

Abstract
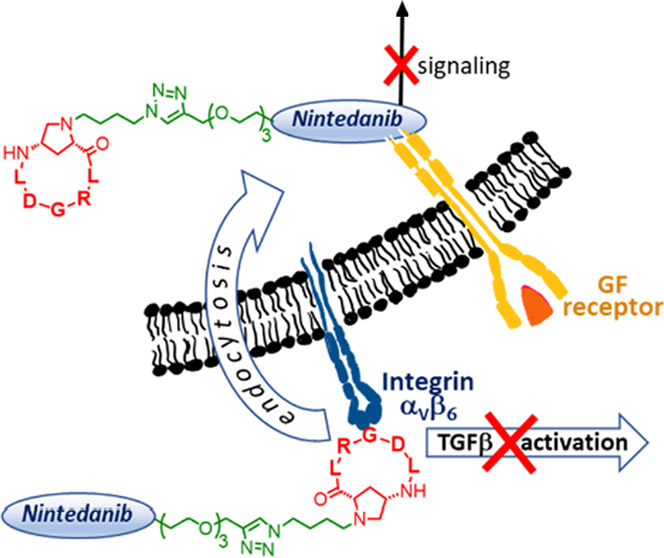
αVβ6 Integrin plays a fundamental role in the activation of transforming growth factor-β (TGF-β), the major profibrotic mediator; for this reason, αVβ6 ligands have recently been forwarded to clinical phases for the therapy of fibrotic diseases. Herein, we report the synthesis and in vitro biological evaluation as antifibrotic agents of three new covalent conjugates, constituted by c(AmpLRGDL), an αVβ6 integrin-recognizing small cyclopeptide, and nintedanib, a tyrosine kinase inhibitor approved for idiopathic pulmonary fibrosis (IPF) treatment. One of these conjugates recapitulates optimal in vitro antifibrotic properties of the two active units. The integrin ligand portion within the conjugate plays a role in inhibiting profibrotic stimuli, potentiating the nintedanib effect and favoring the selective uptake of the conjugate in cells overexpressing αVβ6 integrin. These results may open a new perspective on the development of dual conjugates in the targeted therapy of IPF.
Introduction
Fibrosis denotes an excessive deposition of collagen and other extracellular matrix (ECM) components in a tissue. Deposition of collagen is part of physiological wound healing, but when this process becomes abnormal, connective tissue replaces normal parenchyma, leading to tissue destruction and impairment of organ function. This pathologic process can affect several organs, causing diverse chronic diseases such as, for example, liver cirrhosis, systemic sclerosis, end-stage kidney disease, and idiopathic pulmonary fibrosis (IPF).1 Persistent injurious stimuli including chronic infections and immunological reactions are often triggering events that cause fibrosis disorders. The major profibrotic mediator is the transforming growth factor β (TGF-β),2,3 which is released by macrophages and stimulates the migration and proliferation of fibroblasts and myofibroblasts, as well as the deposition of collagen and other ECM proteins. An intricate network of noncovalent and covalent connections lies behind this key growth factor (Figure 1); TGF-β is expressed in a latent form (L-TGF-β)4 and remains associated by noncovalent bonds with the latency-associated peptide (LAP), forming the small latent complex (SLC), until it is activated. SLC, on the other hand, is covalently linked to another protein (the latent TGF-β binding protein, LTBP), to constitute the large latent complex (LLC).5
Figure 1.

Schematic representation of fibrotic cells carrying GF, TGF-β, and αVβ6 integrin receptors. The small molecules GSK3008348 and PLN-74809 are αVβ6 integrin antagonists that inhibit integrin binding to LAPm thus preventing the active TGF-β release and profibrotic signaling events; nintedanib inhibits the tyrosine kinase activity of GF receptors and prevents downstream intracellular signaling events.
The αV-integrins6 are major activators of L-TGF-β;7,8 in particular, αVβ6 integrins exhibit strong affinity toward the LAP-TGFβ1 complex (Kd = 10.3 nM) by recognizing the key RGD motif present on the LAP moiety.9 The way TGF-β is activated by αVβ6 integrins has been fully elucidated: upon RGD-mediated binding, the actin cytoskeleton generates the force necessary for the elongation of LAP, ultimately allowing for TGF-β release (Figure 1).10
The fact that αVβ6 integrins play such a crucial role in TGF-β activation and exhibit low levels of expression in normal tissues11 but are upregulated in pulmonary tissues of IPF patients has led them to be identified as a therapeutic target for IPF treatment.12−14 GlaxoSmithKline discovered an RGD-mimetic small molecule, GSK3008348 (Figure 1), that binds to αVβ6 integrins with a high affinity in human IPF lungs, and reduces downstream profibrotic TGF-β signaling to normal levels.15,16 This compound has been forwarded to clinical phase 1 on IPF patients and is administered via inhalation. Pliant Therapeutics has developed another small molecule, namely, PLN-74809,17 a dual ligand of both αVβ6 and αVβ1 integrins,18 and in 2020, this compound entered clinical phase 2 for IPF treatment.19 While clinical results from these small-molecule integrin antagonists are expected to have a beneficial outcome, further research in this field is urgent, given the seriousness of IPF disease, the poor prognosis, and the absence of a cure.20
Other growth factors, including platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), and vascular endothelial growth factor (VEGF), are profibrotic mediators21 that stimulate the uncontrolled proliferation of fibroblasts and differentiation of fibroblasts into myofibroblasts, with excessive deposits of ECM components. On these bases, the small molecule nintedanib (Figure 1) has been clinically approved for the treatment of IPF22 since it acts as a multityrosine kinase inhibitor capable of impairing the signaling of PDGFR, bFGFR, and VEGFR by competing with ATP for their intracellular binding sites.23
If the cell surface-exposed αVβ6 integrin on the one hand and the intracellular TK domain of the above growth factors on the other hand are promising therapeutic targets for IPF, then the possibility of tackling both targets with one dual conjugate molecule constitutes uncharted territory worth exploring.
Furthermore, it has been proved that cross-talk between growth factor receptors and αV integrins exerts a critical role in different oncological and nononcological diseases.24 Cross-talk between VEGFR2 and αVβ3 integrins, for example, plays a pivotal role in the development of solid tumors, contributing to tumor progression, metastatic dissemination, and resistance to pharmacological treatment.25,26 In the emerging field of dual-purpose molecules,27 we recently synthesized a series of dual conjugates incorporating both the αVβ3 integrin-targeting cyclopeptidomimetic c(AmpRGD) (Amp being a cis-4-amino-l-proline residue) and the known TKI sunitinib, joined together by covalent and robust linkers. One of these conjugates showed very interesting antiangiogenesis and antitumor activity in melanoma and ovarian carcinomas in vitro and in vivo, demonstrating improved efficacy compared to sunitinib alone and the combined administration of sunitinib and the integrin ligand. On that occasion, cooperation of the two active units within the conjugate was claimed to be operative, with perturbation of cross-talk between the αVβ3 integrin and the VEGFR targets.28−30
Based on these precedents in the dual-targeting strategy, we have herein designed and constructed three new covalent conjugates of general formula I (compounds 1–3, Figure 2) connecting a previously developed Amp-based αVβ6 integrin-targeting cyclohexapeptidomimetic31 namely, c(AmpLRGDL), with a nintedanib portion. These compounds are meant to concomitantly antagonize the αVβ6 integrin overexpressed in fibrotic tissue and inhibit the kinase activity of growth factors, as well as possibly perturbing cross-talk between these two receptors, with the final aim of being used as potential antifibrotic drugs.
Figure 2.

Structure of the dual conjugates 1–3 of general formula I (integrin ligand in red, nintedanib unit in blue, and linker moiety in black).
Considering the localization of the biological targets—outside the cell for the recognition domain of αVβ6 integrins and inside the cell for the TK domain of growth factors—one crucial issue consists in the putative localization of the conjugates. Ideally, after selective interaction of the conjugate with the fibrotic cell via αVβ6 integrin recognition and inhibition of TGF-β release, selective cell internalization via integrin-mediated endocytosis should follow, eventually permitting the nintedanib portion, in the intracellular space, to exert the TKI activity. The chemical synthesis of compounds 1–3 and their biological evaluation on fibroblasts in vitro are herein reported; accordingly, analogies and differences in the structure and biological behavior of the three compounds are underlined.
Results and Discussion
Design of c(AmpLRGDL)-Nintedanib Conjugates
One key step in designing a dual molecular conjugate is the identification of suitable sites on the active units for attaching the linker. These anchoring residues ought to have a negligible impact on biological activity, and therefore they must not be directly involved in the act of drug–target binding. Of course, synthetic feasibility is another indispensable requirement to be kept in mind during design.
As for the integrin ligand, we were aware of the fact that the Nα-nitrogen of the Amp residue was a good anchoring site since in several previous instances the use of similar integrin ligands bearing Nα alkyl or acyl chains of different lengths did not modify the integrin-binding ability of the ligand.28
For the nintedanib portion, we relied on the reported X-ray crystal structure of nintedanib bound to the active site of the FGFR-1 kinase domain, which reveals the N-methyl-piperazinyl moiety of nintedanib pointing outward from the receptor pocket and interacting only weakly with the receptor.23 Moreover, nintedanib analogues lacking the N-methyl-piperazinyl tail were seen to maintain the TK inhibitory activity.32 Thus, the piperazine nucleus was judged to be a good anchoring point, and the substitution of the original methyl chain for a longer alkyl chain was deemed to affect the TK inhibitory activity of the nintedanib unit only marginally.
The three designed conjugates 1–3 differ from each other in the nature of their linkers. Indeed, they all share robust triazole-based linkages, but these differ in chain length, polarity, and monomeric vs dimeric presentation of the integrin ligands. In previous works, we used similar linkers for the construction of sunitinib-carrying dual conjugates and observed how these three types of linkers could profoundly influence both integrin binding and internalization into cells.28 Repurposing such linkers in this new series of conjugates offers us the possibility of comparing the results of this study with our previous ones and elucidating better the linker/activity relationship of the whole conjugate.
Synthesis of Conjugates 1–3
The synthesis began with the preparation of the protected integrin ligand, namely, the c(AmpLRGDL)-azide 5 (Scheme 1). Briefly, the linear precursor 4 was synthesized using the Fmoc-based solid-phase peptide synthesis (SPPS) protocol starting from the preloaded H-Gly-2-ClTrt resin. Each amino acid was sequentially added to the growing sequence, by alternating coupling steps (HATU, HOAt, and collidine) and Fmoc-cleavage procedures (piperidine, DMF). The azide-carrying aminoproline residue 7 was in turn prepared as previously reported by reductive amination using N(4)-Fmoc protected aminoproline and 4-azidobutanal.28 Cleavage from the resin under acidic conditions (AcOH/TFE/DCM) gave linear peptide 4 in a 90% global yield. The cyclization reaction was carried out in-solution (15:1 DCM/DMF) under diluted conditions (2 mM) with HATU, HOAt as coupling reagents, and furnished the protected cyclic peptidomimetic 5 in a 50% yield. Overall, c(AmpLRGDL)-azide 5 was obtained in a 12-step sequence in a 45% overall yield from aminoproline 7. Also, acidic treatment of protected cyclopeptide 5 consigned deprotected cyclohexapeptide c(AmpLRGDL) 6, which was used as a reference compound in the biological assays (vide infra).
Scheme 1. Synthesis of Cyclopeptide 6, and Structure of the Aminoproline Nucleus 7 Used in the SPPS.
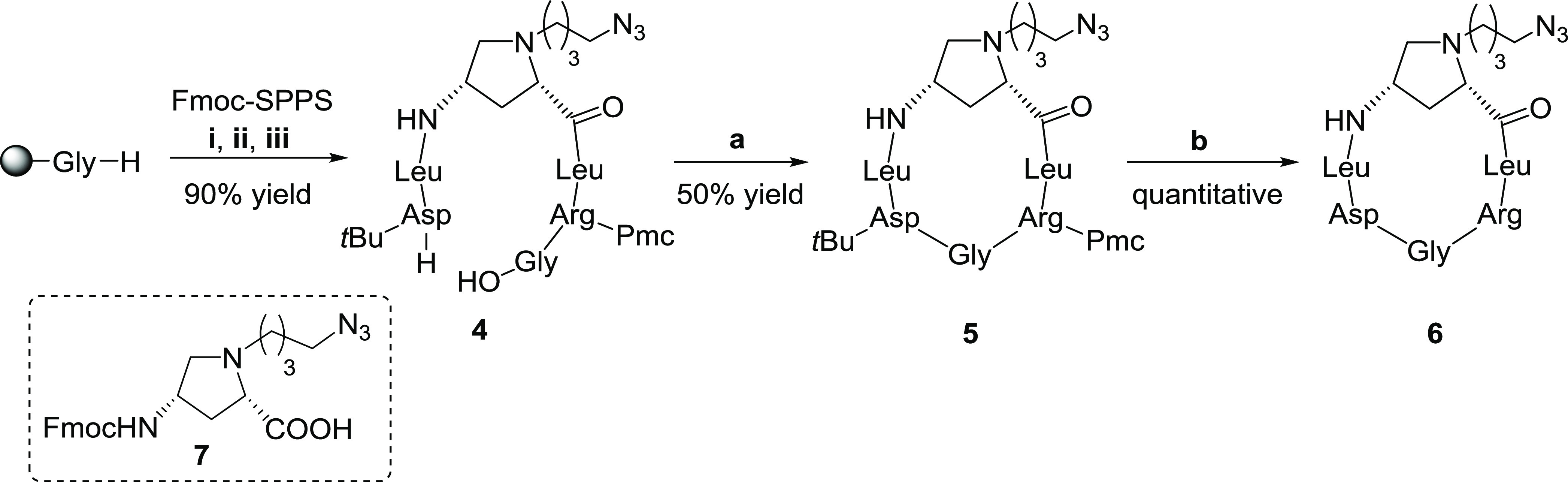
Reagents and conditions: Fmoc-SPPS: (i) Fmoc-Arg(Pmc)-OH, Fmoc-Leu-OH, 7, Fmoc-Leu-OH, Fmoc-Asp(tBu)-OH; HATU, HOAt, collidine, DMF, (ii) piperidine, DMF, (iii) AcOH, TFE, DCM; (a) HATU, HOAt, collidine, DCM/DMF (15:1), 6 h; (b) TFA/TIS/H2O 95:2.5:2.5, rt, 1 h.
The synthesis of compound 12 (Scheme 2), a linker portion in conjugate 1, started with tosylation of 4-azidobutanol 8 under standard conditions to give 9 in a good yield. Nucleophilic substitution of the tosylate group by Boc-piperazine 10 and subsequent palladium-catalyzed hydrogenation of the azido group furnished the primary amine 11 (72% yield, two steps). Finally, the coupling reaction between 11 and 4-pentynoic acid, promoted by HATU coupling reagent, and subsequent Boc removal gave the linker motif 12 (76% yield, two steps), ready for the reaction with chloroacetamide 17.
Scheme 2. Synthesis of the Linker Motives 12, 14, and 16.
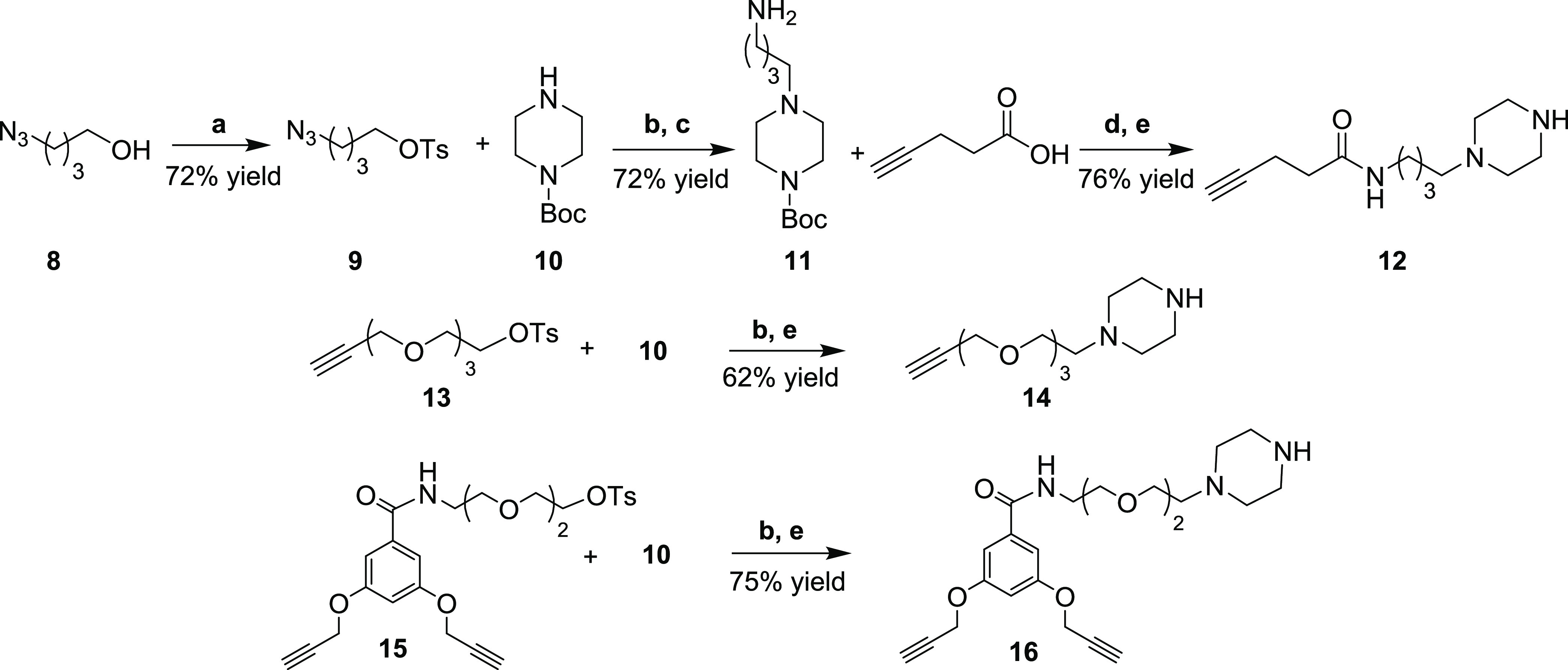
Reagents and conditions: (a) p-TsCl, TEA, DMAP, DCM dry, rt, 22 h; (b) Cs2CO3, ACN dry, N2, 60 °C, 18 h - 3 days; (c) H2, Pd/C, EtOAc, CH3CO2Na, rt, 3 h; (d) HATU, TEA, DCM dry, N2, rt, 3 h; (e) TFA/DCM 1.7:10, rt, 1 h.
The other two piperazine-alkyne moieties 14 and 16, used for the synthesis of compounds 2 and 3, were obtained by nucleophilic substitution of Boc-piperazine 10 on tosyl derivatives 13 and 15, respectively (Scheme 2), which were in turn prepared according to reported procedures.28 The products were obtained in good yields (67 and 75%, respectively), even if the reaction of 10 with 15 required a prolonged reaction time (3 days). Subsequent treatment with TFA in DCM efficiently removed the Boc group, furnishing compounds 14 and 16.
The synthesis of 2-chloro-N-methyl-N-(4-nitrophenyl)acetamide (17) (Scheme 3) was performed by acylation of p-nitroaniline with 2-chloroacetic pivalic anhydride, generated in situ by the reaction of chloroacetic acid with pivaloyl chloride. After methylation of the amide nitrogen (Me2SO4 in acetone), acetamide 17 was recovered in a 40% yield (two steps), which was coupled in parallel with the previous piperazine-carrying compounds 12, 14, and 16 to furnish, after selective reduction of the nitro group (Zn, NH4Cl), aniline derivatives 18, 19, and 20, respectively.
Scheme 3. Synthesis of Acetamide 17 and Aniline Derivatives 18, 19, and 20.

Reagents and conditions: (a) pivaloyl chloride, DCM dry, N2, from 0 °C to rt, 5 h; (b) K2CO3, Me2SO4, acetone, 60 °C, 16 h; (c) K2CO3, acetone, rt, 16–35 h; (d) Zn, NH4Cl, 10% water in MeOH, 70 °C, 5–7 h.
To conclude the construction of the nintedanib portion, oxindole 21 was synthesized from methyl 3-nitrobenzoate and methyl 2-chloroacetate (21% yield, 4 steps), according to procedures published in the literature (not shown).32 All that remained was the assemblage of the three linker-nintedanib portions and subsequent click-mediated anchorage of the RGD-cyclopeptides. Thus, aniline-terminating compounds 18, 19, and 20 reacted with (E)-configured oxindole 21 (Scheme 4) through a stereospecific conjugated nucleophilic substitution, giving the three different linker-nintedanib portions 22, 23, and 24 in moderate to good yields as single (Z)-stereoisomers. Azide-alkyne cycloaddition between protected cyclopeptide 5 and compounds 22, 23, and 24 was performed in parallel according to standard conditions (Cu(OAc)2, sodium ascorbate) in a DMF/water mixture (Scheme 4), providing the corresponding triazole products. The protecting groups of the cyclopeptides were removed by acidic treatment, providing the final conjugates 1–3, which were purified by reversed-phase HPLC and fully characterized by NMR and HRMS.
Scheme 4. Synthesis of the Linker-Nintedanib Nuclei 22, 23, ad 24 and Final Click Reaction/Deprotection Steps for the Synthesis of Covalent Conjugates 1–3.
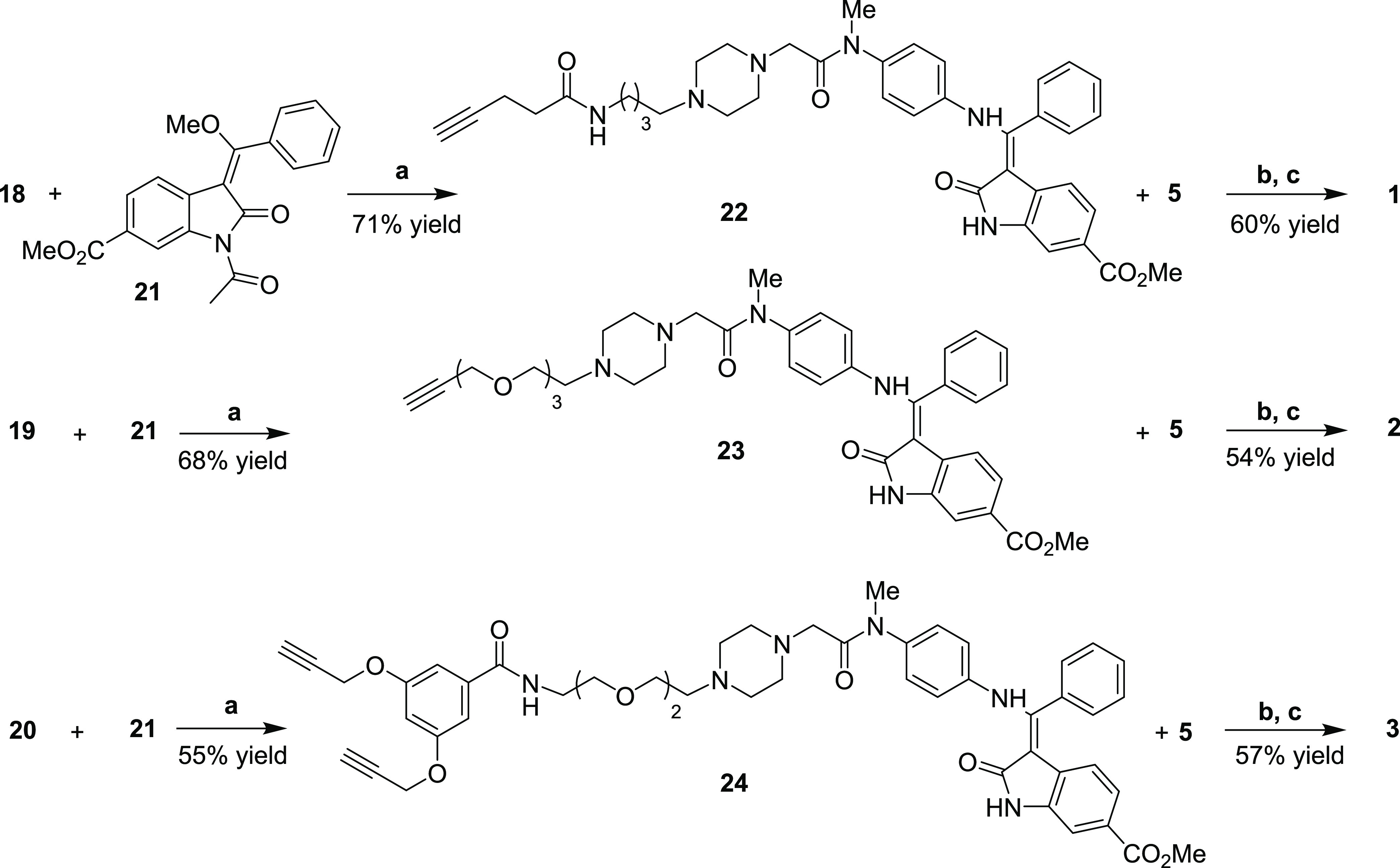
Reagents and conditions: (a) DMF, 80 °C, then piperidine, 24 h; (b) Cu(OAc)2, sodium ascorbate, DMF/H2O 3:7, rt, 12–16 h; (c) TFA/TIS/H2O 95:2.5:2.5, rt, 1 h.
Biological Studies
Kinase Inhibition Activity
To verify whether the tyrosine kinase inhibition activity is retained when the nintedanib moiety is covalently attached within conjugates 1–3, these compounds were tested against human recombinant VEGFR2 by enzymatic radiometric assay, and their activity compared with that of free nintedanib.33 As shown in Table 1, a good TKI activity is maintained in the three conjugates, with IC50 in the nM concentration range, comparable to that of the free drug (for compound 2) or only slightly lowered (for compounds 1 and 3).
Table 1. Tyrosine Kinase Inhibition Activity of Compounds 1–3 and Nintedanib against Human Recombinant VEGFR2a.
| compound | IC50 (nM) |
|---|---|
| nintedanib | 7 |
| 1 | 18 |
| 2 | 10 |
| 3 | 35 |
Reported IC50 values were determined by enzymatic radiometric assay (using [γ-33P]-ATP) and are the average of two independent experiments.
It was concluded that the introduction of varied and bulky linker-cyclopeptide cargoes onto the terminal piperazine nitrogen of nintedanib did not significantly perturb the binding ability of the drug to the receptor kinase, as originally hypothesized in the design step.
Inhibition of Cell Adhesion
With regard to the binding ability of the cyclopeptide portion of conjugates 1–3 to αVβ6 integrins, important preliminary information was taken into account. First, the recognition sequence of the cyclopeptide was chosen based on previous recent work by our group, where a c(AmpLRGDL) cyclopeptide closely related to 6 (in which case, an amide linkage at the Nα-proline was present instead of an amine) was shown to possess both good αVβ6 integrin affinity and good αVβ6vs αVβ3 selectivity in cell-free assays on isolated receptors (IC50 αVβ6 = 8.3 nM; inhibition of biotinylated fibronectin; IC50 αVβ3 = 2120 nM, inhibition of biotinylated vitronectin).31 Second, as stated above, the integrin-binding ability of these Amp-based cyclopeptidomimetics is generally demonstrated not to be affected by the type of connection (amine vs amide) of the alkyl chain to the proline nitrogen.28,31,34 The choice here of using amine-based connections to the proline nitrogen as in 6 was dictated by previous experience on similar TKI drug-peptidomimetic conjugates,28,29 suggesting a possible beneficial influence of basic amine sites on both the cell internalization of the overall construct and the endosomal escape ability into the cytoplasm.
For the new series of nintedanib-based conjugates 1–3, we sought confirmation of good binding affinity/selectivity toward αVβ6 integrins on a cellular level, which could also provide insights into the agonist or antagonist behavior of the constructs. Thus, the ability of conjugates 1–3 and the unconjugated ligand 6 to bind to αVβ6 integrins on the L929 fibroblast cell surface was investigated, through an inhibition cell adhesion assay. L929 cells were proven to express both αVβ6 and αVβ3 integrins as well as αVβ5 albeit to a lesser extent (see Supporting Information Figure S2). L929 fibroblast cell suspensions were exposed to increasing concentrations of compounds 1–3 and 6 for 30 min (at 37 °C in 10% CO2 atmosphere), before plating on a fibronectin-coated surface. Inhibition of adhesion was evaluated after 1 h incubation, under the same conditions, and normalized to that of untreated control cells. The three conjugates and ligand 6 decrease integrin-dependent cell adhesion promoted by fibronectin and therefore they can be referred to as antagonists. In particular, ligand 6 inhibits cell adhesion to fibronectin in a dose-dependent manner, and a concentration of 6 comprised between 1 and 10 μM is necessary for a 50% inhibition (Figure 3A). Conjugate 1 was significantly more potent than ligand 6 in cell adhesion inhibition; indeed, 50% of inhibition was found at doses lower than 1 μM. The inhibition of adhesion of L929 cells exposed to conjugate 2 was clearly dose-dependent, resembling the same trend shown by ligand 6. Interestingly, it seems that at 1 μM, the inhibitory effect of conjugate 2 reaches a sort of plateau. Compound 3 behaves similarly to compound 1, reaching 50% of inhibition at a dose lower than 1 μM and, quite surprisingly, it loses most of its activity at 10 μM, probably because of aggregation phenomena at high concentrations.
Figure 3.

(A) Inhibition of L929 adhesion to fibronectin (FN) in the presence of conjugates 1–3 and c(AmpLRGDL) 6 (0.05−10 μM concentration range). The inhibitory activity was calculated as the percentage of cell adhesion to FN in untreated cells and was expressed as mean ± SD. Experiments were carried out in triplicate. (B) Cytofluorimetric assay of L929 cells, K562 cells, and MDA-MB-231 cells treated for 24 h with free nintedanib and conjugates 1–3 at 5 μM. Percentages of cellular populations showing a fluorescence intensity higher than the threshold value (cellular autofluorescence) are reported. Representative images of at least three independent experiments. (C) Mean fluorescence intensity of L929 cells treated for 24 h with conjugates 1–3 at 5 μM. **p < 0.0001 untreated vs compounds 1 and 2, *p < 0.01 untreated vs compound 3. One-way ANOVA followed by Tukey’s multiple comparison test.
In conclusion, these data confirm our hypothesis formulated during design, revealing that not only does the integrin ligand unit in the conjugates not lose the binding capacity towards αVβ6 but that conjugates 1–3 are also even better antagonists than 6 in the adhesion process.
Cell Internalization Studies of Conjugates 1–3 and Nintedanib
According to our plan, it is the c(AmpLRGDL) portion within the conjugates that should selectively deliver them to αVβ6 integrin-overexpressing cells such as fibroblasts and activated epithelial cells in a fibrotic lesion, inhibit profibrotic integrin-promoted signaling, and exploit integrin-mediated cell internalization to accumulate the conjugates within the target cells. In fact, nintedanib targets the intracellular kinase domain of growth factor receptors, and it may block signaling transduction after cell internalization.
The propensity of conjugates 1–3 toward cellular internalization was evaluated vis-à-vis that of nintedanib in L929 fibroblasts via cytofluorimetric analysis, which drew on the intrinsic fluorescence of the nintedanib moiety (λexc = 405 nm, λem = 421 nm) (Figure 3B). L929 cells were exposed for 24 h to treatment with compounds 1–3 or nintedanib at 5 μM concentration; next, the percentage of positive cells of different treatment populations was determined and compared with the fluorescence intensity of untreated cells (Figure 3B). Differences in compound internalization were also quantified as the mean fluorescence intensity (MFI) of the global population (Figure 3C).
About 83% L929 cells exposed to nintedanib were positive, with MFI higher than 104 (not shown in the figure); as expected, due to its physicochemical properties, the small molecule nintedanib can easily cross the cell membrane and accumulate in the cytoplasm. Compounds 1 and 2 behave similarly to L929 positive cells ranging between 54 and 60% and MFIs of these populations comprised between 250 and 280. Quite surprisingly, compound 3, bearing a dimeric presentation of the cyclopeptide, is much less efficiently internalized, with only 19% of cells positive after 24 h incubation, and MFI markedly reduced. These results diverge from those observed with previously reported sunitinib-RGD conjugates,29 where the dimeric presentation of the integrin ligand increased compound internalization. A possible explanation of the lower cell uptake of conjugate 3 is that its larger size could favor a tendency to aggregate at higher concentrations, as observed in the binding assay (vide supra).
These same experiments were repeated with both K562 and MDA-MB-231 cells. The former cell line expresses neither αVβ6 nor αVβ3 integrins; therefore, it was used as a negative control; the latter cell line, however, expresses low levels of αVβ3 but does not express αVβ6 integrins. The assay showed that nintedanib is efficiently internalized even in these two cell lines, irrespective of integrin expression. On the other hand, the fluorescence of K562 and MDA-MB-231 cells exposed to conjugates 1–3 was very weak in all instances, resembling that of untreated cells.
It was concluded that dual conjugates 1–3 were able to be internalized in L929 fibroblast cells to various extents depending on their structural features, with monomeric compounds 1 and 2 behaving better than the dimeric counterpart 3; in any case, internalization was αVβ6 integrin-mediated. Conversely, nintedanib alone was cell internalized to a large extent in all of the cell types tested, showing no cell selectivity.
Effects of Conjugates 1-3 and Nintedanib on ERK1/2 Phosphorylation
Extracellular signal-regulated kinases (ERKs) are key components of the mitogen-activated protein kinase (MAPK) pathway, the downstream signaling pathway of many different growth factor receptors, as bFGFR, PDGFR, and VEGFR.35 Upregulation of the ERK1/2 phosphorylation cascade promotes cell survival, migration, and proliferation and leads to the fibroblast-to-myofibroblast transition (FMT).36 The concomitant fibroblast proliferation and migration, FMT, and deposition of extracellular matrix proteins are hallmarks of fibrosis.37 Moreover, the MAPK pathway is intertwined with the outside-in αV integrin signaling cascade.24 Accordingly, we reasoned that evaluation of the inhibition of ERK1/2 phosphorylation in the presence of dual conjugates 1–3 would have been indicative of the downstream effect of these molecules on fibroblast signaling promoted by either the integrin ligand or the nintedanib component, or both.
To this end, inhibition of ERK1/2 phosphorylation was assayed in L929 cells exposed to compounds 1–3, nintedanib alone, integrin ligand 6 alone, or nintedanib in combination with ligand 6 (Figure 4A).
Figure 4.
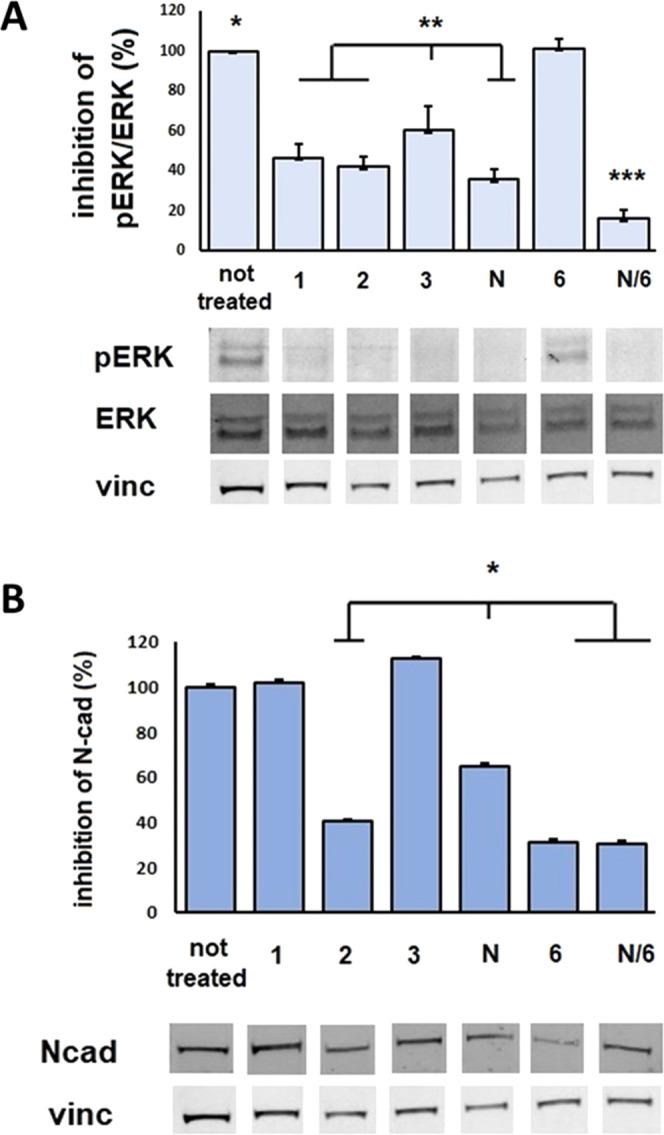
(A) Expression of phosphorylated and total ERK1/2 and vinculin in L929 cells exposed to compounds 1–3, nintedanib (N), c(AmpLRGDL) 6, or 6+nintedanib (N/6) for 24 h (5 μM). Bottom panels show immunoblots, and top panels display respective mean densitometric values ± SEM of three independent experiments of phosphorylated proteins normalized to total protein content. *p < 0.0001 untreated vs treatments, except for compound 6, **p < 0.005 compound 3vs compounds 1, 2, N, and N/6, ***p < 0.0001 N/6vs compounds 1–3 and N by one-way ANOVA followed by Tukey’s multiple comparison test. (B) Expression of N-Cadherin in L929 cells exposed for 24 h to compounds 1–3, nintedanib (N), c(AmpLRGDL) 6, and 6+nintedanib (N/6) at 5 μM. Bottom panels show the representative immunoblots, and top panels display mean densitometric values ± SEM (three independent experiments of N-Cadherin normalized to corresponding total protein contents). *p < 0.05 in comparison with untreated by ANOVA followed by the Newman–Keuls test.
As expected, the TKI nintedanib efficiently inhibits ERK1/2 phosphorylation; interestingly, a similar inhibition is observed with conjugates 1 and 2, even though they are internalized to a lesser extent than free nintedanib (vide supra). Compound 3, the conjugate that was minimally internalized and that exhibited a drop of binding ability at the observed concentration, was also less efficient in ERK1/2 inhibition. Of particular interest is the role of the integrin ligand: when the cells were incubated with c(AmpLRGDL) 6 alone, no inhibition of ERK1/2 phosphorylation was registered, but when 6 was given in combination with nintedanib, maximal inhibition was found, as though occupation of the integrin receptor could potentiate the nintedanib effect. This could also explain the similar inhibition behavior observed for nintedanib and compound 2, irrespective of their different intracellular content (see the different uptake of nintedanib vs2, Figure 3B,C). According to a possible explanation, the integrin ligand portion of the noninternalized fraction of 2 could engage the integrin αVβ6 and enhance the phosphorylation inhibition given by the nintedanib portion of the internalized fraction of 2.
The MTT assay was carried out to evaluate L929 viability after 24 h treatment with conjugates 1–3, nintedanib, compound 6, and a combination of nintedanib with compound 6 (see Supporting Information Figure S3). In the 1–10 μM concentration range, nintedanib and compound 6 showed a mild antiproliferative effect; conjugates 1–3 showed an even lower cytostatic effect.
Fibroblast-to-Myofibroblast Transition
Myofibroblasts, the key components of fibrotic lesions, derive from different cell types, such as circulating mesenchymal cells, endothelial cells, and mainly resident fibroblasts. It was shown that FMT, one of the critical steps in fibrosis, is affected by cell–cell contacts, which in turn depend on N-cadherin surface expression; in fact, blockage of N-cadherin resulted in the inhibition of FMT.38
To examine the possible inhibiting role of profibrotic signaling exerted by nintedanib conjugates of this study, we investigated the ability of compounds 1–3, nintedanib, ligand 6, or the nintedanib/6 combination, to inhibit N-cadherin expression on L929 cells, as a hallmark of FMT inhibition (Figure 4B). N-Cad expression was normalized as a percentage compared to untreated cells. Nintedanib inhibits N-Cad expression on cell surface almost by half; a stronger inhibitory effect is observed after the treatment with free ligand 6, whereas the coadministration of nintedanib and 6 does not give an improved inhibition of N-Cad expression. As for the conjugates, treatment with compound 2 gives a strong inhibition of N-Cad expression, comparable to that of the free integrin ligand, whereas compounds 1 and 3 do not seem to interfere in this mechanism.
Conclusions
Three novel nintedanib-clicked conjugates, compounds 1–3, were efficiently synthesized and characterized, consisting of a nintedanib-like portion, which was joined to one or two cyclic aminoproline-based LRGDL moieties targeting αVβ6 integrins by means of three different and robust covalent linkages. The dual nature of the conjugates as both αVβ6 integrin binders and TKI inhibitors was shown, demonstrating that the covalent conjugation of the two active units within the final constructs did not disturb their respective targeting properties. Cell uptake experiments demonstrated good αVβ6 integrin-mediated internalization properties in fibrotic cells mainly for compounds 1 and 2, while poor uptake was observed for dimeric compound 3 at the tested concentration.
The use of conjugates 1–3 as antifibrotic drugs was investigated in vitro. Both compounds 1 and 2 were able to inhibit ERK1/2 phosphorylation; in addition, conjugate 2 could inhibit fibroblast-to-myofibroblast transition of L929 fibroblasts. The overall biological results indicate that compound 2 seems to better recapitulate optimal in vitro antifibrotic properties; the two active units, namely, the integrin ligand portion and the nintedanib TK inhibiting portion, potentiate each other with favorable synergy. Of note, the simple combination of the two independent active units, namely, ligand 6 and nintedanib, proved beneficial in inhibiting the downstream fibroblast signaling. On the other hand, the covalent association of the two modules as presented in compound 2 could be likewise effective, with the added advantage of being selective toward fibrotic cells, in the context of targeted drug delivery. These results hold promise for future work on the development of selective dual conjugates in the targeted therapy of IPF.
Experimental Procedures
Chemistry
General
All chemicals were of the highest commercially available quality and were used without other purification. Dry solvents were prepared by standard procedures. Anhydrous reactions were performed under N2 or Ar atmosphere. H-Gly-2-ClTrt resin (glycine loading 0.58 mmol/g) was from Novabiochem, and (2S,4S)-Fmoc-4-amino-1-Boc-pyrrolidine-2-carboxylic acid was purchased from PolyPeptide. Silica gel 60 F254 precoated plates were used for analytical TLC, visualized under UV light and/or by dipping in molybdate reagent solution (aqueous H2SO4 solution of Ce(SO4)2(H2O)x and ammonium molybdate) followed by heating. Flash column chromatography was performed using 40–63 μm silica gel. Automated flash column chromatography was performed with the Biotage Isolera One system using Biotage KP-Sil cartridges (direct phase) and KP-C18-HS cartridges (reversed-phase). HPLC purifications were carried out on a Prostar 210 apparatus (UV detection) using C18-10 μm columns (Discovery BIO Wide Pore 10 mm × 250 mm). Purity of the final compounds was checked by HPLC and was in the 96–99% range. ESI-mass spectra were collected on UHPLC/ESI-MS system (ACQUITY ultraperformance LC; ESI, positive ions, single quadrupole analyzer) and are reported in the form of (m/z). High-resolution mass analysis was performed on an LTQ ORBITRAP XL Thermo apparatus. NMR spectra were recorded on an AV400 (Bruker) spectrometer. Chemical shifts (δ) are reported in ppm. Multiplicities of signals are reported as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and b (broad). Coupling constants, J, are in hertz. 1H assignments are corroborated by 1H–1H COSY and 1H–1H TOCSY.
Materials
H-Gly-2-ClTrt resin, Fmoc-Asp(tBu)-OH; Fmoc-Arg(Pmc)-OH, Fmoc-Leu-OH, 2,4,6-collidine, glacial acetic acid, DIPEA, HATU, HOAt, 4-azidobutanol, 3-aminopropanol, and N-Boc-piperazine were commercially available and were used as such without further purification. Nintedanib was purchased by MedChemExpress. Nα-(4-azidobutyl)-4-N-(Fmoc)aminoproline (7), tosyl derivatives 13 and 15, and oxindole nucleus 21 were prepared according to reported procedures.28,32
General Method for HPLC Purification
The final conjugates were purified by reversed-phase HPLC equipped with a preparative column (C18-10 μm, 21.2 mm × 250 mm column), with the solvent system H2O + 0.1% TFA (solvent A) and ACN (solvent B), using a linear gradient from 5% B to 50% B over 23 min, 50% B for 3 min, from 50% B to 5% B over 3 min, (flow rate 8.0 mL/min; detection at 220 nm).
Experimental Synthetic Procedures and Characterization Data
Synthesis of the Covalent Conjugate 1
To a solution of compound 22 (7.7 mg, 0.011 mmol, 1 equiv) and cyclopeptide 5 (16.5 mg, 0.013 mmol, 1.1 equiv) in DMF (1.57 mL), a solution of Cu(OAc)2 (0.64 mg, 0.003 mmol, 0.3 equiv) and sodium ascorbate (1.3 mg, 0.007 mmol, 0.6 equiv) in water (0.67 mL) was added. The reaction was left under stirring under argon atmosphere, after three cycles of argon/vacuum. After 6.5 h, the solvent was removed under reduced pressure and the residue was washed with water (3×) and diethyl ether (3×). The protected intermediate was checked by MS analysis (MS (ES+)m/z 1762.9 [M + H]+) and then deprotected using a solution of TFA:TIS:H2O 95:2.5:2.5 (0.57 mL). The reaction was kept under stirring for 1 h, then the solvent was removed under reduced pressure, and the crude was purified by reversed-phase HPLC, using the described general method (Rt = 22.9 min), giving the final conjugate 1 (13.0 mg, 60% yield). 1H NMR (400 MHz, MeOD) δ 7.77 (s, 1H, CH triazole), 7.67 – 7.56 (m, 5H, ArH), 7.53 (m, 2H, ArH), 7.30 (dd, J = 8.4, 1.9 Hz, 1H, ArH), 7.16 (m, 2H, ArH), 6.97 (m, 2H, ArH), 5.97 (d, J = 8.4 Hz, 1H, ArH), 4.67 (m, 1H, H4Amp), 4.46 – 4.38 (m, 6H, αAsp + αLeu), 4.37 – 4.32 (m, 2H, H2Amp), 4.29 (d, J = 17.1, 1H αGly), 3.86 (s, 3H, CH3), 3.76 (d, J = 17.1, 1H αGly), 3.61 (m, 1H, H5Amp), 3.52 (m, 1H, H5Amp), 3.30-3.09 (m, 16H, δArg + βAsp + CH2), 3.03 – 2.84 (m, 7H, H3Amp + CH2), 2.57 (t, J = 7.1 Hz, 2H, CH2), 2.16 (m, 1H, H3Amp), 2.02 – 1.81 (m, 3H), 1.80 – 1.51 (m, 16H, βLeu + γLeu + βArg + γArg + CH2), 1.01 (d, J = 6.1 Hz, 3H, δLeu), 0.96 (d, J = 6.2 Hz, 6H, δLeu), 0.89 (d, J = 6.1 Hz, 3H, δLeu). HRMS(ES+) C72H101N19O13 calcd for [M + H]+ 1440.7826, found 1440.7854. HRMS(ES+) C72H101N19O13 calcd for [M + H]+ 1440.7826, found 1440.7854.
Synthesis of the Covalent Conjugate 2
Conjugate 2 was synthesized as described for compound 1, using compound 23 (6.9 mg, 0.009 mmol, 1 equiv) and cyclopeptide 5 (14.4 mg, 0.011 mmol, 1.1 equiv). The protected intermediate was checked by MS analysis (MS (ES+)m/z 1717.9 [M + H]+), and then it was deprotected using a solution of TFA/TIS/H2O 95:2.5:2.5 (0.65 mL). The reaction was kept under stirring for 1 h, then the solvent was removed under reduced pressure, and the crude was purified by reversed-phase HPLC, using the described general method (Rt = 22.5 min), giving the final conjugate 2 (9.7 mg, 54% yield). 1H NMR (400 MHz, CDCl3) δ 8.01 (s, 1H, CH triazole), 7.76 – 7.57 (m, 4H, ArH), 7.53 (m, 2H, ArH), 7.30 (dd, J = 8.3, 1.9 Hz, 1H, ArH), 7.17 (m, 2H, ArH), 6.96 (m, 2H, ArH), 5.97 (d, J = 8.3 Hz, 1H, ArH), 4.68 (m, 1H, αAsp), 4.62 (bs, 2H, OCH2-triazole), 4.57 – 4.47 (m, 8H, H2Amp + CH2 + H4Amp + 2αLeu + H5Amp + αArg), 4.28 (d, J = 17.4 Hz, 1H, αGly), 3.86 (bs, 3H, CH3), 3.82 (m, 2H, CH2), 3.76 (d, J = 17.4 Hz, 1H, αGly), 3.67 (s, 10H, CH2), 3.56 (m, 1H, H5b Amp), 3.28 – 3.15 (m, 11H, 2CH2 + δArg + H4′ + CH3), 3.00 – 2.79 (m, 7H, βAsp + H3a Amp + CH2-CON + CH2), 2.17 (d, J = 14.8 Hz, 1H, H3b Amp), 2.03 – 1.57 (m, 18H, H2′ + H3′ + βArg + γArg + γLeu + βLeu + 2CH2), 1.01 (d, J = 6.1 Hz, 3H, δLeu), 0.96 (m, 6H, δLeu), 0.89 (bd, J = 6.1 Hz, 3H, δLeu). HRMS(ES+) C72H103N18O15 calcd for [M + H]+ 1458.7772, found 1459.7800.
Synthesis of the Covalent Conjugate 3
Covalent conjugate 3 was synthesized as described for compound 1 starting from compound 24 (10.0 mg, 0.0115 mmol, 1 equiv) and cyclopeptide 5 (33.4 mg, 0.0253 mmol, 2.3 equiv). The protected intermediate was checked by MS analysis (MS (ES+)m/z 3040.7 [M + H]+), and it was deprotected using a solution of TFA/TIS/H2O 95:2.5:2.5 (0.58 mL). The reaction was kept under stirring for 1 h, then the solvent was removed under reduced pressure, and the crude was purified by reversed-phase HPLC, using the described general method (Rt = 22.1 min), giving the final conjugate 3 (20.1 mg, 56% yield). 1H NMR (400 MHz, MeOD) δ 8.09 (s, 2H, CH triazole), 7.66 – 7.57 (m, 5H, ArH), 7.51 (m, 2H, ArH), 7.29 (dd, J = 8.3, 1.6 Hz, 1H, ArH), 7.12 (m, 4H, ArH), 6.94 (m, 2H, ArH), 5.97 (d, J = 8.3 Hz, 1H, ArH), 5.19 (bs, 4H, Ar-OCH2-traziole), 4.69 (m, 2H, αAsp), 4.50 – 4.31 (m, 14H, H4Amp + αLeu + H2Amp + CH2), 4.28 (d, J = 17.5 Hz, 2H, αGly), 3.86 (s, 3H, CH3), 3.82 – 3.79 (m, 2H, αArg), 3.76 (d, J = 17.5 Hz, 2H, αGly), 3.72 – 3.63 (m, 8H, CH2), 3.62 – 3.51 (m, 4H, H5Amp), 3.29 – 3.08 (m, 16H, δArg + CH2), 2.95 (m, 2H, CH2), 2.85 (m, 4H, βAsp), 2.77 (m, 4H, CH2), 2.18 (m, 2H, H3Amp), 2.05 – 1.94 (m, 4H, CH2), 1.93 – 1.84 (m, 2H, H3Amp), 1.80 – 1.58 (m, 24H, βLeu + βArg + γArg + γLeu + CH2), 1.01 (d, J = 6.2 Hz, 6H, δLeu), 0.96 (d, J = 6.2 Hz, 12H, δLeu), 0.89 (bd, J = 6.1 Hz, 6H, δLeu). HRMS(ES+) C115H167N32O25 calcd for [M + H]+ 2396.2702, found 2395.2753.
Biology
In Vitro Kinase Assay
Evaluation of the effects of compounds 1–3 and nintedanib on the kinase activity of human recombinant VEGFR2 was performed at Eurofins Cerep laboratories, by measuring inhibition of receptor phosphorylation by a radiometric detection method. IC50 values for select inhibitors were calculated from a nine-point dose–response curve generated using the standard assay conditions for the target as described: KDR (Kinase insert domain receptor, Flk-1/VEGFR2, activated by VEGF) was incubated with 8 mM MOPS pH 7.0, 0.2 mM EDTA, 0.33 mg/mL myelin basic protein, 10 mM Mg acetate, and [γ-33P]-ATP. The reaction was initiated by the addition of the Mg/ATP mix. After 40 min incubation at 22 °C with the inhibitor, the reaction was stopped by the addition of phosphoric acid to a concentration of 0.5%. An aliquot of the reaction was spotted onto a filter and washed four times for 4 min in 0.425% phosphoric acid and once in MeOH prior to drying and scintillation counting. https://www.eurofinsdiscoveryservices.com/catalogmanagement/viewItem/KDR-VEGFR2-KDR-Human-RTK-Kinase-Enzymatic-Radiometric-Assay-10-uM-ATP-KinaseProfiler/14-630KP10 (December 22, 2021).
Cell Culture
Murine and human cell lines were obtained from ATCC in this study were used: murine normal fibroblast cell line (L929); human breast cancer cell line (MDA-MB-231), and human erythroleukemia cell line (K562). Dulbecco’s modified Eagle medium, containing 4500 mg/L glucose (Euroclone, MI Italy) supplemented with 10% fetal bovine serum (FBS) was used for L929 and MDA-MB-231. The cells were maintained at 37 °C in a humidified incubator containing 10% CO2. K562 cells were maintained at 37 °C in RPMI-1640 (Euroclone, MI Italy) supplemented with 5% FBS in T25 culture flasks (Sarstedt) in a humidified incubator with 5% CO2. Murine fibroblasts or human breast carcinoma cells (1 × 106) were seeded in 100 mm Sarstedt dishes, and the cells were propagated every 3 days by treatment with trypsin–EDTA solution. K562 cell line was propagated by dilution in warm fresh media reaching a cell density of 2 × 105 cells/mL. L929, MDA-MB-231, and K562 cultures were monitored periodically for mycoplasma contamination.39
Inhibition of Cell Adhesion to Vitronectin
The inhibition of adhesion was determined on integrin αVβ6-positive L929 cells. Briefly, a 96-well plate was coated, at 4 °C, overnight with fibronectin (FN, 2 μg/mL) (sc-29011 Santa Cruz). The plate was washed with PBS solution and incubated at 37 °C for 1 h with 1% bovine serum albumin (BSA) solution in PBS. L929 cells were centrifugated and suspended in serum-free medium (6.0 × 105 cells/mL). The cells were preincubated with different concentrations of compounds 1–3, or c(AmpLRGDL) 6 (ranging from 10 μM to 50 nM) at 37 °C for 30 min to allow reaching ligand–receptor equilibrium. Subsequently, the cells were plated on FN-coated substrata (at 5–6 × 104 cells/well density). Incubation was performed at 37 °C for 1 h. MnCl2 (2 mmol/L) was used to provide a full integrin activation during the experiment. At the end of the incubation, PBS washing was used to remove the nonadherent cells. Adherent cells were fixed, permeabilized, and colored using 200 μL/well of a solution of 0.5% crystal violet in 20% methanol. After 2 h of incubation (4 °C), the plates were washed. Dimethyl sulfoxide (100 μL) was added to each well for dye solubilization. The plates were examined in a counter ELX800 (540 nm) (Bio TEK Instruments). The percentage of cell adhesion inhibition was expressed as a percent of inhibition ± SEM of L929 adhesion compared to that of L929 cells exposed to PBS. Experiments were repeated at least three times and done in triplicate.
Western Blotting Analysis
Subconfluent L929 cell cultures in p100 plates were exposed for 24 h to compounds 1–3, nintedanib (N), c(AmpLRGDL) 6, and 6+nintedanib (N/6) at 5 μM in serum-free medium. After incubation, the cells were washed with ice-cold PBS (1 μM Na4VO3). Cell lysates were obtained using cell RIPA lysis buffer (from Merck Millipore). The inhibitor PMSF (1 μM), sodium orthovanadate (100 μM), and protease inhibitor cocktail set III (Sigma-Aldrich) were mixed with RIPA buffer. Equal amounts of protein (40–60 μg) were prepared in Laemmli buffer and separated on Bolt Bis-Tris Plus gels 4–12. Fractionated proteins were transferred to a PVDF membrane (iBlot-2 system). Membrane blocking was performed for 1 h (RT) with Odyssey blocking buffer. Next, the membranes were probed with primary antibodies (4 °C O/N). Following primary antibodies (1:1000 dilution) were used: rabbit anti-human/mouse/rat phospho-p44/42 (ERK1/2) polyclonal antibody (Cell Signaling #9101), rabbit anti-h/m/r p44/42 (ERK1/2) polyclonal antibody (Cell Signaling #9102), and rabbit anti-h/m/r N-Cadherin polyclonal antibody (Cell Signaling #4061, 140 kDa). After probing, the membranes were washed with T-PBS buffer and then incubated for 1 h (RT) with goat anti-rabbit IgG Alexa Fluor 750 or with goat anti-mouse IgG Alexa Fluor 680 antibodies (Invitrogen, Waltham, MA, 1:10000). Corresponding bands were visualized using Odyssey Infrared Imaging System (LI-COR Bioscience, Lincoln, NE). Monoclonal antibody against vinculin was used to assess an equal amount of protein loaded in each lane (rabbit anti-h/m/r mAb, Cell Signaling #13901, 145 kDa, 1:1000).
Acknowledgments
This research was funded by Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR, PRIN 2020 Protocol n. 2020833Y75_003) and by Ente Cassa di Risparmio di Firenze (BIANCRF181014). Thanks are due to Centro Interdipartimentale Misure “G. Casnati” (University of Parma, Italy) for instrumental facilities.
Glossary
Abbreviations
- Amp
4-amino-l-proline
- Boc
tert-butoxycarbonyl
- DCE
1,2-dichloroethane
- DCM
dichloromethane
- DIPEA
diisopropylethylamine
- DMAP
4-(dimethylamino)pyridine
- Fmoc
9-fluorenylmethoxycarbonyl
- DMF
N,N-dimethylformamide
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HOAt
1-hydroxy-7-azabenzotriazole
- Pmc
2,2,5,7,8-pentamethylchroman-6-sulfonyl
- SPPS
solid-phase peptide synthesis
- TFA
trifluoroacetic acid
- TFE
trifluoroethanol
- TIS
triisopropylsilane
- TLC
thin-layer chromatography
- p-TsCl
p-toluenesulfonyl chloride
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00535.
Synthesis and characterization of all intermediate compounds, cyclopeptidomimetic c(AmpLRGDL) 6, HPLC chromatograms of final compounds 1–3, cytofluorimetric assay of integrin expression, and inhibition of cell proliferation assay (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- King T. E.; Pardo A.; Selman M. Idiopathic Pulmonary Fibrosis. Lancet 2011, 378, 1949–1961. 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- Caja L.; Dituri F.; Mancarella S.; Caballero-Diaz D.; Moustakas A.; Giannelli G.; Fabregat I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. 10.3390/ijms19051294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A.; Abraham D. J. TGF-β Signaling and the Fibrotic Response. FASEB J. 2004, 18, 816–827. 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- Shi M.; Zhu J.; Wang R.; Chen X.; Mi L.; Walz T.; Springer T. A. Latent TGF-β Structure and Activation. Nature 2011, 474, 343–349. 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson I. B.; Rifkin D. B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harbor Perspect. Biol. 2016, 8, a021907 10.1101/cshperspect.a021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barczyk M.; Carracedo S.; Gullberg D. Integrins. Cell Tissue Res. 2010, 339, 269–280. 10.1007/s00441-009-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N. F.; Marshall J. F. Integrin-Mediated TGFβ Activation Modulates the Tumour Microenvironment. Cancers 2019, 11, 1221 10.3390/cancers11091221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthington J. J.; Klementowicz J. E.; Travis M. A. TGFβ: A Sleeping Giant Awoken by Integrins. Trends Biochem. Sci. 2011, 36, 47–54. 10.1016/j.tibs.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Dong X.; Hudson N. E.; Lu C.; Springer T. A. Structural Determinants of Integrin β-Subunit Specificity for Latent TGF-β. Nat. Struct. Mol. Biol. 2014, 21, 1091–1096. 10.1038/nsmb.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X.; Zhao B.; Iacob R. E.; Zhu J.; Koksal A. C.; Lu C.; Engen J. R.; Springer T. A. Force Interacts with Macromolecular Structure in Activation of TGF-β. Nature 2017, 542, 55–59. 10.1038/nature21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuss J. M.; Gillett N.; Lu L.; Sheppard D.; Pytela R. Restricted Distribution of Integrin Beta 6 MRNA in Primate Epithelial Tissues. J. Histochem. Cytochem. 1993, 41, 1521–1527. 10.1177/41.10.8245410. [DOI] [PubMed] [Google Scholar]
- Goodman S. L.; Picard M. Integrins as Therapeutic Targets. Trends Pharmacol. Sci. 2012, 33, 405–412. 10.1016/j.tips.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Henderson N. C.; Arnold T. D.; Katamura Y.; Giacomini M. M.; Rodriguez J. D.; McCarty J. H.; Pellicoro A.; Raschperger E.; Betsholtz C.; Ruminski P. G.; Griggs D. W.; Prinsen M. J.; Maher J. J.; Iredale J. P.; Lacy-Hulbert A.; Adams R. H.; Sheppard D. Targeting of αV Integrin Identifies a Core Molecular Pathway That Regulates Fibrosis in Several Organs. Nat. Med. 2013, 19, 1617–1624. 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack R. J.; Macdonald S. J. F.; Roper J. A.; Jenkins R. G.; Hatley R. J. D. Emerging Therapeutic Opportunities for Integrin Inhibitors. Nat. Rev. Drug Discov. 2022, 1, 60–78. 10.1038/s41573-021-00284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukey P. T.; Coello C.; Gunn R.; Parker C.; Wilson F. J.; Saleem A.; Garman N.; Costa M.; Kendrick S.; Onega M.; Kang’ombe A. R.; Listanco A.; Davies J.; Ramada-Magalhaes J.; Moz S.; Fahy W. A.; Maher T. M.; Jenkins G.; Passchier J.; Marshall R. P. Clinical Quantification of the Integrin αVβ6 by [18F]FB-A20FMDV2 Positron Emission Tomography in Healthy and Fibrotic Human Lung (PETAL Study). Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 967–979. 10.1007/s00259-019-04586-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John A. E.; Graves R. H.; Pun K. T.; Vitulli G.; Forty E. J.; Mercer P. F.; Morrell J. L.; Barrett J. W.; Rogers R. F.; Hafeji M.; Bibby L. I.; Gower E.; Morrison V. S.; Man Y.; Roper J. A.; Luckett J. C.; Borthwick L. A.; Barksby B. S.; Burgoyne R. A.; Barnes R.; Le J.; Flint D. J.; Pyne S.; Habgood A.; Organ L. A.; Joseph C.; Edwards-Pritchard R. C.; Maher T. M.; Fisher A. J.; Gudmann N. S.; Leeming D. J.; Chambers R. C.; Lukey P. T.; Marshall R. P.; Macdonald S. J. F.; Jenkins R. G.; Slack R. J. Translational Pharmacology of an Inhaled Small Molecule αVβ6 Integrin Inhibitor for Idiopathic Pulmonary Fibrosis. Nat. Commun. 2020, 11, 4659 10.1038/s41467-020-18397-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unfortunately, to the best of our knowledge, the molecular structure of this compound has not yet been published.
- Decaris M. L.; Schaub J. R.; Chen C.; Cha J.; Lee G. G.; Rexhepaj M.; Ho S. S.; Rao V.; Marlow M. M.; Kotak P.; Budi E. H.; Hooi L.; Wu J.; Fridlib M.; Martin S. P.; Huang S.; Chen M.; Muñoz M.; Hom T. F.; Wolters P. J.; Desai T. J.; Rock F.; Leftheris K.; Morgans D. J.; Lepist E.-I.; Andre P.; Lefebvre E. A.; Turner S. M. Dual Inhibition of αVβ6 and αVβ1 Reduces Fibrogenesis in Lung Tissue Explants from Patients with IPF. Respir. Res. 2021, 22, 265 10.1186/s12931-021-01863-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- αVβ6 integrin-targeted monoclonal antibodies have also been identified as possible therapeutics of IPF; in particular, BG00011 (formerly STX-100) has entered clinical phase 2 study for IPF but the study was stopped by Biogen due to safety concerns.
- IPF is a progressive chronic disease, that occurs in about 12 per 100,000 people per year, with an average life expectancy of about 5 years following diagnosis.
- Lasky J. A.; Brody A. R. Interstitial Fibrosis and Growth Factors. Environ. Health Perspect. 2000, 108, 751–762. 10.1289/ehp.00108s4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To date, two drugs are approved for IPF treatment namely, nintedanib and pirfenidone.
- Wollin L.; Wex E.; Pautsch A.; Schnapp G.; Hostettler K. E.; Stowasser S.; Kolb M. Mode of Action of Nintedanib in the Treatment of Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. 10.1183/09031936.00174914. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nintedanib is a potent triple angiokinase inhibitor for VEGFR-1,2,3 (IC50 = 34, 13, 13 nM), FGFR-1,2,3 (IC50 = 69, 37, 108 nM), and PDGFR-α,β (IC50 = 59, 65 nM).
- Maldonado H.; Hagood J. S. Cooperative Signaling between Integrins and Growth Factor Receptors in Fibrosis. J. Mol. Med. 2021, 99, 213–224. 10.1007/s00109-020-02026-2. [DOI] [PubMed] [Google Scholar]
- West X. Z.; Meller N.; Malinin N. L.; Deshmukh L.; Meller J.; Mahabeleshwar G. H.; Weber M. E.; Kerr B. A.; Vinogradova O.; Byzova T. V. Integrin β3 Crosstalk with VEGFR Accommodating Tyrosine Phosphorylation as a Regulatory Switch. PLoS One 2012, 7, e31071 10.1371/journal.pone.0031071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanath P. R.; Malinin N. L.; Byzova T. V. Cooperation between Integrin αVβ3 and VEGFR2 in Angiogenesis. Angiogenesis 2009, 12, 177–185. 10.1007/s10456-009-9141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistini L.; Bugatti K.; Sartori A.; Curti C.; Zanardi F. RGD Peptide-Drug Conjugates as Effective Dual Targeting Platforms: Recent Advances. Eur. J. Org. Chem. 2021, 2021, 2506–2528. 10.1002/ejoc.202100240. [DOI] [Google Scholar]
- Sartori A.; Portioli E.; Battistini L.; Calorini L.; Pupi A.; Vacondio F.; Arosio D.; Bianchini F.; Zanardi F. Synthesis of Novel c(AmpRGD)-Sunitinib Dual Conjugates as Molecular Tools Targeting the αVβ3 Integrin/VEGFR2 Couple and Impairing Tumor-Associated Angiogenesis. J. Med. Chem. 2017, 60, 248–262. 10.1021/acs.jmedchem.6b01266. [DOI] [PubMed] [Google Scholar]
- Bianchini F.; Portioli E.; Ferlenghi F.; Vacondio F.; Andreucci E.; Biagioni A.; Ruzzolini J.; Peppicelli S.; Lulli M.; Calorini L.; Battistini L.; Zanardi F.; Sartori A. Cell-Targeted c(AmpRGD)-Sunitinib Molecular Conjugates Impair Tumor Growth of Melanoma. Cancer Lett. 2019, 446, 25–37. 10.1016/j.canlet.2018.12.021. [DOI] [PubMed] [Google Scholar]
- Sartori A.; Corno C.; De Cesare M.; Scanziani E.; Minoli L.; Battistini L.; Zanardi F.; Perego P. Effcacy of a Selective Binder of αVβ3 Integrin Linked to the Tyrosine Kinase Inhibitor Sunitinib in Ovarian Carcinoma Preclinical Models. Cancers 2019, 11, 531 10.3390/cancers11040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugatti K.; Bruno A.; Arosio D.; Sartori A.; Curti C.; Augustijn L.; Zanardi F.; Battistini L. Shifting Towards αVβ6 Integrin Ligands Using Novel Aminoproline-Based Cyclic Peptidomimetics. Chem. - Eur. J. 2020, 26, 13468–13475. 10.1002/chem.202002554. [DOI] [PubMed] [Google Scholar]
- Roth G. J.; Heckel A.; Colbatzky F.; Handschuh S.; Kley J.; Lehmann-Lintz T.; Lotz R.; Tontsch-Grunt U.; Walter R.; Hilberg F. Design, Synthesis, and Evaluation of Indolinones as Triple Angiokinase Inhibitors and the Discovery of a Highly Specific 6-Methoxycarbonyl-Substituted Indolinone (BIBF 1120). J. Med. Chem. 2009, 52, 4466–4480. 10.1021/jm900431g. [DOI] [PubMed] [Google Scholar]
- Determinationof IC50 values of compounds 1–3 and nintedanib against human recombinant VEGFR2 wasperformed at Eurofins Cerep SA laboratories; catalogue on line, https://www.eurofinsdiscoveryservices.com/catalogmanagement/viewItem/KDR-VEGFR2-KDR-Human-RTK-Kinase-Enzymatic-Radiometric-Assay-10-uM-ATP-KinaseProfiler/14-630KP10 (accessed December 22, 2021).
- Zanardi F.; Burreddu P.; Rassu G.; Auzzas L.; Battistini L.; Curti C.; Sartori A.; Nicastro G.; Menchi G.; Cini N.; Bottonocetti A.; Raspanti S.; Casiraghi G. Discovery of Subnanomolar Arginine-Glycine-Aspartate-Based αVβ3/αVβ5 Integrin Binders Embedding 4-Aminoproline Residues. J. Med. Chem. 2008, 51, 1771–1782. 10.1021/jm701214z. [DOI] [PubMed] [Google Scholar]
- Marshall C. Specificity of Receptor Tyrosine Kinase Signaling: Transient versus Sustained Extracellular Signal-Regulated Kinase Activation. Cell 1995, 80, 179–185. 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- D’Urso M.; Kurniawan N. A. Mechanical and Physical Regulation of Fibroblast–Myofibroblast Transition: From Cellular Mechanoresponse to Tissue Pathology. Front. Bioeng. Biotechnol. 2020, 8, 609653 10.3389/fbioe.2020.609653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogal R. K.; Bona C. A. Regulatory Effect of Extracellular Signal-Regulated Kinases (ERK) on Type I Collagen Synthesis in Human Dermal Fibroblasts Stimulated by IL-4 and IL-13. Int. Rev. Immunol. 2008, 27, 472–496. 10.1080/08830180802430974. [DOI] [PubMed] [Google Scholar]
- Michalik M.; Pierzchalska M.; Włodarczyk A.; Wójcik K. A.; Czyż J.; Sanak M.; Madeja Z. Transition of Asthmatic Bronchial Fibroblasts to Myofibroblasts Is Inhibited by Cell–Cell Contacts. Respir. Med. 2011, 105, 1467–1475. 10.1016/j.rmed.2011.04.009. [DOI] [PubMed] [Google Scholar]
- Chen T. R. In Situ Detection of Mycoplasma Contamination in Cell Cultures by Fluorescent Hoechst 33258 Stain. Exp. Cell Res. 1977, 104, 255–262. 10.1016/0014-4827(77)90089-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.