, Zhang Q, et al. MSIsensor: microsatellite instability detection using paired tumor‐normal sequence data. Bioinformatics 2014; 30: 1015–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013; 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Le Tourneau C, Delord JP, Gonçalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open‐label, proof‐of‐concept, randomised, controlled phase 2 trial. Lancet Oncol 2015; 16: 1324–1334. [DOI] [PubMed] [Google Scholar]
- 27. Tuxen IV, Rohrberg KS, Oestrup O, et al. Copenhagen Prospective Personalized Oncology (CoPPO) – clinical utility of using molecular profiling to select patients to phase I trials. Clin Cancer Res 2019; 25: 1239–1247. [DOI] [PubMed] [Google Scholar]
- 28. Ma XJ, Patel R, Wang X, et al. Molecular classification of human cancers using a 92‐gene real‐time quantitative polymerase chain reaction assay. Arch Pathol Lab Med 2006; 130: 465–473. [DOI] [PubMed] [Google Scholar]
- 29. Ramaswamy S, Tamayo P, Rifkin R, et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci U S A 2001; 98: 15149–15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Su AI, Welsh JB, Sapinoso LM, et al. Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res 2001; 61: 7388–7393. [PubMed] [Google Scholar]
- 31. Tothill RW, Kowalczyk A, Rischin D, et al. An expression‐based site of origin diagnostic method designed for clinical application to cancer of unknown origin. Cancer Res 2005; 65: 4031–4040. [DOI] [PubMed] [Google Scholar]
- 32. Varadhachary GR, Talantov D, Raber MN, et al. Molecular profiling of carcinoma of unknown primary and correlation with clinical evaluation. J Clin Oncol 2008; 26: 4442–4448. [DOI] [PubMed] [Google Scholar]
- 33. Hayashi H, Kurata T, Takiguchi Y, et al. Randomized phase II trial comparing site‐specific treatment based on gene expression profiling with carboplatin and paclitaxel for patients with cancer of unknown primary site. J Clin Oncol 2019; 37: 570–579. [DOI] [PubMed] [Google Scholar]
- 34. Hainsworth JD, Rubin MS, Spigel DR, et al. Molecular gene expression profiling to predict the tissue of origin and direct site‐specific therapy in patients with carcinoma of unknown primary site: a prospective trial of the Sarah Cannon research institute. J Clin Oncol 2013; 31: 217–223. [DOI] [PubMed] [Google Scholar]
- 35. Moran S, Martínez‐Cardús A, Sayols S, et al. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol 2016; 17: 1386–1395. [DOI] [PubMed] [Google Scholar]
- 36. Yoon HH, Foster NR, Meyers JP, et al. Gene expression profiling identifies responsive patients with cancer of unknown primary treated with carboplatin, paclitaxel, and everolimus: NCCTG N0871 (alliance). Ann Oncol 2016; 27: 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fizazi K, Greco FA, Pavlidis N, et al. Cancers of unknown primary site: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2015; 26 (Suppl 5): v133–v138. [DOI] [PubMed] [Google Scholar]
- 38. Ross JS, Wang K, Gay L, et al. Comprehensive genomic profiling of carcinoma of unknown primary site: new routes to targeted therapies. JAMA Oncol 2015; 1: 40–49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Impact of cancer type on the ability of an automated RNA‐Seq based classifier (SCOPE) to provide the correct putative diagnosis in the POG cohort
Figure S2. Impact of tumor content on the ability of an automated RNA‐Seq based diagnostic (SCOPE) to provide the correct putative diagnosis in the POG cohort
Table S1. List of actionable mutations detected by WGTA
Table S2. Performance statistics for accurate diagnosis from the automated RNA‐Seq based cancer classifier (SCOPE)
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.