

Abstract
Purpose:
One third of patients with diffuse large B-cell lymphoma (DLBCL) succumb to the disease partly due to rituximab resistance. Rituximab-induced calcium flux is an important inducer of apoptotic cell death and we investigated the potential role of calcium channels in rituximab resistance.
Experimental Design:
The distinctive expression of calcium channel members was compared between patients sensitive and resistant to RCHOP regimen. The observation was further validated through mechanistic in-vitro and in-vivo studies using cell lines and patient-derived xenograft mouse models.
Results:
A significant inverse correlation was observed between CACNA1C expression and RCHOP resistance in two independent DLBCL cohorts and CACNA1C expression was an independent prognostic factor for RCHOP resistance after adjusting for International Prognostic Index, cell of origin classification and MYC/BCL2 double expression. Loss of CACNA1C expression reduced rituximab-induced apoptosis and tumor shrinkage. We further demonstrated direct interaction of CACNA1C with CD20, and its role in CD20 stabilization. Functional modulators of L-type calcium channel showed expected alteration in rituximab-induced apoptosis and tumor suppression. Furthermore, we demonstrated that CACNA1C expression was directly regulated by miRNA-363 whose high expression is associated with worse prognosis in DLBCL.
Conclusions:
We identified the role of CACNA1C in rituximab resistance, and modulating its expression or activity may alter rituximab sensitivity in DLBCL.
Keywords: diffuse large B-cell lymphoma, rituximab, calcium signaling, apoptosis, L-type calcium channel
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common type of non-Hodgkin lymphoma characterized by distinct genetic abnormalities, biological features, and prognosis (1, 2). The addition of rituximab, a humanized chimeric anti-CD20 monoclonal antibody, to the standard CHOP chemotherapy (RCHOP) has significantly improved overall survival (OS) of DLBCL patients (3). However, 30–40% of patients experience incomplete response during treatment or progression after the completion of RCHOP treatment (4). Salvage therapy and/or autologous stem cell transplantation do not improve the dismal prognosis, especially in patients with prior rituximab treatment and early-relapse (5). It is often regarded that rituximab resistance plays a vital role in treatment failure, thus requires further understandings to improve clinical efficacy of rituximab. Rituximab induces cell death via antibody-dependent cell mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), direct apoptotic signaling, and possible vaccinal effects (6). It can inhibit the expression of the anti-apoptotic genes BCL2/BCL-XL by downregulating the survival pathways (7–9) (i.e. p38MAPK, nuclear factor-κB (NF-κB), ERK 1/2 and PI3K-AKT). The resistance to rituximab is either tumor-intrinsic (i.e. loss of CD20 expression) (10) and/or host-associated factors (FcRIII-α polymorphism at amino acid position-158) that inhibit ADCC action (11). Novel anti-CD20 antibodies (e.g. Obinutuzumab) have been developed to exceed rituximab action (12, 13), but with limited improvement for DLBCL prognosis.
CD20 belongs to the MS4A gene family, having a tetraspanning membrane structure with an N- and C-terminal cytoplasmic domain involved in B cell proliferation (14). CD20 has been considered as a component of a cell-surface calcium channel or as a direct regulator of calcium channel activity (15, 16). Calcium channels/ pumps located in the plasma membrane and subcellular organelles can modulate intracellular calcium signaling. Crosslinking of CD20 molecules by rituximab induces a redistribution of CD20 molecules to lipid rafts at the plasma membrane and calcium influx leading to apoptosis (17). The increased intracellular calcium induced by rituximab was demonstrated to depend on the calcium channel at the plasma membrane, rather than those at the endoplasmic reticulum (18). CD20-deficiency significantly reduced ligation-induced intracellular calcium responses (19). However, CD20-deficient B cells presented incomplete blockade of calcium responses (14), which reflect the possibility that other calcium channels may participate in regulating calcium influx.
B lymphocytes express a wide and diverse pool of ion channels, including components of the Cav1 subfamily of voltage-gated L-type calcium channels (VGCC) which are signature molecules of excitable cells (20, 21). L-type calcium channels are composed of five subunits, including α1, α2/δ, β, γ. There are four different pore-forming α1 subunits, Cav1.1 to 1.4, being encoded by calcium channel alpha 1S (CACNA1S), CACNA1C, CACNA1D and CACNA1F genes, which mediate L-type currents and contain drug-binding sites. Its other subunits primarily modify channel gating or affinity, being encoded by CACNA2D1-4, CACNB1-4 and CACNG1-8. These channels in B lymphocytes were proven to play roles on calcium influx and cell proliferation by inhibitors of L-type channels (22). Indeed, L-type calcium channels can be manipulated by small molecules and are capable of mediating long-lasting calcium influx, which are required in triggering apoptosis (23). We hypothesized that enhancing calcium influx of B lymphoma cells may increase clinical efficacy of rituximab. In the current study, we have tested the role of L-type calcium channels in modulating rituximab-induced apoptosis and resistance to initial therapy in DLBCL, and identified critical role of CACNA1C expression in DLBCL prognosis. Furthermore, the biological role of CACNA1C was investigated using in-vitro, in-vivo lymphoma models, which demonstrated direct association of CD20 with CACNA1C in plasma membrane and also regulation of CACNA1C expression by prognostically relevant miRNA-363 in DLBCL(24).
Materials and Methods
Patients and cell lines
The study was carried out in a retrospective series of 48 DLBCL cases with cryopreserved tissues and 63 cases with formalin fixed paraffin-embedded (FFPE) tissues. Basic clinical characteristics of patients are presented in Table 1. The diagnosis of DLBCL was confirmed by at least two pathologists in accordance to the World Health Organization (WHO) classification (25). All patients were treated with the RCHOP regimen and involved-field radiotherapy was performed as consolidation treatment in 5 cases in the primary therapy. Responses to treatment were evaluated by computed tomography (CT) scans or PET/CT following the response criteria for lymphoma as defined by Cheson et al (26). The study was reviewed and approved by hospital review boards with informed consent of the patients and was conducted in accordance with Declaration of Helsinki. Another independent DLBCL cohort (27) was used to validate the findings.
Table 1: Comparison of clinical characteristics between patients sensitive and resistant to RCHOP regimen.
| Characteristics | Resistance (n=43) | Sensitivity (n=68) | P value |
|---|---|---|---|
|
| |||
| Gender | |||
| Male, n (%) | 20 (47) | 32 (47) | 0.955 |
| Age, median (range, ys) | 41 (19–82) | 47 (18–80) | 0.195 |
| Performance status | |||
| ECOG score 0–1, n (%) | 33 (77) | 60 (88) | 0.092 |
| Ann Arbor stage | |||
| Ⅰ/Ⅱ, n (%) | 20 (47) | 42 (62) | 0.115 |
| LDH level | |||
| Normal, n (%) | 23 (53) | 44 (65) | 0.239 |
| Number of extranodal involvement | |||
| More than 1, n (%) | 5 (12) | 6 (9) | 0.630 |
| IPI score | |||
| Low/low-intermediate risk 0–2, n (%) | 30 (70) | 60 (88) | 0.016 |
| Cell origin | |||
| Germinal-center B cell, n (%) | 11 (26) | 20 (29) | 0.661 |
| CACNA1C expression, n (%) | 19 (44) | 52 (76) | 0.001 |
| Double expression, n (%) | 9 (21) | 5 (7) | 0.044 |
OCI-ly7, and OCI-ly8 and OCI-ly3 DLBCL cell lines were cultured in IMDM (Gibco) with 10% fetal bovine serums, 100U/ml penicillin, 100 mg/ml streptomycin, and 0.2% beta mercaptoethanol (Invitrogen). These cell lines were gifted from Department of Pathology and Microbiology, University of Nebraska Medical Center. All of them were genotyped by short tandem repeat analysis and were confirmed to be negative for mycoplasma at the time of testing with the mycoplasma detection kit (Lonza).
RNA isolation and gene expression profiling (GEP)
Total RNA for GEP was extracted using the Qiagen all prep RNA and DNA isolation kit (Qiagen). We used HG-U133 plus 2 arrays (Affymetrix) for GEP, and RNA processing, hybridization, and image processing were performed according to the protocol described previously (28). The expression of miRNA-363 for these cases has been quantitated in a previous study (24).
Quantitative Real Time (qRT)-PCR
For qRT-PCR, total RNA was isolated from tissues and cells using the miRNeasy Mini Kit (Qiagen). 2µg RNA was transcribed with the High Capacity cDNA Reverse Transcription Kits (ABI) according to the manufacturer’s instructions. The qRT-PCR reactions were set up in triplicate using Brilliant II SYBR Green qPCR Master Mix (Toyobo) and ran on an ABI PRISM7300 Real-Time PCR system (Applied Biosciences) with the specific primers (Supplementary Table S1).
Immunohistochemical assay
Indirect immunoperoxidase assays were performed on 5μm thick paraffin sections using antibodies against CACNA1C (Omnimabs, 1:100), CD20 (Abcam, 1:50), BCL2 (Abcam, 1:250) or C-MYC (Abcam, 1:250). Antigen retrieval was performed by high pressure heating for 1 min in PH 6.0 buffer for CACNA1C and in PH 9.0 buffer for CD20, BCL2 and C-MYC. Both CACNA1C and CD20 were located at plasma membrane and the positive one was defined to have more than 30% of lymphoma cells stained.
Flow cytometry assay
The cells were fixed with 80% methanol for 5 min and then permeabilized with PBST (0.2% Tween-20) for 20 min. The cells were incubated in 10% normal goat serum and followed by antibody against CACNA1C (Omnimabs, 1µg/106 cells) for 30 min at 22℃. The goat anti-mouse IgG (H+L) antibody conjugated with FITC (ThermoFisher, 1:200) was added for 30 min at 22℃ in the dark. The fluorochrome-conjugated antibody against CD20 (BD, 1:20) was incubated for 30 min at 22℃ in the dark. Run and analyze on flow cytometry.
Apoptosis assay
After drug treatment, 1×106 cells were washed with PBS and resuspended in the 500µl of 1×binding buffer containing Annexin-V-PE (Calbiochem). After incubation for 15 min at room temperature in the dark, cells were pelleted and resuspended in 500µl binding buffer containing 10µl 7-AAD, and analyzed on a FACScan. The lower right-hand and the upper right-hand quadrant cells were considered apoptotic.
Immunofluorescence assay
After being cultured with or without rituximab (50µg/ml), OCI-ly7 cells were incubated with antibodies against CD20 (Abcam, 1:50) and CACNA1C (Omnimabs, 1:50) at 4℃ overnight. Then the secondary antibodies Alexa Fluor 647 (Thermo Fisher Scientific, 1:1000) and Cy3 (Thermo Fisher Scientific, 1:1000) were incubated in the dark for 1 h at room temperature, following 1μg/ml DAPI being incubated in the dark for 1 min. Finally, samples were added a drop of Dako fluorescence mounting medium (Dako) and mounted with coverslips. Images were collected with Nikon confocal microscope system.
Calcium flux assay
OCI-ly3 and OCI-ly7 cells were suspended at a density of 1×106 cells/ml with the HBSS solution containing 1%BSA(W/V) and 2µM Fluo-4 AM was dispatched into the 5ml Falcon cytometry tubes for 30 min at 37℃ in the dark. After washed twice with HBSS solution, the cells were resuspended in saline solution containing 10mM HEPES, 1mM Na2HPO4, 137mM NaCl, 5mM KCl, 1mM CaCl2, 0.5mM MgCl2, 5mM Glucose, 0.1%BSA(W/V). After adding 50µg/ml rituximab, calcium flux assays were performed on the Becton-Dickinson FACS AriaII flow cytometry. The Fluo-4 bound cytoplasmic free calcium and emitted a green fluorescence(peak at 526 nm)detected using FL1.
Western Blotting assay
Total proteins were extracted with NP40 lysis buffer (Beyotime). Western Blotting assay was carried out using standard protocols with antibodies against CACNA1C (Omnimabs, 1:500), CACNA1D (Omnimabs, 1:200), CACNA1F (Abcam, 1:1000), CACNA1S (Abcam, 1:2000) and CD20 (Abcam, 1:2000). Protein bands were visualized using the enhanced chemiluminescence system (Millipore) according to the manufacturer’s instruction.
Co-immunoprecipitation
Total proteins from OCI-ly7 cells were extracted with NP40 lysis buffer (Beyotime). Anti-CD20 antibody (Abcam, 1:50) or control immunoglobulin (anti-IgG) (Santa Cruz, 1:250) was incubated with cell lysate. Slowly shake antigen-antibody complex on rotating shaker overnight at 4℃ and then be followed by protein A/G PLUS-Agarose (Santa Cruz, 20μl/500μl) incubation for 3 h at 4℃. The pellets were washed three times with NP40 lysis buffer (Beyotime). The pulled-down proteins were examined by Western blotting as described above.
Construction of CACNA1C shRNA and miRNA-363 lentiviral vectors and transduction
The shRNA to suppress the expression of CACNA1C was inserted into the Plk0.1-puro vector digested by AgeI/EcoRI according to the manufacturer’s instruction. The lentiviral vector of miRNA-363 was generated after BamHI/EcoRI cloning and under the control of the Ubc promoter (Supplementary Figure S1A).
293T cells were transfected with 12µg of plasmid containing the desired construct and 8µg of packing vector pSPAX2 and 4µg of envelope vector pMD2G in a 10cm dish for the production of lentivirus. After 48 h, culture supernatant was filtered through the 0.45µm filter and virus concentrated with PEG 6000 method. Lymphoma cells were incubated with the virus preparation overnight, and the culture medium was replaced with 2ml fresh medium. We used puromycin (2µg/ml) to select infected cells.
CRISPR/Cas9 system to knockout miRNA-363
The Single-guide RNA (sgRNA) was designed nearest to the seed region of miRNA-363-3p and inserted into the pSpCas9 (BB)-2A-GFP (PX458) vector. A single-stranded DNA oligonucleotide (ssODN) of 99 bp was also designed in which the seed region of miRNA-363 (ATTGCAC) was replaced by EcoRI sequence (GAATTC). The sgRNA and ssODN were co-transfected into OCI-ly3 cell line using the Lonza Nucleofector™ system (Supplementary Figure S1B). EGFP-positive cells were sorted by Flow Cytometry after transfection for 48 h and then diluted into 96-well to generate single clones. All single clones were examined by DNA sequencing and qRT-PCR before further application.
Xenograft mouse model assay
Five- to seven-week old SCID mice (Charles River) were injected with 1×107 OCI-ly7 cells that had been transduced with e-GFP or CACNA1C shRNA lentiviral vector, or DLBCL cells from patient sample in the posterior flank subcutaneously. When the tumor approached about 100 mm3, mice were randomly divided groups of seven mice in each. Saline (control), rituximab (25µg/g), nimodipine (2µg/g), Bay K8644 (2µg/g), rituximab (25µg/g) plus nimodipine (2µg/g), and rituximab (25µg/g) plus Bay K8644 (2µg/g) were injected intraperitoneally to patient-derived xenograft (PDX) mice once every other day for 10 times, respectively; saline (control) and rituxmab (25µg/g) were injected intraperitoneally to OCI-ly7-derived mouse xenografts once every other day for 7 times. Tumor volume was measured every 3 days with a caliper, and calculated using the formula: (Length×Width2) ×0.5. Animals were maintained in accordance with the principles of laboratory animal care under an Institutional Animal Care and Use Committee-approved protocol.
Statistics
Statistical analyses were performed using the Statistical Software Package for the Social Sciences (SPSS version 13.0 for Windows) and TIGR TM4 software (version 4.8.1). Overall survival (OS) and event-free survival (EFS) were estimated using the Kaplan-Meier method, and differences were assessed using the log-rank test. We used the probe sets (242973_at and 238636_at) for estimating the CACNA1C mRNA expression and rituximab efficacy association (Supplementary Figure S2). Multivariate logistic regression examined the effect of CACNA1C mRNA expression on RCHOP resistance. Differences among groups were considered statistically significant at p-values below 0.05 (*).
Results
Inverse correlation between CACNA1C expression and RCHOP resistance
DLBCL patients treated with RCHOP regimen were divided into sensitive (n=30) and resistant (n=18) groups according to treatment response. Resistant patients were defined as not achieving complete remission (CR) or developing rapid disease progression (less than six months) after 6 to 8 cycles of RCHOP administration. Using GEP, we found that members of L-type calcium channel [i.e., CACNA1 (-C,-D, -F, -S), CACNA2 (-D1, -D2, -D3, -D4), CACNB (-1,-2, -3,-4), CACNG (1 to 8)] were expressed in both sensitive and refractory subgroups. Of them only CACNA1C mRNA expression presented significant difference with lower levels in resistant patients (p=0.009), while CD20 mRNA expression did not show significant difference (Figure 1A) (Supplementary Table S2). The CACNA1C expression was not significantly different between germinal center (GC) B-cell (n=26) and non-GC B-cell (n=22) subtypes (p=0.797). Using immunohistochemistry, we observed that loss of CACNA1C protein was significantly associated with RCHOP resistance (44% vs. 76%, p=0.001) (Table 1). Two cases which represented the distinctive expression were presented in Figure 1B. Univariate analysis revealed the correlation of CACNA1C expression (p=0.001), double expression (p=0.044), and international prognostic index (IPI) score (p=0.017) with resistant disease. Multivariate analysis showed the independent association of CACNA1C expression and IPI score with resistance, excluding double expression.
Figure 1: CACNA1C expression and significance in DLBCL patients and normal B cells.
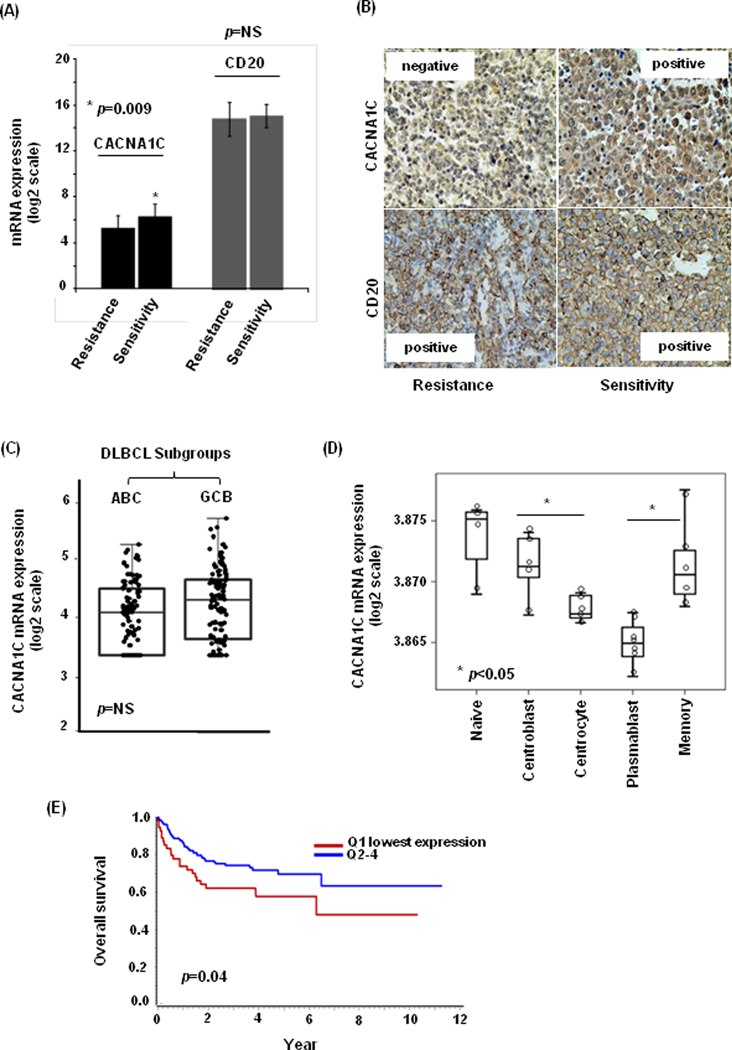
(A) The mRNA expression of CACNA1C was lower in rituximab-resistant patients compared to rituxixmab-sensitive patients (p=0.009), while CD20 mRNA expression showed no correlation (p=NS). (B) The protein expression of CACNA1C was negative in one case with rituximab resistance and positive in the other case with rituximab sensitivity, while CD20 protein expression was positive in both of them. (C) In an independent DLBCL cohort, there was no significant difference in CACNA1C mRNA expression between GCB-DLBCL and ABC-DLCBL subtypes (p=NS). (D) Of the normal B-cell subsets, centrocytes and plasmablast showed comparatively lower expression of CACNA1C. (E) The patients with lowest quartile of CACNA1C expression showed inferiors OS (HR=1.974, p=0.020).
The results were confirmed in an independent DLBCL cohort with RCHOP treatment (27). There was no correlation between CACNA1C expression and cell of origin classification (p=0.94) (Figure1C). Of the normal B-cell subsets, centrocytes and plasmablast cells showed comparatively lower expression of CACNA1C mRNA (Figure 1D). When CACNA1C expression was divided into quartiles, the first quartile (Q1) with the lowest expression showed independent association with RCHOP resistance (OR=2.897, p=0.014). We also demonstrated the independent correlation of the lowest quartile of CACNA1C expression with inferiors OS (HR=1.974, p=0.020) (Figure 1E). Similar trends were observed with EFS analysis (HR=1.686, p=0.053). In patients treated by CHOP regimen, we did not observe the relation of CACNA1C expression with outcome (Supplementary Figure S3).
Loss of CACNA1C expression leading to rituximab resistance
To demonstrate the significance of CACNA1C expression in rituximab resistance, we used three DLBCL cell lines OCI-ly3, OCI-ly7 and OCI-ly8 for in-vitro assays, including qRT-PCR, flow cytometry and western blotting assays. A good concordance was observed between mRNA and protein expression for CACNA1C and CD20 with OCI-ly3 showing the lowest and OCI-ly7 showing the highest expression (Supplementary Figure S4A-E). When these cell lines were treated with rituximab (50µg/ml) for 48 h, we observed a positive relationship between CACNA1C expression and rituximab sensitivity. As shown in Supplementary Figure S4F-G, cell lines with high expression of CACNA1C produced significant apoptosis (27.59±2.41% in OCI-ly7, 18.84±2.63% in OCI-ly8,) compared to low expressing cell line (1.54±1.39% in OCI-ly3; p<0.05).
To further validate mechanistically, we generated CACNA1C deficient stable cell lines using shRNAs (sh1C-1 and sh1C-2) in OCI-ly7 and OCI-ly8 cell lines, which resulted in significant loss of CACNA1C protein expression, and observed significant decrease in percentage of apoptosis in transduced cells after rituximab treatment (OCI-ly7: 23.05±2.12%, sh1C-1 9.85±1.32%, p=0.042; sh1C-2 3.70±1.03%, p=0.022; and OCI-ly8: 17.86±2.32%, sh1C-1 4.80±1.06%, p=0.035; sh1C-2 4.84±1.08%, p=0.038) (Figure 2A-B). To validate the finding in vivo, xenografts mice models were established using transduced OCI-ly7 cell line (with and without sh1C-1 vector), and treated with rituximab. As expected, xenografts with control vector showed significant tumor shrinkage in tumor volume (p=0.035) and weight (p=0.024), whereas those with sh1C vector was not significantly reduced in tumor volume (p=0.273) and weight (p=0.278) (Figure 2C). These data suggested that loss of CACNA1C significantly reduced rituximab-induced tumor inhibition.
Figure 2: The effect of CACNA1C downregulation on rituximab-induced apoptosis and tumor inhibition.
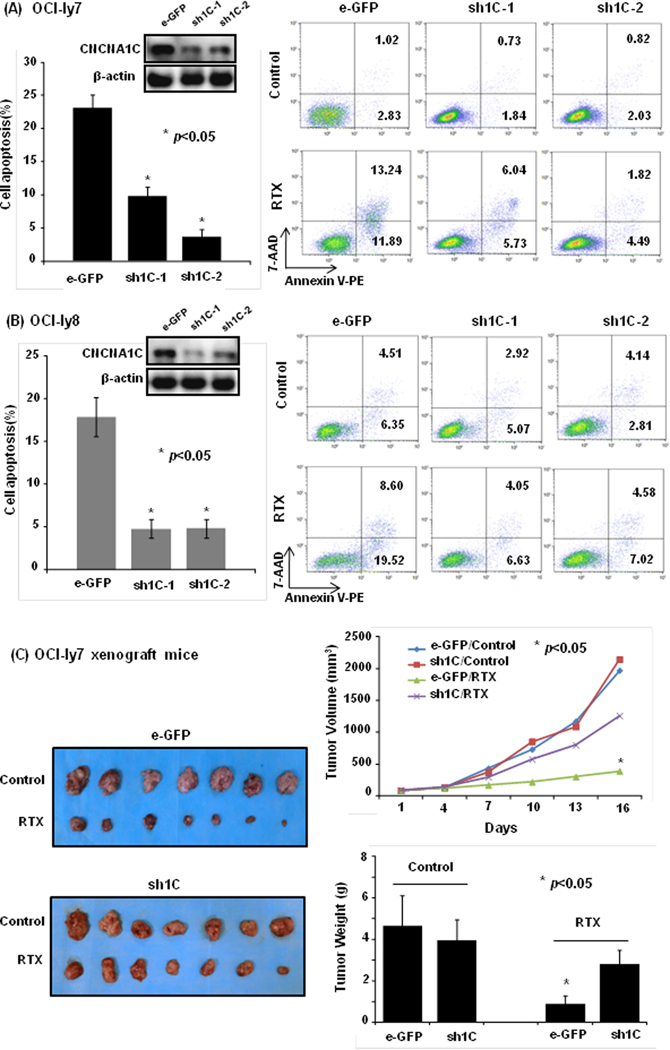
(A and B) The interference with shRNA to CACNA1C reduced the percent of rituximab-induced cell apoptosis (p<0.05) in OCI-ly7 and OCI-Ly8 cell line. (C) In xenograft mice models of OCI-ly7 cell line, downregulation of CACNA1C by its shRNA obviously suppressed rituximab-induced tumor shrinkage in tumor volume and weight (p<0.05).
Direct interaction of CACNA1C with CD20 during rituximab action
Using confocal microscopy we observed that CD20 molecules located in the plasma membrane exhibited a gradual polarization (green light) after rituximab treatment at 0, 2, and 12 h (Figure 3A). CACNA1C molecule mainly confined to the plasma membrane with modest cytoplasmic and nuclear expression, and showed similar polarized distribution (red light) after rituximab treatment. The co-localization of CD20 and CACNA1C was observed in the merged image with overlapping distribution (yellow light) in the plasma membrane, which suggested possible interaction. To validate their interaction, CACNA1C protein was co-immunoprecipitated with either anti-CD20 or anti-IgG antibody after rituximab treatment of 2 h in OCI-ly7 cells. CACNA1C protein was detected in anti-CD20 pull-down proteins, suggesting the association between CD20 and CACNA1C molecules (Figure 3B).
Figure 3: The interaction between CACNA1C and CD20 molecules during rituximab action.
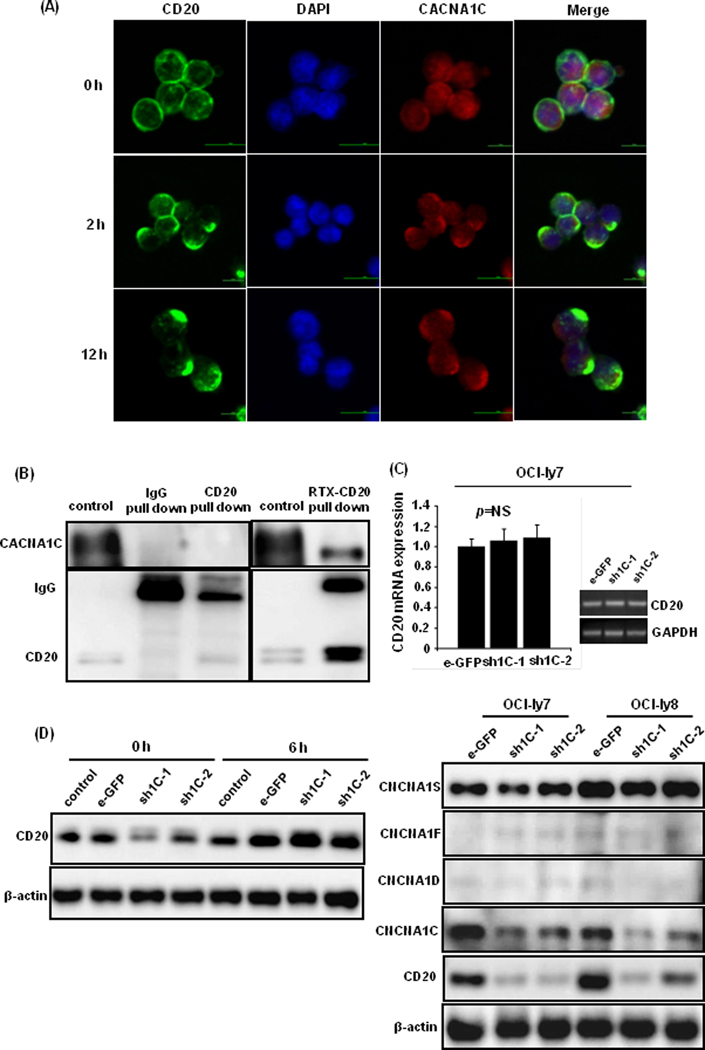
(A) CD20 (green light) and CACNA1C (red light) molecules exhibited a similar polarized distribution after rituximab treatment of 0, 2, and 12 h. Their overlapping distribution (yellow light) was captured in the plasma membrane in the merged phase. (B) CACNA1C protein was detected in anti-CD20 pull-down protein using western blot after rituximab treatment of 2 h in OCI-ly7 cells. (C) Knockdown of CACNA1C expression with transduced shRNAs did not affect CD20 mRNA levels, but downregulated CD20 protein expression. Other isoforms of α1 subunits, such as CACNA1D, CACNA1F, and CACNA1S, showed no obvious alteration. (D) Proteasome inhibitor bortezomib (20nM) restored CD20 expression after administration of 6 h in OCI-ly7 cells.
Since rituximab performs its action via the ligation with cell surface CD20 molecule, we further explored the role of CACNA1C in regulating rituximab action. Knockdown of CACNA1C expression in DLBCL cell lines downregulated CD20 protein expression without any significant change in CD20 mRNA levels (Figure 3C). Other forms of α1 subunits, such as CACNA1D, CACNA1F, and CACNA1S, showed no obvious alteration, indicating the specific effect of CACNA1C on CD20 expression. When these cells were treated with the proteasome inhibitor bortezomib (20nM) for 6 h, CD20 expression was restored suggesting that CACNA1C may play a role in proteasome-dependent CD20 degradation, thus affecting efficacy of rituximab (Figure 3D).
Modulators of L-type calcium channel affecting rituximab efficacy
Since there was not a specific modulator of CACNA1C, antagonist (nimodipine) and agonist (Bay K8644) of pan-L-type calcium channel were used to evaluate the effect of on rituximab efficacy. We observed significant reduction of apoptosis in cell lines when treated with rituximab (50µg/ml) and nimodipine (2μM) for 48 h compared to rituximab alone (OCI-ly7: 26.02±3.12% vs. 10.05±1.25%, p<0.01; OCI-ly8: 17.65±1.54% vs. 1.89±0.98%, p<0.01). When rituximab combined with agonist Bay K8644 (10nM), significant increase in apoptosis was observed in the above cell lines (OCI-ly7: 26.02±3.12% vs. 37.36±3.18%, p<0.01; OCI-ly8: 17.65±1.54% vs. 35.02.±2.58%, p<0.01), and even in OCI-ly3 cell line, which initially showed poor response to rituximab (1.54±1.39% vs. 21.13±4.39%, p<0.01) (Figure 4A).
Figure 4: The influence of L-type calcium channel modulators on rituximab-induced apoptosis and tumor suppression.
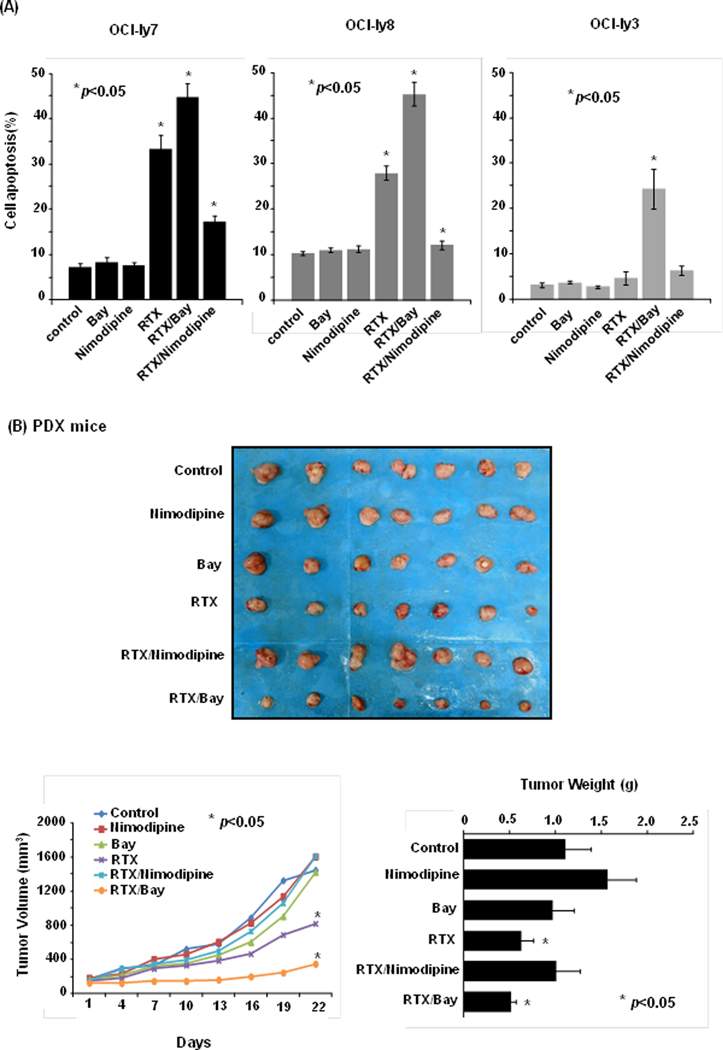
(A) The agonist of L-type calcium (Bay K8644) enhanced the rituximab-induced apoptosis, while the antagonist of L-type calcium channel (nimodipine) attenuated the rituximab-induced apoptosis (p<0.01) in DLBCL cell lines. (B) In PDX mouse models, rituximab combined with agonist (Bay K8644) markedly enhanced rituximab action (p<0.05). Neither its combination with antagonist (nimodipine) nor modulators of L-type calcium channel alone showed obvious effect on tumor growth (p>0.05).
Since rituximab-mediated calcium influx is a sensitive indicator of rituximab action, we measured the calcium influx with fluo-4-AM using flow cytometry after rituximab (50µg/ml) treatment. We observed that the addition of rituximab resulted in significant increase in intracellular calcium ions in OCI-ly3 and OCI-ly7 cell lines. Expectedly, rituximab-mediated intracellular calcium ions was enhanced in cells treated with Bay K8644 for 2 h, whereas it was reduced in cells treated with nimodipine for 2 h. Ionomycin (2μg/ml) was used as a positive control for calcium influx (Supplementary Figure S5A). Neither nimodipine nor Bay K8644 treatment alone led to a significant alteration in intracellular calcium ions. Concurrently, we did not find the influence of nimodipine and Bay K8644 on CD20 expression after treatment of 24 and 48 h in the study (Supplementary Figure S5B).
We tested the action of these compounds in in-vivo DLBCL PDX mice (Figure 4B) and observed that rituximab combined with agonist (Bay K8644) markedly enhanced rituximab action, as estimated by tumor volume (p=0.024) and weight (p=0.012). Neither rituximab combination with antagonist (nimodipine) nor modulators of L-type calcium channel alone showed obvious effect on tumor growth.
MiRNA-363 epigenetically regulating the expression of CACNA1C
Target Scan, a computational algorithm, predicted CACNA1C to be the direct target of miRNA-363. We demonstrated the direct regulation of miRNA-363 on CACNA1C expression using luciferase reporter assay in 293T cells, where we found that luciferase expression was significantly inhibited by miRNA-363 with wild-type 3’-UTR of CACNA1C, but not with mutant 3’-UTR of CACNA1C (Figure 5A). Furthermore, ectopic expression of miRNA-363 in OCI-ly7 and OCI-ly8 cell lines led to significant loss of CACNA1C and as anticipated loss of CD20 protein expression in these cell lines (Figure 5B). The overexpression of miRNA-363 significantly reduced rituximab-induced apoptosis in OCI-ly7 (27.07±2.54% vs. 1.22±1.06%, p=0.018) and OCI-ly8 cell lines (17.23±2.21% vs. 2.94±1.11%, p=0.014)after rituximab treatment of 48 h (Figure 5C). To explore direct regulation, we further knockout miRNA-363 locus in the OCI-ly3 cell line using the CRISPR/Cas9 system, and as expected resulted in the upregulation of CACNA1C (Figure 5D). These edited cells showed significantly increased apoptosis (0.80±0.69% vs. 10.25±1.80%, p=0.001) (Figure 5E), when treated with rituximab.
Figure 5: The association of miRNA-363 with CACNA1C expression and rituximab-induced apoptosis.
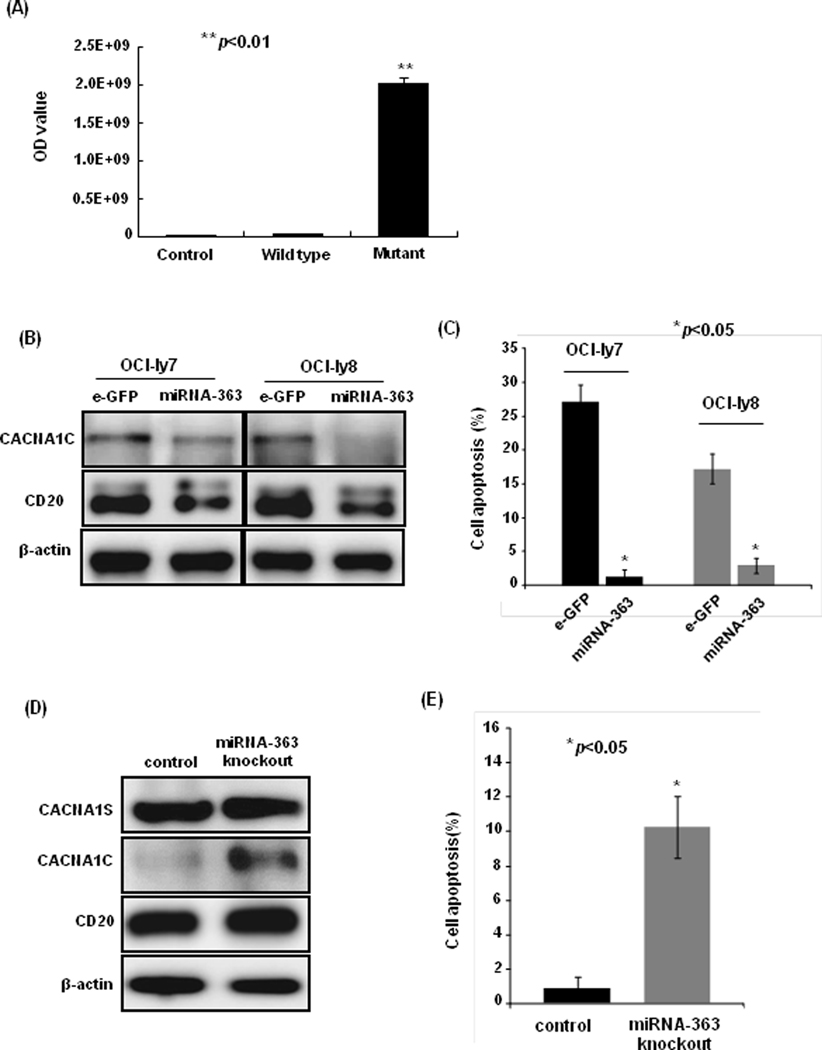
(A) The miRNA-363 inhibited the luciferase activity in 293T cells with wild-type (WT) 3’UTR of CACNA1C gene, but did not affect it in cells with mutant 3’UTR of CACNA1C gene (p<0.01). (B) The ectopic expression of miRNA-363 reduced CACNA1C and CD20 protein expression in OCI-ly7 and OCI-ly8 cell lines. (C) The ectopic expression of miRNA-363 decreased rituximab-induced apoptosis in OCI-ly7 and OCI-ly8 cell lines (p<0.05). (D) The knockout of miRNA-363 led to the upregualtion of CACNA1C expression and (E) the increase of rituximab-induced apoptosis (p<0.05).
Discussion
We investigated the role of L-type calcium channel in rituximab-induced apoptosis, and demonstrated that loss of CACNA1C expression correlates with rituximab-mediated immunochemothrapy resistance in DLBCL. The role of CACNA1C in normal B-cell activation is not known, but several studies showed that L-type calcium channels play a role in B-cell calcium influx and proliferation (22) and increased intracellular calcium correlates with apoptosis (29). Calcium influx can affect the depolarization of mitochondria, which is fundamental in the activation of executioner caspases (30). The increased levels of cytosolic calcium may also activate calcium-dependent proteases, such as calpains, which have been implicated in caspase-independent death (31). The elevated intracellular calcium has been shown to be critical in rituximab-induced apoptosis. Thus, enhancing calcium influx could be an approach to increase rituximab-induced direct cell apoptosis in DLBCL. We attempted to knockout CACNA1C using CRISPR/Cas9 system in DLBCL cell lines, but these knockout cells lost the ability of persistent growth (Supplementary Figure S6), which indicated a critical role of CACNA1C in the long term survival of DLBCL cells.
The independent association of CACNA1C expression with refractory disease and inferior survival was proven in DLBCL patients. We did not find distinctive expression of CACNA1C between GCB and ABC DLBCL subtypes. Of the normal B-cell subsets, centrocytes and plasmablast cells showed comparatively lower CACNA1C expression. Our study further revealed the function of the accessory L-type calcium channels CACNA1C in rituximab-induced apoptosis, and the interaction between CACNA1C and CD20 in cell membrane is critical for this process. We observed direct interaction between CACNA1C and CD20 molecule using confocal immunofluorescent and pulldown assays and demonstrated that loss of CACNA1C expression resulted in lower CD20 stability and also reduced rituximab-induced cell death. This direct relationship between CACNA1C expression and rituximab sensitivity was also demonstrated in DLBCL cell lines and xenograft mouse models.
Rituximab resistance remains to be a clinical challenge. Many studies have explored the regulatory mechanisms of CD20 expression, including transcription factors SPI1/PU.1 (32), Oct2 (33) and Pax5 (34). Other factors, such as epigenetic changes, somatic mutation and non-coding RNA, could also affect CD20 expression (35). A recent study using RNAi screening method identified CREM (cAMP Responsive Element Modulator) as a top candidate for CD20 suppression, CHD4 (Chromodomain Helicase DNA Binding Protein 4) and MBD2 (Methyl-CpG Binding Domain Protein 2) as positive regulators of CD20 expression (36). In addition, CD20 regulation at the post-transcriptional level was also suggested. Bortezomib can sensitize CD20 positive lymphoma cells to rituximab-induced CDC after the incubation of 24 h (37) and rituximab-resistant lymphoma cells were observed to exhibit up-regulation of components of the ubiquitin-proteasome system (UPS) (38, 39). In this study, we found that CACNA1C may perform a critical function in CD20 protein stabilization through proteasome system, since lowered CD20 protein level was restored in cells with CACNA1C shRNAs after bortezomib incubation of 6 h.
Furthermore, we investigated the effect of modulators of L-type calcium channel on rituximab-induced apoptosis and explored the potential approach to raise therapeutic efficacy for DLBCL in the future clinical trials. This study validated that inhibiting L-type calcium channel with nimodipine reduced rituximab action, whereas Bay K8644, an agonist of L-type calcium channel, increased it. These observations provide a rationale that modulating L-type Cav1.2 calcium channel may increase the clinical efficacy of rituximab. Actually, more attention have now been paid on calcium ion channels due to their involvement in the proliferation and invasion of cancer cells and their potential modulation by pharmaceuticals (40, 41). This is the first study showing that modulating Cav1.2 subfamily of L-type calcium channel can affect rituximab-induced cell apoptosis on DLBCL. Blocking L-type calcium channels in human B-cells has been demonstrated to reduce BCR-induced calcium entry (42). Some knowledge gaps on L-type calcium channels remain to be addressed in B lymphocytes and malignancy.
The miRNA-363 is a member of miRNA-106-363 cluster which is also a paralogue of miRNA-17-92 superfamily (43, 44) (45). Although the miRNA-17-92 cluster is a paradigm of oncomirs and cooperates with c-Myc in the formation of mouse B cell lymphoma (46), limited data is available on miRNA-106-363 cluster members. The miRNA-106-363 cluster is mapped to chromosome X (47) and high expression of the miRNA-106-363 cluster has been found in 46% of human T-cell leukemias (48) and colon and prostate cancers (49) suggesting its oncogenic potential. In our previous miRNAs profiling studies (24, 50), miRNA-363 was found to have a higher expression in mantle cell lymphoma (MCL) and was associated with poor response to RCHOP regimen in DLBCL patients (24). However, the mechanism by which miRNA-363 mediates the poor response to RCHOP remains elusive. This study demonstrated that miRNA-363 can directly downregulate the expression of CACNA1C and may thus reduce rituximab-induced apoptosis. The relevance of miRNA-363 to RCHOP resistance will be further addressed in our future studies.
In summary, the alteration of CACNA1C expression and modulators of L-type channel modulates rituximab-induced apoptosis. Notably, the agonist of L-type calcium channel, Bay K8644, has been shown to synergize in rituximab-induced apoptosis and tumor suppression in DLBCL. The optimal dose and safety of Bay K8644 or its analogues are worth evaluating in the future. Reduction of CACNA1C appeared to be also associated with reduced CD20 expression that may reduce target binding by rituximab. Further studies on the interaction between CACNA1C and CD20 in calcium flux in lymphoma may lead to discovery of novel mechanisms of calcium ions transport in DLBCL that may have therapeutic implications. The effect of miRNA-363 on CACNA1C and rituximab efficacy further extended our insight into the mechanisms of refractory disease and it may serve as a prognostic marker or novel therapeutic target in the future.
Supplementary Material
Statement of translational relevance.
B cells express a wide and diverse pool of ion channels, including components of the Cav1 subfamily of voltage-gated L-type calcium channels (VGCC). L-type calcium channels can be manipulated by many small molecules and are capable of mediating calcium influx, which are required to trigger apoptosis. Crosslinking of CD20 molecules by rituximab can induce calcium influx leading to apoptosis. In this study, we identified the role of L-type Cav 1.2 Calcium Channel-α−1C (CACNA1C) in regulating response to rituximab in DLBCL. These observations provide a rationale that modulating L-type Cav1.2 calcium channel may increase the clinical efficacy of rituximab. Actually, more attentions have now been paid on calcium channels due to their involvement in the proliferation and invasion of cancer cells and their potential modulation by pharmaceuticals.
Acknowledgements
YY L is supported by the grants from the National Natural Science Foundation of China (No. 81071938 and No. 81470365). WC C is partially supported by NCI P30 CA033572 and NCI SPORE 2P50 CA107399. J I is supported by the Lymphoma Research Foundation, the Leukemia and Lymphoma Society, and the UNMC Clinical-Translational Research Scholars Program; partial support is from NCI Eppley Cancer Center Support Grant P30CA036727, National Center for Research Resources 5P20RR016469, and National Institute for General Medical Science 8P20GM103427 to GB.
Footnotes
Conflict of interest: All of authors declare no conflict of interest.
Conflict of interest statement: The authors declare no conflict of interest.
Supplementary information is available at Journal’s website.
Reference
- 1.Lossos IS, Morgensztern D. Prognostic biomarkers in diffuse large B-cell lymphoma. J Clin Oncol 2006;24:995–1007. [DOI] [PubMed] [Google Scholar]
- 2.Iqbal J, Naushad H, Bi C, Yu J, Bouska A, Rohr J, et al. Genomic signatures in B-cell lymphoma: How can these improve precision in diagnosis and inform prognosis? Blood Rev 2016;30:73–88. [DOI] [PubMed] [Google Scholar]
- 3.Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood 2010;116:2040–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gisselbrecht C, Glass B, Mounier N, Singh Gill D, Linch DC, Trneny M, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol 2010;28:4184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis TA, Grillo-Lopez AJ, White CA, McLaughlin P, Czuczman MS, Link BK, et al. Rituximab anti-CD20 monoclonal antibody therapy in non-Hodgkin’s lymphoma: safety and efficacy of re-treatment. J Clin Oncol 2000;18:3135–43. [DOI] [PubMed] [Google Scholar]
- 6.Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene 2003;22:7359–68. [DOI] [PubMed] [Google Scholar]
- 7.Bonavida B Rituximab-induced inhibition of antiapoptotic cell survival pathways: implications in chemo/immunoresistance, rituximab unresponsiveness, prognostic and novel therapeutic interventions. Oncogene 2007;26:3629–36. [DOI] [PubMed] [Google Scholar]
- 8.Vega MI, Huerta-Yepaz S, Garban H, Jazirehi A, Emmanouilides C, Bonavida B. Rituximab inhibits p38 MAPK activity in 2F7 B NHL and decreases IL-10 transcription: pivotal role of p38 MAPK in drug resistance. Oncogene 2004;23:3530–40. [DOI] [PubMed] [Google Scholar]
- 9.Camicia R, Winkler HC, Hassa PO. Novel drug targets for personalized precision medicine in relapsed/refractory diffuse large B-cell lymphoma: a comprehensive review. Molecular cancer 2015;14:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terui Y, Mishima Y, Sugimura N, Kojima K, Sakurai T, Mishima Y, et al. Identification of CD20 C-terminal deletion mutations associated with loss of CD20 expression in non-Hodgkin’s lymphoma. Clin Cancer Res 2009;15:2523–30. [DOI] [PubMed] [Google Scholar]
- 11.Ghielmini M, Rufibach K, Salles G, Leoncini-Franscini L, Leger-Falandry C, Cogliatti S, et al. Single agent rituximab in patients with follicular or mantle cell lymphoma: clinical and biological factors that are predictive of response and event-free survival as well as the effect of rituximab on the immune system: a study of the Swiss Group for Clinical Cancer Research (SAKK). Ann Oncol 2005;16:1675–82. [DOI] [PubMed] [Google Scholar]
- 12.Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014;370:1101–10. [DOI] [PubMed] [Google Scholar]
- 13.Hillmen P, Robak T, Janssens A, Babu KG, Kloczko J, Grosicki S, et al. Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COMPLEMENT 1): a randomised, multicentre, open-label phase 3 trial. Lancet 2015;385:1873–83.. [DOI] [PubMed] [Google Scholar]
- 14.Uchida J, Lee Y, Hasegawa M, Liang Y, Bradney A, Oliver JA, et al. Mouse CD20 expression and function. Int Immunol 2004. ;16:119–29. [DOI] [PubMed] [Google Scholar]
- 15.Li H, Ayer LM, Lytton J, Deans JP. Store-operated cation entry mediated by CD20 in membrane rafts. J Biol Chem 2003;278:42427–34. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Ayer LM, Polyak MJ, Mutch CM, Petrie RJ, Gauthier L, et al. The CD20 calcium channel is localized to microvilli and constitutively associated with membrane rafts: antibody binding increases the affinity of the association through an epitope-dependent cross-linking-independent mechanism. J Biol Chem 2004;279:19893–901. [DOI] [PubMed] [Google Scholar]
- 17.Janas E, Priest R, Wilde JI, White JH, Malhotra R. Rituxan (anti-CD20 antibody)-induced translocation of CD20 into lipid rafts is crucial for calcium influx and apoptosis. Clin Exp Immunol 2005;139:439–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniels I, Turzanski J, Haynes AP. A requirement for calcium in the caspase-independent killing of Burkitt lymphoma cell lines by Rituximab. Br J Haematol 2008;142:394–403.. [DOI] [PubMed] [Google Scholar]
- 19.Walshe CA, Beers SA, French RR, Chan CH, Johnson PW, Packham GK, et al. Induction of cytosolic calcium flux by CD20 is dependent upon B Cell antigen receptor signaling. J Biol Chem 2008;283:16971–84. [DOI] [PubMed] [Google Scholar]
- 20.Lo WL, Donermeyer DL, Allen PM. A voltage-gated sodium channel is essential for the positive selection of CD4(+) T cells. Nat Immunol 2012;13:880–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davenport B, Li Y, Heizer JW, Schmitz C, Perraud AL. Signature Channels of Excitability no More: L-Type Channels in Immune Cells. Front Immunol 2015;6:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grafton G, Stokes L, Toellner KM, Gordon J. A non-voltage-gated calcium channel with L-type characteristics activated by B cell receptor ligation. Biochem pharmacol 2003;66:2001–9. [DOI] [PubMed] [Google Scholar]
- 23.Vig M, Kinet JP. Calcium signaling in immune cells. Nat immunol 2009;10:21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iqbal J, Shen Y, Huang X, Liu Y, Wake L, Liu C, et al. Global microRNA expression profiling uncovers molecular markers for classification and prognosis in aggressive B-cell lymphoma. Blood 2015;125:1137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabattini E, Bacci F, Sagramoso C, Pileri SA. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: an overview. Pathologica 2010. ;102:83–7. [PubMed] [Google Scholar]
- 26.Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, et al. Revised response criteria for malignant lymphoma. J Clin Oncol 2007;25:579–86. [DOI] [PubMed] [Google Scholar]
- 27.Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 2008;359:2313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma.N Engl J Med 2002;346:1937–47. [DOI] [PubMed] [Google Scholar]
- 29.Spat A, Szanda G, Csordas G, Hajnoczky G. High- and low-calcium-dependent mechanisms of mitochondrial calcium signalling. Cell calcium 2008;44:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Green DR. Apoptotic pathways: ten minutes to dead. Cell 2005;121:671–4. [DOI] [PubMed] [Google Scholar]
- 31.Emori Y, Kawasaki H, Imajoh S, Imahori K, Suzuki K. Endogenous inhibitor for calcium-dependent cysteine protease contains four internal repeats that could be responsible for its multiple reactive sites. Proc Natl Acad Sci U S A 1987;84:3590–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfreundschuh M, Trumper L, Kloess M, Schmits R, Feller AC, Rudolph C, et al. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of young patients with good-prognosis (normal LDH) aggressive lymphomas: results of the NHL-B1 trial of the DSHNHL. Blood 2004;104:626–33. [DOI] [PubMed] [Google Scholar]
- 33.Thevenin C, Lucas BP, Kozlow EJ, Kehrl JH. Cell type- and stage-specific expression of the CD20/B1 antigen correlates with the activity of a diverged octamer DNA motif present in its promoter. J Biol Chem 1993;268:5949–56. [PubMed] [Google Scholar]
- 34.Fitzsimmons D, Hodsdon W, Wheat W, Maira SM, Wasylyk B, Hagman J. Pax-5 (BSAP) recruits Ets proto-oncogene family proteins to form functional ternary complexes on a B-cell-specific promoter. Gene Dev 1996;10:2198–211. [DOI] [PubMed] [Google Scholar]
- 35.Tomita A. Genetic and Epigenetic Modulation of CD20 Expression in B-Cell Malignancies: Molecular Mechanisms and Significance to Rituximab Resistance. J Clin Exp Hematop 2016;56:89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slabicki M, Lee KS, Jethwa A, Sellner L, Sacco F, Walther T, et al. Dissection of CD20 regulation in lymphoma using RNAi. Leukemia 2016;30:2409–12. [DOI] [PubMed] [Google Scholar]
- 37.Bil J, Winiarska M, Nowis D, Bojarczuk K, Dabrowska-Iwanicka A, Basak GW, et al. Bortezomib modulates surface CD20 in B-cell malignancies and affects rituximab-mediated complement-dependent cytotoxicity. Blood 2010;115:3745–55. [DOI] [PubMed] [Google Scholar]
- 38.Winiarska M, Bil J, Nowis D, Golab J. Proteolytic pathways involved in modulation of CD20 levels. Autophagy 2010;6:810–2. [DOI] [PubMed] [Google Scholar]
- 39.Obrist F, Manic G, Kroemer G, Vitale I, Galluzzi L. Trial Watch: Proteasomal inhibitors for anticancer therapy. Mol Oncol 2015;2:e974463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li M, Xiong ZG. Ion channels as targets for cancer therapy. Int J Physiol Pathophysiol Pharmacol 2011;3:156–66. [PMC free article] [PubMed] [Google Scholar]
- 41.Monteith GR, Davis FM, Roberts-Thomson SJ. Calcium channels and pumps in cancer: changes and consequences. J Biol Chem 2012;287:31666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoek KL, Antony P, Lowe J, Shinners N, Sarmah B, Wente SR, et al. Transitional B cell fate is associated with developmental stage-specific regulation of diacylglycerol and calcium signaling upon B cell receptor engagement. J Immunol 2006;177:5405–13. [DOI] [PubMed] [Google Scholar]
- 43.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003;425:415–9. [DOI] [PubMed] [Google Scholar]
- 44.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001;409:363–6. [DOI] [PubMed] [Google Scholar]
- 45.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008;132:875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature 2005;435:828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Landais S, Quantin R, Rassart E. Radiation leukemia virus common integration at the Kis2 locus: simultaneous overexpression of a novel noncoding RNA and of the proximal Phf6 gene. J Virol 2005;79:11443–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landais S, Landry S, Legault P, Rassart E. Oncogenic potential of the miR-106–363 cluster and its implication in human T-cell leukemia. Cancer Res 2007;67:5699–707. [DOI] [PubMed] [Google Scholar]
- 49.Yu J, Wang F, Yang GH, Wang FL, Ma YN, Du ZW, et al. Human microRNA clusters: genomic organization and expression profile in leukemia cell lines. Biochem Biophys res commun 2006;349:59–68. [DOI] [PubMed] [Google Scholar]
- 50.Iqbal J, Shen Y, Liu Y, Fu K, Jaffe ES, Liu C, et al. Genome-wide miRNA profiling of mantle cell lymphoma reveals a distinct subgroup with poor prognosis. Blood 2012;119:4939–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.