

SUMMARY
Endocannabinoids protect against seizures, but their mechanism of action is still unclear, as they can have effects independent of known cannabinoid receptors. Using Drosophila melanogaster, which lacks canonical cannabinoid receptors, we report that the endocannabinoids anandamide and 2-arachidonoylglycerol protect against seizures in multiple fly seizure models. Surprisingly, inhibition of anandamide catabolism renders flies insensitive to protection by anandamide, indicating that anandamide metabolites are responsible for seizure protection. Consistent with this finding, arachidonic acid, a direct metabolite of anandamide, protects against seizures. To identify downstream effectors, we test for a role of transient receptor potential (TRP) channels and find that the TRPV1 antagonist capsazepine blocks the protective effect of anandamide. Also, a targeted genetic screen of TRP channels identifies water witch as a mediator of protection by anandamide. Using a Drosophila model, we reveal the role of arachidonic acid in seizure protection and identify a cannabinoid-receptor-1/2-independent mechanism of endocannabinoid seizure protection.
Graphical Abstract
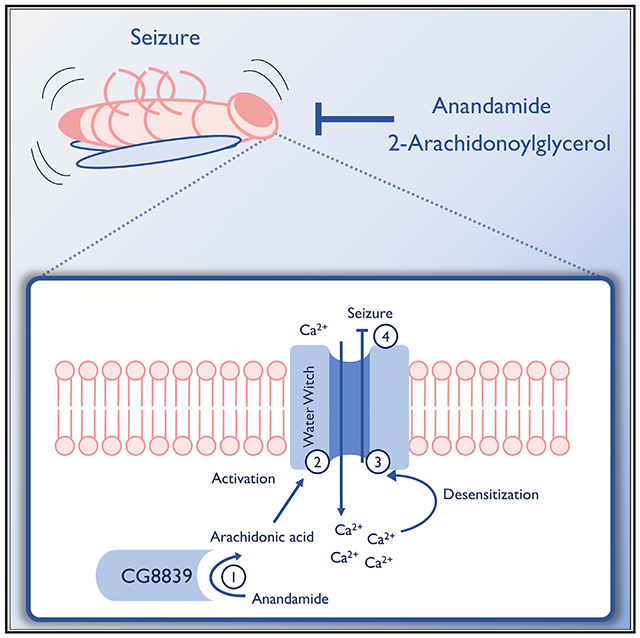
In Brief
Jacobs and Sehgal demonstrate that the endocannabinoids anandamide and 2-arachidonoylglycerol are anticonvulsant in Drosophila melanogaster and that seizure protection by anandamide is mediated by metabolites acting on the TRP channel Water witch.
INTRODUCTION
The endocannabinoid (eCB) system is a major modulator of excitability in the central nervous system, and multiple lines of evidence suggest a link between the eCB system and seizures. First, levels of anandamide (AEA) and 2-arachidonoylglycerol (2-AG), the two major eCBs, are elevated after seizures (Marsicano et al., 2003; Wallace et al., 2003). Second, exogenously applying low doses of eCBs is generally protective against seizures (Deshpande et al., 2007; Wallace et al., 2002). Finally, cannabinoid (CB) receptor levels are altered in epilepsy (Ludányi et al., 2008; Wallace et al., 2003). However, although the involvement of the eCB system in seizures is supported by the literature, mechanisms of eCB action are debatable. CB receptor 1 (CB-1R) has been identified as one of the mediators of eCB anticonvulsant effects (Marsicano et al., 2003; Monory et al., 2006), but eCBs also bind and exert effects through noncanonical CBRs (Muller et al., 2019; O’Sullivan, 2016; Ryberg et al., 2007; Sigel et al., 2011) as do the phytocannabinoids cannabidiol (CBD) and cannabidivarin (CBDV), which possess anticonvulsant effects and have low affinity for CB1R (Hill et al., 2013; Wallace et al., 2001).
The active component of eCBs relevant for seizures is also questionable. Pharmacological inhibition of the eCB catabolic enzymes fatty acid amide hydrolase (FAAH), monoacylglycerol lipase (MAGL), and alpha/beta-hydrolase domain 6 (ABHD6) is generally protective against seizures (Naydenov et al., 2014; Sugaya et al., 2016; Vilela et al., 2013), indicating that eCBs themselves are protective molecules. However, FAAH knockout (KO) mice are more sensitive to induced seizures (Clement et al., 2003). This raises the possibility that products of FAAH are protective, but no such molecules have been identified yet.
Drosophila melanogaster is an excellent model organism for discovering molecular mechanisms, but few studies have used flies to investigate eCB function. This is likely due to the observation that flies lack clear orthologs to CB1/2R (McPartland et al., 2001). On the other hand, Drosophila have detectable levels of eCBs (Jeffries et al., 2014; McPartland et al., 2001) and predicted orthologs of both eCB-metabolizing enzymes and noncanonical CBRs (McPartland et al., 2006). Thus, they could be a useful model to dissect noncanonical mechanisms of eCB action.
We sought to leverage Drosophila’s simple, yet comparative neurobiology, abundance of genetic tools and mutants, short generation time, and availability of established seizure models (Song and Tanouye, 2008) to identify eCB mechanisms of seizure protection. We show that feeding AEA and 2-AG protects against seizures in multiple fly seizure models. Surprisingly, effects of AEA are mediated by its metabolites, which act through the TRP channel Water witch (WTRW) to confer protection from seizures.
RESULTS
AEA and 2-AG Protect against Seizures in Multiple Fly Seizure Models
Easily shocked (eas) flies have a mutation in ethanolamine kinase that makes them sensitive to mechanically induced seizures (Pavlidis et al., 1994). To determine whether eCBs confer protection against mechanically induced seizures, we fed male eas flies AEA (2, 20, and 200 μg/mL) and induced seizures once a day for 10 days with a 10-s vortex (Ganetzky and Wu, 1982). Significant seizure protection was observed with 20 and 200 μg/mL AEA after 2 days and with 2 μg/mL AEA after 3 days (Figure 1A). Significant seizure protection was also observed in female flies when tested after 4 days of drug feeding (Figure 1B). For all subsequent experiments, only male flies were used, and seizures were measured after 4 days of drug feeding.
Figure 1. Endocannabinoids Protect against Seizures.
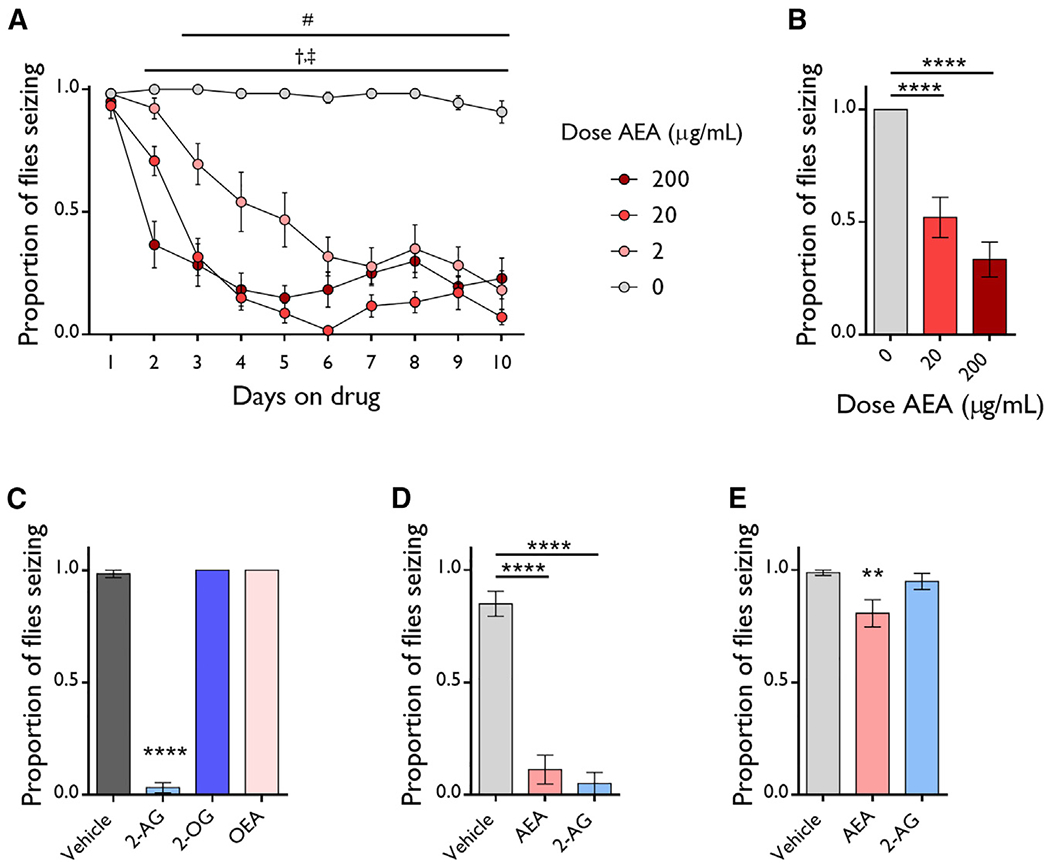
(A) Feeding 2, 20, and 200 μg/mL AEA protects against induced seizures of eas males. n = 11–12 vials/dose. Two-way ANOVA, dose: p ≤ 0.0001, time: p ≤ 0.0001, interaction: p ≤ 0.0001. Dunnett’s multiple comparison test, #: 2 μg/mL versus 0 μg/mL, p ≤ 0.05; †: 20 μg/mL versus 0 μg/mL, p ≤ 0.05; ‡: 200 μg/mL versus 0 μg/mL, p ≤ 0.05.
(B) Doses of 20 and 200 μg/mL AEA protect against seizures in eas females. n = 12 vials/dose. ANOVA with Tukey’s post hoc analysis. ****p ≤ 0.0001.
(C) A dose of 200 μg/mL 2-AG but not 2-OG and OEA protect against seizures in eas males. n = 4–12 vials/dose. ANOVA with Dunnett’s post hoc analysis. ****p ≤ 0.0001.
(D–E) A dose of 200 μg/mL AEA and 2-AG protect against seizures in tko25t flies (n = 4–12 vials/group) (D) and AEA also works in bss1 flies (n = 12–17 vials/group) (E). ANOVA with Dunnett’s post hoc analysis. **p ≤ 0.01, ****p ≤ 0.0001.
All data are presented as mean ± SEM.
To determine if other eCBs also protect against seizures in eas flies, we fed 200 μg/mL 2-AG, 2-oleoylglycerol (2-OG), and oleoylethanolamide (OEA) to eas flies and measured seizures. 2-AG was the only other eCB that protected against seizures (Figure 1C).
Next, we tested whether AEA and 2-AG protect against seizures in two other fly seizure models that are sensitive to mechanically induced seizures: bang senseless (bss1), which has a gain-of-function mutation in the sole Drosophila voltage-gated sodium channel (Parker et al., 2011), and technical knockout (tko25t), which has a mutation in a mitochondrial ribosomal protein (Royden et al., 1987). Notably, 200 μg/mL AEA protected against seizures in both seizure models (Figures 1D and 1E). Also, 200 μg/mL 2-AG protected against seizures in tko25t flies but did not in bss1 flies (Figures 1D and 1E). This suggests that 2-AG and AEA have different mechanisms of action.
Metabolites Are Responsible for Seizure Protection by AEA
Given that exogenous AEA protects flies against seizures, we predicted that increasing endogenous AEA would be protective as well. URB597 inhibits mammalian FAAH, which catabolizes AEA into arachidonic acid (AA) and ethanolamine. We fed 50 μg/mL URB597 to eas flies and found that URB597 feeding alone did not protect against seizures (Figure 2A). We next predicted that eas flies fed URB597 and 20 μg/mL AEA would be more sensitive to the protective effect of AEA. Surprisingly, URB597 completely blocked the protective effect of AEA (Figure 2A). The protective effect of an equimolar dose of 2-AG (58 μM, 22 μg/mL), which is not primarily catabolized by FAAH in mammals, was unaffected by URB597 (Figure 2A). This suggests that the effect of URB597 is specific to AEA and does not lower sensitivity to all anticonvulsants.
Figure 2. Metabolites Mediate the Anticonvulsant Effect of AEA.
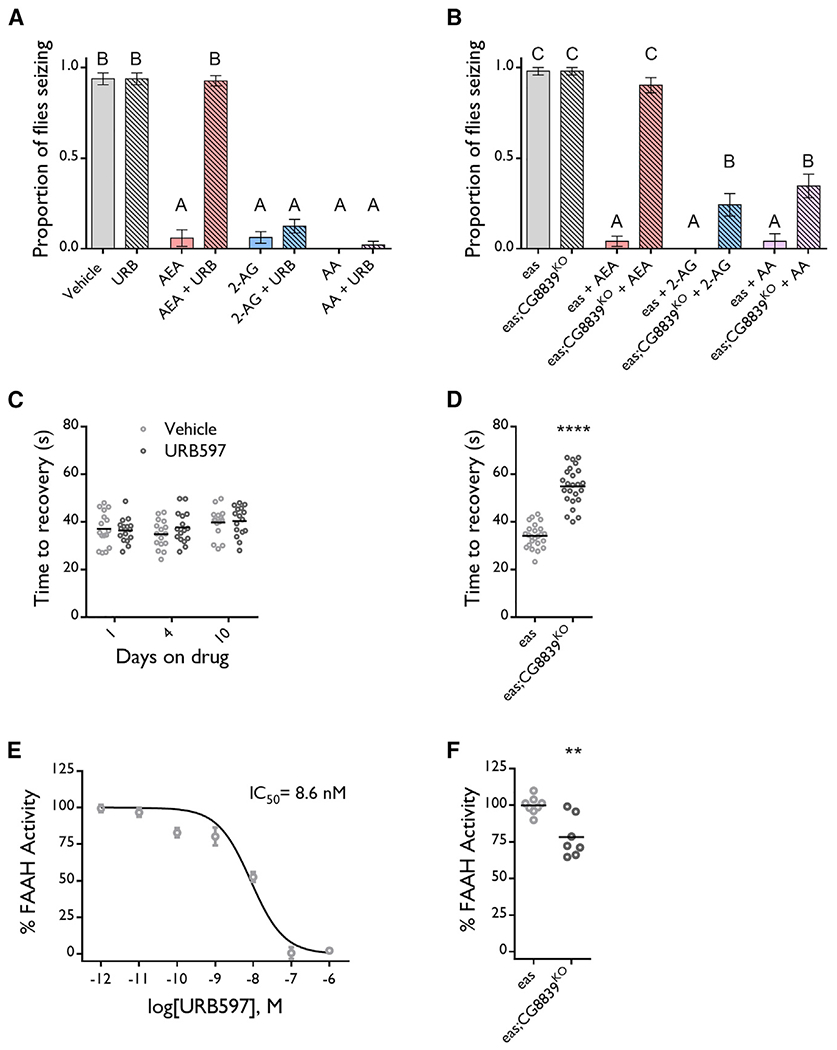
(A) 50 μg/mL URB597 feeding does not protect against seizures, and cofeeding URB597 with 20 μg/mL (58 μM) AEA blocks the protective effect of AEA but not of equimolar doses of 2-AG and AA in eas flies. n = 12–17 vials/group. ANOVA with Tukey’s post hoc analysis. Means with different letters are significantly different (*p ≤ 0.05).
(B) 58 μM AEA does not protect against seizures in eas;CG8839KO flies, but 2-AG and AA are effective. n = 12 vials/group. ANOVA with Tukey’s post hoc analysis. Means with different letters are significantly different (*p ≤ 0.05).
(C) 50 μg/mL URB597 feeding does not increase seizure recovery time in eas flies. n = 15–16 vials/group. Multiple t tests with Holm-Sidak correction.
(D) eas;CG8839KO flies take longer to recover from seizures than eas controls. n = 24 vials/genotype.Unpaired t test. ****p ≤ 0.0001.
(E) URB597 inhibits FAAH activity with an IC50 of 8.6 nM. % FAAH activity normalized to vehicle treated homogenates for each sample. n = 4 samples/dose, 1 technical replicate/sample.
(F) eas;CG8839KO flies have reduced FAAH activity relative to eas flies. The % FAAH activity was normalized to control fly FAAH activity for each experiment. n = 7–8 samples/genotype, 3 technical replicates/sample. Unpaired t test. **p ≤ 0.01.
Bar graph data are presented as mean ± SEM. See also Figure S1.
CG8839 is a previously uncharacterized fly gene that shows high sequence homology with mammalian FAAH2. In order to further test the effects of blocking AEA catabolism, we generated a KO mutant of CG8839 by using the CRISPR-Cas9 system to delete the entire CG8839 coding region and replacing it with a DsRed marker (Figure S1A). The CRISPR KO and insertion of DsRed was confirmed by PCR (Figure S1B). CG8839KO flies were then crossed into the eas background to generate eas;CG8839KO flies. Eas;CG8839KO flies were significantly less protected from seizures by AEA feeding relative to eas controls, which supports our findings with pharmacological inhibition of AEA catabolism (Figure 2B). Eas;CG8839KO flies were also significantly less protected from seizures by 2-AG feeding relative to eas controls (Figure 2B). However, 2-AG was more effective at suppressing seizures in eas;CG8839KO flies than AEA, suggesting that knocking out CG8839 has a stronger effect on AEA sensitivity than 2-AG.
We next sought to determine if eas;CG8839KO flies exhibit more severe seizures relative to eas controls, as do FAAH KO mice (Clement et al., 2003). Because our primary seizure readout of the proportion of flies seizing was saturated (100% seizing), we measured the time of recovery from seizures. We found that eas;CG8839KO flies take 20 s (≈60%) longer than eas controls to recover from seizures, suggesting that eas;CG8839KO have more severe seizures (Figure 2D). We also measured recovery time in flies fed URB597 but found no difference in recovery time relative to vehicle-fed controls (Figure 2C). Although it is unclear why CG8839KO increases recovery time and URB597 does not, these findings are consistent with data in mice (Clement et al., 2003) .
The pharmacological and genetic data above suggest that breakdown products of AEA account for its anticonvulsant effect. To test if AA, a direct metabolite of AEA, is responsible for AEA seizure protection, we fed an equimolar dose of AA (58 μM, 18 μg/mL) to eas flies. We found that AA protects against seizures and that this effect persists with URB597 cofeeding and in eas;CG8839KO flies (Figures 2A and 2B). Together, these results indicate that metabolites are responsible for seizure protection by AEA.
To further test the idea that AA is protective against seizures, we fed eas flies docosatetraenoic acid (DTA) and measured seizures. DTA is metabolized to AA, and feeding DTA increases AA levels in flies (Shen et al., 2010). We found that 50 and 500 μg/mL DTA feeding was protective against seizures, which is consistent with a role of AA in seizure protection, although we cannot rule out the possibility that DTA itself is protective (Figure S1C).
We next tested whether URB597 blocks the effect of AEA in tko25t and bss1 flies. In tko25t flies, URB597 partially blocked the effect of 20 μg/mL AEA after 2 days of drug feeding, but not after 4 days (Figure S1D). This suggests that a lower concentration of metabolites is sufficient for seizure protection in tko25t flies relative to eas flies. A larger dose of AEA (200 μg/mL) confers seizure protection to bss1 flies after 4 days of treatment but not after 2 days (Figure S1E; data not shown). We found that URB597 did not block the effect of 200 μg/mL AEA after 4 days of drug feeding (Figure S1E). Although this finding does not support a role for AEA metabolites in seizure protection in bss1 flies, it may also be the case that, at the higher AEA dose, enough AA is produced for seizure protection despite URB597. We could not increase the dose of URB597 above 50 μg/mL due to solubility.
URB597 and CG8839 KO Reduce Catabolism of 7-Amino, 4-Methyl Coumarin -Arachidonoyl Amide (AMC-AA)
We next sought to verify that URB597, which has never been used in flies, acts by inhibiting FAAH. Thus, we conducted FAAH activity assays with fly homogenates and AMC-AA, a FAAH substrate whose catabolism can be quantified by measuring the production of its fluorescent product, AMC (Ramarao et al., 2005). We incubated fly homogenates with URB597 and found that URB597 reduces the rate of AMC catabolism with a half-maximal inhibitory concentration (IC50) of 8.6 nM (Figures 2E and S1F). This value is in accordance with the IC50 of 33.5 nM obtained from human-FAAH-expressing CHO cells (Ramarao et al., 2005).
We then asked if eas;CG8839KO flies exhibit reduced FAAH activity and found a ≈20% reduction relative to eas controls (Figures 2F and S1G). Although this result directionally supports our hypothesis, we were surprised that knocking out CG8839 did not lead to a larger deficit in FAAH activity. The remaining FAAH activity in eas;CG8839KO flies was sensitive to 1 μM URB597 (Figure S1H), which leads us to speculate that CG8839KO flies upregulate other, FAAH-like enzymes.
Finally, we asked if seizures affect FAAH activity. To test this, we vortexed eas flies and homogenized them 15 min later. This time point was chosen because 2-AG and AEA levels are elevated 15 and 20 min after seizure induction in rodents, respectively (Marsicano et al., 2003; Wallace et al., 2003). We found that seizures did not have a significant effect on FAAH activity (Figure S1I)
AEA Increases Baseline Calcium and Protects against Stimulus-Induced Calcium Elevations
Seizures are characterized by excessive neuronal activity, so we hypothesized that AEA metabolites decrease baseline neuronal activity. To test this, we measured intracellular calcium as a proxy by expressing calcium-dependent nuclear import of Lex A (CaLexA), a genetic tool that drives GFP expression upon calcium binding (Masuyama et al., 2012), in neurons of AEA-fed flies with nsyb-gal4. AEA feeding did not decrease GFP in either wild-type (WT) or eas flies (Figures S2A and S2E). Instead, there was a slight increase in GFP with AEA feeding in WT flies. We also observed that eas flies have higher baseline calcium than WT controls, which could contribute to their susceptibility to seizures.
Mushroom bodies (MBs) are invertebrate brain structures similar to the mammalian hippocampus, in that they play major roles in learning and memory (Heisenberg, 2003) and are believed to be a region important for seizure propagation (Hekmat-Scafe et al., 2010; Saras et al., 2017). Using a MB-specific region of interest (ROI), we found that AEA-fed WT flies and vehicle-fed eas flies had higher MB-specific GFP than vehicle-fed WT flies (Figures S2B and S2E). Thus, as with the entire brain, WT MBs increase calcium upon AEA treatment and eas MBs have high baseline calcium.
We next sought to determine how seizure protection by AEA affects neuronal activity. We provided a seizure-inducing stimulus to CaLexA-expressing flies and imaged brains 90 min after recovery. We found that vortexing eas flies increased GFP relative to control eas flies (Figure S2C). AEA-fed eas flies did not exhibit the same increase in brain GFP, suggesting that AEA protects against seizures by preventing a stimulus-induced rise in calcium. There were no MB-specific differences in GFP between these groups (Figure S2D)
The TRP Channel WTRW Mediates the Protective Effect of AEA
The small increase in calcium with AEA treatment of control flies suggested that it affects the function of cation channels. Indeed, eCBs and their polyunsaturated fatty acid (PUFA) metabolites have been reported to affect TRP channels (Muller et al., 2019). We hypothesized that AEA metabolites confer protection against seizures by activating and subsequently desensitizing TRP channels. To test this, we cofed the TRPV1 antagonist capsazepine (CPZ) with AEA and found that 0.2 and 0.5 mg/mL CPZ blocked the protective effect of 20 μg/mL AEA, suggesting that AEA metabolites signal through a TRPV1-like channel in flies (Figure 3A).
Figure 3. The TRP Channel Water Witch Mediates the Anticonvulsant Effect of AEA.
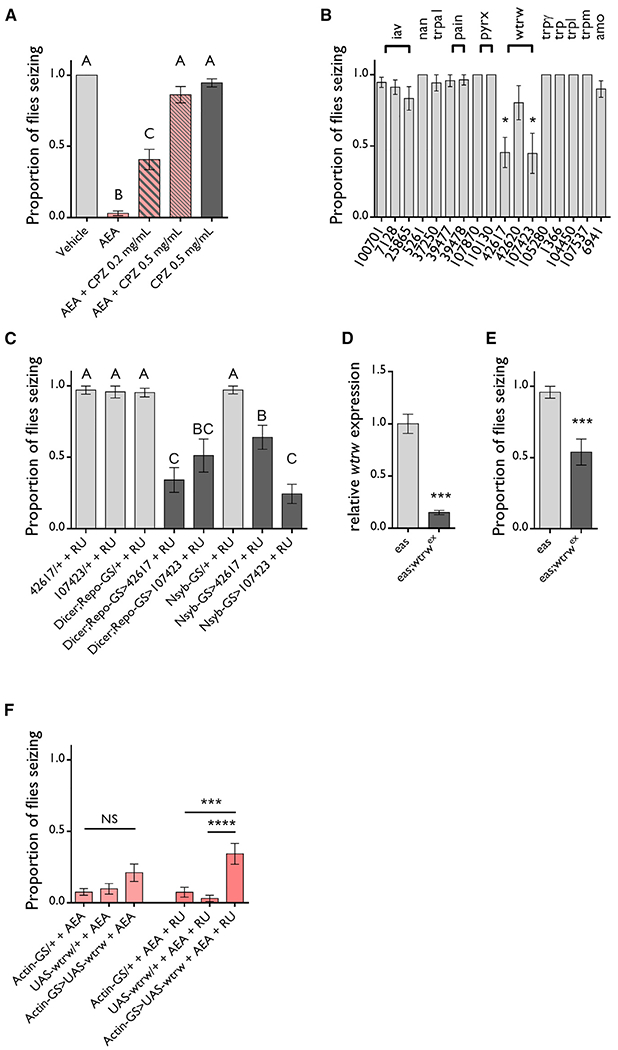
(A) 0.2 mg/mL CPZ partially blocks and 0.5 mg/mL CPZ completely blocks the protective effect of 20 μg/mL AEA on eas flies. n = 12–20 vials/group. ANOVA with Tukey’s post hoc analysis. Means with different letters are significantly different (*p ≤ 0.05).
(B) RNAi screen of TRP channels reveals that two separate wtrw-targeting RNAis protect against seizures when driven by actin-GS after 4 days of feeding 50 μM RU486. All RNAi knockdowns were performed in an eas mutant background. n = 3–13 vials/genotype. One-sample t test. *p ≤ 0.05.
(C) Both wtrw-targeting RNAis protect eas flies against seizures when expressed in neurons or glia by nsyb-GS or dicer;repo-GS, respectively, after 4 days of feeding 50 μM RU486. n = 4–12. ANOVA with Tukey’s post hoc analysis. Means with different letters are significantly different (p ≤ 0.05).
(D) Eas;;wtrwex flies have 3-fold reduced levels of the wtrw transcript relative to eas controls. n = 3 samples/genotype, 4 technical replicates/sample. Unpaired t test. ***p ≤ 0.001.
(E) eas;;wtrwex flies are partially protected against seizures. n = 8 vials/genotype. Unpaired t test. ***p ≤ 0.001.
(F) Overexpression of wtrw driven by actin-GS and 50 μM RU486 feeding makes eas flies less sensitive to the protective effect of 20 μg/mL AEA. n = 20 vials/group. ANOVA with Tukey’s post hoc analysis. ***p ≤ 0.001, ****p ≤ 0.0001.
All data are presented as mean ± SEM. See also Figure S2.
TRP channel antagonists have not been studied in Drosophila, so the specific target of CPZ is not known. To identify the specific TRP channel that mediates the anticonvulsant effect of AEA, we conducted an RNAi screen to knock down TRP channels in flies. Eas;;actin-GS flies were crossed to UAS-RNAi lines targeting different TRP channels, and whole-body RNAi expression was induced in adults by feeding RU486. We found that the expression of two RNAi constructs against water witch (wtrw) (#4217 and #107423) significantly protected against seizures (Figure 3B). Seizure protection was not observed with any other RNAi line.
To determine whether wtrw knockdown is sufficient for seizure protection in either glia or neurons, we expressed wtrw RNAi in both cell types using the repo-GS and nsyb-GS drivers, respectively, and fed RU486. We found that the expression of either wtrw-targeting RNAi conferred partial protection against seizures when expressed in either cell type (Figure 3C).
To confirm our RNAi findings, we crossed the wtrwex allele into the eas background and tested for seizures. We found that eas;;wtrwex flies have 3-fold reduced levels of wtrw transcripts relative to eas controls (Figure 3D). The wtrwex allele partially protected against seizures, which is consistent with a hypomorphic effect and supports our RNAi result (Figure 3E).
Finally, we asked if seizure protection by AEA is reduced in flies overexpressing wtrw. We crossed UAS-wtrw:GFP flies to eas;;actin-GS flies and fed the progeny RU486 and AEA. Eas;;ac-tin-GS > UAS-wtrw:GFP flies were less sensitive to fed AEA relative to eas;;actin-GS/+ and eas;UAS-wtrw:GFP controls, which supports our hypothesis that AEA metabolites signal through WTRW (Figure 3F).
Acute AEA Increases Seizure Recovery Time
If AEA metabolites protect against seizures by first activating and then desensitizing WTRW, one would expect acute AEA to activate WTRW without desensitizing it and, thereby, lead to more severe seizures. To test this, we starved flies for 16 h, transferred them onto food containing 2 μg/mL AEA, and measured seizure recovery time after 1 h AEA exposure. We found that flies exposed to 2 μg/mL AEA for 1 h take 5.6 s (≈15%) longer to recover from seizures than vehicle-fed controls (Figure 4A). This effect was reversed after 48 h of AEA exposure. We confirmed that flies were consuming AEA in our acute exposure protocol in a separate set of experiments by mixing blue dye into their food (Figures S3A and S3B). We also repeated our 1-h acute exposure protocol with an equimolar dose of AA and found that acute AA also increased recovery time (Figure 4B). These findings support our model that AEA metabolites protect against seizures by first activating and then desensitizing WTRW (Figure S4).
Figure 4. Acute Anandamide Increases Time of Recovery from Seizures.
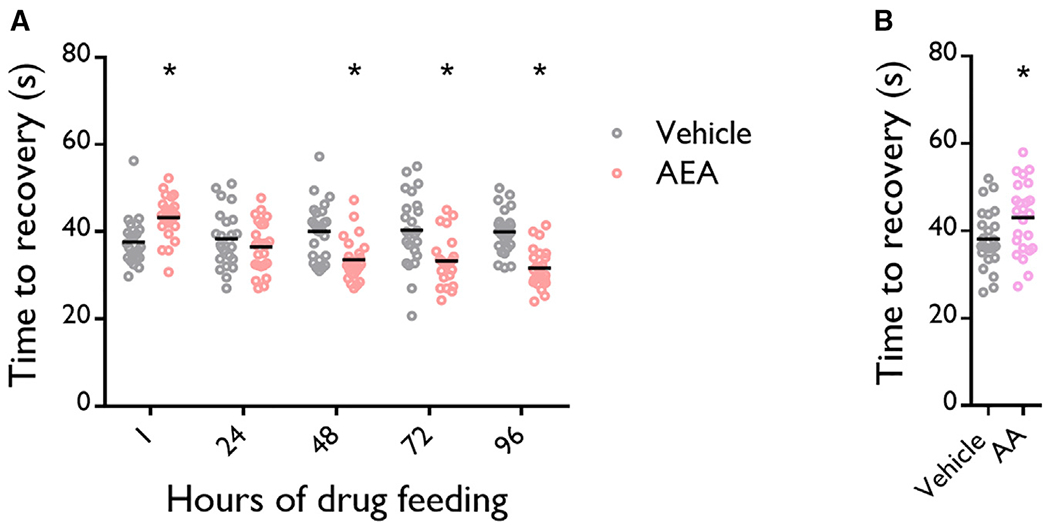
(A) 1 h of 2 μg/mL (5.8 μM) AEA feeding increases seizure recovery time. ≥ 48 h of 2 μg/mL AEA feeding decreases seizure recovery time. n = 24 vials/group. Two-way ANOVA with Sidak’s multiple comparison, *p ≤ 0.05.
(B) 1 h of 5.8 ≤M AA feeding increases seizure recovery time. n = 24 vials/group. Unpaired t test, *p ≤ 0.05.
See also Figure S3.
DISCUSSION
Studies examining the effects of eCBs on seizures and epilepsy have focused primarily on the role of CB1R. Indeed, there is strong evidence suggesting that CB1R mediates some of the protective effects of eCBs (Marsicano et al., 2003; Monory et al., 2006). Our findings that AEA and 2-AG protect against seizures in flies do not discount the role of CB1R in mammals but provide further evidence that signaling through CB1R may not be the sole mechanism of action of eCBs. If the mechanism proposed in this study is used in mammals, AEA mobilization would provide seizure protection through two pathways: CB1 Rs and TRP channels.
Through pharmacological inhibition and genetic ablation, we were able to reduce FAAH activity, which abolished the protective effect of AEA in eas flies. These findings are surprising because several FAAH inhibitors confer seizure protection in mammals (Colangeli et al., 2017; Manna and Umathe, 2012; Naderi et al., 2008; Naidoo et al., 2011; Shubina et al., 2015; Vilela et al., 2013, 2015). However, FAAH KO mice are more sensitive to induced seizures and exhibit more severe seizures when injected with AEA (Clement et al., 2003). Consistent with these findings, we report that flies lacking CG8839 take longer to recover from seizures. Explanations involving biphasic effects of AEA on either CB1R and proconvulsant TRPV1 (Manna and Umathe, 2012) or CB1R on glutamatergic and GABAergic neurons (Lutz, 2004; Marsicano et al., 2003) have been proposed to explain the discrepancy in mice. Our study provides a third possible explanation: acute effects of AEA are mediated through AEA action on CB1Rs, whereas chronic protection requires AEA metabolites such as AA, which are not produced in FAAH KOs.
AA and its metabolites are traditionally thought of as being proinflammatory, which makes our finding that AA is protective against seizures surprising. Interestingly, AA has been shown to inhibit excitatory discharges in granule cells (Lauritzen et al., 2000). Although there is no prior evidence demonstrating that AA is seizure protective, it is elevated after seizures (Bazán, 1970). This suggests that, like AEA and 2-AG, AA or AA metabolites could be endogenous, on-demand protectors against excitotoxicity.
The existence of AA and other long-chain PUFAs in flies remains controversial. Although some groups were unable to detect PUFAs longer than 18 carbons in flies (Shen et al., 2010; Tortoriello et al., 2013; Yoshioka et al., 1985), others were able to measure AEA and 2-AG (Jeffries et al., 2014; McPartland et al., 2001). Additionally, through a collaborative effort with another laboratory, we were able to detect AEA and AA in a sample of fly heads (J.A.J., S. Ghosh, and T. Grosser, unpublished data). A possible explanation for this discrepancy is that flies have low levels of long-chain PUFAs that are difficult to detect. Importantly, 20-carbon PUFAs were detectable in flies fed diets supplemented with 22-carbon PUFAs (Shen et al., 2010), suggesting that flies have the enzymatic machinery necessary to process and use dietary long-chain PUFAs.
In addition to CB1R, eCBs and eCB metabolites are known to bind non-CB receptors, such as TRP channels (Muller et al., 2019), peroxisome proliferator-activated receptors (O’Sullivan, 2016), GABA receptors (Sigel et al., 2011), potassium channels (Carta et al., 2014), and GPR55 (Ryberg et al., 2007). We demonstrate that feeding CPZ, a TRPV1 antagonist, blocks the anticonvulsant effect of AEA. A potential mechanism that would be consistent with our findings is that AEA metabolites protect against seizures through TRP channel activation and subsequent desensitization (Figure S4). Prolonged or repeated administration of both TRPV1 and TRPA1 agonists induces desensitization at their respective targets (Bandell et al., 2004; Motter and Ahern, 2012; Redmond et al., 2014; Sanz-Salvador et al., 2012). Although not demonstrated with respect to seizures, desensitization is proposed to explain paradoxical analgesic effects of TRP channel agonists (Fukushima et al., 2017; McGaraughty et al., 2003; Mitchell et al., 2014; Palazzo et al., 2002).
Wtrw was the only TRP channel whose knockdown affected seizures in our RNAi screen. WTRW has no previously identified ligands but has high sequence homology with mammalian TRPA1, and both AA and its metabolites activate mammalian TRPA1 (Bandell et al., 2004; Motter and Ahern, 2012; Redmond et al., 2014). PUFAs do not appear to affect Drosophila TRPA1 (dTRPA1) (Motter and Ahern, 2012), but it is important to note that WTRW is not the same channel as dTRPA1. Along with AA and its metabolites, AEA itself can activate mammalian TRPV1 and TRPA1 channels (Bandell et al., 2004; Muller et al., 2019; De Petrocellis and Di Marzo, 2009; Redmond et al., 2014; Zygmunt et al., 1999), which suggests that AEA could also activate WTRW. However, we found that AEA did not protect against seizures when its catabolism was blocked. This suggests that AA or its metabolites have a stronger effect on WTRW than AEA.
CPZ feeding alone did not protect against seizures, whereas RNAi silencing of wtrw did. A possible explanation for this finding is that CPZ acts as a neutral antagonist for AA at WTRW, blocking the effects of AA but not affecting WTRW activity in the absence of AA. In support of this idea, previous studies have demonstrated that, at the rat TRPV1 receptor, CPZ only antagonizes capsaicin-evoked TRPV1 activity and does not affect TRPV1 responses to other agonists, such as heat and low pH (McIntyre et al., 2001; Walker et al., 2003).
Modulation of TRP channels is implicated in anticonvulsant properties of CBD (Huizenga et al., 2019; Iannotti et al., 2014) and of a non-CB TRPV1 agonist (Chen et al., 2013) but, to our knowledge, not for eCBs. On the contrary, previous studies have shown that AEA activity at TRP channels is proconvulsant (Bhaskaran and Smith, 2010; Manna and Umathe, 2012). A difference in drug delivery is a likely explanation for these contrary findings. Studies proposing a proconvulsant role delivered AEA acutely, which may be sufficient to activate TRP channels but not to desensitize them. Most paradigms evaluating TRP channel desensitization use prolonged (Sanz-Salvador et al., 2012) or repeated (Bhave et al., 2002; Fukushima et al., 2017) application of TRP agonists. In support of this, AEA displayed biphasic effects in our experiments: acute AEA exposure increased time to recovery and chronic AEA exposure provided seizure protection.
In summary, we provide evidence for a CB1R-independent mechanism of seizure protection by eCBs and validate the use of Drosophila melanogaster as a model organism to study eCB signaling.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Amita Sehgal (amita@mail.med.upenn.edu).
Materials Availability
Fly stocks used in this study are available upon request from the Lead Contact.
Data and Code Availability
Original data have been deposited to Mendeley Data: http://dx.doi.org/10.17632/vmhkt8k9p5.1
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fly stocks and husbandry
Flies were raised and maintained on cornmeal-molasses medium at 25 C. 2-10 day old adult flies were used for experiments. See Method Details for specific fly ages used for each experiment. Male flies were used for all experiments unless specifically stated. iso31 flies were used as wild-type. See the Key Resources Table for genotypes used.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-GFP | Thermo Fisher Scientific | #A-11122; RRID: AB_221569 |
| AlexaFluor 488 goat anti-rabbit | Thermo Fisher Scientific | #A-11034; RRID: AB_2576217 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Arachidonoyl ethanolamide | Cayman Chemical | #90050 |
| 2-arachidonoylglycerol | Cayman Chemical | #62160 |
| Oleoyl ethanolamide | Cayman Chemical | #90265 |
| 2-Oleoylglycerol | Cayman Chemical | #19537 |
| Arachidonic Acid | Cayman Chemical | #90010 |
| 7Z,10Z,13Z,16Z-Docosatetraenoic Acid | Cayman Chemical | #90300 |
| URB597 | Cayman Chemical | #10046 |
| Capsazepine | Cayman Chemical | #10007518 |
| RU486 | Millipore Sigma | M8046 |
| Caffeine | Millipore Sigma | C0750 |
| AMC-arachidonoyl amide | Millipore Sigma | A6855 |
| TRIzol | Thermo Fisher Scientific | #15596026 |
| SYBR Green PCR Master Mix | Thermo Fisher Scientific | #4364346 |
| FD&C Blue | Spectrum Chemical | FD110-25GM |
| Critical Commercial Assays | ||
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | #4368814 |
| BCA Protein Assay Kit | Abcam | ab207003 |
| Deposited Data | ||
| Mendeley Dataset | This Paper | http://dx.doi.org/10.17632/vmhkt8k9p5.1 |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: iso31 | Lab stock | N/A |
| D. melanogaster: easppc80f | Lab stock | Flybase ID: FBal0003489 |
| D. melanogaster: dicer;repo-GeneSwitch | Lab stock | N/A |
| D. melanogaster: nsyb-gal4 | Lab stock | N/A |
| D. melanogaster: nsyb-GeneSwitch | Lab stock | N/A |
| D. melanogaster: actin-GeneSwitch | Lab stock | N/A |
| D. melanogaster: UAS-CaLexA | Lab stock | N/A |
| D. melanogaster: parabss1 | Gift from Dr. Dan Kuebler | Flybase ID: FBal0001325 |
| D. melanogaster: tko25t | Gift from Dr. Dan Kuebler | Flybase ID: FBal0016812 |
| D. melanogaster: wtrwex | Gift from Dr. Marc Freeman | Flybase ID: FBal0246489 |
| D. melanogaster: UAS-wtrw:GFP | Gift from Dr. Marc Freeman | Ma et al., 2016 |
| D. melanogaster: y1 v1; P{TRiP.JF01904}attP2 | BDSC | BDSC ID: 25865 |
| D. melanogaster: w1118; P{GD3260}v7128 | VDRC | VDRC ID: 7128 |
| D. melanogaster: P{KK107960}VIE-260B | VDRC | VDRC ID: 100701 |
| D. melanogaster: w1118; P{GD2456}v5261 | VDRC | VDRC ID: 5261 |
| D. melanogaster: w1118; P{GD2375}v37250/TM3 | VDRC | VDRC ID: 37250 |
| D. melanogaster: w1118; P{GD2221}v39477/TM3 | VDRC | VDRC ID: 39477 |
| D. melanogaster: w1118; P{GD2221}v39478/TM3 | VDRC | VDRC ID: 39478 |
| D. melanogaster: P{KK104597}VIE-260B | VDRC | VDRC ID: 108780 |
| D. melanogaster: P{KK115661}VIE-260B | VDRC | VDRC ID: 110130 |
| D. melanogaster: w1118; P{GD2850}v42617 | VDRC | VDRC ID: 42617 |
| D. melanogaster: w1118; P{GD2850}v42620 | VDRC | VDRC ID: 42620 |
| D. melanogaster: P{KK103625}VIE-260B | VDRC | VDRC ID: 107423 |
| D. melanogaster: P{KK107656}VIE-260B | VDRC | VDRC ID: 105280 |
| D. melanogaster: w1118; P{GD372}v1366 | VDRC | VDRC ID: 1366 |
| D. melanogaster: P{KK106424}VIE-260B | VDRC | VDRC ID: 104450 |
| D. melanogaster: P{KK112299}VIE-260B | VDRC | VDRC ID: 107537 |
| D. melanogaster: w1118; P{GD1101}v6941 | VDRC | VDRC ID: 6941 |
| Recombinant DNA | ||
| Plasmid: pCFD4-U6:1_U6:3tandemgRNAs | Addgene | #49411; RRID:Addgene_49411 |
| Plasmid: pHD-DsRed-attP | Addgene | #51019; RRID:Addgene_51019 |
| Software and Algorithms | ||
| Fiji | Fiji | https://fiji.sc/ |
| Graphpad Prism | Graphpad Software | https://www.graphpad.com/ |
METHOD DETAILS
Seizure assay
2-5 day old flies were used for seizure assays. 4-7 flies were housed in vials containing 2% agar, 5% sucrose and drug. Seizures were induced by banging flies to the bottom of the vial 3 times over the course of 2 s and vortexing for 10 s at high speed. The proportion of flies seizing was recorded. Two vials were recorded at a time and, when possible, experimental and control vials were vortexed together to control for possible variability in vortex strength.
Drugs and the corresponding ethanol volumes were mixed into melted sucrose-agar food for drug and vehicle feeding. Flies were kept on drug containing food for all days of seizure recording.
Seizure recovery time was defined as the time required for a fly to right itself after the 10 s vortex (Ganetzky and Wu, 1982). For these experiments, all seizures were video recorded, and videos were scored at another time. Recovery time was measured for each individual fly in a vial and the average recovery time/vial was used. Flies that did not seize and flies that were identified as outliers through the interquartile method were excluded from average recovery time calculations.
Generation of CG8839KO CRISPR flies
CG8839KO flies were generated using the CRISPR/Cas9 system and homology directed repair (HDR). Guide RNAs (gRNA) targeting the flanks of the CG8839 coding region were generated by https://flycrispr.org/tools and cloned into the pCFD4 plasmid. The following primers were used (gRNA sequences are underlined):
5′ targeting: TATATAGGAAAGATATCCGGGTGAACTTCGATGTCACTGGCGCACGCCCGTTTTAGAGCTAGAAATAGCAAG
3′ targeting: ATTTTAACTTGCTATTTCTAGCTCTAAAACTGGGATGCGTGGGATAGATCGACGTTAAATTGAAAATAGGTC
1 kb homology arms up and downstream of the predicted CRISPR deletion were cloned into the pHD-DsRed-attP plasmid in order to insert a DsRed marker into the deleted region of CG8839 by HDR. The following primers were used:
Homology arm 1
F: GACTCACCTGCTGACTCGCCGAATTTTGGCTGGCGTTGC
R: GACTCACCTGCTCAGCTACCGTGCGCCAGTGACATCATTTC
Homology arm 2
F: GACTGCTCTTCNTATCCACGGTGGCTCCATACCACAATG
R: GCATGCTCTTCNGACCTGCATGACCGACCACATTG
Both plasmids were injected into vasa-cas9 flies by Rainbow Transgenic Flies. F0 male flies were crossed to a balancer line and F1, DsRed positive males were selected. CG8839 KO and HDR were confirmed by PCR. A control line was also established from F1, DsRed negative flies (CRISPR control). The following primers were used:
Correct insertion of DsRed
CG8839 F: GCCGCTCTAAAGTGTAGCTG
DsRed R: TTGGTCACCTTCAGCTTGG
Coding region of CG8839
CG8839 F: GCCGCTCTAAAGTGTAGCTG
CG8839 R: GACTGCTCCTAGAGCCGAAC
FAAH activity assay
2-10 day old eas;CG8839KO and age matched eas flies in groups of 50-80 (pooled male and female flies) were homogenized in ice cold 50 mM Tris-HCl, 1mM EDTA (pH 7.4) with a glass Dounce homogenizer. The homogenate was then centrifuged at 10,000 g for 10 min and the supernatant was diluted in 50 mM Tris-HCl, 1 mM EDTA, 0.1% BSA (pH 7.4). Protein concentration was measured using a BCA protein assay kit (Abcam) following the manufacturer’s protocol.
For URB597 experiments, 50 μL of diluted lysate from eas flies (≈ 0.5-5 μg protein/well) was dispensed into 8 wells/sample of a 96 well plate. 1 μL of escalating doses of URB597 were dispensed into each of the wells and allowed to incubate for 5 min. 50 μL of AMC-AA substrate diluted in 50 mMTris-HCl, 1 mM EDTA, 0.1% BSA (pH 7.4) was then dispensed into each well at a final concentration of 15 uM. Fluorescence was immediately measured at excitation/emission: 360/460 using a Victor 3V (Perkin Elmer) plate reader in continuous measurement mode. Fluorescence was plotted against time and slopes were fit over the linear portion of the graphs for each sample. AMC production rates for each URB597 dose was normalized to the rate of the vehicle treated well. A similar protocol was used for eas versus eas;CG8839KO and eas versus eas + vortex experiments.
Immunofluorescence microscopy
2-5 day old nsyb-gal4>UAS-CaLexA and eas;;nsyb-gal4 > UAS-CaLexA flies were maintained on either vehicle or 20 μg/mL AEA for 4 days. Flies were vortexed for 10 s, 90 min prior to brain dissection for vortexing experiments. Brains were dissected in ice cold PBS with 0.1% Triton-X (PBST) and fixed in 4% paraformaldehyde for 20 min. Brains were then rinsed 3 × 10 min in PBST, blocked with 5% normal goat serum for 30 min, and incubated in 1:1000 rabbit anti-GFP overnight. Brains were then rinsed 3 × 10 min in PBST, incubated in 1:200 Alexa Fluor 488 goat anti-rabbit for 2 h, and rinsed 3 × 10 min in PBST. Brains were then mounted with Vectashield.
Brains were imaged on a Leica SP5 confocal microscope with a 40x oil immersion objective. 40 image slices at 2 μm resolution were captured for each brain.
ImageJ was used for image analysis. A constant minimum threshold was used for all brains to exclude background signal. Mean GFP was measured for each slice and then summed for each brain. The summed mean GFP for each brain was normalized to either nsyb-gal4 > CaLexA + vehicle or eas;;nsyb-gal4 > CaLexA + vehicle + no vortex for vortexing experiments. Outliers were identified by the interquartile method and excluded. MB GFP was quantified by drawing a MB ROI, and measuring mean GFP in that ROI over the summed slices of each brain. The same ROI was used for all brains. Mean MB GFP was then normalized to either nsyb-gal4 > CaLexA + vehicle or eas;;nsyb-gal4 > CaLexA + vehicle + no vortex for vortexing experiments, and divided by the normalized overall brain GFP for each brain. Representative images were created in ImageJ by Z-projecting all slices in the brain.
Quantitative PCR
RNA was extracted from 5-10 whole flies per genotype with TRIzol and reverse transcribed to cDNA with High-Capacity cDNA Reverse Transcription Kit. qPCR was conducted with SYBR Green PCR Master Mix on a ViiA7 Real Time PCR machine. Relative gene expression was calculated by the △△Ct method and normalized to actin. Relative wtrw expression in eas;;’wtrwex flies was normalized to age matched eas controls. The following primers were used.
Wtrw
F: GCTATAAGGAGGGCAGCACC
R: CCATCAAGTTGGGTGGAATCG
Actin
F: GCGCGGTTACTCTTTCACCA
R: ATGTCACGGACGATTTCACG
Blue dye feeding assay
Blue dye assays were conducted generally following the protocol described by Deshpande et al. (2014). 2% (w/v) FD&C Blue no. 1 was mixed into food containing 2 μg/mL AEA and vehicle. Flies in groups of 7-8 were starved for 16 h and then flipped onto blue dye food. Flies were anesthetized with CO2, and the proportion of flies with blue dye visible in their stomachs was recorded. Flies were then homogenized in PBST (0.1% Triton-X) and homogenate absorbance was measured at 620 nm. Volume consumed was calculated using a standard curve.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments can be found in figure legends. All statistical tests were conducted with Graphpad Prism. Unpaired t tests were used for statistical comparisons between two groups. ANOVA with Tukey’s multiple comparisons tests were used for statistical comparisons between more than two groups. ANOVA with Dunnett’s multiple comparisons tests were used for statistical comparisons between more than two groups and one control group. Multiple t tests with a Holm-Sidak correction were used for statistical comparisons of two groups repeated over consecutive days. A two-way ANOVA with Dunnett’s multiple comparisons test was used for comparing different AEA doses to vehicle in Figure 1A. A one sample t test comparing against a hypothetical mean of 1 (100% seizing) was used for the TRP RNAi screen in Figure 3B. ANOVA with Sidak’s multiple comparisons tests were used for statistical comparisons between selected groups in Figure S2.
Supplementary Material
Highlights.
Anandamide and 2-arachidonoylglycerol are protective in several fly seizure models
Metabolites are responsible for seizure protection by anandamide
Seizure protection by anandamide is mediated by the TRP channel Water witch
ACKNOWLEDGMENTS
We thank Dr. Soumita Ghosh and Dr. Tilo Grosser for helping us measure eCBs in our fly samples. This work is supported by the National Institutes of Health (NIH) (T32 HL07953) and the Howard Hughes Medical Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107710.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, and Patapoutian A (2004). Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41, 849–857. [DOI] [PubMed] [Google Scholar]
- Bazán NG Jr. (1970). Effects of ischemia and electroconvulsive shock on free fatty acid pool in the brain. Biochim. Biophys. Acta 218, 1–10. [DOI] [PubMed] [Google Scholar]
- Bhaskaran MD, and Smith BN (2010). Effects of TRPV1 activation on synaptic excitation in the dentate gyrus of a mouse model of temporal lobe epilepsy. Exp. Neurol 223, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, and Gereau RW IV. (2002). cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 35, 721–731. [DOI] [PubMed] [Google Scholar]
- Carta M, Lanore F, Rebola N, Szabo Z, Da Silva SV, Lourenço J, Verraes A, Nadler A, Schultz C, Blanchet C, and Mulle C (2014). Membrane lipids tune synaptic transmission by direct modulation of presynaptic potassium channels. Neuron 81, 787–799. [DOI] [PubMed] [Google Scholar]
- Chen CY, Li W, Qu KP, and Chen CR (2013). Piperine exertsanti-seizure effects via the TRPV1 receptor in mice. Eur. J. Pharmacol 714, 288–294. [DOI] [PubMed] [Google Scholar]
- Clement AB, Hawkins EG, Lichtman AH, and Cravatt BF (2003). Increased seizure susceptibility and proconvulsant activity of anandamide in mice lacking fatty acid amide hydrolase. J. Neurosci 23, 3916–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colangeli R, Pierucci M, Benigno A, Campiani G, Butini S, and Di Giovanni G (2017). The FAAH inhibitor URB597 suppresses hippocampal maximal dentate afterdischarges and restores seizure-induced impairment of short and long-term synaptic plasticity. Sci. Rep 7, 11152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, and Di Marzo V (2009). Role of endocannabinoids and endovanilloids in Ca2+ signalling. Cell Calcium 45, 611–624. [DOI] [PubMed] [Google Scholar]
- Deshpande LS, Blair RE, Ziobro JM, Sombati S, Martin BR, and DeLorenzo RJ (2007). Endocannabinoids block status epilepticus in cultured hippocampal neurons. Eur. J. Pharmacol 558, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande SA, Carvalho GB, Amador A, Phillips AM, Hoxha S, Lizotte KJ, and Ja WW (2014). Quantifying Drosophila food intake: comparative analysis of current methodology. Nat. Methods 11, 535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima A, Mamada K, Iimura A, and Ono H (2017). Supraspinal-selective TRPV1 desensitization induced by intracerebroventricular treatment with resiniferatoxin. Sci. Rep 7, 12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganetzky B, and Wu CF (1982). Indirect Suppression Involving Behavioral Mutants with Altered Nerve Excitability in DROSOPHILA MELANOGASTER. Genetics 100, 597–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisenberg M (2003). Mushroom body memoir: from maps to models. Nat. Rev. Neurosci 4, 266–275. [DOI] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Mercado A, Fajilan AA, Lee AW, Hsu R, Mount DB, and Tanouye MA (2010). Seizure sensitivity is ameliorated by targeted expression of K+-Cl- cotransporter function in the mushroom body of the Drosophila brain. Genetics 184, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill TDM, Cascio MG, Romano B, Duncan M, Pertwee RG, Williams CM, Whalley BJ, and Hill AJ (2013). Cannabidivarin-rich cannabis extracts are anticonvulsant in mouse and rat via a CB1 receptor-independent mechanism. Br. J. Pharmacol 170, 679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizenga MN, Sepulveda-Rodriguez A, and Forcelli PA (2019). Preclinical safety and efficacy of cannabidivarin for early life seizures. Neuropharmacology 148, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannotti FA, Hill CL, Leo A, Alhusaini A, Soubrane C, Mazzarella E, Russo E, Whalley BJ, Di Marzo V, and Stephens GJ (2014). Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: potential for the treatment of neuronal hyperexcitability. ACS Chem. Neurosci 5, 1131–1141. [DOI] [PubMed] [Google Scholar]
- Jeffries KA, Dempsey DR, Behari AL, Anderson RL, and Merkler DJ (2014). Drosophila melanogaster as a model system to study long-chain fatty acid amide metabolism. FEBS Lett. 588, 1596–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen I, Blondeau N, Heurteaux C, Widmann C, Romey G, and Lazdunski M (2000). Polyunsaturated fatty acids are potent neuroprotectors. EMBOJ. 19, 1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludányi A, Eross L, Czirják S, Vajda J, Halász P, Watanabe M, Palkovits M, Maglóczky Z, Freund TF, and Katona I (2008). Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J. Neurosci 28, 2976–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz B (2004). On-demand activation of the endocannabinoid system in the control of neuronal excitability and epileptic form seizures. Biochem. Pharmacol 68, 1691–1698. [DOI] [PubMed] [Google Scholar]
- Ma Z, Stork T, Bergles DE, and Freeman MR (2016). Neuromodulators signal through astrocytes to alter neural circuit activity and behaviour. Nature 539, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna SSS, and Umathe SN (2012). Involvement of transient receptor potential vanilloid type 1 channels in the pro-convulsant effect of anandamide in pentylenetetrazole-induced seizures. Epilepsy Res. 100, 113–124. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Ortega-Gutiérrez S, Van der Stelt M, et al. (2003). CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302, 84–88. [DOI] [PubMed] [Google Scholar]
- Masuyama K, Rao Y, Zhang Y, and Wang JW (2012). Mapping neural circuits with activity-dependent nuclear import of a transcription factor. J. Neurogenet 26, 89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaraughty S, Chu KL, Bitner RS, Martino B, El Kouhen R, Han P, Nikkel AL, Burgard EC, Faltynek CR, and Jarvis MF (2003). Capsaicin infused into the PAG affects rat tail flick responses to noxious heat and alters neuronal firing in the RVM. J. Neurophysiol 90, 2702–2710. [DOI] [PubMed] [Google Scholar]
- McIntyre P, McLatchie LM, Chambers A, Phillips E, Clarke M, Savidge J, Toms C, Peacock M, Shah K, Winter J, et al. (2001). Pharmacological differences between the human and rat vanilloid receptor 1 (VR1). Br. J. Pharmacol 132, 1084–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPartland J, Di Marzo V, De Petrocellis L, Mercer A, and Glass M (2001). Cannabinoid receptors are absent in insects. J. Comp. Neurol 436, 423–429. [DOI] [PubMed] [Google Scholar]
- McPartland JM, Matias I, Di Marzo V, and Glass M (2006). Evolutionary origins of the endocannabinoid system. Gene 370, 64–74. [DOI] [PubMed] [Google Scholar]
- Mitchell K, Lebovitz EE, Keller JM, Mannes AJ, Nemenov MI, and ladarola MJ (2014). Nociception and inflammatory hyperalgesia evaluated in rodents using infrared laser stimulation after Trpv1 gene knockout or resiniferatoxin lesion. Pain 155, 733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertová M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, et al. (2006). The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron 51, 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motter AL, and Ahern GP (2012). TRPA1 is a polyunsaturated fatty acid sensor in mammals. PLoS One 7, e38439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller C, Morales P, and Reggio PH (2019). Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci 11, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naderi N, Aziz Ahari F, Shafaghi B, Najarkolaei AH, and Motamedi F (2008). Evaluation of interactions between cannabinoid compounds and diazepam in electroshock-induced seizure model in mice. J. Neural Transm. (Vienna) 115, 1501–1511. [DOI] [PubMed] [Google Scholar]
- Naidoo V, Nikas SP, Karanian DA, Hwang J, Zhao J, Wood JT, Alapafuja SO, Vadivel SK, Butler D, Makriyannis A, and Bahr BA (2011). A new generation fatty acid amide hydrolase inhibitor protects against kainate-induced excitotoxicity. J. Mol. Neurosci 43, 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naydenov AV, Horne EA, Cheah CS, Swinney K, Hsu KL, Cao JK, Marrs W, Blankman JL, Tu S, Cherry AE, et al. (2014). ABHD6 blockade exerts antiepileptic activity in PTZ-induced seizures and in spontaneous seizures in R6/2 mice. Neuron 83, 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan SE (2016). An update on PPAR activation by cannabinoids. Br. J. Pharmacol 173, 1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo E, de Novellis V, Marabese I, Cuomo D, Rossi F, Berrino L, Rossi F, and Maione S (2002). Interaction between vanilloid and glutamate receptors in the central modulation of nociception. Eur. J. Pharmacol 439, 69–75. [DOI] [PubMed] [Google Scholar]
- Parker L, Padilla M, Du Y, Dong K, and Tanouye MA (2011). Drosophila as a model for epilepsy: bss is a gain-of-function mutation in the para sodium channel gene that leads to seizures. Genetics 187, 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Ramaswami M, and Tanouye MA (1994). The Drosophila easily shocked gene: a mutation in a phospholipid synthetic pathway causes seizure, neuronal failure, and paralysis. Cell 79, 23–33. [DOI] [PubMed] [Google Scholar]
- Ramarao MK, Murphy EA, Shen MWH, Wang Y, Bushell KN, Huang N, Pan N, Williams C, and Clark JD (2005). A fluorescence-based assay for fatty acid amide hydrolase compatible with high-throughput screening. Anal. Biochem 343, 143–151. [DOI] [PubMed] [Google Scholar]
- Redmond WJ, Gu L, Camo M, McIntyre P, and Connor M (2014). Ligand determinants of fatty acid activation of the pronociceptive ion channel TRPA1. PeerJ 2, e248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royden CS, Pirrotta V, and Jan LY (1987). The tko locus, site of a behavioral mutation in D. melanogaster, codes for a protein homologous to prokaryotic ribosomal protein S12. Cell 51, 165–173. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J, Elebring T, Nilsson K, Drmota T, and Greasley PJ (2007). The orphan receptorGPR55 is a novel cannabinoid receptor. Br. J. Pharmacol 152,1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Salvador L, Andrés-Borderia A, Ferrer-Montiel A, and Planells-Cases R (2012). Agonist- and Ca2+-dependent desensitization of TRPV1 channel targets the receptor to lysosomes for degradation. J. Biol. Chem 287, 19462–19471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saras A, Wu VV, Brawer HJ, and Tanouye MA (2017). Investigation of seizure-susceptibility in a Drosophila melanogaster model of human epilepsy with optogenetic stimulation. Genetics 206, 1739–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen LR, Lai CQ, Feng X, Parnell LD, Wan JB, Wang JD, Li D, Ordovas JM, and Kang JX (2010). Drosophila lacks C20 and C22 PUFAs. J. Lipid Res 51, 2985–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubina L, Aliev R, and Kitchigina V (2015). Attenuation of kainic acid-induced status epilepticus by inhibition of endocannabinoid transport and degradation in guinea pigs. Epilepsy Res. 111, 33–44. [DOI] [PubMed] [Google Scholar]
- Sigel E, Baur R, Rácz I, Marazzi J, Smart TG, Zimmer A, and Gertsch J (2011). The major central endocannabinoid directly acts at GABA(A) receptors. Proc. Natl. Acad. Sci. USA 108, 18150–18155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, and Tanouye MA (2008). From bench to drug: human seizure modeling using Drosophila. Prog. Neurobiol 84, 182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugaya Y, Yamazaki M, Uchigashima M, Kobayashi K, Watanabe M, Sakimura K, and Kano M (2016). Crucial Roles of the Endocannabinoid 2-Arachidonoylglycerol in the Suppression of Epileptic Seizures. Cell Rep. 16, 1405–1415. [DOI] [PubMed] [Google Scholar]
- Tortoriello G, Rhodes BP, Takacs SM, Stuart JM, Basnet A, Raboune S, Widlanski TS, Doherty P, Harkany T, and Bradshaw HB (2013). Targeted lipidomics in Drosophila melanogaster identifies novel 2-monoacylglycerols and N-acyl amides. PLoS One 8, e67865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilela LR, Gobira PH, Viana TG, Medeiros DC, Ferreira-Vieira TH, Doria JG, Rodrigues F, Aguiar DC, Pereira GS, Massessini AR, et al. (2015). Enhancement of endocannabinoid signaling protects against cocaine-induced neurotoxicity. Toxicol. Appl. Pharmacol 286, 178–187. [DOI] [PubMed] [Google Scholar]
- Vilela LR, Medeiros DC, Rezende GHS, de Oliveira ACP, Moraes MFD, and Moreira FA (2013). Effects of cannabinoids and endocannabinoid hydrolysis inhibition on pentylenetetrazole-induced seizure and electroencephalographic activity in rats. Epilepsy Res. 104, 195–202. [DOI] [PubMed] [Google Scholar]
- Walker KM, Urban L, Medhurst SJ, Patel S, Panesar M, Fox AJ, and McIntyre P (2003). The VR1 antagonist capsazepine reverses mechanical hyperalgesia in models of inflammatory and neuropathic pain. J. Pharmacol. Exp. Ther 304, 56–62. [DOI] [PubMed] [Google Scholar]
- Wallace MJ, Blair RE, Falenski KW, Martin BR, and DeLorenzo RJ (2003). The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. J. Pharmacol. Exp. Ther 307, 129–137. [DOI] [PubMed] [Google Scholar]
- Wallace MJ, Martin BR, and DeLorenzo RJ (2002). Evidence for a physiological role of endocannabinoids in the modulation of seizure threshold and severity. Eur. J. Pharmacol 452, 295–301. [DOI] [PubMed] [Google Scholar]
- Wallace MJ, Wiley JL, Martin BR, and DeLorenzo RJ (2001). Assessment of the role of CB1 receptors in cannabinoid anticonvulsant effects. Eur. J. Pharmacol 428, 51–57. [DOI] [PubMed] [Google Scholar]
- Yoshioka T, Inoue H, Kasama T, Seyama Y, Nakashima S, Nozawa Y, and Hotta Y (1985). Evidence that arachidonic acid is deficient in phosphatidylinositol of Drosophila heads. J. Biochem 98, 657–662. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V, Julius D, and Högestätt ED (1999). Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400, 452–457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data have been deposited to Mendeley Data: http://dx.doi.org/10.17632/vmhkt8k9p5.1