

SUMMARY
A rich diversity of transmembrane G protein-coupled receptors (GPCRs) are used by eukaryotes to sense physical and chemical signals. In humans alone, 800 GPCRs comprise the largest and most therapeutically targeted receptor class. Recent advances in GPCR structural biology have produced hundreds of GPCR structures solved by X-ray diffraction and increasingly, cryo-electron microscopy (cryo-EM). Many of these structures are stabilized by site-specific cholesterol binding, but it is unclear whether these interactions are a product of recurring cholesterol-binding motifs and if observed patterns of cholesterol binding differ by experimental technique. Here, we comprehensively analyze the location and composition of cholesterol binding sites in the current set of 473 human GPCR structural chains. Our findings establish that cholesterol binds similarly in cryo-EM and X-ray structures and show that 92% of cholesterol molecules on GPCR surfaces reside in predictable locations that lack discernable cholesterol-binding motifs.
In Brief
Cholesterol (CLR) regulates G protein-coupled receptor (GPCR) function, but it is unclear if this is due to binding conserved CLR motifs. Taghon et al. collect, validate, and cluster all GPCR-bound CLR in the PDB and show that 92% reside in 12 distinct GPCR regions that lack consensus CLR-binding sites.
Graphical Abstract

INTRODUCTION
G protein-coupled receptors (GPCRs) comprise a superfamily of 7-transmembrane receptors that detect and transduce diverse physical and chemical signals across the cell membrane. In humans, more than 800 GPCRs activate four subtypes of Gα proteins to control the production of cytosolic second messengers, such as cyclic adenosine monophosphate (Gi/o and Gs subtypes) and Ca2+ (Gq subtypes), as well as the activation of Rho small GTPases (G12/13 subtypes) (Sutkeviciute and Vilardaga, 2020). GPCR signaling responses can also be transduced by the Gβγ heterodimer, or diverted from Gα-driven responses via biased signaling through arrestin-mediated signaling pathways (Shukla and Dwivedi-Agnihotri, 2020; Smrcka and Fisher, 2019). Given their numerous physiologic and regulatory functions, over 130 GPCRs are also therapeutically targeted by more than one-third of approved drugs (Sriram and Insel, 2018).
As with all transmembrane proteins, GPCR structure and function are influenced by the composition of the membrane bilayer. In humans, membranes are composed mainly of glycerophospholipids and cholesterol (CLR), the latter of which is known to bind specific sites in GPCRs to regulate conformation, stability, and function (Duncan et al., 2020; Genheden et al., 2017). For example, CLR is a known stabilizer of the β2 adrenergic receptor (ADRB2) (Yao and Kobilka, 2005) and allosteric modulator of GPCRs from a diversity of subfamilies, such as the cholecystokinin A (CCK1), cannabinoid CB1 (CNR1), μ-opioid (OPRM1), and oxytocin (OXTR) receptors (Muth et al., 2011; Oddi et al., 2011; Potter et al., 2012; Qiu et al., 2011). The first structural evidence for site-specific CLR binding in GPCRs was provided by early X-ray models of ADRB2 and adenosine A2A receptor (ADORA2A), in which CLR was observed bound in distinct transmembrane locations (Cherezov et al., 2007; Lovera et al., 2019). These initial observations gave rise to the concept of CLR-binding motifs and to the expectation that such motifs pervade GPCR surfaces.
The first site-specific CLR-binding motif was identified in ADRB2 and named the cholesterol consensus motif (CCM). This CCM comprises four side chains distant in primary sequence (W158, I154, R151, and Y70) that originate from an inner leaflet surface concavity formed by transmembrane helices 2, 3, and 4 (Hanson et al., 2008). Multiple sequence alignments suggest that this CCM extends far beyond ADRB2 to include as many as 44% of human GPCRs (Hanson et al., 2008). In addition to CCMs, many GPCRs contain putative Cholesterol Recognition Amino Acid Consensus (CRAC) motifs identifiable as contiguous residue sequences localized to single transmembrane helices (Fantini et al., 2016; Kiriakidi et al., 2019). These motifs are proposed to bind CLR longitudinally from tail to hydroxyl head (CRAC: L/V–(X)1–5–Y/F–(X)1–5–R/K), or in reverse orientation as “CARC” motifs (CARC: R/K–(X)1–5–Y/F–(X)1–5–L/V) to stabilize GPCRs in the bilayer. Although site-specific CLR binding in GPCRs is routinely attributed to both CCMs and CRAC/CARC motifs (Di Scala et al., 2017; Fantini et al., 2016; Jafurulla et al., 2011; Jones et al., 2020), such assertions have yet to be systematically assessed and validated. As such, it remains to be established whether CLR binding in GPCRs can be explained by recurring CLR occupancy at distinct, readily identifiable side chain motifs.
Much progress has been made in GPCR structural biology since the first CLR-binding sites were identified and nascent CLR-binding motifs were defined (Garcia-Nafria and Tate, 2020). There are now hundreds of GPCR structures solved by both X-ray diffraction and increasingly cryo-electron microscopy (cryo-EM). Given this volume of information, and the differences intrinsic to each structural method, we sought to reassess CLR binding patterns in GPCRs and to interrogate the notion that CLR binds to predictive, well-defined structural motifs on GPCR surfaces. Here, we provide this insight using our structural informatics program known as pHinder (Isom and Dohlman, 2015; Isom et al., 2013, 2016) to evaluate CLR binding patterns in the current set of 473 human GPCR structures available in the Protein DataBank (PDB). Our calculations show that there is excellent agreement between the locations of CLR-binding sites modeled in X-ray and cryo-EM structures, and that 92% of CLRs map to 12 distinct Cholesterol Network Clusters (CNCs) on GPCR surfaces. Furthermore, by triangulating, extracting, and clustering the interaction shells of hundreds of aligned CLRs, we show that CLR binding to GPCRs cannot be explained by recurring, generalizable motifs. Last, we report a cross-species analysis showing that most human GPCRs capable of functioning in CLR-free yeast lack bound CLR in their structural models, suggesting there may be CLR-dependent and -independent classes of human GPCRs.
RESULTS
Rapid growth of GPCR structures solved by cryo-electron microscopy
As of November 2020, there were 473 human GPCR structural chains representing 81 unique receptors deposited in the PDB (Table S1). Although the majority of these structures have been solved by X-ray diffraction (81%) (Figure 1A), rapid advances in cryo-EM have led to a recent surge in GPCR structures. In only 3 years, the share of cryo-EM structures has grown to 18% (Figure 1A). This shift reflects several methodological advantages, including the avoidance of crystal growth and optimization, fewer restrictions on detergents and additives, and the ability to assemble high-resolution structures from smaller amounts of purified protein (Garcia-Nafria and Tate, 2020). We used this structural information to assess lipid-binding consistency between GPCR structures solved by X-ray diffraction of ordered crystal lattices and single-particle cryo-EM reconstructions.
Figure 1. Structural informatics of lipids bound to human GPCR surfaces.
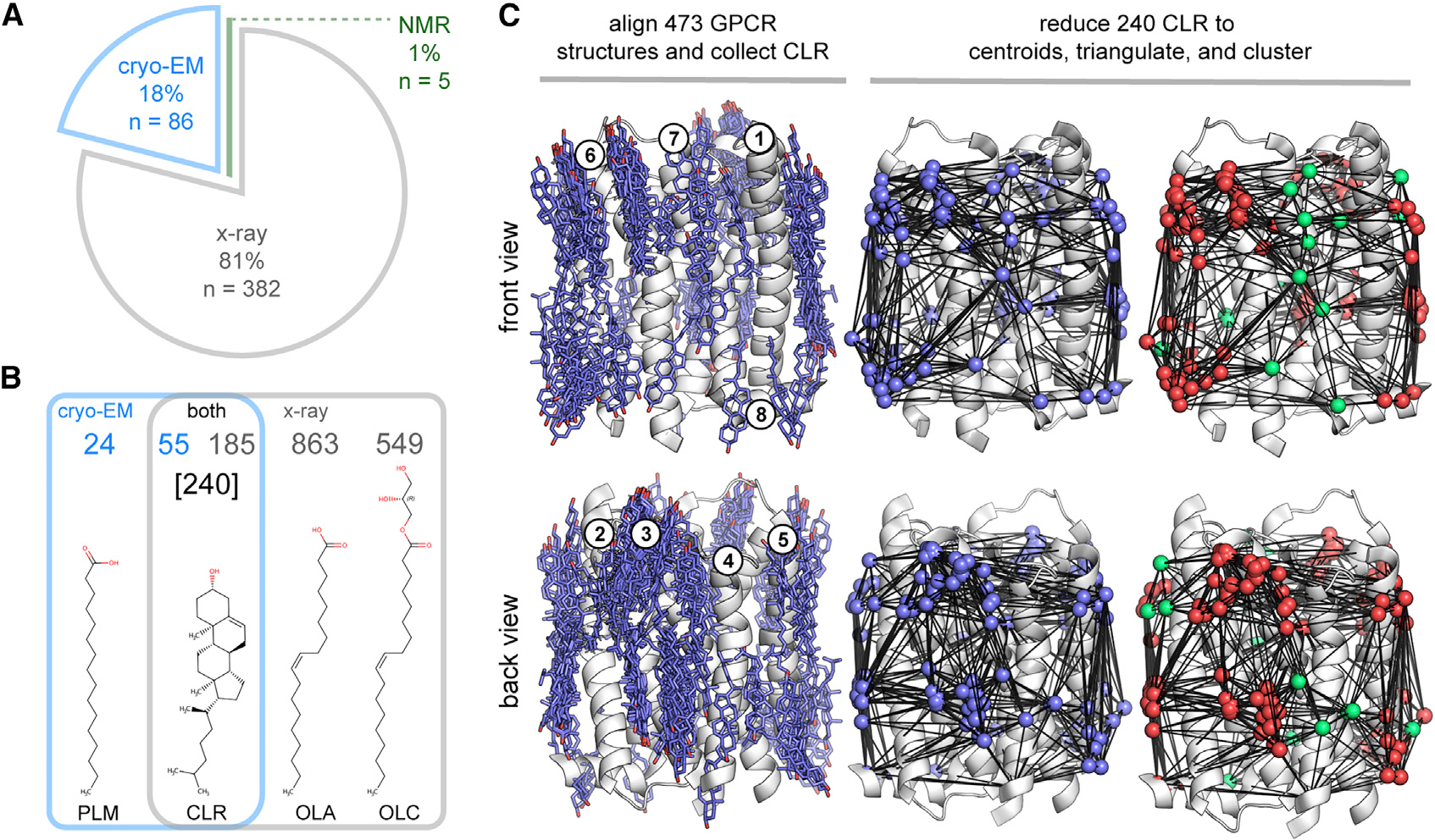
(A–C) Breakdown of the current set of 473 human GPCR structural chains by method (A) and their most prevalent bound lipids (B). Overview of the pHinder algorithm for calculating CNCs (C). First, 240 surface-bound CLRs were collected from 473 GPCR structures aligned to the reference receptor bovine rhodopsin (RHO, PDB code 1F88, chain A) (C, left). The set of CLRs were then reduced to geometric centroids, triangulated, and clustered (C, right). The full set of 240 CLR centroids is colored purple. The subset of CLR centroids clustered in CNCs (221 of 240 nodes) are colored red, and the subset of CLR centroids that did not cluster to CNCs (19 of 240 nodes) are colored green. Additional details regarding the set of 473 human GPCR structures, CLR collection procedure, and CAN algorithm are available in STAR methods and Table S1.
Lipids and cholesterol are the most prevalent molecules observed in GPCR structures
As shown in Figure 1B, lipids, including CLR, are the most frequently observed molecules bound to GPCR surfaces in both X-ray and cryo-EM structures. Our analysis shows that oleic acid (OLA), (R)-1-monoolein (OLC), and CLR are the most prevalent heteroatom molecules in the subset of 382 GPCR X-ray structures (863, 549, and 185 instances, respectively), and that palmitate (PLM) and CLR prevail in the subset of 86 cryo-EM structures (24 and 55 instances, respectively). As shown in Figure 1B, OLA and OLC lipids are exclusive to X-ray structures, and PLM is predominantly found in cryo-EM GPCR structures. In contrast, CLR is found in a significant proportion of both X-ray and cryo-EM GPCR structures. These observed CLRs could correspond to molecules that originate from the membranes of host expression systems, but are more likely to be exogenously added CLR or the soluble GPCR-stabilizing additive cholesterol hemisuccinate (CHS) (Kulig et al., 2014), which is often modeled as CLR in PDB files. Given that surface-bound CLRs were common to both X-ray and cryo-EM GPCR structures, we next sought to determine the similarity between the collective CLR binding patterns observed by each technique
GPCR-cholesterol interactions map to 12 CNCs
As illustrated in Figure 1C, CLR collected from the combined set of 473 X-ray and cryo-EM human GPCR structures are observed over most of the transmembrane receptor surface. However, the density of these CLR molecules is not evenly distributed, indicating that there are preferred CLR-binding sites. To identify regions of highest CLR density, we used Consensus Network Analysis (CNA) (Isom and Dohlman, 2015; Isom et al., 2016), an approach we developed for quantifying the spatial conservation of amino acid side chains and heteroatom molecules across large sets of aligned protein structures. As shown in Figure 1C, the set of CLR molecules is reduced to geometric centroids, triangulated, and clustered by the CNA algorithm. Clustering identified 12 distinct CNCs that accounted for 92% (221 of 240) of CLR observed on 24 unique GPCRs (Figure 2A). Although some well-studied receptors, such as ADORA2A and ADRB2, contributed many redundant structural chains, all CNCs were composed of CLRs from at least two unique receptors. Remarkably, only 19 CLR centroids from 4 GPCRs (ADRA2C, CCR9, P2RY12, SMO) did not cluster to a CNC, some of which are visible in Figure 1C as triangulated nodes that are colored green. Of the 12 CNCs, only two were exclusive to cryo-EM structures (CNC56o and CNC18i) and one was limited to X-ray structures (CNC1i). Apart from a noticeable CNC void between transmembrane helices 1 and 7 (Figure 2, front view), CLR sampled a variety of CNC locations split between the inner (4 CNCs) and outer (5 CNCs) bilayer leaflets. Based on these observations, we concluded that a wide variety of GPCRs repeatedly bind CLR in the same specific locations in both X-ray and cryo-EM structures.
Figure 2. Cholesterol Network Clusters in human GPCRs.
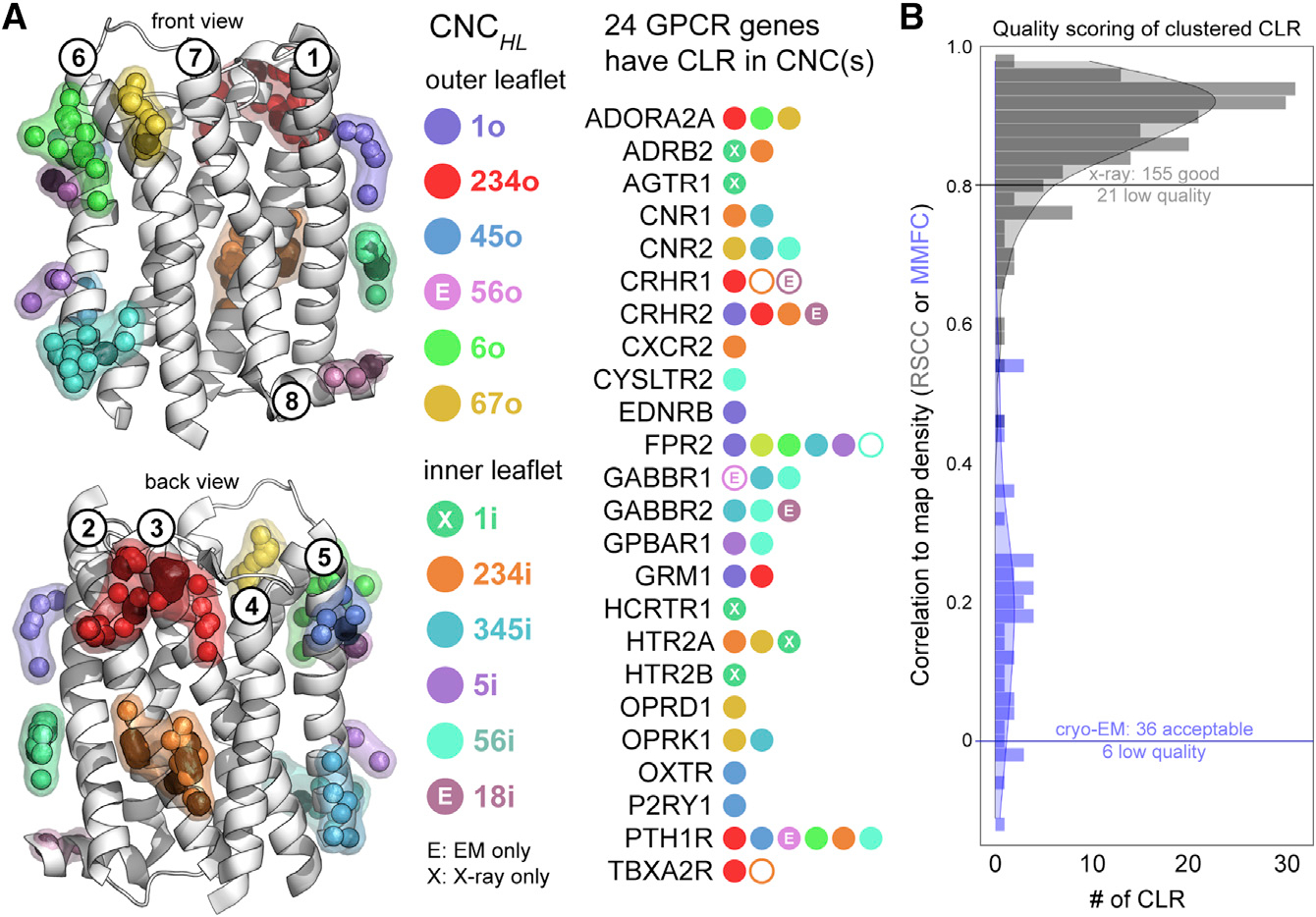
(A and B) Two hundred and twenty-one of 240 (92%) CLR molecules in human GPCR structures map to one of 12 spatially distinct CNCs. Of the 81 unique GPCR genes that contribute the set of 473 GPCR structures, 24 have CLR(s) located in CNCs described using the nomenclature CNCHL, where the H subscript corresponds to the transmembrane helix or helices contacted by the CNC, and the L subscript indicates whether the CNC is located in the inner or outer leaflet of the membrane bilayer. CNCs labeled with an X or E correspond to CLR clusters observed exclusively in X-ray or cryo-EM structures, respectively (A). RSCC (gray) and MMFC (blue) scoring for the set of 221 CLR that mapped to CNCs (B). Low-quality CLRs that mapped to CNCs are indicated as black centroids (A, left) and empty circles in the list of 24 GPCR genes (A, right).
Quality of modeled CLR and implications for CNCs
Given the subjective nature of modeling ligands into X-ray and cryo-EM density maps, we assessed the quality of the 221 CLRs that mapped to CNCs using two objective structural metrics. As shown in Figure 2B, using Real Space Correlation Coefficients (RSCC) we analyzed the 179 X-ray CLRs that mapped to CNCs and identified only 21 that were low quality (RSCC <0.8) (Smart et al., 2018). In addition, using Map-in-Map Fit Coefficients (MMFC) calculated in UCSF Chimera (Goddard et al., 2007; Pintilie et al., 2010), we scored the quality of the 42 cryo-EM CLRs that mapped to CNCs and eliminated six that had MMFC values ≤0 (also Figure 2B). These 27 low-quality X-ray and cryo-EM CLRs are indicated in Figure 2A by black spheres within the CNCs. Based on these findings, we concluded that the vast majority of CLR in available GPCR structures was of sufficient quality for CNC classification.
CRAC/CARC motifs are not predictive of CLR binding in GPCRs
As shown in Figure 3, we exhaustively identified, extracted, and mapped putative CRAC/CARC motifs in the set of 473 GPCR structures. In addition, we used innovative geometric algorithms available in our pHinder informatics suite to ensure the proper orientation and surface accessibility of each potential CRAC/CARC motif. This process is summarized in Figure 3A for putative CRAC and CARC motifs in the adenosine A1 receptor (ADORA1). Our procedure identified 46 and 57 geometrically valid CRAC and CARC motifs in 164 and 186 GPCR structures, respectively (Figure 3B). Using the same CNA procedure as for CLR, the CRAC/CARC motifs were reduced to geometric centroids, triangulated, and clustered. As expected, these motifs mapped to the leading (7 CARC clusters) and trailing (6 CRAC clusters) ends of transmembrane helices. Although several CRAC/CARC clusters coincided with CNCs, we were able to document only six cases in which structural CLRs appear to engage validated CRAC (AGTR1, GABBR1, GPBAR1) and CARC (CNR2, P2RY1, PTH1R) motifs. Based on these observations, we concluded that CRAC/CARC motifs are rarely associated with experimentally observed CLR and are not predictive of site-specific CLR binding in GPCRs.
Figure 3. CRAC/CARC motifs are not predictive of cholesterol binding in human GPCRs.
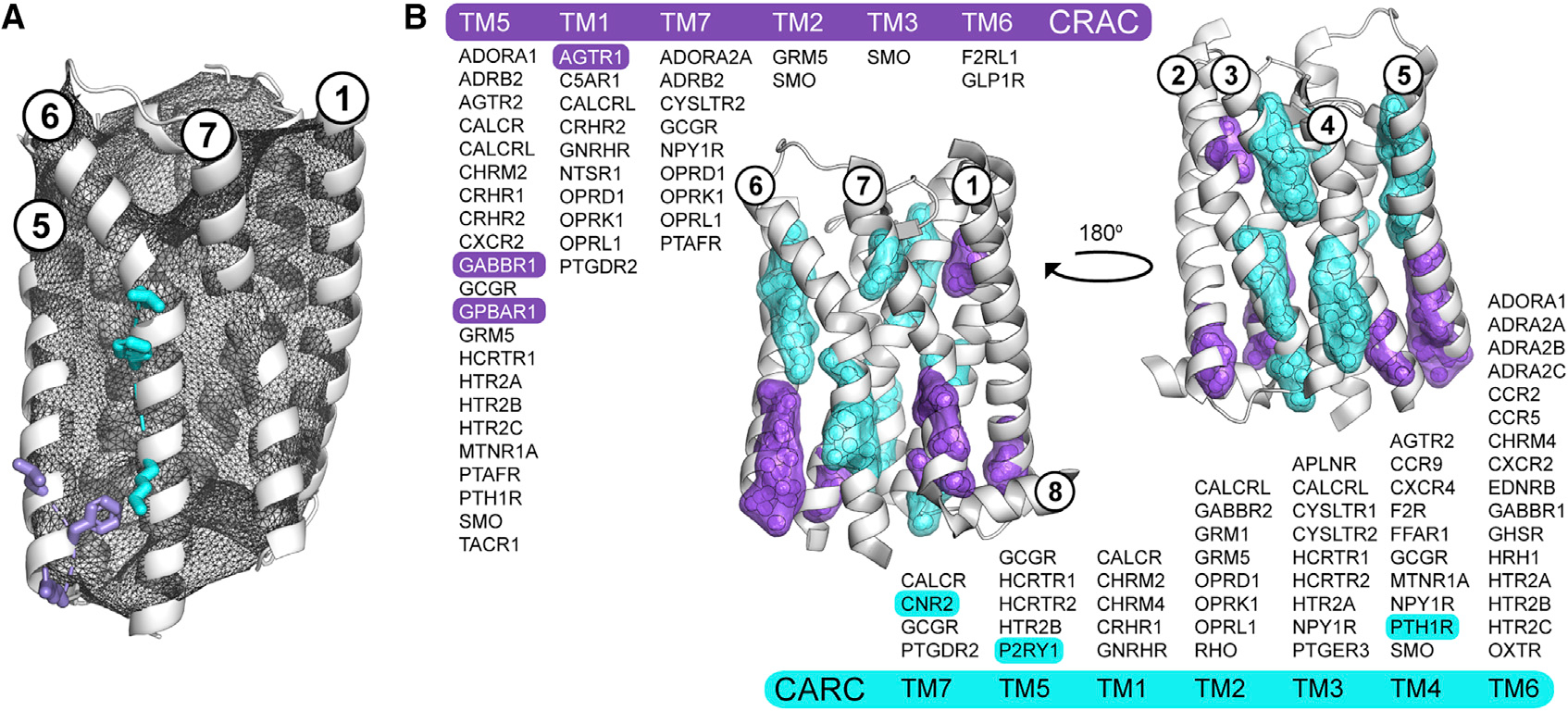
(A and B) Overview of the procedure for validating the location and directionality of CRAC/CARC motifs in GPCRs using the adenosine A1 receptor (ADORA1, PDB code 5N2S, chain A) and two of its putative CRAC (L201, F204, R208; purple) and CARC (K234, F241, L245; cyan) motifs (A). Valid CRAC/CARC motifs met two geometric criteria: the three side chains comprising the motif are located outside the pHinder surface (shown in black mesh) and oriented in the same general direction. Triangulation and clustering of the 164 and 186 putative CRAC (purple) and CARC (cyan) motifs in 473 GPCR structural chains (B). Each motif was first converted to a geometric centroid prior to triangulating and clustering with a minimum cluster size of five. The subset of GPCRs with validated CRAC/CARC motifs are listed by transmembrane helix (TM). Only six of these motifs (highlighted) had CLR observed nearby in a structure. CRAC clusters containing contributions from TM3 (SMO) and TM6 (F2RL1, GLP1R), and CARC clusters containing contributions from TM1 (CACLR, CHRM2, CHRM4, GNRHR) are not shown because they did not meet the minimum cluster size.
There is no structural evidence for site-specific cholesterol-binding motifs in GPCRs
After observing that 92% of all CLRs are found in 12 CNCs, we exhaustively analyzed the side chain microenvironments of CLR to validate (e.g., CCM) and identify new CLR-binding motifs. For this analysis, we removed the low-quality CLR identified in Figure 2B. Despite our extensive efforts and the large amount of available structural data, we could not detect recurring CCMs or new, previously unidentified, motif patterns. In our comprehensive analysis, which is summarized in Figure 4, we extracted, triangulated, and aligned the terminal side chain (TSC) atom (see STAR methods) microenvironments of the filtered set of 217 higher-quality CLRs. This process is shown for a CLR bound in the aforementioned CCM of ADRB2 (Figure 4A) (Hanson et al., 2008). As a result of this triangulation procedure, only the first geometric shell of CLR-ADRB2 contacts are extracted (Figure 4A). Structural alignment of the CLR set along with their individual TSC atom microenvironments results in a distributed point cloud that represents the TSC atoms of 2,663 residues comprising all CLR-binding sites in human GPCRs (Figure 4B). After removing contributions from highly redundant GPCR structures, such as ADORA2A and ADRB2, we used our CNA algorithm to triangulate and cluster the resultant nonredundant subset of 435 TSC atoms. As shown in Figure 4C, only 19% (84 of 435) of nonredundant TSC atoms were found in four distinct clusters that mapped exclusively to the b face of CLR. None of the independent clusters identified a single consensus residue, and collectively, none of the clusters detected CCM or CRAC/CARC patterns. Based on these results, and on the number of CLR and GPCR structures analyzed, we concluded that it is unlikely CLR interactions with GPCRs can be explained by site-specific CLR-binding motifs.
Figure 4. Clustered CLR microenvironments in GPCRs lack distinct CLR-binding motifs.
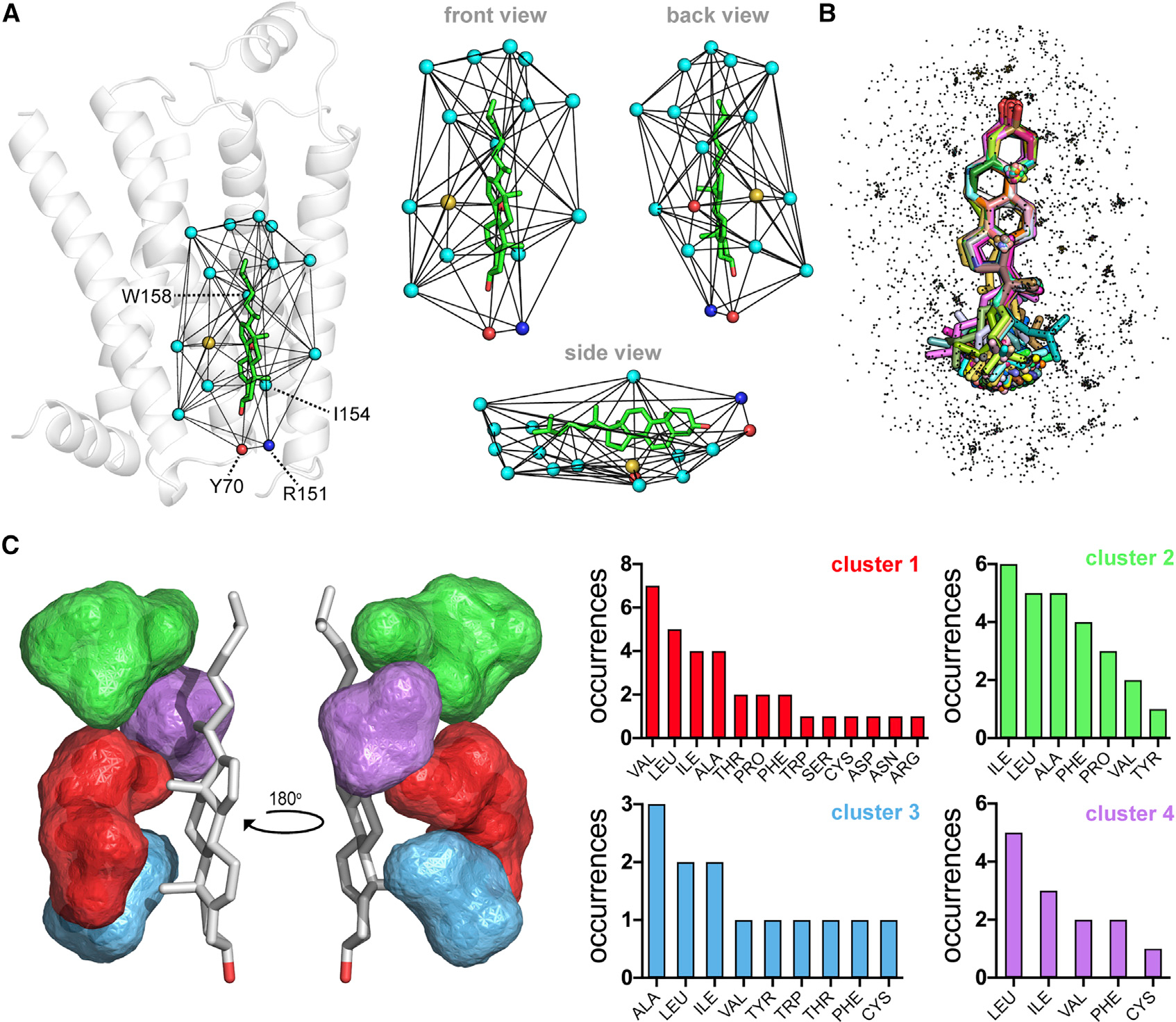
(A–C) Overview of the triangulation and extraction procedure for collecting CLR microenvironments (A). Triangulation of the CLR (CLR 402, PDB code 3D4S, chain A) used to classify the CCM, indicated by the residue labels Y70, R151, I154, and W158, in an early structure of the prototypical β2-adrenergic receptor. Spheres represent the TSC atom of each residue in the first CLR triangulation shell and are colored by atom type (cyan, carbon; yellow, sulfur; blue, nitrogen; red, oxygen). Composite overlay of 217 aligned CLR (sticks) and their TSC atom microenvironments (dots) (B). Triangulation and clustering of the combined CLR microenvironment data shown in (B), but with redundant structural information removed (C). Cluster locations and composition bar charts are matched by color. The low-quality CLR indicated in Figure 2B were removed from this analysis, reducing the number of CLRs in the set from 240 to 217 (B and C).
DISCUSSION
Cholesterol binding patterns are similar in x-ray and cryo-EM GPCR structures
Heteroatom molecules are a common feature of most high-resolution protein structural models. In structures of soluble and membrane proteins, water molecules from bulk solvent are frequently observed in recurring, site-specific locations (Orban et al., 2010; Venkatakrishnan et al., 2019). These “structural waters” typically display favorable hydrogen-bonding geometries that play a role in the local stabilization of protein structure. Similarly, structures of membrane proteins contain surface-bound lipids and sterols derived from the bulk lipid and lipid-detergent solvent phases used for sample preparation, and possibly the membranes of host expression systems. As such, the CLR entities used in our analysis originated from (1) bulk CLR in a lipidic cubic preparation, (2) the amphiphilic CLR analog and structural stabilizer CHS, which accounts for many “structural CLRs” (Kulig et al., 2014), or (3) the host expression membrane. In the latter case, structural CLRs must survive the extraction and solubilization of the target GPCR with various detergents (Miljus et al., 2020). Depending on the detergent mixture used, there is precedent for some tightly bound “cofactor” lipids, including host CLR, surviving this process (Milic and Veprintsev, 2015). However, we presume the vast majority of structural CLRs used in our analysis were derived from either bulk CLR or the CHS commonly used in both methods.
Unlike X-ray structures, which are derived from ordered crystal lattices formed in detergents or the lipidic cubic phase (Caffrey, 2015; Gacasan et al., 2017), cryo-EM structures are derived from reconstructions of many single GPCR particles (Garcia-Nafria and Tate, 2020). These fundamental differences could alter observed ligand and lipid binding patterns. However, our analysis shows that at least in the case of CLR, both X-ray and cryo-EM structures exhibit highly conserved binding patterns within CNCs. As more cryo-EM structures become available and the confidence of CLR modeling increases, we anticipate that additional CNCs could emerge, most likely in the void we identified between transmembrane helices 1 and 7. However, given the large number of structures and CLR molecules used in this analysis, we conclude that the majority of CLR CNCs in GPCRs have been elucidated by this study and have predictive value moving forward.
The concept of site-specific cholesterol-binding motifs is misleading and not generalizable
The lack of widespread site-specific CLR-binding motifs in GPCRs is best exemplified by the first proposed CCM in ADRB2. The GPCR structure, CLR, and interaction shell used to define this CCM are shown in Figure 4A. At the time of this discovery, multiple sequence alignments suggested that CCMs would be found in as many as 96 GPCRs (Hanson et al., 2008), 35 of which have known structures. However, this prediction has not come to fruition, as only one other GPCR (HTR2A) has a high-quality CLR in the CCM. Furthermore, in our cluster-based analysis, CNC234i coincides with the CCM site yet contains CLRs from four GPCRs that lack CCM residues. In light of these findings, and in agreement with the previous observation that most CCMs lack bound CLR (Gimpl, 2016), we conclude that the CCM has little predictive power and that CNCs represent generalized and predictable CLR-binding sites shared by many GPCRs. Similar to CCMs, CRAC/CARC motifs are poor indicators of site-specific CLR binding in GPCRs. Given their composition and location at the ends of transmembrane helices, CRAC/CARC motifs in GPCRs instead appear to reflect the general architecture of transmembrane helices having their polar/apolar side chains positioned outside/inside the membrane bilayer. When considering the presence of CRAC/CARC motifs in GPCRs, it is critical to consider the transmembrane accessibility and linear geometry of the branched (L/V), aromatic (Y/F), and basic (R/K) side chains that interact with the CLR tail, rings, and hydroxyl, respectively. Simply identifying a putative CRAC/CARC motif from primary sequence is not sufficient. As a consequence, several GPCRs have been reported to contain CRAC/CARC motifs that we can invalidate on closer structural and geometrical examination (Di Scala et al., 2017; Fantini and Barrantes, 2013; Fantini et al., 2016; Jafurulla et al., 2011; Oddi et al., 2011). In a few of these cases (e.g., ADORA1, CNR1, and RHO), structural information was available but not used to visually interrogate and disqualify the proposed motifs. By considering primary sequence, structure, and motif geometry, our calculations convincingly establish that CRAC/CARC motifs are rarely predictive of site-specific CLR binding in GPCRs.
Outside of putative CCM and CRAC/CARC motifs, we could not identify unequivocal site-specific CLR-binding motifs in the set of 473 human GPCRs. Despite our exhaustive effort to collect, align, triangulate, and cluster side chain interactions with CLR binding shells, there is no indication that CLR binding in GPCRs can be predicted by distinct, recurring three-dimensional arrangements of specific amino acid side chains. Rather, our clustering results suggest that CLR binds to GPCRs in readily identifiable, sequence-independent locations. While there are limits to this analysis regarding model confidence, we took steps to identify low-quality CLRs. Using RSCCs, we confirmed that most CLRs modeled in X-ray structures and clustered in CNCs were of high quality. In contrast, CLR modeling was less certain in lower-resolution cryo-EM maps, as indicated by MMFC analysis. In some cases, reliance on previous X-ray observations to guide CLR modeling could artificially enrich CNCs. However, we believe the number of high-quality CLRs used in this study and the diversity of GPCR structures independently solved by many different labs makes our analysis as comprehensive and generalizable as possible.
Evidence for CLR-dependent and CLR-independent GPCR function
While it is known that CLR is important for the function of some GPCRs (Kiriakidi et al., 2019), CLR is not observed in the majority of GPCRs with known structures (53 of 81). This could reflect differences in sample preparation and structure refinement, but also suggests that some receptors do not require interactions with CLR to be structurally stable and functional. This is consistent with our recent observation that many human GPCRs function properly in the CLR-free yeast species Saccharomyces cerevisiae (Kapolka and Isom, 2020; Kapolka et al., 2020; Rowe et al., 2020). Instead of CLR, yeasts use the sterol ergosterol to regulate the physical properties of their membranes and stabilize membrane proteins (Liu et al., 2019). Out of a set of 30 human GPCRs functional in yeast, 13 have published structures. With the exception of ADORA2A and CNR2, which contain multiple CLRs in multiple CNCs (Figure 2A), structures for the remaining 11 GPCRs lack any surface-bound CLR, even outside of CNCs: ADORA1, ADRA2A, ADRA2B, CHRM1, CHRM3, LPAR1, MTNR1A, MTNR1B, PTAFR, PTGER3, and S1PR1. While it is possible that ergosterol in yeast is able to fulfill some of the structural and functional roles of CLR, this observation suggests that for a large subset of GPCRs, CLR-specific interactions are dispensable for proper receptor function. As such, structures of GPCRs lacking CLR in CNCs may eventually prove to be predictive of receptors capable of functioning in CLR-depleted or CLR-free membranes.
Exploiting GPCR-sterol binding for therapeutic development
In most contexts, CLR is thought to act as a positive or negative allosteric modulator of GPCR function because it binds outside the orthosteric receptor binding site (Duncan et al., 2020). Such allosteric modulators are often attractive drug candidates because they can be used to regulate GPCR activity by potentiating endogenous orthosteric signaling (Dunn et al., 2019; Felder, 2019). Here we show that the vast majority of CLRs in GPCRs are found in 12 CNCs located within spatially distinct allosteric binding pockets. Given the diversity of CNC residue composition across receptor space, these locations might serve as targetable sites for receptor-specific sterol-based therapeutics and pharmacological tools for studying the allosteric modulation of receptors in vivo. Furthermore, the four-ring sterol scaffold of CLR is highly amenable to chemistry, with there being over 1,200 natural sterol-derived molecules produced in humans alone (Wishart et al., 2018). Using medicinal chemistry to expand the sterol scaffold further into chemical space could usher in a new generation of allosteric modulators that target CNCs.
The future predictability of CLR binding to GPCRs
In summary, we have shown that the overwhelming majority of cholesterol molecules in GPCRs are found in 12 distinct CLR network clusters, and that these CNCs lack site-specific motifs. Based on this finding, it is now possible to predict that more than 90% of CLRs in future GPCR structures will be found in CNCs. While we anticipate the possibility of identifying additional CNCs, especially as the surge in cryo-EM GPCR structures continues, it is likely that the majority of CNCs that will ultimately be observed in GPCRs were identified from the 473 X-ray and cryo-EM structural chains used in this work.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
All requests should be directed to and will be fulfilled by the Lead Contact, Daniel Isom (disom@miami.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All computer code, PDB files, and datasets associated with this study are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
No experimental models were used. All analyzed structural data was provided by the PDB (see Table S1).
METHOD DETAILS
Collecting human GPCR structures
Sequences of 164,174 structures were downloaded from the PDB (http://www.rcsb.org) using their custom report web service and query parameters: PDB ID: *; Report Name: Sequence. These sequences were then filtered for human GPCR structures using our local library of 373 human GPCR protein sequences and BLAST+ (National Center for Biotechnological Information). The Uniprot (http://www.uniprot.org) code associated with each GPCR sequence/structure was then queried using the Uniprot identifier mapping widget to cross reference each structural chain with its species of origin. The resultant query file was downloaded in .csv format, with each file line corresponding to the PDB-Uniprot mapping information for a given GPCR structural chain. Using the query term “Homo sapiens”, we parsed all human GPCR structural chains from this file. This procedure identified 605 human GPCR structural chains, many of which corresponded to isolated extracellular GPCR domains. To obtain a final set of only complete, 7-TM GPCR chains, we used a three-step heuristic procedure. First, we calculated the secondary structure of each structural chain using the program DSSP (Kabsch and Sander, 1983) and identified helices at least 30 residues in length with custom Python scripts. Second, we calculated the fractional hydropathy of these helices by counting their hydrophobic (I, F, L, V) and hydrophilic (E, D, H, K, R) residues. Third, using a cutoff filter of 0.15 for the average fractional helix hydropathy, we identified the subset of 473 GPCR structural chains used in this study, which corresponded to 81 unique human GPCR genes as of November 2020 (Table S1).
GPCR structural alignments
The set of 473 GPCR structures and their heteroatom molecules were aligned to the reference structure of bovine rhodopsin (RHO, PDB code 1F88, chain A) using PyMOL (Schrödinger, New York, NY) and custom Python scripts.
Molecular surfaces and triangulations
All molecular surfaces and triangulations in this study were calculated using our pHinder suite of structural informatics algorithms (Isom and Dohlman, 2015; Isom et al., 2013, 2016), written in Python.
Terminal side chain atoms
In our triangulations we used a reduced representation of amino acid residues that we call the terminal side chain (TSC) atom. Using this approach, each amino acid residue is represented by a single TSC atom. In the case of linear side chains, the TSC atom is the last atom in the residue side chain (e.g. NZ for the lysine side chain). In the case of branched and aromatic side chains, the TSC atom is calculated as a geometric centroid using a subset of two or more relevant side atoms (e.g. OD1 and OD2 of the aspartic acid side chain).
Counting human GPCR heteroatom molecules
The heteroatom molecules associated with the aligned set of 473 GPCR structures were collected, converted to geometric centroids, filtered by their proximity (8Å cutoff) to the surface atoms of the reference RHO structure (see GPCR structural alignment), and tallied. This procedure was used to count the number of OLA, OLC, and PLM molecules presented in Figure 1B, while the number of CLR molecules was counted using the procedure described below.
Collecting human GPCR-bound cholesterol
CLR molecules associated with the aligned set of 473 GPCR structures were collected and filtered using a two-step heuristic procedure. First, CLRs residing outside of the membrane bilayer were identified and discarded. This filtering process was accomplished using two transmembrane-bounding planes defined by Cα atoms of the reference RHO structure, where the extracellular plane was defined by Cα atoms for RHO residues W35, E201, and T277, and the intracellular plane was defined by Cα atoms for RHO residues V61, A153, and L226. Using a distance-to-plane calculation, all CLRs residing between both transmembrane-bounding planes, or less than 8Å outside of either bounding plane, were retained. Next, using a 10Å cutoff, the TSC atom microenvironment of each remaining CLR was extracted using two waypoints: the C23 atom in the CLR tail and a geometric centroid calculated from the CLR π-ring comprised of the C5 through C10 atoms. The CLR waypoint atoms and extracted TSC atoms were then subjected to two rounds of triangulation to identify the immediate GPCR shell of each CLR. Any CLR with an interaction shell of less than two TSC nodes was considered distant from the GPCR surface and was discarded. After filtering, the total CLR set was reduced from 246 to 240 CLR molecules.
Cholesterol quality scoring
To assess CLR quality, we chose to numerically score each CLR using one of two methods. All x-ray CLR with Real Space Correlation Coefficients (RSCC) ≥ 0.8, as reported by the wwPDB validation server (https://www.wwpdb.org/validation/validation-servers), were considered high quality. Three CLR entries for ADRB2, PDB code 2RH1, were not available on the wwPDB server, but were confirmed to correlate with observed density by visual inspection. All cryo-EM CLR were scored using the Fit in Map tool available in UCSF Chimera (Goddard et al., 2007; Pintilie et al., 2010) using the options: Selected atoms; Use map simulated from atoms; resolution (= reported resolution of structure); correlation calculated about mean data value; Update. During this procedure, pseudo-maps are generated for each CLR with resolution equal to the respective PDB entry’s reported EM map resolution. The CLR pseudo-maps are then compared to the actual EM map to derive a Map-in-Map Fit Correlation (MMFC) between −1 (anticorrelated) and 1 (perfect map overlap at the stated resolution). Applying a cutoff of 0.8 would eliminate all cryo-EM CLR (highest MMFC = 0.54) and preclude a comparison of the two structural methods. Therefore, we chose to retain all cryo-EM CLR with MMFC > 0. Histograms and density plots of CLR correlation in Figure 2B were generated using R (R Foundation for Statistical Computing, Vienna, Austria).
Identifying CNCs
The set of 240 vetted CLR were triangulated using pHinder and the CNA algorithm was applied to identify spatially distinct CLR clusters that define CNCs. Briefly, the CNA algorithm is used after triangulation to trim away network edges longer than a user-defined distance limit (6Å in this calculation). As a result of this trimming process, spatially independent CLR clusters emerge, with each cluster having node-to-node CLR distances less than the user-defined distance limit. Additionally, a user-defined minimum cluster size (3 in this calculation) was used to avoid creating clusters for distant outliers.
Clustering geometrically valid CRAC/CARC motifs
We first calculated the secondary structure of each structural chain using the program DSSP (Kabsch and Sander, 1983) and custom Python scripts to identify and collect the 7 TMs of each GPCR. We next developed Python scripts to exhaustively scan each TM for CRAC (L/V–(X)1–5–Y/F–(X)1–5–R/K) and CARC (R/K–(X)1–5–Y/F–(X)1–5–L/V) sequences. We then vetted the accessibility and geometric validity of each putative CRAC/CARC sequence using a two-step procedure. First, we used the pHinder-calculated surface of each GPCR to confirm that the essential residues (ERs) of each potential CRAC (L/V, Y/F, R/K) and CARC (R/K, Y/F, L/V) motif were exposed to the membrane bilayer (see Figure 3 for an example). Second, we calculated the pairwise angles between sequential ERs using vectors defined by their Cα and TSC atoms. Putative motifs having sequential ER pairwise angles less than 70° were classified as geometrically valid, as this ensured the ERs pointed in the same general direction. Each predicted motif was then visually inspected and a small fraction of false positives (those with buried ERs or ERs that extended from TMs into hydrophilic loops) were identified and discarded. For clustering, each putative motif was reduced to a geometric centroid and the CNA algorithm was applied using a network edge distance limit of 4Å, and minimum cluster size of 5.
Clustering cholesterol microenvironments
Using a 10Å cutoff, the TSC microenvironment of 217 vetted CLR was extracted using two CLR waypoints: the C23 atom in the CLR tail and the geometric centroid calculated from the CLR π-ring comprised of the C5 through C10 atoms. The CLR waypoint atoms and extracted TSC atoms were then subjected to two rounds of triangulation to identify the immediate side chain shell of each CLR, which were then combined and clustered using a network edge distance limit of 1Å, and minimum cluster size of 10. Redundant contributions from GPCRs with many nearly identical structures were removed from each cluster, and the resultant set of non-redundant TSC atoms was recombined and clustered again using a network edge distance limit of 3Å, and minimum cluster size of 10 to give the clusters presented in Figure 4.
QUANTIFICATION AND STATISTICAL ANALYSIS
Map-in-map fit correlations for cryo-EM originating CLR molecules were quantified with UCSF Chimera.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Deposited Data | ||
|
| ||
| GPCR structural chains-see Table S1 | Protein Data Bank (PDB) | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| NCBI BLAST+ | NIH National Center for Biotechnology Information (NCBI) | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| DSSP | Kabsch and Sander, 1983 | https://swift.cmbi.umcn.nl/gv/dssp/ |
| pHinder | Isom et al., 2013 | https://doi.org/10.1016/j.molcel.2013.07.012 |
| PyMol | Schrödinger, Inc. | https://www.pymol.org/ |
| UCSF Chimera | UCSF Resource for Biocomputing, Visualization, and Informatics (RVBI) | https://www.cgl.ucsf.edu/chimera/ |
Highlights.
A rapidly growing number of GPCR structures have been solved by cryo-EM
Cholesterol-binding patterns are similar in cryo-EM and X-ray GPCR structures
92% of cholesterol in GPCR structures map to 12 Cholesterol Network Clusters (CNCs)
There are no broadly recurring cholesterol-binding motifs in GPCRs
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health through the National Institute of General Medical Sciences (R35GM119518) to D.G.I. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Dr. Santiago Vilar for his critical review of the manuscript.
Footnotes
DECLARATION OF INTERESTS
The authors declare that they have no conflicts of interest with the contents of this article.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.str.2021.01.004.
REFERENCES
- Caffrey M (2015). A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Crystallogr. F Struct. Biol. Commun. 71, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. (2007). High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Scala C, Baier CJ, Evans LS, Williamson PTF, Fantini J, and Barrantes FJ (2017). Relevance of CARC and CRAC cholesterol-recognition motifs in the nicotinic acetylcholine receptor and other membrane-bound receptors. Curr. Top. Membr. 80, 3–23. [DOI] [PubMed] [Google Scholar]
- Duncan AL, Song W, and Sansom MSP (2020). Lipid-dependent regulation of ion channels and G protein-coupled receptors: insights from structures and simulations. Annu. Rev. Pharmacol. Toxicol. 60, 31–50. [DOI] [PubMed] [Google Scholar]
- Dunn HA, Orlandi C, and Martemyanov KA (2019). Beyond the ligand: extracellular and transcellular G protein-coupled receptor complexes in physiology and pharmacology. Pharmacol. Rev. 71, 503–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantini J, and Barrantes FJ (2013). How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 4, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantini J, Di Scala C, Baier CJ, and Barrantes FJ (2016). Molecular mechanisms of protein-cholesterol interactions in plasma membranes: functional distinction between topological (tilted) and consensus (CARC/CRAC) domains. Chem. Phys. Lipids 199, 52–60. [DOI] [PubMed] [Google Scholar]
- Felder CC (2019). GPCR drug discovery-moving beyond the orthosteric to the allosteric domain. Adv. Pharmacol. 86, 1–20. [DOI] [PubMed] [Google Scholar]
- Gacasan SB, Baker DL, and Parrill AL (2017). G protein-coupled receptors: the evolution of structural insight. AIMS Biophys. 4, 491–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Nafria J, and Tate CG (2020). Cryo-electron microscopy: moving beyond X-ray crystal structures for drug receptors and drug development. Annu. Rev. Pharmacol. Toxicol. 60, 51–71. [DOI] [PubMed] [Google Scholar]
- Genheden S, Essex JW, and Lee AG (2017). G protein coupled receptor interactions with cholesterol deep in the membrane. Biochim. Biophys. Acta Biomembr 1859, 268–281. [DOI] [PubMed] [Google Scholar]
- Gimpl G (2016). Interaction of G protein coupled receptors and cholesterol. Chem. Phys. Lipids 199, 61–73. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, and Ferrin TE (2007). Visualizing density maps with UCSF Chimera. J. Struct. Biol. 157, 281–287. [DOI] [PubMed] [Google Scholar]
- Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, and Stevens RC (2008). A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 16, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom DG, and Dohlman HG (2015). Buried ionizable networks are an ancient hallmark of G protein-coupled receptor activation. Proc. Natl. Acad. Sci. U S A 112, 5702–5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom DG, Sridharan V, Baker R, Clement ST, Smalley DM, and Dohlman HG (2013). Protons as second messenger regulators of G protein signaling. Mol. Cell 51, 531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom DG, Sridharan V, and Dohlman HG (2016). Regulation of ras paralog thermostability by networks of buried ionizable groups. Biochemistry 55, 534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafurulla M, Tiwari S, and Chattopadhyay A (2011). Identification of cholesterol recognition amino acid consensus (CRAC) motif in G-protein coupled receptors. Biochem. Biophys. Res. Commun. 404, 569–573. [DOI] [PubMed] [Google Scholar]
- Jones AJY, Gabriel F, Tandale A, and Nietlispach D (2020). Structure and dynamics of GPCRs in lipid membranes: physical principles and experimental approaches. Molecules 25:4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, and Sander C (1983). Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kapolka NJ, and Isom DG (2020). HCAR3: an underexplored metabolite sensor. Nat. Rev. Drug Discov. 19, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapolka NJ, Taghon GJ, Rowe JB, Morgan WM, Enten JF, Lambert NA, and Isom DG (2020). DCyFIR: a high-throughput CRISPR platform for multiplexed G protein-coupled receptor profiling and ligand discovery. Proc. Natl. Acad. Sci. U S A 117, 13117–13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriakidi S, Kolocouris A, Liapakis G, Ikram S, Durdagi S, and Mavromoustakos T (2019). Effects of cholesterol on GPCR function: insights from computational and experimental studies. Adv. Exp. Med. Biol.1135, 89–103. [DOI] [PubMed] [Google Scholar]
- Kulig W, Tynkkynen J, Javanainen M, Manna M, Rog T, Vattulainen I, and Jungwirth P (2014). How well does cholesteryl hemisuccinate mimic cholesterol in saturated phospholipid bilayers? J. Mol. Model. 20, 2121. [DOI] [PubMed] [Google Scholar]
- Liu JF, Xia JJ, Nie KL, Wang F, and Deng L (2019). Outline of the biosynthesis and regulation of ergosterol in yeast. World J. Microbiol. Biotechnol. 35, 98. [DOI] [PubMed] [Google Scholar]
- Lovera S, Cuzzolin A, Kelm S, De Fabritiis G, and Sands ZA (2019). Reconstruction of apo A2A receptor activation pathways reveal ligand-competent intermediates and state-dependent cholesterol hotspots. Sci. Rep. 9, 14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milic D, and Veprintsev DB (2015). Large-scale production and protein engineering of G protein-coupled receptors for structural studies. Front. Pharmacol. 6, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljus T, Sykes DA, Harwood CR, Vuckovic Z, and Veprintsev DB (2020). GPCR solubilization and quality control. Methods Mol. Biol. 2127, 105–127. [DOI] [PubMed] [Google Scholar]
- Muth S, Fries A, and Gimpl G (2011). Cholesterol-induced conformational changes in the oxytocin receptor. Biochem. J. 437, 541–553. [DOI] [PubMed] [Google Scholar]
- Oddi S, Dainese E, Fezza F, Lanuti M, Barcaroli D, De Laurenzi V, Centonze D, and Maccarrone M (2011). Functional characterization of putative cholesterol binding sequence (CRAC) in human type-1 cannabinoid receptor. J. Neurochem. 116, 858–865. [DOI] [PubMed] [Google Scholar]
- Orban T, Gupta S, Palczewski K, and Chance MR (2010). Visualizing water molecules in transmembrane proteins using radiolytic labeling methods. Biochemistry 49, 827–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintilie GD, Zhang J, Goddard TD, Chiu W, and Gossard DC (2010). Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 170, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter RM, Harikumar KG, Wu SV, and Miller LJ (2012). Differential sensitivity of types 1 and 2 cholecystokinin receptors to membrane cholesterol. J. Lipid Res. 53, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Wang Y, Law PY, Chen HZ, and Loh HH (2011). Cholesterol regulates micro-opioid receptor-induced beta-arrestin 2 translocation to membrane lipid rafts. Mol. Pharmacol. 80, 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe JB, Taghon GJ, Kapolka NJ, Morgan WM, and Isom DG (2020). CRISPR-addressable yeast strains with applications in human G protein-coupled receptor profiling and synthetic biology. J. Biol. Chem. 295, 8262–8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, and Dwivedi-Agnihotri H (2020). Structure and function of beta-arrestins, their emerging role in breast cancer, and potential opportunities for therapeutic manipulation. Adv. Cancer Res. 145, 139–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart OS, Horsky V, Gore S, Svobodova Varekova R, Bendova V, Kleywegt GJ, and Velankar S (2018). Validation of ligands in macromolecular structures determined by X-ray crystallography. Acta Crystallogr. D Struct. Biol. 74, 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV, and Fisher I (2019). G-protein betagamma subunits as multifunctional scaffolds and transducers in G-protein-coupled receptor signaling. Cell Mol. Life Sci. 76, 4447–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, and Insel PA (2018). G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol. 93, 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutkeviciute I, and Vilardaga JP (2020). Structural insights into emergent signaling modes of G protein-coupled receptors. J. Biol. Chem. 295, 11626–11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Ma AK, Fonseca R, Latorraca NR, Kelly B, Betz RM, Asawa C, Kobilka BK, and Dror RO (2019). Diverse GPCRs exhibit conserved water networks for stabilization and activation. Proc. Natl. Acad. Sci. U S A 116, 3288–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vazquez-Fresno R, Sajed T, Johnson D, Li C, Karu N, et al. (2018). Hmdb 4.0: the human metabolome database for 2018. Nucleic Acids Res. 46, D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, and Kobilka B (2005). Using synthetic lipids to stabilize purified beta2 adrenoceptor in detergent micelles. Anal. Biochem. 343, 344–346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All computer code, PDB files, and datasets associated with this study are available upon request.