

Abstract
Organic compounds having significant nonlinear optical (NLO) applications are being employed in the optoelectronics field. In the current work, a series of non-fullerene acceptor (NFA) based compounds are designed by modifying the acceptors with different substituents using DTS(FBTTh2)2R1 as a reference compound. To study the NLO responses to the tuning of various acceptors, DFT and TD-DFT based parameters were calculated at the M06 level along with the 6-31G(d,p) basis set. The designed compounds (MSTD2–MSTD7) showed smaller values of the energy gap in comparison to the reference compound. The energy gaps of the title compounds were linked to global reactivity insights; MSTD7 provided a lower band gap, with smaller and larger quantities for hardness and softness characteristics, respectively. Further, UV–vis analyses were performed for all of the designed compounds, displaying wavelengths red-shifted from that of DTS(FBTTh2)2R1. The intraelectron transfer (ICT) process and stability of the title compounds were explored via frontier molecular orbital (FMO) and natural bond orbital (NBO) studies, respectively. Out of all the designed compounds, the highest value of linear polarizability ⟨α⟩ of 3.485 × 10–22 esu, first hyperpolarizability (βtotal) of 13.44 × 10–27 esu and second-order hyperpolarizability ⟨γ⟩ of 3.66 × 10–31 esu were exhibited by MSTD7. In short, all of the designed compounds exhibited promising NLO properties because of their low charge transport resistance. These NLO properties may be useful for experimental researchers to uncover NLO materials for modern applications.
Introduction
Nonlinear optically active chromophores have received more consideration in view of their importance in many technological applications in recent years. Organic-based scaffolds have a broad band spectrum, nonlinearity, and fast response time features. They are also extensively adapted for the storage of optical data, processing of images, and optical switching applications.1−8 A comparative study between the organic and inorganic nonlinear chromophores reveals that organic systems have many preferable properties such as a short response time, large molecular plasticity, outstanding processing performance, and high nonlinear polarization rate.9−12 To achieve the required nonlinear optical characteristics, researchers have concentrated on synthesizing organic molecules with certain geometries and electronic molecular characteristics.13−15
The delocalization of π electrons in the primary framework causes polarization in organic NLO based materials.16−18 In an organic molecule, electrons flow toward the donor via a π linker to the acceptor, consequently leading to intermolecular charge transfer (ICT).19,20 Indeed, changing the molecular structure to generate high-performance nonlinear optical materials is a smart method.21−23 As a result, organic compounds with a large conjugated delocalized π electronic density may be employed as the preferred species for second- and third-order nonlinear optics materials.24−26 It has also been observed from the data reported earlier that D (donor) and A (acceptor) moieties are responsible for providing the required ground-state charge asymmetry.27−29
Among the different series of delocalized electronic conjugated frameworks, fullerene and its conjugated derivatives are good candidates as has been reported by their NLO output.30−33 Fullerenes exhibit a reasonable NLO response due to π conjugation a general charge delocalization.33,34 Despite the fact that fullerene acceptors have been the most popular materials for two decades, the restricted electrical characteristics and moderate absorbing capacity of fullerene derivatives in the visible region has influenced the evolution of organic photovoltaic devices.35
In the past decade, non-fullerene acceptors (NFAs) have received a great deal of attention, primarily for research into the improvement of organic solar cells. To a large extent, in this area they have taken the spotlight away from fullerene derivatives. The organic acceptors, which replace the typical fullerene acceptors present in the photoactive layers of standard organic solar cells, have various advantages, including light absorption, donor–acceptor combination variety, and massive acceptor material manufacturing.36 For this, a great number of NFA species have been studied in the past decade for an exploration of their electro- and photoelectrochemical properties due to the strong electron transfer abilities of such derivatives.37 On the basis these fascinating features of π-conjugated NF-based compounds, we selected NF-based compounds in this work to explore their NLO properties from a theoretical perspective. To our knowledge, NLO research on the title NF-based molecule and its developed compounds has yet to be published. Hence, to overcome the research gap, calculations based on density functional theory (DFT) have been carried out to explore NLO properties. The NFA molecule DTS(FBTTh2)2R1(36) has been taken as a reference, and different NF-based acceptor compounds have been designed via coupling of various acceptors and π-conjugated linkers. The current work is extremely important for estimating the NLO applications of organic compounds. In addition to this, it helps to realize the consequences of the donor, various acceptors, and π linkers of non-fullerene-based compounds. In order to understand the NLO properties, DFT and TD-DFT calculations were performed to calculate the natural bonding orbitals (NBOs), global reactivity parameters (GRPs), frontier molecular orbitals (FMOs), densities of states (DOSs), and UV–vis absorption spectra. NFA molecules are considered to play a substantial role in the arena of NLO.
Computational Study
The current work was completed employing the Gaussian 09 program38 at the M06 level of theory and the 6-31G(d,p) basis set. In dichloromethane (DCM) solvent, the DFT calculations were carried out to determine the data for FMO, NBO, the density of state (DOS), absorption spectra, and NLO properties of fullerene-free molecules having a D−π–D–A−π–A configuration. An FMO analysis was performed to determine the band gap which enables an estimation of the least amount of energy needed for the transition from the HOMO to the LUMO. An NBO analysis was employed to determine the hyperconjugative interactions and intramolecular charge transfers. The DOS calculations were used to determine the scattering of energy states. UV–visible absorption spectra were obtained to investigate electronic transitions. The dipole moment (μ), linear polarizability ⟨α⟩, first-hyperpolarizability (β), and second-hyperpolarizability ⟨γ⟩ values were calculated by employing eqs 1–4.39,40
| 1 |
| 2 |
| 3 |
Tensors of β in three directions are described in eqs S1–S3 in the Supporting Information.
| 4 |
The results were obtained by utilizing Gauss View,41 Avogadro,42 and ChemCraft43 programs from the output files.
Results and Discussion
The present work focuses on the NLO characteristics of non-fullerene-based organic scaffolds. The no-nfullerene-based reference compound DTS(FBTTh2)2R1 has a D−π–A–D–A−π–D type configuration that contains three fragments: donors, π spacers, and acceptors. The branches C8H17 (n-octyl) and C6H13 (n-hexyl) in DTS(FBTTh2)2R1 have been altered with CH3 (methyl) at the donor part to overcome the computational cost, as shown in Figure 1. The theoretically designed compounds MSTD2–MSTD7 derived from DTS(FBTTh2)2R1 having a D−π–D–A−π–A type configuration with various acceptors and π spacers are depicted in Scheme 1. As we have already mentioned, that 7,7′-(4,4-dimethyl-4H-silolo[3,2-b:4,5-b′]dithiophene-2,6-diyl)bis(6-fluoro-4-(5′-methyl-[2,2′-bithiophen]-5-yl)benzo[c][1,2,5]thiadiazole) DTS(FBTTh2)2R1 is considered as a reference moiety. First of all, we designed MSTD2 from DTS(FBTTh2)2R1 that comprises 4-methyl-3a,4-dihydrocyclopenta[b]indole (MDCI) as the first donor moiety, 7,7-dimethyl-7H-3,4-dithia-7-silacyclopenta[a]pentalene (MTSCP) as the second donor moiety, 5-phenyl[2,2′]bithiophenyl (PTP) as the first π spacer, a bithiophenyl ring (BTP) as the second π-spacer, 5-fluorobenzo[1,2,5]thiadiazole (FBT) as the first acceptor moiety, and 3-dinitromethylene-2-methyleneindan-1-one (DNMI) as the second acceptor moiety. In the designed compounds MSTD3–MSTD7, the first donor moiety (MDCI), second donor moiety (MTSCP), first π spacer (PTP), second π spacer (BTP), and the first acceptor moiety (FBT) are retained for design purposes but the second acceptor is changed with alternative acceptors, as indicated in Scheme 1 and Figure 1. In this context, the NLO properties (i) polarizability ⟨α⟩ and (ii) hyperpolarizability (β) by DFT and (iii) absorption wavelength by TD-DFT were determined to offer information on how different acceptors interact and fixed MDCI, MTSCP, PTP, BTP, and FBT units affect the NLO responses.
Figure 1.

Modification of DTS(FBTTh2)2 to DTS(FBTTh2)2R1.
Scheme 1. Sketch of the Designed Compounds.

The molecular structures and optimized geometries of DTS(FBTTh2)2R1 and MSTD2-MSTD87 are provided in Figures S1 and S2 in the Supporting Information. The Cartesian coordinates are also given in Tables S1–S7.
Electronic Structure
The study of frontier molecular orbitals (FMOs) is extremely significant for an understanding of the optical and electric characteristics of species, and it is also utilized to investigate their chemical stability.44−46 The highest occupied molecular orbitals are referred to as HOMOs, while the lowest unoccupied molecular orbitals are referred to as LUMOs, which are very significant FMOs.47−52 An FMO analysis enables us to determine significant quantum chemistry parameters, including electronic properties, chemical stabilities, electron transfer properties, and reactivities, of the investigated compounds.53 The energy difference (Egap = ELUMO – EHOMO) of compounds is directly associated with their kinetic stability and chemical reactivity.54,55 The Egap values also controls factors, including electronegativity, electrophilicity, delicate quality, hardness, reactivity, and softness. The lower the value of energy difference (ΔE), the higher the value of polarizability, resulting in a magnificent NLO value.55−58Table 1 gives the Egap values for the compounds under investigation.
Table 1. EHOMO, ELUMO, and the Energy Gap ΔE (ELUMO – EHOMO) of the Designed Compounds in eV.
| compound | EHOMO | ELUMO | ΔE |
|---|---|---|---|
| DTS(FBTTh2)2R1 | –5.119 | 2.788 | 2.331 |
| MSTD2 | –5.238 | –3.403 | 1.835 |
| MSTD3 | –5.248 | –3.528 | 1.720 |
| MSTD4 | –5.240 | –3.463 | 1.777 |
| MSTD5 | –5.234 | –3.416 | 1.818 |
| MSTD6 | –5.265 | –3.774 | 1.491 |
| MSTD7 | –5.250 | –3.668 | 1.582 |
It is concluded from the data presented in Table 1 that DTS(FBTTh2)2R1 has a theoretically estimated energy difference (ΔE) value, i.e., 2.331 eV, greater than the Egap values obtained in the designed molecules. The donors, acceptors, π spacers, and end chain acceptors are involved in the configuration of the designed compounds, which are the reasons for the reduction of their energy difference (ΔE) values. The Egap value for MSTD2 decreased to 1.835 eV because of the structural modification with 7-([2,2′-bithiophen]-5-yl)-4-(4,4-dimethyl-6-(4-(5′-(9-methyl-9H-carbazol-2-yl)[2,2′-bithiophen]-5-yl)phenyl)-4H-silolo[3,2-b:4,5-b′]dithiophen-2-yl)-5-fluorobenzo[c][1,2,5]thiadiazole as donors and π spacers and 3-dinitromethylene-indan-1-one (DNMI) as an acceptor unit. In MSTD3 and MSTD4, Egap values further decreased to 1.720 and 1.777 eV by the introduction of two and one chloro species on the acceptor unit DNMI, respectively, in which two chloro groups were found to be more effective in decreasing the band gap in comparison to one chloro grouop. This Egap value in MSTD5 slightly increased to 1.818 eV due to the introduction of one fluoro group on the acceptor part (DNMI). This increase in the Egap value was perhaps due to less enhancement of the resonance effect induced due to a fluoro group as compared to a chloro group. As the fluoro and chloro groups are electron donating due to resonance effects, the compounds MSTD3, MSTD4, and MSTD5 have decreases in Egap values by enhancement of resonance and electron withdrawal toward the acceptor region.59,60
Furthermore, MSTD6 and MSTD7 were also found to have smaller Egap values in comparison to DTS(FBTTh2)2R1 and MSTD1–MSTD5 due to the presence of nitro groups in the DNMI acceptor. The presence of two and three nitro groups in MSTD6 and MSTD7, respectively, reduced the Egap values to 1.491 and 1.582 eV. The nitro group has a strongly electron attracting character, and its electron-attracting ability is a consequence of a strong negative inductive activity.61 The observed lowest Egap valueof MSTD6 among all designed compounds is because of the extensive conjugation involved in the biphenyl rings and the end-capped acceptor part comprising nitro units.
The ascending order of Egap of the title compounds is MSTD6 < MSTD7 < MSTD3 < MSTD4 < MSTD5 < MSTD2 < DTS(FBTTh2)2R1. Overall, MSTD2–MSTD7 have small energy gaps in comparison to DTS(FBTTh2)2R1. This decrease in band gap can be attributed to the presence of electronegative species (fluoro, chloro, and nitro groups) on the acceptor units, which can effectively remove electronic charge density from other portions, lowering the band gap (Table 1). The potential of the designed compounds to perform intramolecular charge transfer (ICT) from donor to acceptor units through π linkers is further effectively found in frontier molecular orbital studies as coupled with the Egap values.
The Egap values define the ICT experience from the donor to the acceptor component, which is aided by the π spacer, which gives a deep insight into the related functional associations of the NLO structures.62,63 The contour surfaces of FMOs are utilized to define the transfer of electronic charges, as shown in Figure 2. In the reference compound (DTS(FBTTh2)2R1), the highest occupied molecular orbital electronic density is mostly present over the entire molecule, donors, acceptors, and π spacers, while the LUMO is mostly located on acceptors and π spacers and to some extent on the donor unit. In MSTD2–MSTD5, the electronic density of the LUMO is only found on the acceptor unit and partially on the second π linker. The electronic population in the HOMO is found mostly on 4-(6-(4-([2,2′-bithiophen]-5-yl)-4,4-dimethyl-4H-silolo[3,2-b:4,5-b′]dithiophen-2-yl)-5-fluorobenzo[c][1,2,5]thiadiazole (on the first π spacer, second donor, and first acceptor ). In addition to this, in MSTD6 and MSTD7 the electronic density of the LUMO is located perfectly over the various terminal acceptor moieties, while in the HOMO, the charge density is largely positioned over 4-(6-(4-([2,2′-bithiophen]-5-yl)-4,4-dimethyl-4H-silolo[3,2-b:4,5-b′]dithiophen-2-yl)-5-fluorobenzo[c][1,2,5]thiadiazole (on the first π-spacer, second donor, and first acceptor). The NLO response is assumed to be dependent on charge transfer from the donor to the acceptor via the π spacer. MSTD2–MSTD7 may be considered as suitable NLO candidates as a result of this effect.
Figure 2.

HOMO–LUMO structures of the reference DTS(FBTTh2)2R1 and its derivatives.
Global Reactivity Parameters
The HOMO–LUMO energy gap may be used to describe the reactivity and stability of the designed compounds using global reactivity parameters (GRP).14 The energy gap of FMOs (Egap = ELUMO – EHOMO) may be used to calculate global reactivity parameters such as ionization potential (IP), electron affinity (EA), electronegativity (X),64,65 global hardness (η),66 electrophilicity index (ω), chemical potential (μ), and global softness (σ).67 The ionization potential refers to a compound’s ability to donate electrons, while the electronegativity refers to its ability to attract electrons. Compounds with a higher value of chemical potential (μ) and hardness (η) are considered to be kinetically stable molecules. Additionally, these factors both have a direct relationship with the Egap values of orbitals and are inversely linked with the global softness (σ).66−68 GRPs can be calculated by applying eqs 5 and 6(64,65)
| 5 |
| 6 |
where IP describes the ionization potential (in au) and EA describes the electron affinity (in au).
The electronegativity (X),64,65 chemical hardness (η),66 and chemical potential (μ) have been calculated with the help of the Koopmans theorem67 and presented in the form eqs 7–9.
| 7 |
| 8 |
| 9 |
The global softness (σ) can be calculated with the help of eq 10.67
| 10 |
Parr et al.68 presented an electrophilicity index that can be designated by eq 11.
| 11 |
MSTD7 has the highest IP value among the proposed compounds, 0.1866 Eh (Table 2), indicating that the donor moiety efficiently distributes the electronic charge density to the acceptor, while MSTD2 has the lowest IP value of 0.1753 Eh. Overall, the ascending order of the ionization potential is MSTD2 < MSTD3 < MSTD5 < MSTD4 < MSTD6 < MSTD7 < DTS(FBTTh2)2R1. The highest value of chemical hardness (η) is shown by DTS(FBTTh2)2R1 (0.0428 Eh) which decreases to 0.0324 Eh in MSTD7. The descending order of chemical hardness is represented as MSTD7 < MSTD3 < MSTD2 < MSTD6 < MSTD5 < MSTD4 < DTS(FBTTh2)2R1.
Table 2. Ionization Potential (IP), Electron Affinity (A), Electronegativity (X), Global Hardness (η), Chemical Potential (μ), Global Electrophilicity (ω), and Global Softness (σ)a.
| compound | IP | EA | X | η | μ | ω | σ |
|---|---|---|---|---|---|---|---|
| DTS(FBTTh2)2R1 | 0.1881 | 0.1025 | 0.1453 | 0.0428 | –0.1453 | 0.2465 | 11.6775 |
| MSTD2 | 0.1753 | 0.1056 | 0.1405 | 0.0349 | –0.1405 | 0.2828 | 14.3339 |
| MSTD3 | 0.1792 | 0.1042 | 0.1417 | 0.0337 | –0.1417 | 0.2677 | 13.3295 |
| MSTD4 | 0.1819 | 0.1033 | 0.1423 | 0.0389 | –0.1423 | 0.2600 | 12.8473 |
| MSTD5 | 0.1794 | 0.1038 | 0.1416 | 0.0378 | –0.1416 | 0.2652 | 13.2288 |
| MSTD6 | 0.1845 | 0.1143 | 0.1494 | 0.0351 | –0.1494 | 0.3181 | 14.2489 |
| MSTD7 | 0.1866 | 0.1218 | 0.1542 | 0.0324 | –0.1541 | 0.3669 | 15.4371 |
All values are given in hartree (Eh) units.
The investigated compounds have lower global softness values (σ) in comparison to their chemical hardness (η) and electrophilicity index (ω) values. The highest σ value is exhibited by MSTD7, i.e., 15.4371 Eh, whereas DTS(FBTTh2)2R1 has the lowest σ value of 11.677 Eh. In all examined compounds, the increasing trend of global softness (σ) values is reported as DTS(FBTTh2)2R1 < MSTD4 < MSTD5 < MSTD3 < MSTD6 < MSTD2 < MSTD7.
Natural Bond Orbital (NBO) Analysis
An NBO analysis helps in the detection of hydrogen bonds, originating from hyperconjugated interactions in the designed molecules.52,69 It provides a true image for investigating the intramolecular delocalization and the shifting of electronic charge density from the donor to the acceptor region of the system in D−π–A architectures.56,58 With a larger value of the energy of stabilization, the interaction between the donor and acceptor moieties is very significant.70Equation 12 may be used to represent the stablization energy formula using a second-order perturbation approach71
| 12 |
where E(2) describes the stabilization energy, qi represents the occupancy of the donor orbital, Fi,j shows the off-diagonal NBO Fock matrix elements, and εj and εi signify the diagonal elements.72 For the discovery of intra-atomic and subatomic holding and connection among bonds, an NBO examination is an effective procedure and a helpful premise for researching conjugative collaboration or charge movement in subatomic frameworks.70 An NBO analysis for DTS(FBTTh2)2R1 and MSTD2–MSTD7 has been carried out and elaborated in Tables S10–S16 in the Supporting Information, and some selected values are given in Table 3.
Table 3. Selected Values of NBO Analysis for DTS(FBTTh2)2R1 and MSTD2–MSTD7a.
| compound | donor (i) | type | acceptor (j) | type | E(2) | E(j)E(i) (au) | Fi,j (au) |
|---|---|---|---|---|---|---|---|
| DTS(FBTTh2)2R1 | C11–C12 | π | C13–N20 | π* | 26.73 | 0.27 | 0.080 |
| C5–C6 | π | C5–C6 | π* | 0.68 | 0.30 | 0.013 | |
| N20–S51 | σ | C12–C13 | σ* | 8.32 | 1.27 | 0.092 | |
| C7–C8 | σ | C6–S9 | σ* | 0.50 | 0.95 | 0.020 | |
| S9 | LP(2) | C5–C6 | π* | 27.05 | 0.28 | 0.078 | |
| N19 | LP(1) | C13–C14 | σ* | 7.67 | 0.94 | 0.076 | |
| MSTD2 | C1–C2 | π | C83–C113 | π* | 26.69 | 0.31 | 0.082 |
| N115–O117 | π | C90–C91 | π* | 0.53 | 0.33 | 0.012 | |
| C1–C2 | σ | C36–C37 | σ* | 8.35 | 1.27 | 0.092 | |
| N115–O117 | σ | C29–S32 | σ* | 0.51 | 0.95 | 0.02 | |
| S9 | LP(1) | N115–O116 | π* | 21.34 | 0.74 | 0.113 | |
| S9 | LP(2) | N115–O117 | σ* | 177.19 | 0.16 | 0.152 | |
| MSTD3 | C60–C61 | π | C83–C113 | π* | 27.99 | 0.3 | 0.083 |
| N115–O117 | π | C88–C89 | π* | 0.66 | 0.51 | 0.018 | |
| S50–N51 | σ | C36–C37 | σ* | 8.35 | 1.27 | 0.092 | |
| C26–C27 | σ | C29–S32 | σ* | 0.51 | 0.95 | 0.02 | |
| O116 | LP (3) | N115–O117 | π* | 177.93 | 0.16 | 0.153 | |
| O87 | LP (2) | C77–C86 | σ* | 21.89 | 0.75 | 0.116 | |
| MSTD4 | C60–C61 | π | C83–C116 | π* | 27.48 | 0.3 | 0.082 |
| C87–C88 | π | C87–C88 | π* | 0.52 | 0.33 | 0.012 | |
| S50–N51 | σ | C36–C37 | σ* | 8.34 | 1.27 | 0.092 | |
| C1–C2 | σ | C4–S10 | σ* | 0.5 | 0.95 | 0.02 | |
| O94 | LP(3) | N92–O93 | π* | 177.6 | 0.16 | 0.153 | |
| O86 | LP(2) | C77–C85 | σ* | 21.76 | 0.75 | 0.116 | |
| MSTD5 | C60–C61 | π | C83–C116 | π* | 27.18 | 0.3 | 0.082 |
| C87–C88 | π | C87–C88 | π* | 0.58 | 0.33 | 0.013 | |
| S50–N51 | σ | C36–C37 | σ* | 8.34 | 1.27 | 0.092 | |
| C1–C2 | σ | C4–S10 | σ* | 0.5 | 0.95 | 0.02 | |
| O94 | LP(3) | N92–O93 | π* | 177.56 | 0.16 | 0.152 | |
| O90 | LP(2) | C88–N89 | σ* | 14.35 | 0.61 | 0.084 | |
| MSTD6 | C60–C61 | π | C83–C117 | π* | 30.34 | 0.3 | 0.085 |
| C37–C38 | π | C37–C38 | π* | 0.62 | 0.31 | 0.013 | |
| S50–N51 | σ | C36–C37 | σ* | 8.36 | 1.27 | 0.092 | |
| C21–C22 | σ | C20–S25 | σ* | 0.51 | 0.94 | 0.02 | |
| O105 | LP(3) | N104–O106 | π* | 186.34 | 0.17 | 0.16 | |
| O102 | LP(2) | C81–N101 | σ* | 15.08 | 0.58 | 0.083 | |
| MSTD7 | C60–C61 | π | C83–C118 | π* | 32.77 | 0.3 | 0.09 |
| C110–C111 | π | C110–C111 | π* | 0.71 | 0.28 | 0.013 | |
| C118–H119 | σ | C60–S66 | σ* | 9.48 | 0.73 | 0.074 | |
| C26–C27 | σ | C29–S32 | σ* | 0.50 | 0.95 | 0.019 | |
| O90 | LP(3) | N89–O91 | π* | 179.75 | 0.16 | 0.154 | |
| O90 | LP(2) | C88–N89 | σ* | 14.68 | 0.58 | 0.082 |
LP denotes the lone pair (i) donor and (j) acceptor; E(2) denotes the energy of a hyperconjugative interaction (stabilization energy). E(j) – E(i) is the energy difference between the donor (i) and acceptor (j) NBO orbitals. Fi,j is the Fock matrix element between i and j NBO orbitals.
Results cited in Table 3 indicate that the maximum value of a probable transition was observed in DTS(FBTTh2)2R1: i.e., π(C11–C12) → π*(C13–N20) at 26.73 kcal/mol has been detected to be the most reliable transition in directing the maximum stabilization energy and a powerful association of donor (π) and acceptor (π*) units.
With a stabilization energy value of 0.68 kcal/mol, the transition from π(C5–C6) to π*(C5–C6) was termed as the least stable. Similarly, transitions such as σ(N20–S51) → σ*(C12–C13) and σ(C7–C8) → σ*(C6–S9) have larger and smaller stabilization energy values of 8.32 and 0.50 kcal/mol, respectively. A weak contact between an electron donor (σ) and acceptor (σ*) causes the outputs of these stabilization energies.
When the phenomenon of resonance is taken into account, electronic interactions such as LP2(S9) → π*(C5–C6) with the highest value of the energy of stabilization, 27.05 kcal/mol and LP1(N19) → σ*(C13–C14) with the lowest energy value of 7.67 kcal/mol were detected in the reference molecule DTS(FBTTh2)2R1, and the data are presented in Table 3.
In compound MSTD2, the transition π(C1–C2) → π*(C83–C113) has the highest stabilization energy of 26.69 kcal/mol while the transition π(N115–O117) → π*(C90–C91) has the lowest energy of 0.53 kcal/mol. Hence, σ(C1–C2) → σ*(C36–C37) and σ(N115–O117) → σ*(C29–S32) transitions in MSTD2 had the largest stabilization energy of 8.35 kcal/mol and the lowest stabilization energy of 0.51 kcal/mol, respectively. As indicated in Table 3, the LP2(S9) → σ*(N115–O117) transition due to resonance has an energy of stabilization of 177.19 kcal/mol, whereas the LP(1)(S9) → π*(N115–O116) transition in MSTD2 has a stabilization energy of 21.34 kcal/mol.
The compound MSTD3 showed the transition π(C60–C61) → π*(C83–C113) had the maximum value of the stabilization energy (27.99 kcal/mol) and the π(N115–O117) → π*(C88–C89) transition had the lowest value of stabilization energy (0.66 kcal/mol).
For the σ(C26–C27) → σ*(C29–S32) transition, MSTD3 has the lowest stabilization energy value of 0.51 kcal/mol because of the weaker degree of association between σ(donor) and σ*(acceptor). In addition to this, LP(3)(O116) → π*(N115–O117) and LP2(O87) → σ*(C77–C86) also have high and low stabilization energies of 177.93 and 21.89 kcal/mol, respectively.
For MSTD4, the transition π(C60–C61) → π*(C83–C116) has a maximum value of stabilization energy of 27.48 kcal/mol, while π(C87–C88) → π*(C87–C88) has the lowest value of stabilization energy of 0.52 kcal/mol. The highest and lowest energy values, 8.34 and 0.5 kcal/mol, exhibited by MSTD4 are due to σ(S50–N51) → σ*(C36–C37) and σ(C1–C2) → σ*(C4–S10) transitions, respectively. The highest stabilization energy values (177.6 and 21.76 kcal/mol) are because of resonance, which is shown in LP(3) O94) → π*(N92–O93) and LP2(O86) → σ*(C77–C85) transitions, respectively (Table 3).
The most prominent transition for π → π* exhibited by MSTD5 is π(C60–C61) → π*(C83–C116) with a stabilization energy of 27.18 kcal/mol, while the transition π(C87–C88) → π*(C87–C88) with an energy value of 0.58 kcal/mol is shown to have the lowest value of stabilization energy. Furthermore, the σ(S50–N51) → σ*(C36–C37) transition exhibited the maximum stabilization energy, 8.34 kcal/mol; on the other hand, σ(C1–C2) → σ*(C4–S10) with an energy value of 0.5 kcal/mol is found to have the minimum value of the stabilization energy. However, the highest 177.56 kcal/mol and the lowest 14.35 kcal/mol stabilization energy values are shown by LP(3)(O94) → π*(N92–O93) and LP2(O90) → σ*(C88–N89) transitions because of resonance phenomena.
A similar trend of stabilization energies is observed in MSTD6, with the maximum probable π → π* transition being π(C60–C61) → π*(C83–C117) with a stability value of 30.34 kcal/mol. Furthermore, the transition π(C37–C38) → π*(C37–C38) with an energy value of 0.62 kcal/mol is shown to have the lowest stability. The transitions σ(S50–N51) → σ*(C36–C37) and σ(C21–C22) → σ*(C20–S25) have the highest and lowest energy values of 8.36 and 0.51 kcal/mol, respectively, because of enervated connections between σ(donor) and σ*(acceptor). In addition to this, other electronic transitions such as LP(3)(O105) → π*(N104–O106) and LP2(O102) → σ*(C81–N101) had stabilization energies of 186.34 and 15.08 kcal/mol, respectively, due to resonance, as shown in Table 3.
The highest stabilization energy value for the π(C60–C61) → π*(C83–C118) transition is 32.77 kcal/mol, whereas the lowest stabilizationb energy value for the π(C110–C111) → π*(C110–C111) transition is 0.71 kcal/mol in MSTD7. Due to the presence of weak interactions, 9.48 and 0.50 kcal/mol are found for the transitions σ(C118–H119) → σ*(C60–S66) and σ(C26–C27) → σ*(C29–S32), respectively.
Due to resonance, the transitions LP(3)(O90) → π*(N89–O91) and LP2(O90) → σ*(C88–N89) have the highest stabilization energy values of 179.75 and 14.68 kcal/mol. Tables S10–S16 demonstrate that more transitions have been investigated in DTS(FBTTh2)2R1 and MSTD2–MSTD7. Furthermore, the results showed that the π electron bonding orbitals of C–C delocalized antibonding orbitals play a significant role in the ring stability and overall structural design. As a result, it is safe to say that extended conjugation is found in MSTD2–MSTD7 and that it is necessary for the stabilization of these systems and giving matchless NLO features.
Density of State (DOS)
The distinct kinds of states inhabited by electrons at a quantized energy level, i.e., the different kinds of electronic states per unit volume per unit energy, are known as the densities of state (DOSs). An extraordinary high value of the DOS reveals that different kinds of states are accessible for energy levels. The zero value along the axis illustrates that no state is available for electron excitation. DOS calculations may be used to determine the general scattering of energy levels as a function of energy, as well as the energy gap.73 The DOS plots of DTS(FBTTh2)2R1 and MSTD2–MSTRD7 at the M06/6-31G(d,p) level of theory are given in Figure 3.
Figure 3.

DOS plots of DTS(FBTTh2)2R1 and MSTD2–MSTD7.
For reference compound DTS(FBTTh2)2R1 the contribution by the acceptor group seems to be 43.2% to the HOMO and 14.7% for the LUMO. Here, donors contribute 44.1%, 44.2%, 44.1%, 44.4%, 43.3%, and 43.7% to the HOMO and 2.0%, 1.7%, 2.5%, 2.6%, 4.3% and 9.2% to the LUMO in MSTD2–MSTD7, respectively. The acceptor contributes 10.9%, 10.6%, 10.5%, 10.7%, 10.0%, and 11.6% to the HOMO and 76.1%, 76.4%, 75.8%, 74.9%, 77.1%, and 83.2% to the LUMO for MSTD2–MSTD7, respectively. The π linkers contribute 45%, 45.2%, 45.3%, 44.8%, 46.7%, and 44.9% to the HOMO and 21.9%, 21.9%, 21.7%, 22.6%, 18.6%, and 16.4% for the LUMO for MSTD2–MSTD7, respectively. These contributions show that the design of different compounds by varying different efficient acceptor moieties has a leading role in the transmittance of an electronic charge cloud in different ways. Each compound is split into three respective segments: i.e., donor, π spacer, and acceptor. For compound MSTD2, the highest charge density of the HOMO is found to be between −7 and −12 eV and it seems to be on the π spacer and the highest density of the LUMO is found to be −1.5 eV by the donor group, which confirms the efficient charge transfer from the donor to the acceptor group through the π spacer. In the case of MSTD3, the highest HOMO density is found to be −7 eV, which is 45.2% on the π spacer, while the highest LUMO density is found to be on the acceptor which makes a 76.4% charge contribution. For MSTD4 and onward, a maximum HOMO density of around −7 to −14 eV is due to the acceptor and the maximum LUMO density of around 2–4 eV is restricted by the π spacer, which increases the electron-withdrawing nature and also extends the conjugation. All of the results from a DOS analysis strongly support the same results computed from an FMO analysis.
UV–Vis Analysis
UV–vis spectroscopy offers further evidence with regard to different types of transitions in the compounds, such as maximum absorption wavelength (λmax), transition energy (E), and oscillator strength (fos).74 These parameters are determined by TD-DFT calculations at the M06/6-31G(d,p) level in the gaseous phase and are given in Table 4, while further findings are presented in Tables S21–S27. A pictographic illustration of the absorption shifts of DTS(FBTTh2)2R1 and MSTD2–MSTD7 is given in Figure 4.
Table 4. TD-DFT-Calculated Transition Energies (eV), Maximum Absorption Wavelengths (λmax, nm), Oscillator Strengths (fos) and Transition Natures of the Designed Compounds.
| Compounds | λ | E | fos | MO contributions |
|---|---|---|---|---|
| DTS(FBTTh2)2R1 | 711.408 | 1.743 | 2.092 | H → L (94%), H-1 → L+1 (5%) |
| MSTD2 | 810.190 | 1.530 | 1.589 | H → L (83%), H-1 → L (8%) |
| MSTD3 | 859.563 | 1.442 | 1.396 | H → L (87%), H-1 → L (7%) |
| MSTD4 | 833.726 | 1.487 | 1.479 | H → L (85%), H-1 → L (8%) |
| MSTD5 | 818.264 | 1.515 | 1.592 | H → L (84%), H-1 → L (8%) |
| MSTD6 | 780.997 | 1.588 | 1.228 | H-1 → L (27%), H → L+1 (52%) |
| MSTD7 | 930.247 | 1.333 | 1.009 | H → L (91%), H-1 → L (5%) |
Figure 4.
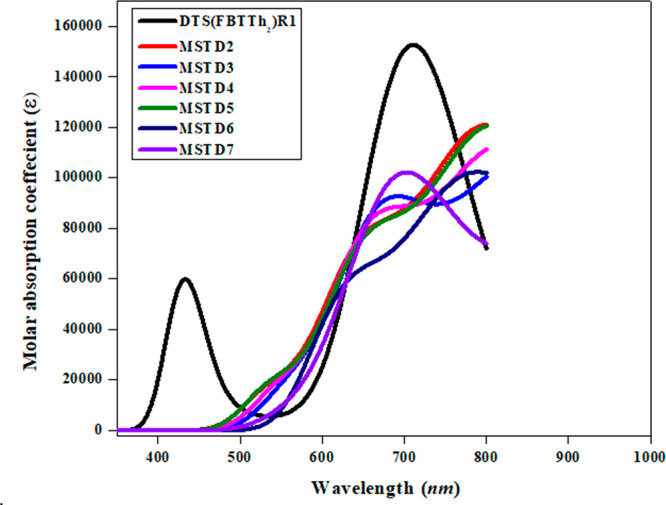
Absorption plots of DTS(FBTTh2)2R1 and MSTD2–MSTD7.
Table 4 shows that the energy and maximum absorption wavelengths (λmax) are inversely proportional to each other.75 The calculations of absorption spectra of designed compounds (MSTD2–MSTD7) and reference compound DTS(FBTTh2)2R1 at M06/6-31G(d,p) level of theory were performed. The maximum absorption wavelength (λmax), oscillator strength (fos), and excitation energy, calculated in dichloromethaneare presented in Table 4. The λmax values of the compounds under investigation range from 711.408 to 930.247 nm. The maximum absorption was observed at 711.408 nm for the reference compound, whereas the absorption values (λmax) of MSTD2–MSTD7 are 810.190, 859.563, 833.726, 818.264, 780.997, and 930.247 nm, respectively. MSTD7 has a λmax value of 930.247 nm, and there is a small value of the energy gap (1.582 eV) from DTS(FBTTh2)2R1 because of the presence of an end-capped electron-withdrawing acceptor group.
The increasing order of the designed molecules on the basis of λmax is MSTD6 < MSTD2 < MSTD5 < MSTD4 < MSTD3 < MSTD7. It is observed that the reference molecule DTS(FBTTh2)2R1 has the lowest λmax value among the designed molecules. Experimentally, the absorption spectra of reference molecule obtained correlate with the simulated results. It is obvious from the results that electron-withdrawing end-capped acceptor moieties have a significant effect on λmax values. These electron-deficient moieties are responsible for the red shift in absorption spectra. Absorption band lengths of MSTD2–MSTD7 ranges from 780.997 to 930.247 nmm which are red-shifted relative to the reference molecule DTS(FBTTh2)2R1. The designed compounds MSTD3 and MSTD7 have a greater red shift with the lowest excitation energies of 1.442 and 1.333 eV, respectively. Due to the small energy gap, charge transfer is feasible. The λmax values of MSTD2, MSTD4, and MSTD5 are closely related, having an oscillation strength of ∼1.5. The excitation energies of MSTD2-MSTD7 are 1.530, 1.442, 1.487, 1.515, 1.588, and 1.333 eV, respectively, which are lower in comparison to that of the reference compound DTS(FBTTh2)2R1: i.e., 1.743 eV.
As a result of the above explanation, it can be inferred that the newly proposed molecules have better optoelectronic characteristics in comparison to the reference molecule. It is believed that all of the newly designed compounds have good excitation energies, oscillator strengths, and absorptions. Thus, MSTD2–MSTD7 can be used as significant non-fullerene acceptor moieties in photovoltaic devices.
Nonlinear Optical (NLO) Properties
The nonlinear optical (NLO) response of organic compounds with adequate electronic communication across their various moieties is rather good.76,77 In research communities, several experimental and computational scientists have focused on the fields of material science, physics, and chemistry.78−80 Because of the increasing need for optical modulation, harmonic production, frequency mixing, and associated statistics, interdisciplinary efforts have been made to produce NLO materials.81−84 The intensity of the optical reactions to record a progressive linear response (polarizability, α) and the nonlinear responses (hyperpolarizabilities; β, γ, etc.) was predicted to be based on the electronic properties.85,86 Herein, Table 5 shows the computed μtotal, ⟨α⟩, βtotal and ⟨γ⟩ values of the designed compounds. Tables S17–S20 provide the findings for all tensors.
Table 5. Dipole Moment, Average Polarizability, First Hyperpolarizability, Second-Order Hyperpolarizability, and Major Contributing Tensors (esu).
| compound | μtotal | ⟨α⟩ × 10–22 | βtotal × 10–27 | ⟨γ⟩ × 10–31 |
|---|---|---|---|---|
| DTS(FBTTh2)2R1 | 0.3 | 2.123 | 0.008118 | 0.403 |
| MSTD2 | 5.26 | 3.098 | 6.763 | 1.45 |
| MSTD3 | 6.13 | 3.277 | 8.730 | 2.02 |
| MSTD4 | 5.20 | 3.184 | 7.684 | 1.71 |
| MSTD5 | 4.92 | 3.131 | 7.221 | 1.57 |
| MSTD6 | 8.72 | 3.485 | 13.44 | 3.66 |
| MSTD7 | 6.39 | 3.296 | 10.39 | 2.67 |
Urea was employed as a reference material for a comparison of the initial hyperpolarizability and dipole moment values of MSTD2–MSTD7, and they were found to be 5.26, 6.13, 5.20, 4.92, 8.72, and 6.39 D, respectively, according to a review of the literature. It is found that the value of dipole momenst of all designed compounds are greater relative to urea (1.373 D).87 The dipole moment of MSTD6 is the highest (8.72 D) among all of the title compounds. The overall decreasing order of values of dipole moments is MSTD6 > MSTD7 > MSTD3 > MSTD2 > MSTD4 > MSTD5 > DTS(FBTTh2)2R1. The decreasing order of average linear polarizability values of the title compounds was observed to be MSTD6 > MSTD7 > MSTD3 > MSTD4 > MSTD5 > MSTD2 > DTS(FBTTh2)2R1. For the proposed compound MSTD6, the average polarizability was found to be the greatest. An extraordinary increment in its value was found, i.e., 3.485 × 10–22 esu. This was because of the presence of three nitro groups that enriched the electron density near the acceptor unit and enhanced the electron-withdrawing ability. The β factor generally increases with an increase in the strength of the substituents such as fluoro, chloro, and cyano groups attached to the acceptor unit 3-dinitromethyleneindan-1-one (DNMI), which affects the nonlinearity of a compound. Furthermore, as the substitutions take place, the contribution of the conjugated system is extended and β is more dominant. The decreasing order of βtotal value for all designed compounds was as follows: MSTD6 > MSTD7 > MSTD3 > MSTD4 > MSTD5 > MSTD2 > DTS(FBTTh2)2R1. Additionally, MSTD6 has highest value of βtotal (13.34 × 10–27 esu) in comparison to the other designed compounds. In addition to this, a relative study was also carried out by using urea as a standard compound, whose βtotal value is found to be 3.714 × 10–31 esu.88 The first-hyperpolarizability values of MSTD2–MSTD7 are 8.33 × 10–25, 1.07 × 10–24, 9.46 × 10–25, 8.89 × 10–25, 1.65 × 10–25, and 1.27 × 10–25 times greater, respectively, in comparison to the reference compound DTS(FBTTh2)2R1 and 1.82 × 104, 2.35 × 104, 2.06 × 104, 1.94 × 104, 3.61 × 104, and 2.79 × 104 times greater, respectively, in comparison to the βtotal value of urea. Briefly, for MSTD2–MSTD7 because of the incorporation of stronger electron-withdrawing species in the acceptor part, the NLO response exposure is higher ,which might minimize the HOMO–LUMO energy gap, increase the ICT between the orbitals, and make the molecule more polarized.
The reverse order of the Egap between the HOMO and LUMO orbitals revealed the second-order hyperpolarizability ⟨γ⟩ values of all proposed compounds. MSTD6 has the highest value of ⟨γ⟩ (3.66 × 10–31 esu). The following is the descending order of all of the created compounds: MSTD6 > MSTD7 > MSTD3 > MSTD4 > STD5 > MSTD2 > DTS(FBTTh2)2R1. The current research provides insights into the fascinating NLO data of the aforementioned π-conjugated push–pull organic compounds for modern NLO applications. In addition, due to their high NLO response, the designed compounds can be used as targets for further research.
Transition Density Matrices (TDMs) and Exciton Binding Energy (Eb)
The transition density matrix (TDM) is mainly used to elucidate the electronic charge transfer in the designed compounds MSTD2–MSTD7 and the reference compound DTS(FBTTh2)2R1.89 TDM analysis is achieved by using the data obtained by the transference of charges from the donor to acceptor moiety via a π linker.90 The TDMs of investigated compounds were computed at the M06 level of theory and the 6-31G(d,p) basis set. The TDM diagrams of all the investigated compounds show the nature of transitions in these compounds. The needed atoms are divided into sections according to their contribution: i.e., donor (D), π spacer (π), and acceptor (A). Hydrogen atoms have been excluded due to their low susceptibility in effective charge transfer phenomena. All of the results for DTS(FBTTh2)2R1 and MSTD2–MSTD7 are shown in Figure 5.
Figure 5.

TDM graphs of DTS(FBTTh2)2R1 and MSTD2–MSTD7.
Figure 5 shows that the electron density of all molecules is mostly on the diagonal of the donor as well as on π linkers. A diagonal charge transfer is identified in all non-fullerene-based compounds that have been studied. The π linker acts as a bridge and assists in the relocation of electrons from the donor toward the non-fullerene acceptor units. The diagonal charge is efficiently transferred through a π bridge from the donor to acceptor region, which transfers the charge without or with less charge trapping.
The binding energy (Eb) is the difference between electrical (EH-L) and optical (Eopt) band gap energies and is an important parameter for an evaluation of the optoelectronic properties of the studied compounds. Equation 13 is used for the theoretical calculation of the binding energy of reference and designed compounds.
| 13 |
In eq 13, Eb is the energy gap, EH-L is the band gap, and Eopt is the first excitation energy.91 All calculated values of binding energy are given in Table 6.
Table 6. ELUMO – EHOMO (Energy Gap), Eopt, and Eb Values of the Investigated Compounds.
| compound | EH-L (eV) | Eopt (eV) | Eb (eV) |
|---|---|---|---|
| DTS(FBTTh2)2R1 | 2.331 | 1.743 | 0.588 |
| MSTD2 | 1.835 | 1.530 | 0.305 |
| MSTD3 | 1.720 | 1.442 | 0.278 |
| MSTD4 | 1.777 | 1.487 | 0.290 |
| MSTD5 | 1.818 | 1.515 | 0.303 |
| MSTD6 | 1.491 | 1.588 | –0.097 |
| MSTD7 | 1.582 | 1.333 | 0.249 |
The binding energy of the reference compound was found to be 0.588 eV. The binding energy values of compounds MSTD2–MSTD7 are 0.305, 0.278, 0.290, 0.303, −0.097, and 0.249, respectively. The binding energy values are noted to be in ascending order as MSTD6 < MSTD7 < MSTD3 < MSTD4 < MSTD5 < MSTD2 < DTS(FBTTh2)2R1. All of the designed MSTD2–MSTD7 molecules give lower values of Eb (eV) in comparison to rthe eference compound DTS(FBTTh2)2R1. The NF-based designed compounds show promising binding energy valuesand so may also be used in a vast range of NLO applications.
Conclusion
Herein, six novel organic molecules (MSTD2–MSTD7) were theoretically designed from DTS(FBTTh2)2R1 by the modification of the different substituted acceptors. The influence of different acceptors is explored for nonlinear amplitudes. FMO results revealed that all of the designed compounds have a small band gap in the range of 1.491–1.835 eV relative to DTS(FBTTh2)2R1 (2.331 eV). Global reactivity parameters were associated with a lower band gap having lower values of hardness with higher values of softness. Moreover, all of the developed compounds had higher λmax values and lower transition energies in the UV–vis region in comparison to the reference molecule (711.408 nm). The development of charge separation between the donor (D) and acceptor (A) species in the molecules was shown by an NBO analysis. Subsequently, this charge separation could be the reason for the great NLO response because of charge transfer from D to A. Interestingly, unusually large ⟨α⟩ , βtotal, and ⟨γ⟩ values computed to be 3.485× 10–22, 13.44 × 10–27, and 3.66 × 10–31 esu, respectively, were found for MSTD6. The first-hyperpolarizability values of MSTD2–MSTD7 are 8.33 × 10–25, 1.07 × 10–24, 9.46 × 10–25, 8.89 × 10–25, 1.65 × 10–25, and 1.27 × 10–25 times greater than that of the reference compound DTS(FBTTh2)2R1 and 1.82 × 104, 2.35 × 104, 2.06 × 104, 1.94 × 104, 3.61 × 104, and 2.79 × 104 times greater than the βtotal value of urea. Similarly, MSTD6 has the highest value of ⟨γ⟩ (3.66 × 10–31 esu). The following is the descending order of all of the created compounds: MSTD6 > MSTD7 > MSTD3 > MSTD4 > STD5 > MSTD2 > DTS(FBTTh2)2R1. We expect that our research will aid in the development of organic compounds with the desired characteristics for improving optical device performance.
Acknowledgments
The authors acknowledge the financial support of the Taif University Researchers Supporting Project (No. TURSP-2020/14), Taif University, Taif, Saudi Arabia. M.I. expresses appreciation to the Deanship of Scientific Research at King Khalid University Saudi Arabia through rthe esearch groups program under grant number R.G.P. 2/40/43.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01474.
Cartesian coordinates, calculated energies (E) and energy gaps (ΔE), global reactivity parameters, natural bond orbital (NBO) analysis, dipole polarizabilities, dipole moments, computed first hyperpolarizabilities (βtot), second hyperpolarizabilities, UV–vis data (wavelengths, excitation energiesm and oscillator strengths), and structures of DTS(FBTTh2)2R1 and its derivatives (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Geethakrishnan T.; Palanisamy P. Z-scan determination of the third-order optical nonlinearity of a triphenylmethane dye using 633 nm He–Ne laser. Opt. Commun. 2007, 270, 424–428. 10.1016/j.optcom.2006.09.035. [DOI] [Google Scholar]
- Han P.; Wang D.; Gao H.; Zhang J.; Xing Y.; Yang Z.; Cao H.; He W. Third-order nonlinear optical properties of cyanine dyes with click chemistry modification. Dyes Pigm. 2018, 149, 8–15. 10.1016/j.dyepig.2017.09.052. [DOI] [Google Scholar]
- Loh K. P.; Bao Q.; Eda G.; Chhowalla M. Graphene oxide as a chemically tunable platform for optical applications. Nat. Chem. 2010, 2, 1015–1024. 10.1038/nchem.907. [DOI] [PubMed] [Google Scholar]
- Helmchen F.; Denk W. Deep tissue two-photon microscopy. Nat. Methods 2005, 2, 932–940. 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- Jia J.; Li Y.; Gao J. A series of novel ferrocenyl derivatives: Schiff bases-like push-pull systems with large third-order optical responses. Dyes Pigm. 2017, 137, 342–351. 10.1016/j.dyepig.2016.11.008. [DOI] [Google Scholar]
- Ma X.; Lin C.-S.; Zhang H.; Lin Y.-J.; Hu S.-M.; Sheng T.-L.; Wu X.-T. Synthesis, spectral and redox switchable cubic NLO properties of chiral dinuclear iron cyanide/isocyanide-bridged complexes. Dalton Trans. 2013, 42, 12452–12459. 10.1039/c3dt51197a. [DOI] [PubMed] [Google Scholar]
- Feng Q.; Li Y.; Shi G.; Wang L.; Zhang W.; Li K.; Hou H.; Song Y. A photo-controllable third-order nonlinear optical (NLO) switch based on a rhodamine B salicylaldehyde hydrazone metal complex. J. Mater. Chem. C 2016, 4, 8552–8558. 10.1039/C6TC01549B. [DOI] [Google Scholar]
- Luan Y.; Yang M.; Ma Q.; Qi Y.; Gao H.; Wu Z.; Wang G. Introduction of an organic acid phase changing material into metal–organic frameworks and the study of its thermal properties. J. Mater. Chem. A 2016, 4, 7641–7649. 10.1039/C6TA01676F. [DOI] [Google Scholar]
- Li M.; Li Y.; Zhang H.; Wang S.; Ao Y.; Cui Z. Molecular engineering of organic chromophores and polymers for enhanced bulk second-order optical nonlinearity. J. Mater. Chem. C 2017, 5, 4111–4122. 10.1039/C7TC00713B. [DOI] [Google Scholar]
- Zhao Y.; Li H.; Shao Z.; Xu W.; Meng X.; Song Y.; Hou H. Investigation of regulating third-order nonlinear optical property by coordination interaction. Inorg. Chem. 2019, 58, 4792–4801. 10.1021/acs.inorgchem.8b03154. [DOI] [PubMed] [Google Scholar]
- Jia J.-H.; Tao X.-M.; Li Y.-J.; Sheng W.-J.; Han L.; Gao J.-R.; Zheng Y.-F. Synthesis and third-order optical nonlinearities of ferrocenyl Schiff base. Chem. Phys. Lett. 2011, 514, 114–118. 10.1016/j.cplett.2011.08.035. [DOI] [Google Scholar]
- Makhal K.; Arora S.; Kaur P.; Goswami D.; Singh K. Third-order nonlinear optical response and ultrafast dynamics of tetraoxa [22] porphyrin (2.1. 2.1) s. J. Mater. Chem. C 2016, 4, 9445–9453. 10.1039/C6TC03239G. [DOI] [Google Scholar]
- Makowska-Janusik M., Influence of the Polymeric Matrix on the NLO Molecular Response in Guest-Host Materials. Nonlinear Optics, Quantum Optics: Concepts in Modern Optics 2007, 37 ( (1-3), ), 75-85. [Google Scholar]
- Ghiasuddin; Akram M.; Adeel M.; Khalid M.; Tahir M. N.; Khan M. U.; Asghar M. A.; Ullah M. A.; Iqbal M. A combined experimental and computational study of 3-bromo-5-(2, 5-difluorophenyl) pyridine and 3, 5-bis (naphthalen-1-yl) pyridine: Insight into the synthesis, spectroscopic, single crystal XRD, electronic, nonlinear optical and biological properties. J. Mol. Struct. 2018, 1160, 129–141. 10.1016/j.molstruc.2018.01.100. [DOI] [Google Scholar]
- Ali Z.; Shafiq M.; Asadabadi S. J.; Aliabad H. R.; Khan I.; Ahmad I. Magneto-electronic studies of anti-perovskites NiNMn3 and ZnNMn3. Computational materials science 2014, 81, 141–145. 10.1016/j.commatsci.2013.07.040. [DOI] [Google Scholar]
- Asokan P.; Kalainathan S. Bulk crystal growth, optical, electrical, thermal, and third order NLO properties of 2-[4-(Diethylamino) benzylidene] malononitrile (DEBM) single crystal. J. Phys. Chem. C 2017, 121, 22384–22395. 10.1021/acs.jpcc.7b07805. [DOI] [Google Scholar]
- Jerca F. A.; Jerca V. V.; Kajzar F.; Manea A. M.; Rau I.; Vuluga D. M. Simultaneous two and three photon resonant enhancement of third-order NLO susceptibility in an azo-dye functionalized polymer film. Phys. Chem. Chem. Phys. 2013, 15, 7060–7063. 10.1039/c3cp50547b. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S.; Biswas C.; Raavi S. S. K.; Venkata Suman Krishna J.; Vamsi Krishna N.; Giribabu L.; Soma V. R. Synthesis, optical, electrochemical, DFT studies, NLO properties, and ultrafast excited state dynamics of carbazole-induced phthalocyanine derivatives. J. Phys. Chem. C 2019, 123, 11118–11133. 10.1021/acs.jpcc.9b01531. [DOI] [Google Scholar]
- Janjua M. R. S. A. Computational Study on Non-linear Optical and Absorption Properties of Benzothiazole based Dyes: Tunable Electron-Withdrawing Strength and Reverse Polarity. Open Chemistry 2017, 15, 139–146. 10.1515/chem-2017-0017. [DOI] [Google Scholar]
- Janjua M. R. S. A. Non-linear Optical response of Phenoxazine-based Dyes: Molecular Engineering of Thiadiazole Derivatives as π-spacers. Journal of the Mexican Chemical Society 2017, 61, 260–265. [Google Scholar]
- Hou H.; Song Y.; Fan Y.; Zhang L.; Du C.; Zhu Y. A novel coordination polymer [Co (NCS) 2 (bpms) 2] n (bpms= 1, 2-bis (4-pyridylmethyl) disulfenyl): synthesis, crystal structure and third-order nonlinear optical properties. Inorg. Chim. Acta 2001, 316, 140–144. 10.1016/S0020-1693(01)00391-7. [DOI] [Google Scholar]
- Jia J.; Li T.; Cui Y.; Li Y.; Wang W.; Han L.; Li Y.; Gao J. Study on the synthesis and third-order nonlinear optical properties of DA poly-quinacridone optical materials. Dyes Pigm. 2019, 162, 26–35. 10.1016/j.dyepig.2018.09.038. [DOI] [Google Scholar]
- Fuks-Janczarek I.; Luc J.; Sahraoui B.; Dumur F.; Hudhomme P.; Berdowski J.; Kityk I. Third-order nonlinear optical figure of merits for conjugated TTF- quinone molecules. J. Phys. Chem. B 2005, 109, 10179–10183. 10.1021/jp0508711. [DOI] [PubMed] [Google Scholar]
- Biswal B. P.; Valligatla S.; Wang M.; Banerjee T.; Saad N. A.; Mariserla B. M. K.; Chandrasekhar N.; Becker D.; Addicoat M.; Senkovska I.; et al. Nonlinear optical switching in regioregular porphyrin covalent organic frameworks. Angew. Chem. 2019, 131, 6970–6974. 10.1002/ange.201814412. [DOI] [PubMed] [Google Scholar]
- Chen B.; Ni S.; Sun L.; Luo X.; Zhang Q.; Song Y.; Zhong Q.; Fang Y.; Huang C.; Chen S.; et al. Intramolecular charge transfer tuning of azo dyes: spectroscopic characteristic and third-order nonlinear optical properties. Dyes Pigm. 2018, 158, 474–481. 10.1016/j.dyepig.2018.05.063. [DOI] [Google Scholar]
- Bundulis A.; Nitiss E.; Mihailovs I.; Busenbergs J.; Rutkis M. Study of structure–third-order susceptibility relation of indandione derivatives. J. Phys. Chem. C 2016, 120, 27515–27522. 10.1021/acs.jpcc.6b07003. [DOI] [Google Scholar]
- Wielopolski M.; Kim J.-H.; Jung Y.-S.; Yu Y.-J.; Kay K.-Y.; Holcombe T. W.; Zakeeruddin S. M.; Grätzel M.; Moser J.-E. Position-dependent extension of π-conjugation in D-π-A dye sensitizers and the impact on the charge-transfer properties. J. Phys. Chem. C 2013, 117, 13805–13815. 10.1021/jp402411h. [DOI] [Google Scholar]
- Katono M.; Wielopolski M.; Marszalek M.; Bessho T.; Moser J.-E.; Humphry-Baker R.; Zakeeruddin S. M.; Grätzel M. Effect of extended π-conjugation of the donor structure of organic D–A- π–A dyes on the photovoltaic performance of dye-Sensitized solar cells. J. Phys. Chem. C 2014, 118, 16486–16493. 10.1021/jp411504p. [DOI] [Google Scholar]
- Panneerselvam M.; Kathiravan A.; Solomon R. V.; Jaccob M. The role of π-linkers in tuning the optoelectronic properties of triphenylamine derivatives for solar cell applications–A DFT/TDDFT study. Phys. Chem. Chem. Phys. 2017, 19, 6153–6163. 10.1039/C6CP07768D. [DOI] [PubMed] [Google Scholar]
- Nalwa H. S., Organic materials for third-order nonlinear optics. In Nonlinear Optics of Organic Molecules and Polymers; CRC Press: 2020; pp 611–797. [Google Scholar]
- Kato S.-i.; Diederich F. Non-planar push–pull chromophores. Chem. Commun. 2010, 46, 1994–2006. 10.1039/b926601a. [DOI] [PubMed] [Google Scholar]
- Pinna A.; Malfatti L.; Piccinini M.; Falcaro P.; Innocenzi P. Hybrid materials with an increased resistance to hard X-rays using fullerenes as radical sponges. Journal of Synchrotron Radiation 2012, 19, 586–590. 10.1107/S0909049512012848. [DOI] [PubMed] [Google Scholar]
- Guldi D. M.; Illescas B. M.; Atienza C. M.; Wielopolski M.; Martín N. Fullerene for organic electronics. Chem. Soc. Rev. 2009, 38, 1587–1597. 10.1039/b900402p. [DOI] [PubMed] [Google Scholar]
- Couris S.; Koudoumas E.; Ruth A.; Leach S. Concentration and wavelength dependence of the effective third-order susceptibility and optical limiting of C60 in toluene solution. Journal of Physics B: Atomic, Molecular and Optical Physics 1995, 28, 4537. 10.1088/0953-4075/28/20/015. [DOI] [Google Scholar]
- Dai S.; Xiao Y.; Xue P.; James Rech J.; Liu K.; Li Z.; Lu X.; You W.; Zhan X. Effect of core size on performance of fused-ring electron acceptors. Chem. Mater. 2018, 30, 5390–5396. 10.1021/acs.chemmater.8b02222. [DOI] [Google Scholar]
- Zhan C.; Zhang X.; Yao J. New advances in non-fullerene acceptor based organic solar cells. RSC Adv. 2015, 5, 93002–93026. 10.1039/C5RA17715D. [DOI] [Google Scholar]
- Guldi D. M.; Maggini M.; Scorrano G.; Prato M. Intramolecular electron transfer in fullerene/ferrocene based donor- bridge- acceptor dyads. J. Am. Chem. Soc. 1997, 119, 974–980. 10.1021/ja960070z. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G., et al. Gaussian 09, Rev. D.01; Gaussian Inc.: 2009.
- Valverde C.; de Lima e Castro S. A.; Vaz G. R.; de Almeida Ferreira J. L.; Baseia B.; Osorio F. A. P. Third-order nonlinear optical properties of a carboxylic acid derivative. Acta Chim. Slov. 2018, 65, 739–749. 10.17344/acsi.2018.4462. [DOI] [PubMed] [Google Scholar]
- Ullah F.; Ayub K.; Mahmood T. Remarkable second and third order nonlinear optical properties of organometallic C 6 Li 6–M 3 O electrides. New J. Chem. 2020, 44, 9822–9829. 10.1039/D0NJ01670E. [DOI] [Google Scholar]
- Dennington R. D.; Keith T. A.; Millam J. M.. GaussView 5.0.8.; Gaussian Inc.: 2008.
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrienko G. A.Chemcraft: Graphical Software for Visualization of Quantum Chemistry Computations, 2010.
- Haroon M.; Al-Saadi A. A.; Janjua M. R. S. A. Insights into end-capped modifications effect on the photovoltaic and optoelectronic properties of S-shaped fullerene-free acceptor molecules: A density functional theory computational study for organic solar cells. J. Phys. Org. Chem. 2022, e4314 10.1002/poc.4314. [DOI] [Google Scholar]
- Janjua M. R. S. A. Prediction and Understanding: Quantum Chemical Framework of Transition Metals Enclosed in a B12N12 Inorganic Nanocluster for Adsorption and Removal of DDT from the Environment. Inorg. Chem. 2021, 60, 10837–10847. 10.1021/acs.inorgchem.1c01760. [DOI] [PubMed] [Google Scholar]
- Janjua M. R. S. A. How does bridging core modification alter the photovoltaic characteristics of triphenylamine-based hole transport materials? Theoretical understanding and prediction. Chem - Eur. J. 2021, 27, 4197–4210. 10.1002/chem.202004299. [DOI] [PubMed] [Google Scholar]
- Srnec M.; Solomon E. I. Frontier molecular orbital contributions to chlorination versus hydroxylation selectivity in the non-heme iron halogenase SyrB2. J. Am. Chem. Soc. 2017, 139, 2396–2407. 10.1021/jacs.6b11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid M.; Ali M.; Aslam M.; Sumrra S. H.; Khan M. U.; Raza N.; Kumar N.; Imran M. Frontier molecular, Natural bond orbital, UV-Vis spectral stduy, Solvent influence on geometric parameters, Vibrational frequencies and solvation energies of 8-Hydroxyquinoline. Int. J. Pharm. Sci. Res. 2017, 8, 13040. 10.13040/IJPSR.0975-82328(2).457-69. [DOI] [Google Scholar]
- Jawaria R.; Hussain M.; Khalid M.; Khan M. U.; Tahir M. N.; Naseer M. M.; Braga A. A. C.; Shafiq Z. Synthesis, crystal structure analysis, spectral characterization and nonlinear optical exploration of potent thiosemicarbazones based compounds: A DFT refine experimental study. Inorg. Chim. Acta 2019, 486, 162–171. 10.1016/j.ica.2018.10.035. [DOI] [Google Scholar]
- Haroon M.; Khalid M.; Akhtar T.; Tahir M. N.; Khan M. U.; Saleem M.; Jawaria R. Synthesis, spectroscopic, SC-XRD characterizations and DFT based studies of ethyl2-(substituted-(2-benzylidenehydrazinyl)) thiazole-4-carboxylate derivatives. J. Mol. Struct. 2019, 1187, 164–171. 10.1016/j.molstruc.2019.03.075. [DOI] [Google Scholar]
- Shahid M.; Salim M.; Khalid M.; Tahir M. N.; Khan M. U.; Braga A. A. C. Synthetic, XRD, non-covalent interactions and solvent dependent nonlinear optical studies of Sulfadiazine-Ortho-Vanillin Schiff base:(E)-4-((2-hydroxy-3-methoxy-benzylidene) amino)-N-(pyrimidin-2-yl) benzene-sulfonamide. J. Mol. Struct. 2018, 1161, 66–75. 10.1016/j.molstruc.2018.02.043. [DOI] [Google Scholar]
- Naseem S.; Khalid M.; Tahir M. N.; Halim M. A.; Braga A. A.; Naseer M. M.; Shafiq Z. Synthesis, structural, DFT studies, docking and antibacterial activity of a xanthene based hydrazone ligand. J. Mol. Struct. 2017, 1143, 235–244. 10.1016/j.molstruc.2017.04.093. [DOI] [Google Scholar]
- Soleimani Amiri S.; Makarem S.; Ahmar H.; Ashenagar S. Theoretical studies and spectroscopic characterization of novel 4-methyl-5-((5-phenyl-1, 3, 4-oxadiazol-2-yl) thio) benzene-1, 2-diol. J. Mol. Struct. 2016, 1119, 18–24. 10.1016/j.molstruc.2016.04.053. [DOI] [Google Scholar]
- Harit T.; Bellaouchi R.; Asehraou A.; Rahal M.; Bouabdallah I.; Malek F. Synthesis, characterization, antimicrobial activity and theoretical studies of new thiophene-based tripodal ligands. J. Mol. Struct. 2017, 1133, 74–79. 10.1016/j.molstruc.2016.11.051. [DOI] [Google Scholar]
- Arshad M. N.; Al-Dies A.-A. M.; Asiri A. M.; Khalid M.; Birinji A. S.; Al-Amry K. A.; Braga A. A. Synthesis, crystal structures, spectroscopic and nonlinear optical properties of chalcone derivatives: a combined experimental and theoretical study. J. Mol. Struct. 2017, 1141, 142–156. 10.1016/j.molstruc.2017.03.090. [DOI] [Google Scholar]
- Tahir M. N.; Khalid M.; Islam A.; Mashhadi S. M. A.; Braga A. A. Facile synthesis, single crystal analysis, and computational studies of sulfanilamide derivatives. J. Mol. Struct. 2017, 1127, 766–776. 10.1016/j.molstruc.2016.08.032. [DOI] [Google Scholar]
- Gunasekaran S.; Balaji R. A.; Kumeresan S.; Anand G.; Srinivasan S. Experimental and theoretical investigations of spectroscopic properties of N-acetyl-5-methoxytryptamine. Can. J. Anal. Sci. Spectrosc 2008, 53, 149–162. [Google Scholar]
- Adeel M.; Braga A. A.; Tahir M. N.; Haq F.; Khalid M.; Halim M. A. Synthesis, X-ray crystallographic, spectroscopic and computational studies of aminothiazole derivatives. J. Mol. Struct. 2017, 1131, 136–148. 10.1016/j.molstruc.2016.11.046. [DOI] [Google Scholar]
- Rahmalia W.; Fabre J.-F.; Usman T.; Mouloungui Z. Aprotic solvents effect on the UV–visible absorption spectra of bixin. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2014, 131, 455–460. 10.1016/j.saa.2014.03.119. [DOI] [PubMed] [Google Scholar]
- Namuangruk S.; Fukuda R.; Ehara M.; Meeprasert J.; Khanasa T.; Morada S.; Kaewin T.; Jungsuttiwong S.; Sudyoadsuk T.; Promarak V. D–D- π–A-Type organic dyes for dye-sensitized solar cells with a potential for direct electron injection and a high extinction coefficient: synthesis, characterization, and theoretical investigation. J. Phys. Chem. C 2012, 116, 25653–25663. 10.1021/jp304489t. [DOI] [Google Scholar]
- Jezuita A.; Ejsmont K.; Szatylowicz H. Substituent effects of nitro group in cyclic compounds. Struct. Chem. 2021, 32, 179–203. 10.1007/s11224-020-01612-x. [DOI] [Google Scholar]
- Pham P.-T.; Xia Y.; Frisbie C. D.; Bader M. M. Single crystal field effect transistor of a Y-shaped ladder-type oligomer. J. Phys. Chem. C 2008, 112, 7968–7971. 10.1021/jp712044u. [DOI] [Google Scholar]
- Cai X.; Burand M. W.; Newman C. R.; da Silva Filho D. A.; Pappenfus T. M.; Bader M. M.; Brédas J.-L.; Mann K. R.; Frisbie C. D. N-and P-channel transport behavior in thin film transistors based on tricyanovinyl-capped oligothiophenes. J. Phys. Chem. B 2006, 110, 14590–14597. 10.1021/jp061168v. [DOI] [PubMed] [Google Scholar]
- Pearson R. G. Absolute electronegativity and absolute hardness of Lewis acids and bases. J. Am. Chem. Soc. 1985, 107, 6801–6806. 10.1021/ja00310a009. [DOI] [Google Scholar]
- Parr R. G.; Pearson R. G. Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. 10.1021/ja00364a005. [DOI] [Google Scholar]
- Parr R. G.; Chattaraj P. K. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. 10.1021/ja00005a072. [DOI] [Google Scholar]
- Koopmans T. Ordering of wave functions and eigenenergies to the individual electrons of an atom. Physica 1934, 1, 104–113. 10.1016/S0031-8914(34)90011-2. [DOI] [Google Scholar]
- Parr R. G.; Szentpály L. v.; Liu S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. 10.1021/ja983494x. [DOI] [Google Scholar]
- Pasha A. R.; Khalid M.; Shafiq Z.; Khan M. U.; Naseer M. M.; Tahir M. N.; Hussain R.; Braga A. A. C.; Jawaria R. A comprehensive study of structural, non-covalent interactions and electronic insights into N-aryl substituted thiosemicarbazones via SC-XRD and first-principles DFT approach. J. Mol. Struct. 2021, 1230, 129852. 10.1016/j.molstruc.2020.129852. [DOI] [Google Scholar]
- Subashchandrabose S.; Krishnan A. R.; Saleem H.; Parameswari R.; Sundaraganesan N.; Thanikachalam V.; Manikandan G. Vibrational spectroscopic study and NBO analysis on bis (4-amino-5-mercapto-1, 2, 4-triazol-3-yl) methane using DFT method. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2010, 77, 877–884. 10.1016/j.saa.2010.08.023. [DOI] [PubMed] [Google Scholar]
- Muthu S.; Ramachandran G. Spectroscopic studies (FTIR, FT-Raman and UV–Visible), normal coordinate analysis, NBO analysis, first order hyper polarizability, HOMO and LUMO analysis of (1R)-N-(Prop-2-yn-1-yl)-2, 3-dihydro-1H-inden-1-amine molecule by ab initio HF and density functional methods. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2014, 121, 394–403. 10.1016/j.saa.2013.10.093. [DOI] [PubMed] [Google Scholar]
- Liu J.-n.; Chen Z.-r.; Yuan S.-F. Study on the prediction of visible absorption maxima of azobenzene compounds. Journal of Zhejiang University. Science. B 2005, 6B, 584. 10.1631/jzus.2005.B0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goszczycki P.; Stadnicka K.; Brela M. Z.; Grolik J.; Ostrowska K. Synthesis, crystal structures, and optical properties of the π-π interacting pyrrolo [2, 3-b] quinoxaline derivatives containing 2-thienyl substituent. J. Mol. Struct. 2017, 1146, 337–346. 10.1016/j.molstruc.2017.06.008. [DOI] [Google Scholar]
- Mahmood A.; Khan S. U. D.; Rana U. A.; Janjua M. R. S. A.; Tahir M. H.; Nazar M. F.; Song Y. Effect of thiophene rings on UV/visible spectra and non-linear optical (NLO) properties of triphenylamine based dyes: a quantum chemical perspective. J. Phys. Org. Chem. 2015, 28, 418–422. 10.1002/poc.3427. [DOI] [Google Scholar]
- Sadlej-Sosnowska N. Application of natural bond orbital analysis to delocalization and aromaticity in C-substituted tetrazoles. J. Org. Chem. 2001, 66, 8737–8743. 10.1021/jo010062n. [DOI] [PubMed] [Google Scholar]
- Janjua M. R. S. A.; Yamani Z. H.; Jamil S.; Mahmood A.; Ahmad I.; Haroon M.; Tahir M. H.; Yang Z.; Pan S. First principle study of electronic and non-linear optical (NLO) properties of triphenylamine dyes: interactive design computation of new NLO compounds. Aust. J. Chem. 2016, 69, 467–472. 10.1071/CH15402. [DOI] [Google Scholar]
- Haroon M.; Janjua M. R. S. A. Prediction of NLO response of substituted organoimido hexamolybedate: First theoretical framework based on p-anisidine adduct [Mo6O18 (p-MeOC6H4N)] 2. Materials Today Communications 2021, 26, 101880. 10.1016/j.mtcomm.2020.101880. [DOI] [Google Scholar]
- Haroon M.; Khalid M.; Shafiq Z.; Khan M. U.; Janjua M. R. S. A. High-throughput calculations and experimental insights towards the development of potent thiazoline based functional materials. Materials Today Communications 2021, 27, 102485. 10.1016/j.mtcomm.2021.102485. [DOI] [Google Scholar]
- Janjua M. R. S. A.; Liu C.-G.; Guan W.; Zhuang J.; Muhammad S.; Yan L.-K.; Su Z.-M. Prediction of remarkably large second-order nonlinear optical properties of organoimido-substituted hexamolybdates. J. Phys. Chem. A 2009, 113, 3576–3587. 10.1021/jp808707q. [DOI] [PubMed] [Google Scholar]
- Janjua M. R. S. A. Quantum mechanical design of efficient second-order nonlinear optical materials based on heteroaromatic imido-substituted hexamolybdates: First theoretical framework of POM-based heterocyclic aromatic rings. Inorg. Chem. 2012, 51, 11306–11314. 10.1021/ic3002652. [DOI] [PubMed] [Google Scholar]
- Muhammad S.; Janjua M. R. S. A.; Su Z. Investigation of dibenzoboroles having π-electrons: toward a new type of two-dimensional NLO molecular switch?. J. Phys. Chem. C 2009, 113, 12551–12557. 10.1021/jp903075s. [DOI] [Google Scholar]
- Liu C.-G.; Su Z.-M.; Guan X.-H.; Muhammad S. Redox and photoisomerization switching the second-order nonlinear optical properties of a tetrathiafulvalene derivative across six states: a DFT study. J. Phys. Chem. C 2011, 115, 23946–23954. 10.1021/jp2049958. [DOI] [Google Scholar]
- Muhammad S.; Irfan A.; Al-Sehemi A. G.; Al-Assiri M.; Kalam A.; Chaudhry A. R. Quantum chemical investigation of spectroscopic studies and hydrogen bonding interactions between water and methoxybenzeylidene-based humidity sensor. J. Theor. Comput. Chem. 2015, 14, 1550029. 10.1142/S0219633615500297. [DOI] [Google Scholar]
- Muhammad S.; Liu C.; Zhao L.; Wu S.; Su Z. A theoretical investigation of intermolecular interaction of a phthalimide based “on–off” sensor with different halide ions: tuning its efficiency and electro-optical properties. Theor. Chem. Acc. 2009, 122, 77–86. 10.1007/s00214-008-0486-8. [DOI] [Google Scholar]
- Peng Z.; Yu L. Second-order nonlinear optical polyimide with high-temperature stability. Macromolecules 1994, 27, 2638–2640. 10.1021/ma00087a039. [DOI] [Google Scholar]
- Breitung E. M.; Shu C.-F.; McMahon R. J. Thiazole and thiophene analogues of donor- acceptor stilbenes: molecular hyperpolarizabilities and structure- property relationships. J. Am. Chem. Soc. 2000, 122, 1154–1160. 10.1021/ja9930364. [DOI] [Google Scholar]
- Adant C.; Dupuis M.; Bredas J. Ab initio study of the nonlinear optical properties of urea: Electron correlation and dispersion effects. Int. J. Quantum Chem. 1995, 56, 497–507. 10.1002/qua.560560853. [DOI] [Google Scholar]
- Kanis D. R.; Ratner M. A.; Marks T. J. Design and construction of molecular assemblies with large second-order optical nonlinearities. Quantum chemical aspects. Chem. Rev. 1994, 94, 195–242. 10.1021/cr00025a007. [DOI] [Google Scholar]
- Martin R. L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. 10.1063/1.1558471. [DOI] [Google Scholar]
- Dennler G.; Scharber M. C.; Ameri T.; Denk P.; Forberich K.; Waldauf C.; Brabec C. J. Design Rules for Donors in Bulk-Heterojunction Tandem Solar Cells Towards 15% Energy-Conversion Efficiency. Adv. Mater. 2008, 20, 579–583. 10.1002/adma.200702337. [DOI] [Google Scholar]
- Dkhissi A. Excitons in organic semiconductors. Synth. Met. 2011, 161, 1441–1443. 10.1016/j.synthmet.2011.04.003. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.