

Abstract
Purpose:
Cancer-associated fibroblasts (CAF) have been implicated as potential mediators of checkpoint immunotherapy response. However, the extensive heterogeneity of these cells has precluded rigorous understanding of their immunoregulatory role in the tumor microenvironment.
Experimental Design:
We performed high dimensional single-cell RNA sequencing (scRNA-Seq) on four patient tumors pre- and post-treatment from a neoadjuvant trial of advanced-stage head and neck squamous cell carcinoma (HNSCC) patients that were treated with the αPD-1 therapy, nivolumab. The head and neck CAF (HNCAF) protein activity profiles, derived from this cohort of paired scRNA-Seq, were used to perform protein activity enrichment analysis on the 28-patient parental cohort of clinically annotated bulk transcriptomic profiles. Ex vivo coculture assays were used to test functional relevance of HNCAF subtypes.
Results:
Fourteen distinct cell types were identified with the fibroblast population showing significant changes in abundance following nivolumab treatment. Among the fibroblast subtypes, HNCAF-0/3 emerged as predictive of nivolumab response, while HNCAF-1 was associated with immunosuppression. Functionally, HNCAF-0/3 were found to reduce TGFβ-dependent PD-1+TIM-3+ exhaustion of CD8 T cells, increase CD103+NKG2A+ resident memory phenotypes, and enhance the overall cytolytic profile of T cells.
Conclusions:
Our findings demonstrate the functional importance of distinct HNCAF subsets in modulating the immunoregulatory milieu of human HNSCC. Additionally, we have identified clinically actionable HNCAF subtypes that can be used as a biomarker of response and resistance in future clinical trials.
Keywords: Cancer associated fibroblasts, head and neck cancer, immunotherapy, T cell regulation, biomarker
Introduction
Anti-PD-1 immune checkpoint inhibitors (ICI) are currently the first line therapy for recurrent/metastatic head and neck squamous cell carcinoma (HNSCC) (1, 2). Yet, overall response rates can be as low as 20%, with increased responses in tumors with elevated PD-L1 expression and tumor infiltrating T cells (1, 3, 4). While this is partly a T cell dependent mechanism, there may be additional cellular subpopulations in the tumor microenvironment (TME) responsible for mediating response to ICI (5–8). Recent studies have suggested that cancer associated fibroblasts (CAF) are associated with this resistance, but their role in T cell immunomodulation is still unclear in the human TME (9–11).
In human breast cancer, four CAF subtypes (CAF-S1 to S4), were identified based on the expression of six fibroblast markers — fibroblast activation protein (FAP), integrin β1 (CD29), ⍺-smooth muscle actin (⍺-SMA), fibroblast-specific protein-1 (FSP-1), platelet-derived growth factor receptor β (PDGFRβ), and caveolin-1 (CAV1) (12). In their follow-up study, single-cell RNA sequencing (scRNA-Seq) further divided CAF-S1 into eight subtypes with the majority of these subtypes linked to immunosuppression and resistance to immunotherapy (11). In contrast, only three molecularly and phenotypically distinct CAF subpopulations were identified in pancreatic cancer, based on spatial location and imputed function, as defined by cytokine and surface marker expression. These include inflammatory CAF (iCAF), myofibroblastic CAF (myCAF) and antigen-presenting CAF (apCAF) (13, 14). In head and neck, three CAF types were previously identified by single-cell RNA sequencing corresponding to myCAF and two undefined CAF subtypes (CAF1 and CAF2), but the functionality of these subtypes and their association with immunotherapy response remains unknown (15). In general, these various orthogonal approaches to cancer specific CAF characterizations have not provided a concordant classification of this important stromal host cell type.
To assess whether CAF-related or other TME subpopulations can regulate clinical responses to nivolumab, we leveraged single-cell RNA sequencing from a neoadjuvant trial to longitudinally profile pre- and post-treatment human head and neck squamous cell carcinoma and generate a dynamic atlas of the human HNSCC TME. Our novel bioinformatic approach uses the VIPER algorithm (16, 17) to address limitations imposed by high noise and significant gene dropout rates in most scRNA-Seq analysis platforms. Specifically, VIPER leverages knowledge of regulatory networks to allow full quantitative characterization of protein activity by assessing the enrichment of their transcriptional targets in differentially expressed genes. On average, the resulting protein activity profiles outperform antibody-based measurements and dramatically outperform gene expression-based analyses in terms of identifying and characterizing molecularly distinct TME subpopulations (18), thus enabling mechanistic dissection of the HNSCC microenvironment at hitherto unattained resolution. We present the results of these protein activity-based analyses on clinical biospecimens to generate a high-resolution atlas of the human HNSCC immune and stromal micro-environment under ICI pressures.
Methods
Clinical Design and Tissue Collection
Biospecimens were harvested from a window of opportunity trial of locally advanced HNSCC patients (oral cavity, oropharynx, larynx, hypopharynx) who were candidates for primary surgical intervention with curative intent (NCT03238365). This study was conducted in accordance with the Declaration of Helsinki guidelines and approved by the Vanderbilt institutional review board (IRB #171883, IRB #030062). Informed written consent was obtained from all patients. All enrolled patients were treated with 1 month of 240mg nivolumab every 2 weeks for 2 doses prior to definitive surgery (N=50). Half of the patients received tadalafil, and no differences were noted in response rates between the two cohorts (43). Consented patients were required to have fresh pre-nivolumab biopsy as well pre- and post-imaging. Meta-clinical annotation using pathological criteria was used to delineate paired subject specimens as responders vs. non-responders. For both pre- and post-treatment timepoints, fresh specimens were collected for frozen fixation, paraffin embedded fixation, and processed for both bulk and single-cell transcriptomic sequencing. Due to insufficient RNA or data quality, only 32 of the 50 patients were used for bulk and single-cell RNA sequencing analyses.
Tissue Dissociation
Fresh head and neck squamous cell carcinoma tumor specimens were collected in DMEM supplemented with streptomycin (200 mg/ml), penicillin (200 U/ml), and amphotericin B (250 mg/mL). Tumor specimens were minced to 2–4 mm sized pieces in separate 6-cm dishes and digested to single cell suspension using the Miltenyi Biotec human tumor dissociation kit (Miltenyi Biotec #130–095-929) on the Miltenyi gentleMACS Octo dissociator (Miltenyi Biotec #130–096-427) following manufacturer’s instructions. Dissociated cells were aliquoted for single-cell sequencing, flow cytometry analysis, or CAF culture.
Single-Cell RNA Sequencing
Samples were processed to generate single-cell gene expression profiles (scRNA-Seq) using the 10X Chromium 3’ Library and Gel Bead Kit (10X Genomics), following the manufacturer’s user guide. After GelBead in-Emulsion reverse transcription (GEM-RT) reaction, 12–15 cycles of polymerase chain reaction (PCR) amplification were performed to obtain cDNAs used for RNAseq library generation. Libraries were prepared following the manufacturer’s user guide and sequenced on the Illumina NovaSeq 6000 Sequencing System. Gene expression data were processed with “cellranger count” on the pre-built human reference set of 30,727 genes to generate counts matrices. Cell Ranger performed default filtering for quality control, and produced for each sample a barcodes.tsv, genes.tsv, and matrix.mts file containing counts of transcripts for each sample, such that the expression of each gene is in terms of the number of unique molecular identifiers (UMIs) tagged to cDNA molecules corresponding to that gene. These data were loaded into the R version 3.6.1 programming environment, where the publicly available Seurat package v3.0 was used to further quality-control filter cells to those with fewer than 10% mitochondrial RNA content, more than 1,500 unique UMI counts, and fewer than 15,000 unique UMI counts.
Single-Cell Data Processing
Gene Expression UMI count matrices for each sample were normalized and scaled in R using the Seurat SCTransform command to perform a regularized negative binomial regression based on the 3000 most variable genes. Scaled data from each patient were batch-corrected by Seurat using the functions FindIntegrationAnchors and IntegrateData, with default parameters. The resulting dataset included 22906 high-quality cells (mean UMI count 4802) across four patients, including both pre-treatment and post-treatment time points for each patient (Patient1: 5857 pre-treatment, 7360 post-treatment, Patient2: 4938 pre-treatment, 1550 post-treatment, Patient3: 487 pre-treatment, 1741 post-treatment, Patient4: 401 pre-treatment, 572 post-treatment). The batch-corrected dataset was projected into its first 50 principal components using the RunPCA function in Seurat, and further reduced into a 2-dimensional visualization space using the RunUMAP function with method umap-learn and Pearson correlation as the distance metric between cells. The data were clustered by the Louvain algorithm with silhouette score resolution-optimization selecting the resolution with maximum bootstrapped silhouette score in the range of resolution from 0.01 to 1.0 incremented by 0.01 (18). This resulted in an initial coarse clustering on gene expression. Within each cluster metaCells were computed for downstream regulatory network inference by summing SCTransform-corrected template counts for the 10 nearest neighbors of each cell by Pearson correlation distance.
For each single cell, inference of cell type was performed using the SingleR package and the preloaded Blueprint-ENCODE reference, which includes normalized gene expression values for 259 bulkRNA-Seq samples generated by Blueprint and ENCODE from 43 distinct cell types representing pure populations of stroma and immune cells (Martens and Stunnenberg, 2013; the ENCODE Consortium, 2012). The SingleR algorithm computes correlation between each individual cell and each of the 259 reference samples, and then assigns both a label of the cell type with highest average correlation to the individual cell and a p-value computed by wilcox test of correlation to that cell type compared to all other cell types. Highest-Frequency SingleR labels within each cluster among labels with p<0.05 are projected into the Gene Expression UMAP space, such that localization of SingleR labels is highly concordant with the unsupervised clustering. Unsupervised Clusters determined by the resolution-optimized Louvain algorithm are therefore labelled as a particular cell type based on the most-represented SingleR cell type label within that cluster.
Differential gene expression analysis between single cell clusters throughout the manuscript is computed by the MAST hurdle model, as implemented in the Seurat FindAllMarkers command, with a log-fold change threshold of 0.5 and minimum fractional expression threshold of 0.25, indicating that the resulting gene markers for each cluster are restricted to those with log fold change greater than 0 and non-zero expression in at least 25% of the cells in the cluster.
Regulatory Network Inference
From the integrated dataset, metaCells were computed within each gene expression-inferred cluster by summing SCTransform-corrected template counts for the 10 nearest neighbors of each cell by Pearson correlation distance. 200 metaCells per cluster were sampled to compute a regulatory network from each cluster. All regulatory networks were reverse engineered by the ARACNe algorithm. ARACNe was run with 100 bootstrap iterations using 1785 transcription factors (genes annotated in gene ontology molecular function database as GO:0003700, “transcription factor activity”, or as GO:0003677, “DNA binding” and GO:0030528, “transcription regulator activity”, or as GO:0003677 and GO:0045449, “regulation of transcription”), 668 transcriptional cofactors (a manually curated list, not overlapping with the transcription factor list, built upon genes annotated as GO:0003712, “transcription cofactor activity”, or GO:0030528 or GO:0045449), 3455 signaling pathway related genes (annotated in GO biological process database as GO:0007165, “signal transduction” and in GO cellular component database as GO:0005622, “intracellular” or GO:0005886, “plasma membrane”), and 3620 surface markers (annotated as GO:0005886 or as GO:0009986, “cell surface”). ARACNe is only run on these gene sets so as to limit protein activity inference to proteins with biologically meaningful downstream regulatory targets, and we do not apply ARACNe to infer regulatory networks for proteins with no known signaling or transcriptional activity for which protein activity may be difficult to biologically interpret. Parameters were set to zero DPI (Data Processing Inequality) tolerance and MI (Mutual Information) p-value threshold of 10−8, computed by permuting the original dataset as a null model. Each gene list used to run ARACNe is available on github (18).
Protein Activity Inference
Protein activity was inferred for all cells by running the metaVIPER algorithm, using all cluster-specific ARACNe networks, on the SCTransform-scaled and Anchor-Integrated gene expression signature of single cells from each patient. Because the SCTransform Anchor-Integrated scaled gene expression signature is already normalized as an internal signature comparing all cells to the mean expression in the dataset, VIPER normalization parameter was set to “none.” The resulting VIPER matrix included 1239 proteins with activity successfully inferred from ARACNe gene regulatory networks. VIPER-Inferred Protein Activity matrix was loaded into a Seurat Object with CreateSeuratObject, then projected into its first 50 principal components using the RunPCA function in Seurat, and further reduced into a 2-dimensional visualization space using the RunUMAP function with method umap-learn and Pearson correlation as the distance metric between cells. Clustering was performed by resolution-optimized Louvain algorithm, as for gene expression, and SingleR-inferred cell type labels were carried over to identify cluster-by-cluster cell type labels. Differential Protein Activity between clusters identified by resolution-optimized Louvain was computed using bootstrapped t-test, run with 100 bootstraps, and top proteins for each cluster were ranked by p-value. The entire pipeline is implemented as in Obradovic et al. (18). Cluster cell counts were normalized to a fraction of the total sample separately for each patient and separately for pre-treatment and post-treatment samples, with differences in pre-treatment vs post-treatment frequency distribution.
Association of HNCAF Signatures with Response to Immunotherapy
Fibroblast clusters including 5,414 cells from overall VIPER clustering of all cells were further isolated and sub-clustered, with differential protein activity and frequency pre-treatment vs post-treatment compared as in the analysis of initial clustering for all cells. A proteomic gene set for each head and neck cancer-associated fibroblast (HNCAF) cluster was defined based on proteins differentially upregulated in each cluster. In the dataset of clinical trial patients profiled by bulkRNA-Seq that had been annotated with subsequent response (n=9) or non-response (n=19) to ⍺PD-1 immunotherapy (43), we applied VIPER transformation using the single-cell ARACNe networks on z-score scaled log10(TPM) counts from pretreatment bulkRNA-Seq data, and computed a differential protein activity signature ranking proteins by most upregulated in responders to most downregulated in responders. Enrichment of each HNCAF cluster marker set in the VIPER-transformed signature of responders vs nonresponders from bulkRNA-Seq was computed by Gene Set Enrichment Analysis (GSEA), with normalized enrichment score and p-value determined by 1000 random permutations of gene labels. Insufficient number of HPV+ samples prevented CAF enrichment analysis in HPV+ vs HPV− groups.
Clinical association of HNCAF signatures with outcome was further tested in TCGA head and neck cancer cohort processed by ARACNe and VIPER as above. Sample-by-Sample Normalized Enrichment Scores were computed ranking VIPER-inferred protein activity in each patient sample from highest to lowest activity and then applying GSEA. Normalized Enrichment scores for HNCAF cluster signatures were binarized to less than zero (low) or greater than zero (high), and Kaplan-Meier curve showing association with Overall Survival time was plotted along with the log-rank p-value, such that HNCAF-0 enrichment is associated with improved overall survival (p=0.0095, median survival time = 602 days vs 1671 days) and HNCAF-1 enrichment is associated with worse overall survival (p=0.0041, median survival time = 1718 days vs 773 days). We further plotted the sample-by-sample enrichment of these HNCAF populations among different TCGA tumor types with high stromal involvement (HNSC, PAAD, SARC, UCS, BRCA, CHOL, LIHC) and plotted the distribution of these enrichment scores by tumor type to assess relative tumor-type specificity of the identified HNCAF signatures.
Digital Spatial Profiling
NanoString GeoMX Digital Spatial profiling was further applied, profiling IO360 immune gene panel expression among three regions of interest (ROIs) from one patient and four ROIs from another. Anti-CD8, anti-⍺SMA, anti-PanCK, and DAPI stains were used for morphology identification and ROIs were selected based on high abundance of tumor (PanCK), cytolytic T cells (CD8), and fibroblasts (⍺SMA). ROIs were split into PanCK-positive and PanCK-negative components, with gene expression evaluated separately in each. In order to further assess spatial co-localization of HNCAF subtypes with functionally exhausted T-cells, we applied segment-by-segment gene set enrichment of HNCAF-0 and HNCAF-1 markers as well as enrichment of a published T-cell exhaustion signature (44), and correlate normalized enrichment scores for these populations between spatial segments.
CAF Phenotyping
In order to assess phenotypic concordance between prior fibroblast categorizations, including CAF-S1/S2/S3/S4 subtypes described in the setting of breast cancer (11, 12) and iCAF/myCAF subtypes described in the setting of pancreatic cancer (13), we have performed pairwise gene set enrichment of fibroblast phenotype marker gene sets among our HNCAF clusters identified by scRNA-Seq. For CAF-S1/S2/S3/S4 phenotype-matching, we sorted S1/S2/S3/S4 cells by FACS using the gating strategy described by Costa et al. (12) and subsequently performed bulk RNA Sequencing of each sorted population, applied VIPER using single-cell derived ARACNe networks, and computed differential protein activity of each population against the mean to define population-specific signatures, with genes ranked from most differentially-upregulated protein activity to most differentially-downregulated protein activity in CAF-S1/S2/S3/S4. We then performed pairwise Gene Set Enrichment Analysis of HNCAF cluster marker gene sets (by VIPER protein activity) among CAF-S1/S2/S3/S4 gene signatures. We highlight the findings that CAF-S1 gene signature was significantly enriched for the gene sets of HNCAF-0 (NES=7.43, p=1.1 × 10−13), HNCAF-1 (NES=6.54, p=6 × 10−11), and HNCAF-3 (NES=6.24, p=4.4 × 10−10), CAF-S2 gene signature was not significantly enriched for any HNCAF population, CAF-S3 signature was significantly enriched for HNCAF-4 gene set (NES=3.09, p=2 × 10−3), and CAF-S4 signature was significantly enriched for HNCAF-2 gene set (NES=6.7, p=2.2 × 10−11). Published CAF-S1 subtype marker gene sets (11) were directly tested by GSEA for enrichment in each single-cell, with resulting enrichment scores plotted by HNCAF cell cluster. Published iCAF/myCAF VIPER-inferred marker gene sets (13) were directly tested by GSEA for enrichment in each single-cell, with resulting enrichment scores plotted by HNCAF cell cluster, such that cells in HNCAF-1 are enriched for iCAF signature and cells in HNCAF-2 are enriched for myCAF signature. This phenotypic classification scheme highlights the distinction between our HNCAF categorization observed from scRNA-Seq and prior CAF classification paradigms.
Receptor-Ligand Interactions
Receptor-Ligand Interactions were inferred between coarse-grained cell types using 2,557 high-quality receptor-ligand interactions reported in the RIKEN FANTOM5 database (45). This list of receptor-ligand pairs was filtered to identify pairs where the ligand was significantly upregulated, at the gene expression level, in at least one subpopulation, across patients, and the corresponding receptor was significantly activated in another subtype, based on VIPER protein activity analysis, as proposed in (18). We further filtered these interactions to those detected in cancer-associated fibroblasts and plotted the number of unique receptor-ligand interaction pairs inferred between fibroblasts and each other detected subpopulation.
CAF isolation and culture
Fresh head and neck squamous cell carcinoma tumor specimens were processed to single cell suspension as described above. For HNCAF-0/3, tumor single cell suspension was cultured in DMEM supplemented with 10% FBS, streptomycin (100 μg/ml), and penicillin (100 U/ml) for 2–3 weeks at 37°C until fibroblasts grew out. For HNCAF-1, tumor single cell suspension was cultured in pericyte medium (ScienCell #1201) supplemented with 2% FBS, streptomycin (100 μg/ml), and penicillin (100 U/ml) for 2–3 weeks at 37°C until fibroblasts grew out. To verify CAF identity, RNA was isolated from CAF lysates using TRIzol (Invitrogen #10296010) and sent for bulk RNA sequencing. Gene set enrichment analyses for the HNCAF subtype protein activity signatures were then performed on the bulk sequencing data, along with inference of cell type proportions by CIBERSORTx. Fibroblasts were passaged when cultures reached ~80% confluence and all experiments were performed with CAF under 10 passages.
T cell isolation
CD3+ T lymphocytes were isolated from the peripheral blood of healthy human donors. Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Paque Plus, following manufacturer’s instructions. CD3+ T cells were isolated from PBMCs using magnetic bead sort with the MojoSort Human CD3 T Cell Isolation Kit (Biolegend #480022) according to manufacturer’s instructions. For isolation of CD3+ tumor-infiltrating lymphocytes (TILs), fresh head and neck squamous cell carcinoma tumor specimens were processed to single cell suspension as described above. CD3+ tumor-infiltrating lymphocytes were isolated from the tumor single cell suspension using magnetic bead sort with the MojoSort Human CD3 T Cell Isolation Kit.
T cell and CAF Coculture Assays
25,000 primary CAF were plated in DMEM supplemented with 10% FBS in 96 well plates. After CAF had attached to the plate, 50,000 CD3+ T cells were added to the CAF with or without CD3/CD28 activation beads (Gibco # 11131D) and cocultured at 37°C for 5–7 days with or without 20 ng/mL TGFβ. Media was renewed on days 3 and 5. Cocultures with tumor-infiltrating lymphocytes were only cultured for 3 days to preserve TIL viability. Following incubation, T cells were harvested and stained with Live/Dead Aqua (1:1600) for 15 minutes in PBS. Cells were then washed and stained for 15 minutes with an antibody cocktail containing anti-CD4-APC/Fire 810 (1:1000, SK3), anti-CD8-BB515 (1:200, RPA-T8), anti-PD-1-BV421 (1:100, EH12.2H7), anti-TIM-3-BV786 (1:100, F38–2E2), anti-NKG2A-PE (1:200, S19004C), and anti-CD103 (1:400, Ber-ACT8). Cells were then washed, fixed, permeabilized and stained with an intracellular antibody cocktail containing anti-Perforin-PerCP/Cy5.5 (1:100, B-D48) and anti-Granzyme B-Alex Fluor 700 (1:100, QA16A02). Cells were subsequently analyzed by flow cytometry using the Cytek Aurora.
Transwell T cell and CAF Coculture Assays
100,000 primary CAF were plated in DMEM supplemented with 10% FBS in the lower chamber of the transwell (0.4 μm pore size, Corning Polycarbonate Membrane Transwells #3401). 200,000 CD3+ T cells were plated in DMEM supplemented with 10% FBS in the upper chamber of the transwell. Cells were incubated at 37°C for 7 days. Media was renewed on days 3 and 5. Following incubation, T cells were stained and analyzed by flow cytometry using the Cytek Aurora as described above.
ELISA
The level of IFNɣ in cell culture supernatants was measured using an ELISA MAX Deluxe kit (Biolegend #430104) following manufacturer’s instructions. Supernatants were collected from CAF-T cell cocultures as described above.
Statistical Analysis
All quantitative and statistical analyses were performed using the R computational environment and packages described above with the exception of CAF composition and co-culture experiments. Statistical analyses of these assays were performed using Prism 9 software (GraphPad). Differential gene expression was assessed at the single-cell level by the MAST single-cell statistical framework as implemented in Seurat v3 (46), and differential VIPER activity was assessed by t-test, each with Benjamini-Hochberg multiple-testing correction. Comparisons of cell frequencies were performed by non-parametric Wilcox rank-sum test, and survival analyses were performed by log-rank test. In all cases, statistical significance was defined as an adjusted p-value less than 0.05. Details of all statistical tests used can be found in the corresponding figure legends.
Data Availability
All sequencing data are deposited and publicly available in a Mendeley data repository (doi:10.17632/yk8wj7xgdg.1) as well as the GEO database (GSE195832).
Results
Proteomic Master Regulatory Network Analysis of Longitudinal Single-Cell Transcriptomic Profile Identifies Functionally Unique CAF Populations in The HNSCC Microenvironment.
Longitudinal scRNA-Seq of patient tumors, pre- and post-treatment with nivolumab, and gene expression clustering with Seurat revealed 12 broadly distinct cellular populations, consistently expressed across all the tumors sequenced (Fig. S1A–S1B). To achieve higher resolution of cellular subpopulation characterization, we used scRNA-Seq profiles from each cluster to infer subpopulation-specific gene regulatory networks with the ARACNe algorithm (19), followed by protein activity analysis using the Virtual Inference of Protein-activity by Enriched Regulon (VIPER). Protein activity-based re-clustering identified two additional, previously undetected clusters for a total of 14 distinct cellular populations which were also consistently expressed across all four patients (Fig. 1A). To visualize key differences between these cellular populations, we generated a heatmap showing the activity of the five proteins most differentially activated in each cluster (Fig. 1B). The full list of differentially active proteins (p<0.05, FDR corrected) and differentially expressed genes (p<0.05, FDR corrected) are included in the Supplementary Data (Table S1). We first assessed the ability of these VIPER generated populations to accurately respond to expected treatment-induced changes. As expected, both gene expression and protein activity analyses revealed increased T cells and interferon-gamma protein activity following nivolumab treatment (Fig. 1C (clusters 1 and 5), Fig. S1C–S1D). When we interrogated changes in the abundance of other cell populations, VIPER clustering revealed heterogeneity among fibroblast cells not discoverable from gene expression clustering alone, with two clusters (clusters 4 and 9) presenting highly statistically significant post-treatment cellular fraction increase (Fig. 1C). Cell lineage inference, using SingleR (20), identified both clusters as fibroblasts, suggesting that PD-1 targeted immunotherapy in head and neck cancer was associated with CAF upregulation (Fig. 1D). Accordingly, imputed receptor-ligand interactions between cell types (18) suggested strong interplay between CAF and CD8 T cells (Fig. S1E). Furthermore, two additional clusters (clusters 6 and 7), also characterized as fibroblasts by SingleR (Fig. 1D), did not show significant fractional representation differences following immunotherapy (Fig. 1C), thus suggesting the existence of functionally distinct CAF subpopulations within the HNSCC TME.
Figure 1. VIPER analysis of longitudinal single-cell transcriptomic profiles of HNSCC show CAF changes associated with immunotherapy.
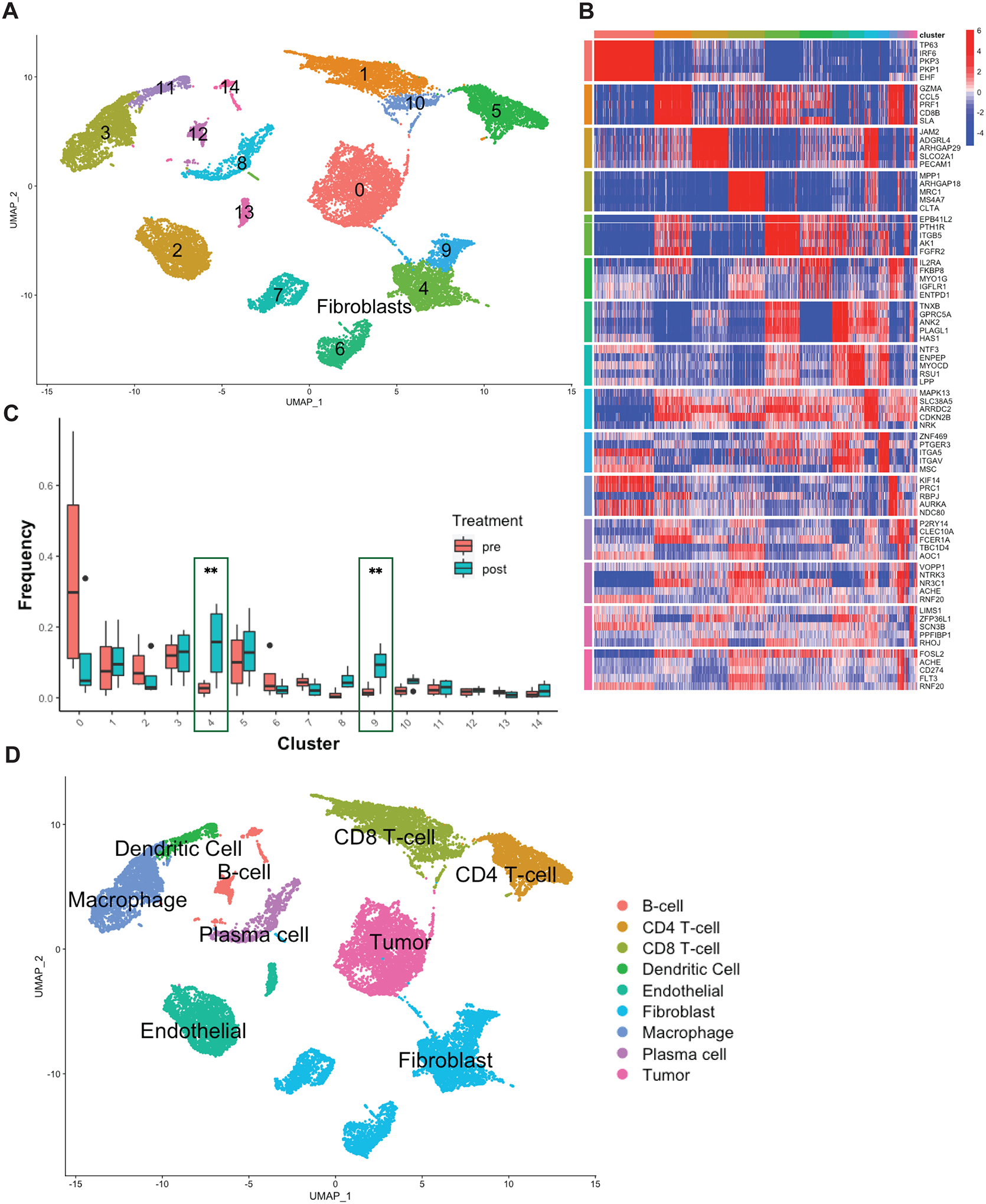
A) 2-dimensional UMAP projection of cells across all samples (n=4), processed by VIPER and clustered by resolution-optimized Louvain. Cells are colored by unsupervised cluster number, with fibroblast clusters (4,6,7,9) further labelled by cell type. B) Heatmap of top 5 most differentially upregulated proteins per cluster for each cell population in A. C) Boxplot of population frequency at baseline and following ⍺PD-1 immunotherapy for each cell type cluster in A. CAF subtypes increasing in response to immunotherapy are emphasized with green outline, ** indicates p<0.01. D) SingleR cell type inference projected on UMAP plot. Each cluster is assigned a lineage cell type based on its majority SingleR-inferred label.
VIPER Fibroblast Clustering Identifies Unique Sub-Populations Associated with Response and Resistance to Immunotherapy
To further evaluate functional differences between the distinct CAF sub-populations in the HNSCC TME, we performed protein activity-based sub-clustering focusing only on fibroblast cells using ARACNe and VIPER. The analysis identified five molecularly-distinct CAF clusters, preliminarily termed HNCAF-0 – HNCAF-4 (Fig. 2A). Importantly, gene expression-based sub-clustering of fibroblasts only identified two distinct CAF populations corresponding to the two fibroblast populations seen in Fig. S1A (Clusters 3 and 6). As expected, these two clusters phenotypically match the two CAF populations previously identified in HNSCC such that Cluster 3 corresponds to CAF1 and Cluster 6 corresponds to CAF2 (Fig. S1F) (15). Among the five HNCAF populations identified by protein activity-based clustering, cell fractional representation increased for HNCAF-0 and HNCAF-3, decreased for HNCAF-1 and HNCAF-2, and was unaffected for HNCAF-4 (Fig. 2B). The top ten most differentially active proteins, presented as a ranked list of differentially active transcription factors and signaling molecules, in each of the five clusters highlight their potential functional properties (Fig. 2C) to help define the molecular biology of each HNCAF phenotype (Table S2). To assess the associations of each HNCAF subpopulation with clinical response to ⍺PD-1 immunotherapy, we used the HNCAF molecular signatures to analyze the bulk RNA sequencing (bulkRNA-Seq) profiles from the 28-patient parental cohort annotated with clinical response to nivolumab. For this purpose, we first used VIPER to generate protein activity profiles from each bulk profile, using fibroblast specific regulatory networks generated at the single-cell level, and then evaluated the enrichment of the most differentially active proteins in each HNCAF subpopulation (marker protein sets) among proteins differentially active in responders vs non-responders. The analysis revealed statistically significant enrichment of HNCAF-0 and HNCAF-3 marker genes in pre-treatment samples of patients who subsequently responded to ⍺PD-1 immunotherapy (Fig. 2D). This result indicates that the HNCAF-0 and HNCAF-3 populations, which also expand following nivolumab treatment, may be predictive of clinical response in human HNSCC patients. By contrast, HNCAF-1, HNCAF-2, and HNCAF-4 cells did not expand following therapy and their markers were not significantly enriched in responders vs non-responders.
Figure 2. Fibroblast sub-clustering reveals distinct populations associated with differential responses to αPD-1-based immunotherapy with the potential to predict clinical outcome.
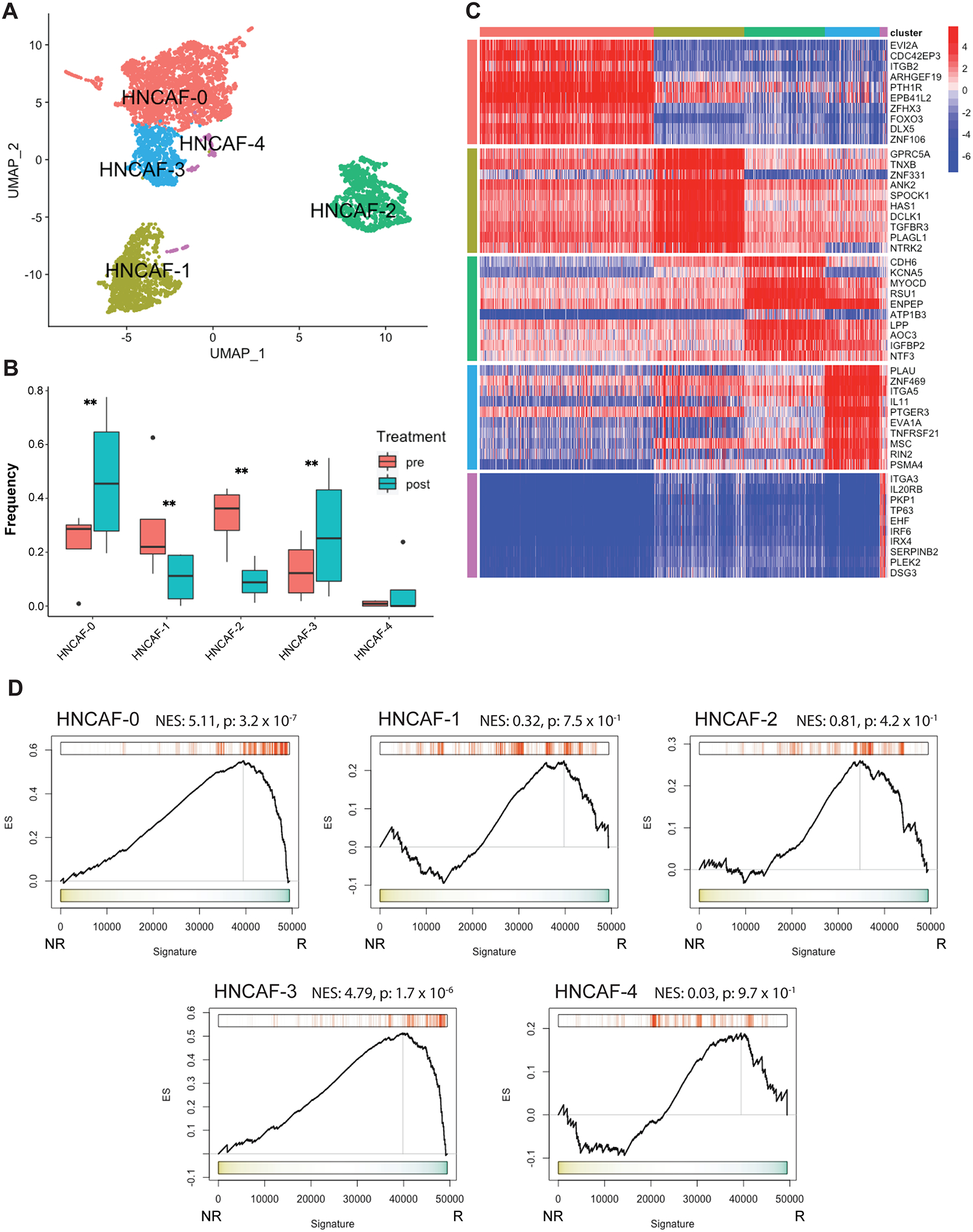
A) 2-dimensional UMAP projection of CAF across all samples (n=4), re-clustered by resolution-optimized Louvain and colored by cluster identity. B) Boxplot of cluster frequencies of each HNCAF type pre- vs post-nivolumab therapy, ** indicates p<0.01. C) Heatmap of top 10 most differentially upregulated proteins per cluster for each CAF population. D) Protein Activity Profile Enrichment plots of single-cell protein population markers for each HNCAF cluster (Supplemental Table 1) in bulkRNA-Seq signature of immunotherapy responders (R, n=9) vs non-responders (NR, n=19), profiled pre-treatment. In each plot, ES represents raw enrichment score and NES represents the normalized enrichment score, as computed by GSEA using the set of statistically significant differentially active proteins for each HNCAF population at the single-cell level as a gene set, and testing for enrichment in the ranked list of proteins most to least enriched in therapy responders vs non-responders (ranked 0–5000). P-values for Normalized Enrichment Score are assessed by 1000 random permutations of gene ranking.
Fibroblast Subtype Analysis Reveals Novel Classification Paradigm in HNSCC
Due to scant literature on CAF in human HNSCC, we next quantified the extent of CAF infiltration from surgical HNSCC specimens using flow cytometry. CAF abundance — as defined by CD45− EpCAM− CD31−— ranged between 12% and 58% of the total live cells (Fig. 3A). Having confirmed significant abundance of CAF in human HNSCC, we proceeded to assess the presence of novel HNCAF subpopulations predicted by the VIPER analysis. Distinct CAF subpopulations termed CAF-S1, CAF-S2, CAF-S3, and CAF-S4 have been previously identified in breast cancer based on the expression of CD29 and fibroblast activation protein (FAP) by flow cytometry (12). Kieffer et. al. showed that CAF-S1 can be found in HNSCC but the presence of other CAF-S populations in HNSCC remains unknown (11). Hence this protein-based framework was initially used to assess how the VIPER imputed HNCAF align with breast cancer CAF scheme. Following the same gating strategy employed by Costa et al. (Fig. S2), we confirmed the presence of all four breast cancer CAF-S populations in HNSCC (Fig. 3B) (12). Interestingly, CAF-S1 and CAF-S2 were most abundant, while CAF-S3 and CAF-S4 abundance was quite minimal (Fig. 3C). We then sorted CAF-S1 – S4 cells from HNSCC tumors (Fig. S2) and performed bulk RNA sequencing of each subpopulation to assign these sorted cells to the VIPER generated HNCAF populations. Pairwise gene set enrichment analysis of the HNCAF protein activity signatures in the bulk transcriptome indicated that the gene sets representative of HNCAF-0, HNCAF-1, and HNCAF-3 were all enriched in the same breast subtype (CAF-S1), while HNCAF-2 and HNCAF-4 were both enriched in CAF-S4 with HNCAF-4 also enriched in CAF-S3 (Fig. S3A). CAF-S2—primarily defined as double-negative for FAP and CD29—did not significantly align with any HNCAF. These analyses importantly showed that VIPER-clustered HNCAF provide much greater resolution of functionally distinct CAF phenotypes compared to the flow-based CAF-S1/S2/S3/S4 framework. Specifically, the CAF-S1 subtype matched three distinct HNCAF subtypes, which have opposing association with clinical responses to immunotherapy. Additionally, the HNCAF subtypes did not clearly correlate with any of the CAF-S1 subclusters later identified by gene expression in breast cancer apart from ecm-myCAF (Fig. S3B). HNCAF-0,1,2 and 3 were all significantly enriched for the ecm-myCAF signature, with HNCAF-0 and HNCAF-3 being more enriched than HNCAF-1 and HNCAF-2. However, ecm-myCAF are associated with immunosuppression and resistance to immunotherapy leading us to believe that HNCAF-0 and HNCAF-3 differ from ecm-myCAF in terms of functionality (11).
Figure 3. Prognostically associated HNCAF sub-populations provide greater resolution than previously characterized CAF phenotypes.
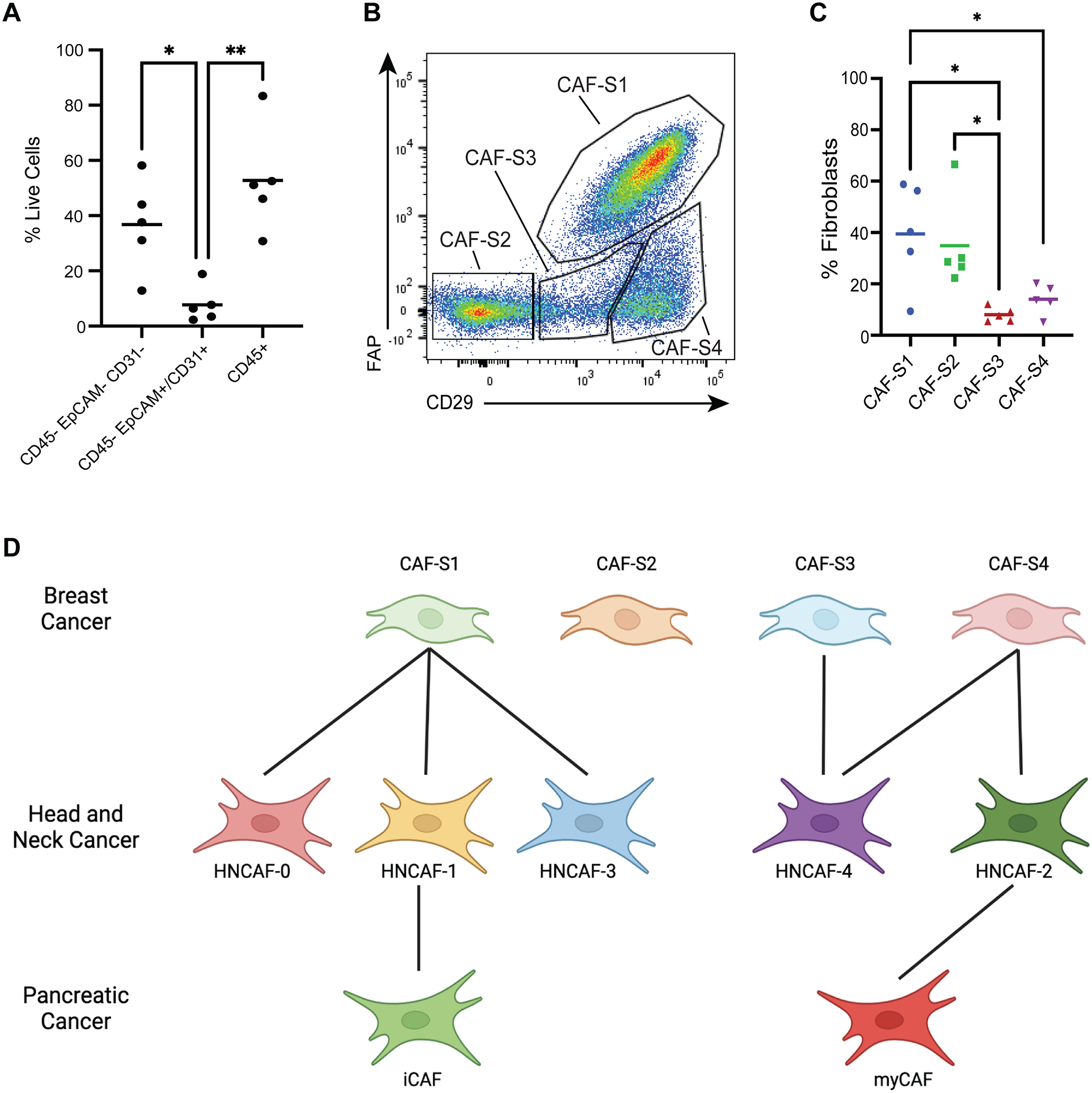
A) Relative frequencies of stromal (CD45-Epcam-CD31-), epithelial/endothelial (CD45-Epcam+/CD31+) and immune (CD45+) cell components across HNSCC patients quantified by flow cytometry (n=5). B) Flow cytometry gating strategy to isolate previously described CAF phenotypes from HNSCC specimens. C) Relative frequency for each CAF subtype from B among total CAF quantified by flow cytometry (n=5). D) Phenotype-matching between unsupervised HNCAF clusters from scRNA-Seq and previously defined CAF-S1 to CAF-S4, as well as iCAF and myCAF. Each single-cell HNCAF population is labelled as the sorted CAF-S1 to CAF-S4 population with highest gene set enrichment, as shown in Figure S3. The data in A and C were analyzed using one-way ANOVA and * indicates p<0.05, ** indicates p<0.01.
We also tested for concordance of our classification schema with previously defined gene set markers of inflammatory CAF (iCAF) and myofibroblastic CAF (myCAF), as first described in pancreatic cancer (13). Cell-by-cell enrichment of iCAF and myCAF gene signatures revealed an enrichment for the iCAF signature in HNCAF-1 cells and for myCAF in HNCAF-2 cells (Fig. S3C–S3D). Correlations between our HNCAF populations and the breast or pancreatic CAF phenotypes is summarized in the classification scheme shown in Fig. 3D. While presenting some similarity to CAF-S1 cells, we conclude that HNCAF-0 and HNCAF-3 represent novel, molecularly distinct fibroblast subpopulations, potentially unique to head and neck cancer and predictive of patient outcome (Fig. 2D), which do not completely match the iCAF/myCAF classification. Furthermore, in contrast to the HNCAF-0 and HNCAF-3 subtypes, iCAF/myCAF and CAF-S1/S2/S3/S4 are not significantly enriched in responder cohorts, suggesting that these classification schemes do not accurately depict CAF function in HNSCC (Fig. S4). Cumulatively, these data underscore the greater resolution of HNCAF from our VIPER analysis compared to previous efforts, and more critically, the HNCAF populations identified through VIPER allow prognostic correlations of CAF cells in HNSCC.
HNCAF-0 Predicts Favorable Disease Course in TCGA, in Contrast to HNCAF-1.
To evaluate the prognostic relevance of the CAF populations identified in a setting free from external immunotherapeutic pressures, we quantified the enrichment of HNCAF protein activity signatures in The Cancer Genome Atlas (TCGA) HNSCC cohort (Fig. 4, Fig. S5). Gene set enrichment (GSEA) analysis (21), on a patient-by-patient basis, revealed significant enrichment of the HNCAF-0 signature in patients with better overall survival in TCGA (Fig. 4A), suggesting that these cells may not only be important regulators of immunotherapy response but may also play a key role in mounting clinically relevant, endogenous immune responses against HNSCC. In contrast, the HNCAF-1 protein activity signature was associated with early worse overall survival when there were sufficient number of patients (Fig. 4B). Prognosis in HNSCC has been associated with higher TIL level in the TCGA cohort (22), and these results were consistent with the differential immunomodulatory functional roles of distinct VIPER derived HNCAF cells.
Figure 4. HNCAF-0 and HNCAF-1 have contrasting prognostic associations.
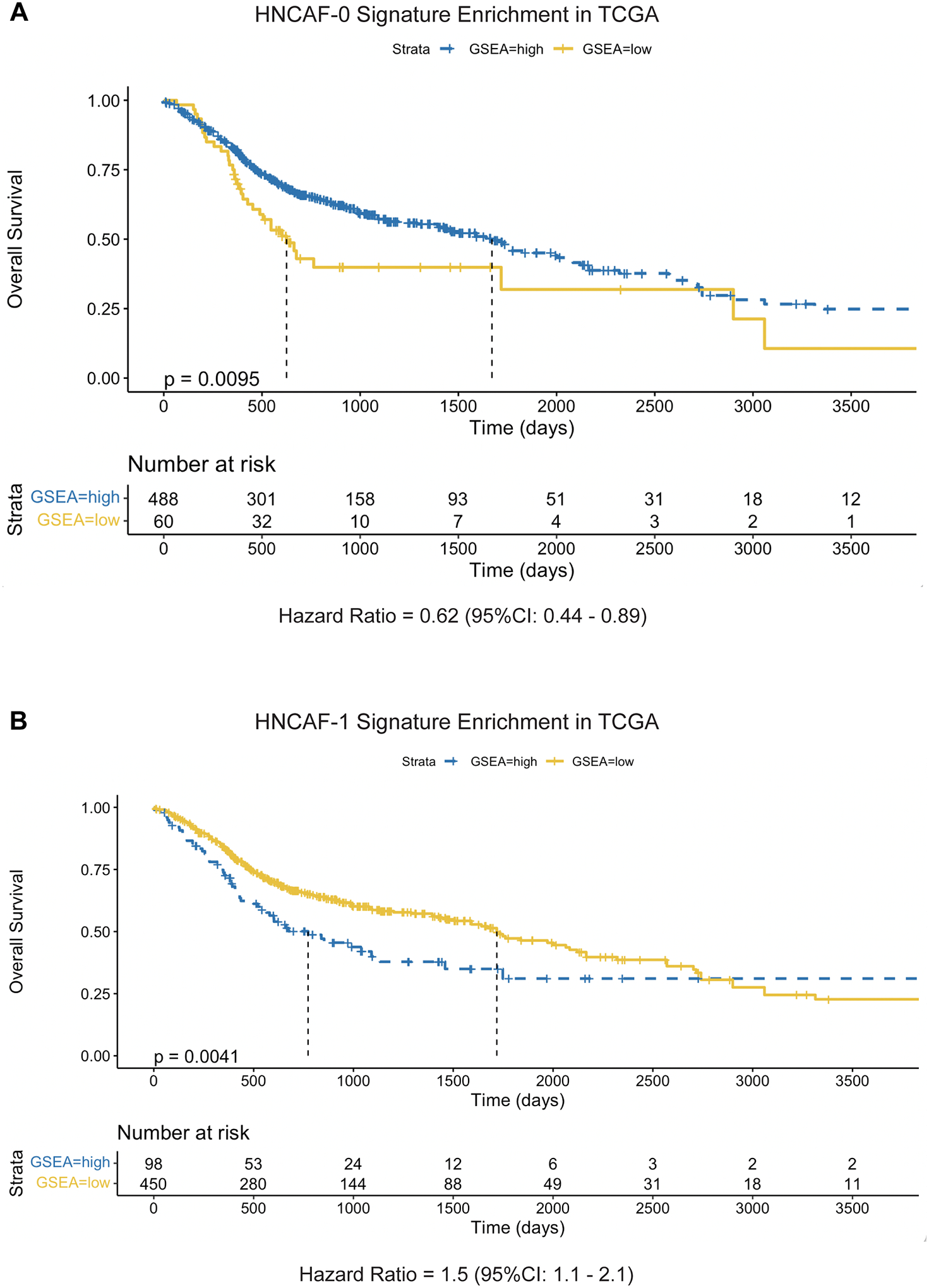
A) Kaplan-Meier plot of HNCAF-0 Protein Activity Gene Set Enrichment among TCGA dataset of head and neck squamous cell carcinoma patients in association with overall survival time, limited to patients with under 10 years of follow-up. Enrichment scores were binarized by log-rank maximization to “high HNCAF-0” and “low HNCAF-0” and showed significant association with improved survival (p=0.0095, median survival time = 602 days vs 1671 days). Hazard ratio is shown below the plot, with 95% confidence interval. B) Kaplan-Meier plot of HNCAF-1 Protein Activity Gene Set Enrichment among TCGA head and neck squamous cell carcinoma patients in association with overall survival time, limited to patients with under 10 years of follow-up, as in A. HNCAF-1 enrichment shows significant association with worse survival (p=0.0041, median survival time = 1718 days vs 773 days). Hazard ratio is shown below the plot, with 95% confidence interval.
HNCAF-0/3 cells inhibit TGFβ dependent T-cell exhaustion in functional co-culture experiments.
Prompted by these intriguing clinical findings (Fig. 2D), we studied the potential interactions of HNCAF-0 and HNCAF-3 cells with other TME subpopulations. Interactome analysis showed that HNCAF have more receptor-ligand interactions with CD8 T cells than with any other cell subtype in the TME (see methods) (Fig. S1E). Therefore, we next interrogated the relationship between HNCAF-0 and human CD8 T cells in situ and in vitro. Digital spatial profiling (DSP, NanoString) of immune-related transcripts and protein markers was performed on HNSCC tissue from patients prior to nivolumab treatment. We first analyzed global CAF infiltration pattern in HNSCC tissue using aSMA, aCD8, and acytokeratin antibodies. These multiplexed immunofluorescent images exhibited colocalization of CAF with CD8+ T cells in the stromal compartment (Fig. 5A, white arrow). Reliable validated antibodies for each of the VIPER derived HNCAF subpopulations are currently unavailable to prevent histological analysis of each HNCAF population at this time.
Figure 5: HNCAF spatially co-localizes with CD8 T cells and HNCAF-0/3 functionally decrease TGFb dependent T cell exhaustion in vitro.
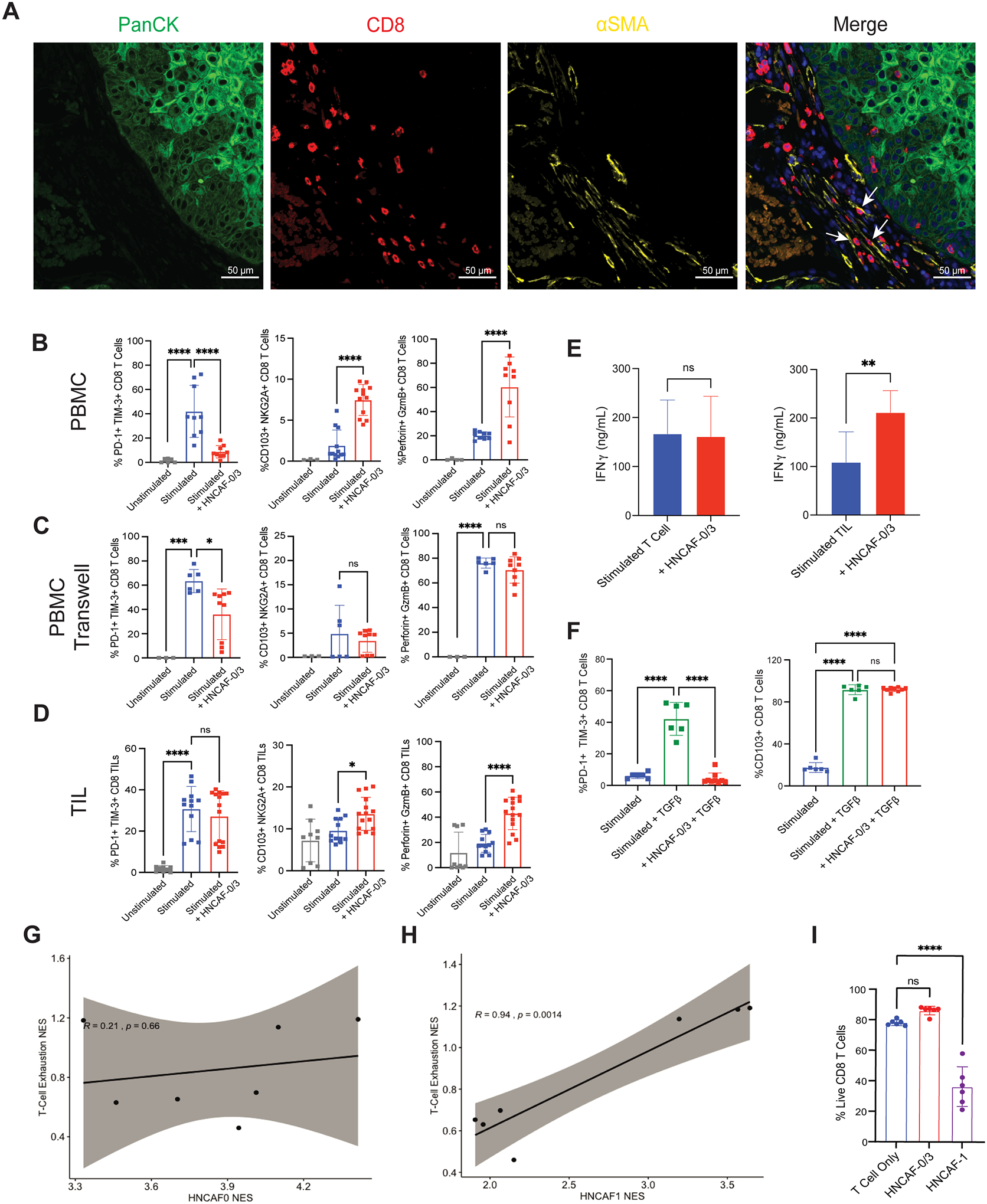
A) Pre-treatment DSP immunofluorescence imaging from a representative patient treated with ⍺PD-1 immunotherapy, such that tumor cell localization is indicated by panCK staining (green), Cytotoxic T cell localization by CD8 staining (red), fibroblast localization by ⍺SMA staining (yellow), and nucleated cells by DAPI staining (blue). Arrows indicate interactions between ⍺SMA+ fibroblasts and CD8+ T cells. B) Co-culture experiment of naïve T cells derived from peripheral blood mononuclear cells (PBMC) with CD3/CD28 stimulation and isolated HNCAF-0/3 cells, showing T cell exhaustion markers (%PD-1+ TIM-3+ cells), tissue residency memory markers (%CD103+ NKG2A+ cells), and cytotoxicity (%Perforin+ GzmB+ cells) (n=3–12). C) Co-culture experiment of T cells with HNCAF-0/3 cells as in B except in contact-isolating transwell culture (n=3–9). D) Co-culture experiment of Tumor-Infiltrating Lymphocytes (TIL) with CD3/CD28 stimulation and isolated HNCAF-0/3 cells (n=9–15). E) Interferon gamma levels in co-culture of naïve T cells derived from PBMC and TIL with HNCAF-0/3 cells determined by ELISA (n=3 for PBMCs; n=6 for TIL). F) Rescue experiment of T cells derived from PBMCs with CD3/CD28 stimulation and TGFβ with or without HNCAF-0/3 (n=6–9). G) Spatial enrichment of HNCAF-0/3 gene set vs enrichment of T-cell exhaustion signature in Nanostring DSP of tissue slices across patients (n=2). H) Spatial enrichment of HNCAF-1 gene set vs enrichment of T-cell exhaustion signature in Nanostring DSP of tissue slices across patients (n=2). Signatures are positively correlated with respect to spatial co-localization (correlation coefficient=0.94, p=0.0014). I) Quantitation of live cells out of total CD8 T cells determined by flow cytometry from co-culture with HNCAF-0/3 or HNCAF-1. B-D, F) Percentages were quantified by flow cytometry. Results are shown as mean ± SD and are representative of at least three independent experiments. The data were analyzed using one-way ANOVA (B-D, F, I) or the Student t test (E) and * indicates p<0.05, ** indicates p<0.01, *** indicates p<0.001, and **** indicates p<0.0001.
To test whether HNCAF-0 cells can directly affect the biology of the CD8 T cells, we performed in vitro co-culture assays with HNCAF harvested from surgical resection mixed with either naïve T cells or tumor-infiltrating T cells. Primary fibroblasts enriched for HNCAF-0/3 were isolated from human HNSCC samples and their transcriptional identity was verified by RNA-Sequencing and protein activity analysis (Fig. S6). Due to the phenotypic similarity between HNCAF-0 and HNCAF-3, we were unable to enrich solely for HNCAF-0 and proceeded with a heterogeneous population enriched for both HNCAF-0 and HNCAF-3. When co-cultured with T cells isolated from peripheral blood mononuclear cells of healthy donors, HNCAF-0/3 cells reduced the PD-1+TIM-3+ exhaustion phenotype among exogenously activated CD8 T cells and increased the CD103+NKG2A+ tissue resident memory phenotype, as well as their cytolytic function, based on Perforin and Granzyme B expression (Fig. 5B). It is important to note that reduced exhaustion and increased tissue resident memory phenotypes caused by HNCAF-0/3 was not due to PD-1:PD-L1 signaling (Fig. S7A–S7B). Transwell co-culture assays revealed that HNCAF-0/3-mediated T cell activation increase and induction of tissue resident memory phenotypes—but not T cell exhaustion phenotype mitigation—depends on cell-to-cell contact (Fig. 5C). Additionally, coculture of HNCAF-0/3 cells with tumor-infiltrating T cells isolated directly from human HNSCC specimens resulted in an equivalent increase in tissue resident memory phenotype and cytotoxicity among CD8 T cells, but HNCAF-0/3 cells could not rescue the exhaustion phenotype of terminally exhausted tumor-infiltrating T cells (Fig. 5D). However, the duration of coculture for these experiments was shorter than with PBMCs due to issues with tumor-infiltrating T cell viability and downregulation of PD-1+TIM-3+ exhaustion phenotypes likely requires longer interaction times. Regardless, HNCAF-0/3 cells strongly promoted production of the activation markers, Perforin, Granzyme B, and IFNɣ, in tumor-infiltrating T cells (Fig. 5D–5E), suggesting that HNCAF-0/3 may prevent terminal exhaustion in early activated T cells as well as reinvigorate already exhausted T cells in the TME. Notably, we found that HNCAF-0/3 completely rescued TGFβ-mediated PD-1/TIM-3 induction in culture, without inhibiting total TGFβ signaling, as defined by CD103 induction (Fig. 5F).
To evaluate CAF influences on T cell exhaustion in situ without a validated antibody for each HNCAF populations, we leveraged the digital spatial profiling data to evaluate colocalization of HNCAF-0 and HNCAF-1 protein activity signatures in regions enriched for the T-cell functional exhaustion signature. Indeed, the HNCAF-1 signature enrichment significantly correlated with increased T cell exhaustion signature enrichment (r = 0.94, p = 0.0014). In sharp contrast, the HNCAF-0 signature was not significantly associated with the T-cell exhaustion signature in the TME region of interest (Fig. 5G–5H). Given the association of HNCAF-1 cells with an immunosuppressive environment, we aimed to evaluate the direct impacts of isolated HNCAF-1 cells on T cell phenotypes in co-culture, as performed for HNCAF-0/3 cells. However, despite repeated experiments, T cells co-cultured with HNCAF-1 rapidly died, leaving an insufficient number of viable cells for further analyses (Fig. 5I, Fig. S7C–S7E). T cell death induction was not observed when T cells were cultured in isolation or with HNCAF-0/3 cells, suggesting HNCAF-1-mediated accelerated T cell apoptosis ex vivo.
HNCAF-0 can predict clinical outcome to aPD-1 blocking antibodies.
To test for the potential generalizability of these HNCAF subpopulations, we next performed enrichment of HNCAF protein activity signatures across TCGA, by tumor type, focusing on tumors with high stromal cell content. Enrichment analyses revealed that HNCAF-0 enrichment is relatively specific to HNSCC while HNCAF-1 enrichment is more broadly observed (Fig. S8A–S8B). Intriguingly, HNCAF-1 enrichment is highest in pancreatic adenocarcinoma, which is known to be unresponsive to PD-1 based immunotherapy (Fig. S8B). HNCAF-1 phenotypically matches the previously defined iCAF population from pancreatic cancer (Fig. 3D).
To externally validate our HNCAF-0/3’s potential for clinical response prediction, we tested for enrichment of protein activity signatures in another cohort of HNSCC patients treated with αPD-1 immunotherapy, pembrolizumab (23). The analysis revealed statistically significant enrichment of HNCAF-0 and HNCAF-3 marker genes in pre-treatment samples of patients who subsequently responded to pembrolizumab, validating the association of HNCAF-0/3 with immunotherapy response in an independent cohort (Fig. 6A). Although not as enriched as HNCAF-0 and HNCAF-3, analysis of this cohort also revealed a significant enrichment of HNCAF-2 in responders pre-treatment (Fig. 6A). HNCAF-3, which showed the strongest overall enrichment in responders compared to non-responders (Fig. 6A), also showed the highest predictive Area Under Receiver Operating Characteristic (AUROC), with an AUROC of 0.8 (Fig. 6B), an improvement as compared to reported AUROC of PD-L1 Tumor Proportion Score (AUROC = 0.66) and PD-L1 Combined Positive Score (AUROC = 0.61) (24).
Figure 6: HNCAF-0 and HNCAF-3 are also predictive of favorable responses to pembrolizumab.
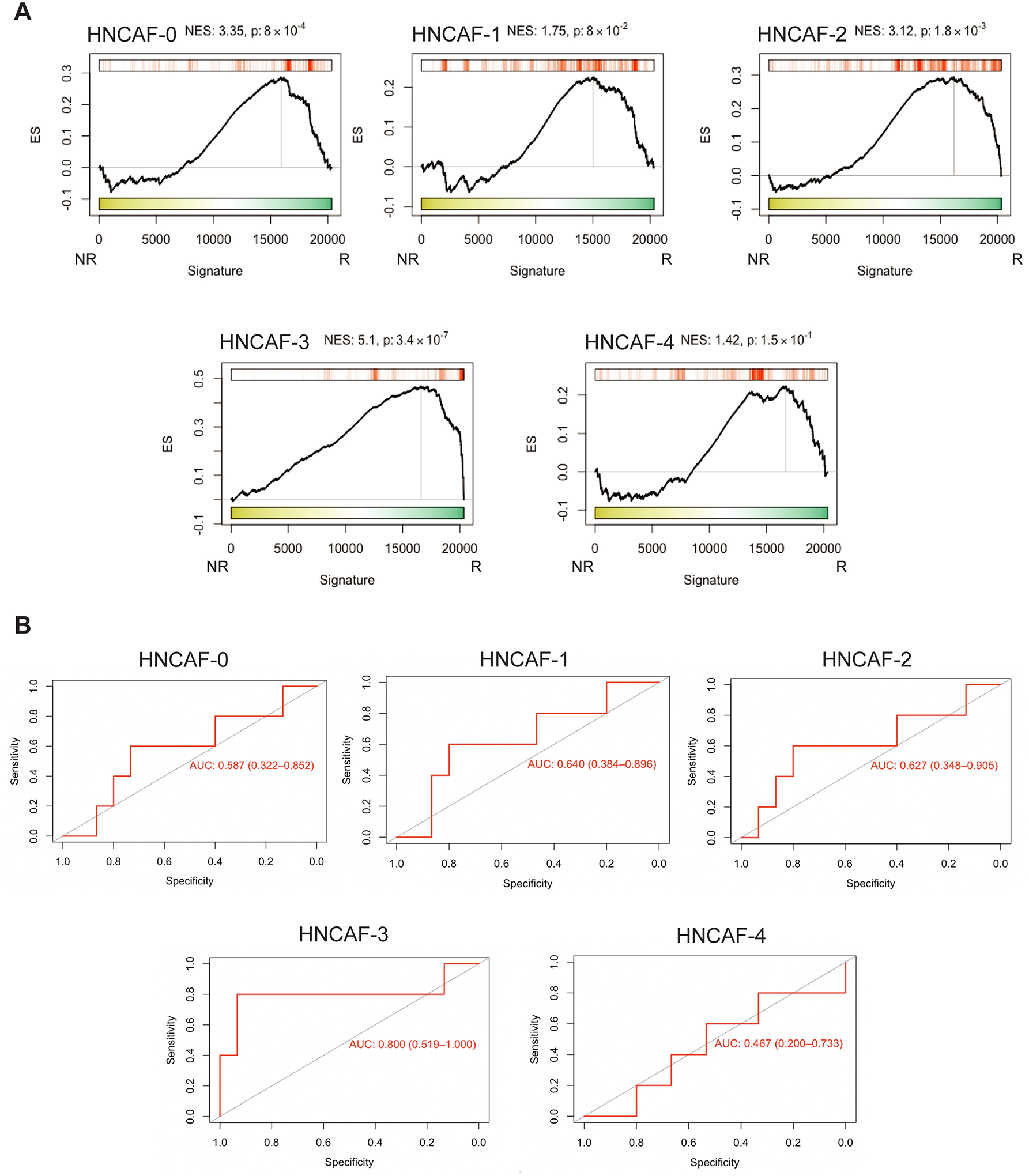
A) Protein Activity Profile Enrichment plots of single-cell protein population markers for each HNCAF cluster (Supplemental Table 1) in bulkRNA-Seq signature of pembrolizumab immunotherapy responders (R, n=5) vs non-responders (NR, n=15), profiled pre-treatment (23). B) Area Under the Receiver Operating Characteristic (AUROC) plots corresponding to pre-treatment predictive power of patient-by-patient protein activity profile enrichment for each HNCAF population show in A. 95% confidence intervals are included.
Discussion
In this study, we used protein activity profiles, as measured by the VIPER algorithm analysis of a longitudinal single-cell transcriptomics HNSCC dataset, to identify five molecularly distinct CAF subtypes. We took advantage of the longitudinally harvested biospecimens from a neoadjuvant clinical trial to show that two subtypes, HNCAF-0 and HNCAF-3, are predictive of favorable clinical responses to PD-1 checkpoint blockade therapy. Moreover, we discovered HNCAF-0/3 cells have an immunostimulatory effect on CD8 T cells while HNCAF-1 cells are associated with immunosuppression. From a functional perspective, we have shown that HNCAF-0/3 fibroblasts prevent induction of an exhausted T cell phenotype and are associated with increased CD8 T cell cytotoxicity. HNCAF-1 fibroblasts, in contrast, correlate with increased T cell exhaustion in situ and T cell apoptosis in vitro, suggesting contrasting roles for these CAF subtypes. Functional significance of HNCAF-2 and HNCAF-4 subtypes have yet to be defined.
Protein activity-based clustering with VIPER has previously been shown to outperform gene expression-based clustering and we have corroborated this with the identification of five HNCAF subtypes compared to two with gene expression-based clustering (18). Although we are not the first to identify CAF in HNSCC by single-cell sequencing, we were able to achieve greater resolution with VIPER allowing for a more granular picture of the CAF composition in HNSCC (11, 15). We were also able to integrate the major subclasses of CAF identified in breast cancer and pancreatic cancer into our HNSCC CAF data. We showed through GSEA that CAF-S1, identified in breast cancer and in our HNSCC samples as CD29+FAP+ fibroblasts, correspond to three of the HNCAF groups we identified, HNCAF-0, HNCAF-1 and HNCAF-3 (12). Kieffer et al. further dissected CAF-S1 in breast cancer and identified 8 subgroups but only focused on the 5 most abundant groups (11). We performed GSEA of these 5 subgroups but were only able to identify one with significant enrichment in our HNCAF subpopulations - ecm-myCAF. We did find that the ecm-myCAF signature was significantly enriched on 4 of our 5 HNCAF groups. The discrepancies between our sub-clustering of CAF-S1 and Kieffer et al. may be due to tumor specific differences in the CAF heterogeneity between breast and HNSCC as well as the use of a protein activity-based algorithm in contrast to gene expression. Additionally, we have found a CAF population (HNCAF-0) that is relatively specific to HNSCC amidst the other CAF types seen across different cancers. At this time, we hypothesize that tumor intrinsic factors may be influencing the differentiation of tumor specific CAF subpopulations (HNCAF-0) while nonspecific CAF subpopulations like HNCAF-1 could be derived from mesenchymal stem cells (25–29).
Despite the incomplete biological understanding of the HNCAF, our work introduces a novel clinically actionable biomarker for HNSCC. Immune checkpoint inhibitors (ICI) have revolutionized the field of cancer immunotherapy with monoclonal antibodies targeted against CTLA-4, PD-1, and PD-L1 being recently approved for use as frontline therapies for HNSCC and other cancer types (1, 3, 30). The factors driving resistance to ICI remain largely unknown, making it difficult to select those who will respond and who will not. Accordingly, there remains an unmet need for reliable biomarkers predictive of response to guide patient selection and optimization of ICI treatment. In recent studies, CAF have been implicated to influence resistance to checkpoint immunotherapy. A preclinical model of pancreatic ductal adenocarcinoma showed that depletion of CAF expressing high levels of FAP improves response to ⍺PD-L1 blockade (9). Similarly, single-cell RNA sequencing revealed a LRRC15+ CAF population associated with worse response to ⍺PD-L1 immunotherapy in a clinical trial for bladder cancer (10). Furthermore, distinct CAF populations identified in breast cancer were also shown to be associated with poor ⍺PD-1 immunotherapy response in melanoma and lung cancer (11). All these studies have implicated CAF primarily as contributors to resistance; however, the precise nature of molecularly distinct CAF subtypes and their role in mediating the effect of immunotherapy remains poorly investigated. With our ability to provide a higher resolution CAF repertoire, we show that the presence of two unique HNSCC-specific CAF subtypes is predictive of clinical response to immunotherapy in HNSCC. In particular, our functional findings suggest that HNCAF-0/3 fibroblasts are active participants in the immune response elicited by PD-1 directed immunotherapy through T cell modulation. For HNSCC, we are currently evaluating whether these CAF subtypes behave differently in virally associated HPV+ HNSCC vs. HPV− tumors.
Previous studies have typically shown CAF as promoters of immunosuppression. CAF have been shown to prevent T cell infiltration and to kill T cells in an antigen-dependent manner, via PD-L2 and FasL (9, 31). CAF have also been shown to suppress T cells through upregulation of PD-L1 and PD-L2 and through recruitment of regulatory T cells (12, 32). While confirming the immunosuppressive role of some HNCAF subtypes, our work has also established a novel pro-inflammatory role for distinct CAF subtypes, which act as a promoter of T cell activation and cytotoxicity. In light of this immunostimulatory function, we have termed the HNCAF-0/3 phenotype as T cell-stimulating CAF (tsCAF). Based on previous studies identifying TGFβ1 signaling through SMAD3 as a regulator of PD-1 expression, we hypothesize that tsCAF may repress SMAD3 to transcriptionally inhibit PD-1/TIM-3 expression (33). Although our data strongly suggests that HNCAF-0/3 are immunostimulatory, the inability to sort these cells to obtain pure populations is a major limitation of this study as the CAF cells used in our functional studies do not encompass a pure population at this point. Proteomic based approach to select and sort CD29+FAP+ CAF into tsCAF and HNCAF-1 is an active area of investigation. Recently, tumor restrictive CD105− CAF have been demonstrated in murine models of PDAC which are mediated by the adaptive immune system (34). CD105− CAF highly overlap with the myCAF gene signature, which has also previously been demonstrated to be tumor constraining (35). Since the tsCAF we describe here are distinct from the myCAF population both molecularly and by surface marker expression, we believe they represent a distinct CAF population from CD105− CAF.
Interestingly, we found that co-culture of HNCAF-0/3 with CD8 T cells induced a tissue-resident memory (Trm) phenotype that co-expressed NKG2A. NKG2A is an inhibitory receptor that we and others found to be highly enriched in tumor-infiltrating Trm+ CD8 T cells in HNSCC (36, 37). NKG2A ligation with its ligand HLA-E reduces cytotoxicity and effector function and is therefore a novel immunotherapy target (38). Clinical trials combining NKG2A blockade (monalizumab) and other checkpoint inhibitors have shown promising results in HNSCC (36). While it is not clear why NKG2A is upregulated on tumor-infiltrating CD8 T cells, our findings suggest HNCAF characterization may also inform clinical development of NKG2A inhibition along with other immune checkpoint inhibitors. We found that upregulation of NKG2A required contact between the HNCAF-0/3 and activated CD8 T cells, which suggests that induction is mediated by either a surface ligand on HNCAF-0/3 or a component in the extracellular matrix produced by the CAF. To our knowledge, NKG2A expression on CD8 T cells has never been associated with fibroblasts and our finding here provides a clear link between intra-tumoral NKG2A expression on CD8 T cells and the tumor stroma. Future studies will need to be performed on HNSCC specimens to determine if NKG2A expression on CD8 T cells is localized to stromal regions or associated with increased HNCAF-0/3 cells.
Plasticity of CAF subtypes have also been well demonstrated (14, 39), and our CAF atlas provides an excellent framework to develop strategies to force CAF differentiation towards the pro-inflammatory tsCAF phenotype rather than the immunosuppressive HNCAF-1 phenotype, to be combined with immunotherapy. Our characterization of each CAF subpopulation characterization through their master regulatory network lends itself towards this strategy. Further investigation of the signals that induce tsCAF formation and activation, possibly by targeting master regulators of the two subtypes, either genetically (40, 41) or pharmacologically, via the OncoTreat algorithm (42) is currently underway. Critically, this study highlights a much greater molecular heterogeneity of CAF subtypes than previously appreciated and demonstrates the critical need to functionally characterize their pleiotropic effects in terms of cancer progression, outcome, and response to immunotherapy and other cancer treatments.
Supplementary Material
Translational Relevance:
Our utilization of a systems biology-based master regulator analysis of longitudinal single-cell transcriptomic profiles unveiled novel cancer associated fibroblast (CAF) subtypes that can potentially predict clinical responses to αPD-1 blocking antibodies in head and neck cancer patients. While PD-L1 expression in the tumor is one biomarker of response, there is a clear clinical need to improve upon this imperfect test. Our report underscores this need by demonstrating that important stromal cells can regulate immune cells in the human tumor microenvironment. These results also highlight the strength of systems biology-based analysis at the single-cell resolution to uncover unique human cancer biology that can be extracted from appropriately curated clinical trial specimens. The fibroblast populations identified may also serve as therapeutic targets in future combinatorial trials.
Acknowledgements
This work was supported by the Barry Baker Biorepository Fund (YJK), James Rowen Fund (YJK, PH), NIH R01 CA178613 (YJK), R01 DE027749 (YJK), Dept. of Defense CDMRP Breakthrough Award (YJK), the American Cancer Society 131356-RSG-17-160-01-CSM (PH), and NIH RO1CA211695-01A1 (PH). AC was supported by an NIH/NCI Outstanding Investor Award (R35 CA197745) to AC, and two NIH Instrumentation grants (S10 OD012351 and S10 OD021764) to AC. Fresh HNSCC tumor tissue was harvested and processed by Drs. James Netterville, Kyle Mannion, Sara Rohde, Robert Sinard, and Brandee Brown.
Conflicts of Interest
Dr. Kim is co-inventor of patents licensed to Aduro Biotech, and has served as paid scientific advisor to AZ Medimmune, Sanofi, Takeda, and Mersanna. Dr. Drake is a co-inventor on patents licensed from JHU to BMS and Janssen, has served as a paid consultant to AZ Medimmune, BMS, Pfizer, Roche, Sanofi Aventis, Genentech, Merck, and Janssen, and has received sponsored research funding to his institution from BMS IIoN and Janssen. Dr. Califano is founder, equity holder, consultant, and director of DarwinHealth Inc., which has licensed IP related to these algorithms from Columbia University. Columbia University is an equity holder in DarwinHealth Inc.
References
- 1.Ferris RL, et al. , Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med, 2016. 375(19): p. 1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seiwert TY, et al. , Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol, 2016. 17(7): p. 956–965. [DOI] [PubMed] [Google Scholar]
- 3.Mehra R, et al. , Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: pooled analyses after long-term follow-up in KEYNOTE-012. Br J Cancer, 2018. 119(2): p. 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burtness B, et al. , Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet, 2019. 394(10212): p. 1915–1928. [DOI] [PubMed] [Google Scholar]
- 5.Strauss L, et al. , Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol, 2020. 5(43). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chow MT, et al. , Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity, 2019. 50(6): p. 1498–1512 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garris CS, et al. , Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity, 2018. 49(6): p. 1148–1161 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broz ML, et al. , Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell, 2014. 26(6): p. 938. [DOI] [PubMed] [Google Scholar]
- 9.Feig C, et al. , Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A, 2013. 110(50): p. 20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dominguez CX, et al. , Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov, 2020. 10(2): p. 232–253. [DOI] [PubMed] [Google Scholar]
- 11.Kieffer Y, et al. , Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov, 2020. 10(9): p. 1330–1351. [DOI] [PubMed] [Google Scholar]
- 12.Costa A, et al. , Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell, 2018. 33(3): p. 463–479 e10. [DOI] [PubMed] [Google Scholar]
- 13.Elyada E, et al. , Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov, 2019. 9(8): p. 1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohlund D, et al. , Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med, 2017. 214(3): p. 579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puram SV, et al. , Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell, 2017. 171(7): p. 1611–1624 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alvarez MJ, et al. , Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet, 2016. 48(8): p. 838–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding H, et al. , Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm. Nat Commun, 2018. 9(1): p. 1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Obradovic A, et al. , Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell, 2021. 184(11): p. 2988–3005 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basso K, et al. , Reverse engineering of regulatory networks in human B cells. Nat Genet, 2005. 37(4): p. 382–90. [DOI] [PubMed] [Google Scholar]
- 20.Aran D, et al. , Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol, 2019. 20(2): p. 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanian A, et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A, 2005. 102(43): p. 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spector ME, et al. , Prognostic Value of Tumor-Infiltrating Lymphocytes in Head and Neck Squamous Cell Carcinoma. JAMA Otolaryngol Head Neck Surg, 2019. 145(11): p. 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uppaluri R, et al. , Neoadjuvant and Adjuvant Pembrolizumab in Resectable Locally Advanced, Human Papillomavirus-Unrelated Head and Neck Cancer: A Multicenter, Phase II Trial. Clin Cancer Res, 2020. 26(19): p. 5140–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emancipator K, et al. , Comparing programmed death ligand 1 scores for predicting pembrolizumab efficacy in head and neck cancer. Mod Pathol, 2021. 34(3): p. 532–541. [DOI] [PubMed] [Google Scholar]
- 25.Direkze NC, et al. , Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res, 2004. 64(23): p. 8492–5. [DOI] [PubMed] [Google Scholar]
- 26.Karnoub AE, et al. , Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature, 2007. 449(7162): p. 557–63. [DOI] [PubMed] [Google Scholar]
- 27.Mishra PJ, et al. , Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res, 2008. 68(11): p. 4331–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kojima Y, et al. , Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A, 2010. 107(46): p. 20009–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan HX, et al. , TGFbeta1 is essential for MSCs-CAFs differentiation and promotes HCT116 cells migration and invasion via JAK/STAT3 signaling. Onco Targets Ther, 2019. 12: p. 5323–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larkin J, et al. , Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med, 2015. 373(1): p. 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lakins MA, et al. , Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat Commun, 2018. 9(1): p. 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorchs L, et al. , Human Pancreatic Carcinoma-Associated Fibroblasts Promote Expression of Co-inhibitory Markers on CD4(+) and CD8(+) T-Cells. Front Immunol, 2019. 10: p. 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park BV, et al. , TGFbeta1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov, 2016. 6(12): p. 1366–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hutton C, et al. , Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell, 2021. 39(9): p. 1227–1244 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozdemir BC, et al. , Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell, 2014. 25(6): p. 719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andre P, et al. , Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell, 2018. 175(7): p. 1731–1743 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Montfoort N, et al. , NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell, 2018. 175(7): p. 1744–1755 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee N, et al. , HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A, 1998. 95(9): p. 5199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biffi G, et al. , IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov, 2019. 9(2): p. 282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arumugam K, et al. , The Master Regulator Protein BAZ2B Can Reprogram Human Hematopoietic Lineage-Committed Progenitors into a Multipotent State. Cell Rep, 2020. 33(10): p. 108474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Talos F, et al. , A computational systems approach identifies synergistic specification genes that facilitate lineage conversion to prostate tissue. Nat Commun, 2017. 8: p. 14662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alvarez MJ, et al. , A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat Genet, 2018. 50(7): p. 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luginbuhl A, et al. , Discordant treatment response in primary tumors and lymph node metastases after four weeks of preoperative PD-1 blockade in head and neck squamous cell carcinoma (HNSCC). Journal of Clinical Oncology, 2019. 37(15_suppl): p. 6016–6016. [Google Scholar]
- 44.Zhang L, et al. , Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature, 2018. 564(7735): p. 268–272. [DOI] [PubMed] [Google Scholar]
- 45.Lizio M, et al. , Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol, 2015. 16: p. 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finak G, et al. , MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol, 2015. 16: p. 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data are deposited and publicly available in a Mendeley data repository (doi:10.17632/yk8wj7xgdg.1) as well as the GEO database (GSE195832).