

Abstract
The diagnostic utility of somatic mutations in the context of cytopenias is unclear: clonal hematopoiesis can be found in healthy individuals, patients with aplastic anemia (AA), clonal cytopenia of undetermined significance (CCUS) and myelodysplastic syndrome (MDS). We examined a cohort of 207 well-characterized cytopenic patients with a 640-gene next generation sequencing (NGS) panel and compared its diagnostic utility with a “virtual” 41 gene panel. The TET2, SF3B1, ASXL1, and TP53 were the most commonly mutated genes (frequency > 10%). Mutations in the 640-gene panel show high sensitivity (98.3%) but low specificity (47.6%) for diagnosis of MDS. Notably, mutations of splicing factors and genes in the RAS pathway are relatively specific to MDS. Furthermore, high variant allele frequency (VAF) predicts MDS: when the VAF is set at 20%, the positive predictive value (PPV) for MDS is 95.9%, with a specificity of 95.3%. The presence of two or more somatic mutations with ≥10% VAF showed a PPV of 95.2%. While the “virtual” 41-gene panel showed a mild decrease in sensitivity (95.7% vs 98.3%), 100% specificity was observed when either VAF was set at ≥20% (100% vs 95.3%), or two or more somatic mutations had VAFs ≥ 10%. Our study shows targeted gene panel sequencing improves the diagnostic approach and accuracy for unexplained cytopenia, with its high sensitivity and high PPV for MDS when applying VAF cutoffs. Furthermore, a 41-gene panel was shown to have at least comparable performance characteristics to the large 640-gene panel.
1 ∣. INTRODUCTION
The presence of cytopenias can be diagnostically challenging.1,2 There are many non-neoplastic causes of cytopenias, including vitamin deficiency, drug, infection, inflammation, immunologic disorders and others.3 Distinguishing MDS from other causes of cytopenias remains a clinical conundrum in some patients. Prevalence of both MDS and unexplained cytopenia increases with age, reaching 20% at age 85 and older for the latter.4,5 While one of the important features of MDS is morphologic dysplasia, assessment of dysplasia is subjective. Diagnostic criteria of 10% or over the dysplasia threshold by the World Health Organization (WHO) for MDS is controversial,6 and dysplasia can be seen in non-neoplastic conditions.7,8 Evidence of clonality, another important and more objective feature of MDS, is commonly provided by cytogenetics. However, recurrent chromosomal abnormalities are only detected in about 50% of patients9-11 with MDS, limiting the utility of cytogenetics.
In recent years, targeted NGS panels have been used in oncology for early detection of tumor, risk stratification and prognosis, disease monitoring, and targeted therapy.12-16 Mutational analysis for patients with unexplained cytopenia is becoming common practice, especially in large academic medical centers.17,18 Detection of somatic mutations provides clonal evidence for MDS, but for the most part, the diagnostic utility of somatic mutations in patients with cytopenia is not well-defined. Somatic mutations can be detected in over 30% of patients with unexplained cytopenia who do not meet the diagnostic criteria of MDS,1 healthy older individuals without evidence of disease,19-21 and patients with AA.22 It is important to study whether the size of the clones harboring mutation, the number of mutations, the genes being mutated, or the specific mutations have diagnostic utility to point to a specific etiology.2 Moreover, an increasing number of somatic mutations have been identified in MDS.23-28 Most published studies used NGS panels with 30 to 50 genes,29,30 and the optimal target gene panel and the impact of panel size are not well-defined.
In this study, with over 200 clinical cases of unexplained cytopenia, we assessed the clinical utility of mutational analysis for cytopenia workup with a large NGS panel (640 genes), and also assessed and compared different NGS panel sizes.
2 ∣. METHODS
2.1 ∣. Patients and samples
This study was approved by the Institutional Review Board (IRB) at the Johns Hopkins Medical Institutions. A total of 207 consecutive patients with documented peripheral blood unilineage, bilineage and trilineage cytopenias, who had bone marrow and peripheral blood samples submitted to a CLIA-certified molecular pathology laboratory for NGS. This was at the discretion of the treating clinician, and they were enrolled from the Johns Hopkins Hospital between September 2015 and March 2018. The list of 640 genes in the panel is shown in Table S1. All patients underwent routine diagnostic evaluation for their hematologic abnormalities, with a comprehensive bone marrow evaluation, including morphologic review of bone marrow aspirate smears and bone marrow biopsy, flow cytometry and cytogenetic studies. Cytogenetic findings were interpreted according to the WHO classification.31,32 All patients underwent NGS for diagnostic workup of cytopenia. As part of the diagnostic process for these patients, a thorough work up for non-marrow based (or non-clonal) causes of cytopenias was performed before NGS testing. We ruled out vitamin deficiencies, autoimmune causes, peripheral destruction and other marrow suppressive issues (such as medication induced). Patients with a diagnosis of acute leukemia, myeloproliferative neoplasms (MPN), lymphoma, other neoplasms or history of cytotoxic therapy for other malignancies were excluded from the study. Cytopenia was defined as a hemoglobin level <10 g/dL, absolute neutrophil count <1.8 × 109/L, or platelet count <1.00 × 1011/L in at least one lineage. The gold standard for patient classification was a combination of blood cell counts, blood and marrow morphology and cytogenetic studies. The diagnosis of MDS was based on the criteria of the WHO classification of myeloid neoplasms and its revision.31-33 The MDS patients were classified as either low grade (bone marrow blast<5% and peripheral blood blasts <1%), or high grade MDS (bone marrow blast 5%-19% and/or peripheral blood blasts 2%-19%).32,34
2.2 ∣. Targeted gene panel sequencing
Libraries were prepared using the Agilent SureSelect-XT Target Enrichment Kit. Briefly, between 0.2 and 1 μg of DNA was fragmented to a size of 250 to 300 bp, using a Covaris M220 sonicator. The DNA fragments were end-repaired and A-tailed, then adaptors were added by ligation and the fragments were enriched by PCR (six cycles). Each library was then hybridized to a SureSelect custom panel 2.8 M bait set (Agilent) according to the manufacturer's protocol. The custom panel was designed as a comprehensive cancer gene panel, and covered 640 genes important in oncogenesis. After stringent washing the captured DNA was amplified with 12 cycles of PCR per manufacturer's protocol. The size and concentration of captured DNA was assessed using a Tapestation 2200 (Agilent). All captured samples were clustered on a cBOT system, and sequenced on a single lane of a PE-flow cell on a HiSeq2500 (Illumina), using a 2 × 150 bp PE protocol. All reads were aligned to the human genome (GRCh37/hg19), using the Burrows-Wheeler alignment (BWA) algorithm. The final BAM file was used for variant calling with a custom variant caller pipeline, which called variants directly from the BAM file with a 1% VAF filter, a filter of >3 mutant read in both directions, and a strand bias filter. The 1% VAF filter was chosen from limit of blank experiments. Our variant caller pipeline integrated information of strand bias scoring, coverage, VAF, dbSNP annotation, COSMIC annotation, mutation region, and mutation types. Two strand bias scores were calculated: the first one (SB1) considered only strand bias in variant reads: MAX (Var+, Var−)/ (Var+, Var−); the second (SB2) adjusted variant calls for inherent strand bias: (Var+/Ref+)/ (Var−/Ref−). A variant call passed the strand bias filter if either SB1 ≥ 0.7 and/or 2.0 ≥ SB2 ≥ 0.5. An additional filter which compares the VAF of the variant detected, and the background noise of the same loci based on a pool of normal samples was also applied. The mean sequencing coverage was around 850 reads, and exceeded 300 reads in more than 94% of all regions. After common SNPs (minor allele population frequency ≥1%), synonymous mutations, and intronic mutations were filtered, presumptive somatic mutations were identified based on the following criteria: VAF ≤ 25% which cannot be explained by cytogenetic change, loss of function mutations (frame-shift mutations or nonsense mutations), or ≥10 samples in Catalogue of Somatic Mutations in Cancer (COSMIC) database with the same mutations. All mutations were confirmed by manual inspection with integrative genomics viewer (IGV) (http://software.broadinstitute.org/software/igv/). A virtual panel of 41 genes was selected based on the most common mutated genes in MDS 35 (Figure 1 of the reference) and AA22 (Figure 1 of the reference), with the genes whose mutations are absent in our cohort excluded.
FIGURE 1.

Spectrum of the mutated gene identified in 201 patients with cytopenias. A, The frequency and patterns of 79 recurrent mutated gene identified in 201 cases, which are shown in indicated colors (640-gene NGS panel). B, Distribution of mutations of 41 genes ordered by gene function which are commonly mutated in MDS and AA as a “virtual panel” in 201 cases. Aplastic anemia (AA); myelodysplastic syndrome (MDS)
2.3 ∣. Statistical analysis
All of the data were given as mean ± SD or median (range). Statistical significance of mutated genes in different groups was determined by ANOVA, t test and Chi-Square Test with a contingency table. A P value less than .05 was considered statistically significant.
3 ∣. RESULTS
3.1 ∣. Demographic and clinical features of patients with cytopenia
In this study, as shown in Table 1, 34 of 207 patients were diagnosed with AA, 68 patients were diagnosed as low grade MDS, 47 patients were diagnosed as high grade MDS, 6 patients were suspicious for but not diagnostic for MDS, and the remaining 52 patients showed no evidence of a primary marrow disorder (defined as cytopenia group in this study). The AA patients were mostly <50 years old, while MDS patients were mostly 60 to 80 years old. The demographic and clinical features of 207 patients with cytopenia are shown in Table 1, and the demographic and clinical features of six suspected MDS patients without a definite diagnosis are shown in Table S2.
TABLE 1.
Demographic and clinical features of patients with cytopenia
| Patient numbers | 207 | ||
|---|---|---|---|
| Sex, Males: females | 113/94 | ||
| Age, mean ± SD (years) | 59.98 ± 17.75 (4-91) years | ||
| AA | 43.15 ± 17.84 (11-75) | ||
| Cytopenia | 57.23 ± 19.65 (21-91) | ||
| MDS-low | 65.54 ± 12.52 (31-83) | ||
| MDS-high | 66.53 ± 14.32 (4-87) | ||
| Uncertain | 64.67 ± 9.65 (53-74) | ||
| Diagnosis | |||
| AA | 34 (16.4%) | ||
| Cytopenia | 52 (25.1%) | ||
| MDS-low grade | 68 (32.8%) | ||
| MDS-high grade | 47 (22.7%) | ||
| Uncertain diagnosis | 6 (3%) | ||
| Cytogenetics | Normal karyotype | Abnormal karyotype | Failed/not done |
| AA | 21 (61.8%) | 2 (5.9%) | 11 (32.3%) |
| Cytopenia | 31 (59.6%) | 4 (7.7%) | 17 (32.7%) |
| MDS-Low | 25 (36.8%) | 33 (48.5%) | 10 (14.7%) |
| MDS-High | 14 (29.8%) | 29 (61.7%) | 4 (8.5%) |
| Uncertain | 5 (83.3%) | 0 (0%) | 1 (16.7%) |
3.2 ∣. Frequency and spectrum of somatic mutations
Of all 207 patients, 588 somatic mutations involving 181 genes were detected, with 79 genes showing recurrent mutations (Figure 1A); the other 102 genes had only one mutation each (Table S3). As shown in Figure 1A, TET2, ASXL1, SF3B1 and TP53 showed the highest mutation frequencies, each involving more than 10% of all patients. The SF3B1 mutations occur predominantly in low-grade MDS (MDS with ring sideroblasts), and TP53 mutations occur predominantly in high-grade MDS. The MAGI1 mutations occurred exclusively in cytopenia patients, and PIGA mutations occurred exclusively in AA patients. Consistently, somatic mutations in TP53, RUNX1 and ASXL1 predict high grade MDS (P < .001), whereas spliceosome gene SF3B1 mutations predict low grade MDS (P < .001). We then selected 41 genes which are commonly mutated in MDS35 and AA22 as a “virtual panel”, and studied the mutation distribution based on gene function (Figure 1B). Mutations of splicing factors including SF3B1, SRSF2, ZRSR1, U2AF1 and U2AF2, as well as those in RAS pathways, occur almost exclusively in MDS. In contrast, mutations of factors involving DNA methylation, especially TET2 and DNMT3A, do occur in both AA and cytopenia groups, supporting the presence of clonal hematopoiesis of indeterminate potential (CHIP).20,21,36-38 Furthermore, in this cohort, somatic mutations involving SRSF2, RUNX1, U2AF1, ZRSR2, CBL, IDH2, NRAS, NF1, IDH1, PHF6, ETV6, NCOR2, SMC3, FLT3, CEBPA, NPM1, KIT, GATA2, U2AF2, SETBP1, WT1, CUX1 and RIT1 were only found in the MDS group, not in AA and cytopenia groups. The PIGA (12%) was the most frequently mutated gene in AA cases, and TET2 (10%) was the most frequently mutated gene in the cytopenia group.
3.3 ∣. Predictive value of variant allele frequency
The 588 somatic mutations identified in the 207 samples have VAFs ranging from 1.0% to 88.1% (Figure 2A). The MDS patients had a higher mean VAF than AA and cytopenia groups (Figure S1A). Overall, 67 of 68 (98.5%) low grade MDS patients carried somatic mutations with a mean VAF of 32.8% (n = 67), 46 of 47 (97.9%) high grade MDS patients carried somatic mutations with a mean VAF of 38.9% (n = 46). Twenty of 34 (58.8%) AA patients showed somatic mutations with a mean VAF of 11.2% (n = 20); 25 of 52 (48.1%) cytopenic patients without evidence of a primary marrow disorder showed somatic mutations with a mean VAF of 8.4% (n = 25). With the 41 gene NGS panel, a total of 363 somatic mutations were identified with VAFs ranging from 1.1% to 84.2% (Figure 2B). Similar to the 640-gene NGS panel, MDS patients showed higher VAFs than AA and cytopenia patients (Figure S1B). Among them, 64 of 68 (94.1%) low grade MDS patients carried somatic mutations with a mean VAF of 31.8% (n = 64), 46 of 47 (97.9%) high grade MDS patients carried somatic mutations with a mean VAF of 38.0% (n = 46), while 12 of 34 (35.3%) AA patients carried somatic mutations with a mean VAF of 7.7% (n = 12). Thirteen of 52 (25.0%) patients in the cytopenia group carried somatic mutations with a mean VAF of 4.5% (n = 13). With both 640-gene and 41-gene panels, the percentages of MDS patients with mutations and mean VAFs are significantly higher than patients from both AA and cytopenia groups, while there are no significant difference between AA and cytopenia groups (by chi-square test).
FIGURE 2.
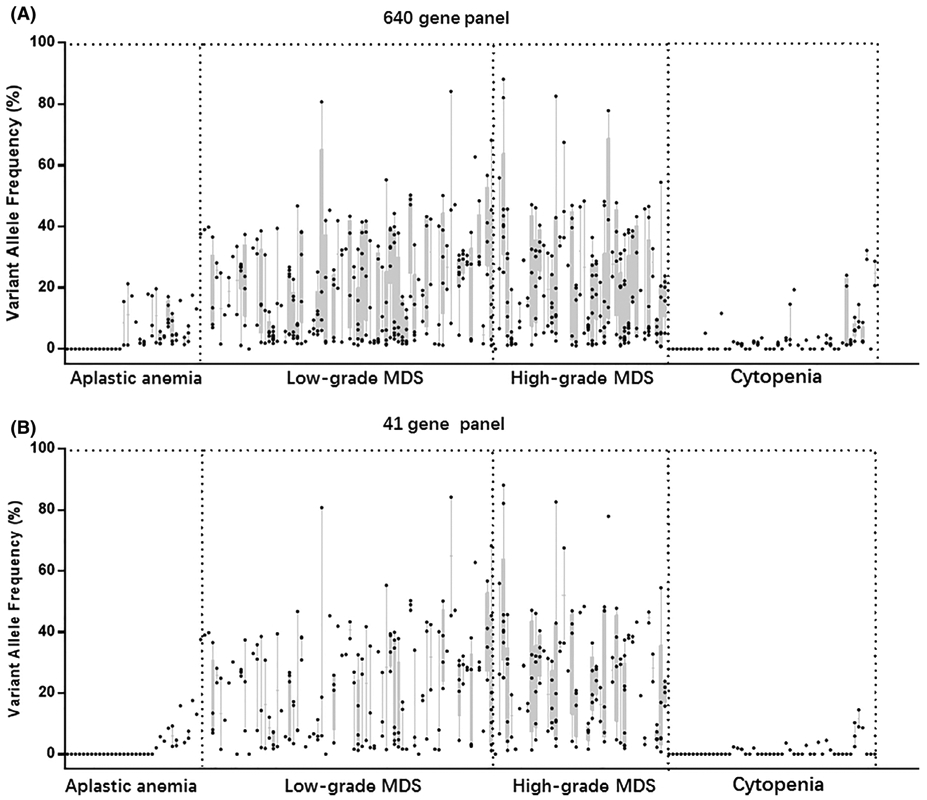
Range plot of the variant allele frequencies of 201 cases with different NGS panels Patients are in columns, sorted by “gold standard” diagnosis. VAFs are shown as dots within columns; a dot at VAF = 0 indicates that the patient had no somatic mutations. The min and max VAFs of the cases are the boundaries of each column. A, Range plot of the VAFs by the 640 gene panel. B, Range plot of the VAFs by the 41 gene panel. VAF, variant allele frequency
We next assessed diagnostic utility of NGS for MDS diagnosis based on VAF. As shown in Tables 2 and S4A, the sensitivity of the 640-gene NGS panel for MDS was 98.3%, and the negative predictive value (NPV) was 95.3%. We first performed Receiver Operator Characteristic analysis for VAF, designating MDS as “positive”. As shown in Figure S2, VAF is a significant predictor for MDS based on both 640-gene and 41-gene panels. Using 20% as a cutoff shows maximum sensitivity and specificity with the 640-gene panel, and shows 100% specificity for MDS with the 41-gene panel. When the VAF for one or more variants was set at 20% the PPV for MDS was 95.9% with a specificity of 95.3% (Tables 2 and S4B). While the presence of two or more somatic mutations at any VAF in a specimen predicts MDS with a PPV of 76.3% (Tables 2 and S4C), the PPV was increased to 95.2% when the VAF cutoff is set to 10% (Tables 2 and S4D). On the other hand, with the 41-gene NGS panel the sensitivity for MDS was 95.7% (Tables 2 and S4E). When the VAF was set at 20%, the PPV for MDS reached 100% with a specificity of 100% (Tables 2 and S4F). While presence of two or more somatic mutations at any VAF in a specimen predicts MDS with a PPV of 91.9% (Tables 2 and S4G), the PPV was increased to 100% when the VAF cutoff was set to 10% (Tables 2 and S4H). Overall mutational profiling with both 640-gene and 41-gene NGS panels was sensitive in predicting MDS, while the 41 gene-NGS panel was more specific, and a mutation with ≥20% VAF, or ≥2 mutations with ≥10% VAF highly predicts MDS.
TABLE 2.
Diagnostic performance of mutations for MDS with different cutoffs. The sensitivity, specificity, positive predictive value and negative predictive value with different VAF cutoffs and number of mutations for MDS in different NGS panels
| Any mutations (VAF ≥ 1%) | VAF ≥ 20% | ≥2 mutations | VAF ≥ 10% and ≥2 mutations | |
|---|---|---|---|---|
| 640 gene panel | ||||
| Sensitivity | 98.3% | 81.7% | 84.3% | 68.7% |
| Specificity | 47.6% | 95.3% | 65.1% | 95.3% |
| NPV | 95.3% | 79.6% | 75.7% | 69.5% |
| PPV | 71.5% | 95.9% | 76.3% | 95.2% |
| 41 gene panel | ||||
| Sensitivity | 95.7% | 75.7% | 69.6% | 53.0% |
| Specificity | 72.1% | 100% | 91.9% | 100% |
| NPV | 92.4% | 75.4% | 69.3% | 61.4% |
| PPV | 81.5% | 100% | 91.9% | 100% |
Abbreviations: NPV: negative predictive value; PPV: positive predictive value.
3.4 ∣. The predictive value of the quantity and types of mutations
We also analyzed the diagnostic value of the number of mutated genes (Table S5 and Figure S4). The mean mutation number of low-grade and high-grade MDS patients (3.6 and 4.8 respectively by the 640-gene panel, and 2.3 and 3.4 respectively by the 41-gene panel) was significantly higher than AA and cytopenia patients (1.5 and 1.3 respectively by the 640-gene panel, and 0.5 and 0.4 respectively by 41-gene panel) (P < .05 by t test). However, there are no significant differences in the types of mutations (single nucleotide variants vs insertions vs deletions) in each group (Figure S3A,B). Interestingly, 64% (158/247) and 71% (157/221) of the mutations detected by the 640-gene panel in low-grade and high-grade MDS patients, respectively, were also detected in the 41-gene panel. However, only 36% (16/44) and 32% (17/53) of mutated genes by the 640-gene panel in AA and cytopenia patients (Table S5), respectively, were on the list of 41 genes, suggesting most of the mutated genes detected by the 640-gene panel in the non-MDS patients were not common driver genes. As shown in Figure S4A,B, if we only consider mutations with significant clone size (VAF ≥ 10%), non-MDS patients have either zero or one mutation, while myelodysplastic syndrome patients have a broader range of mutation numbers, with up to eight in some high-grade MDS patients. Greater than five mutations are only observed in high-grade MDS patients.
3.5 ∣. Next-generation sequencing improves sensitivity of cytogenetic study for MDS
In the 201 patients with a definitive diagnosis, 91 showed normal karyotype, 68 showed abnormal karyotype and 42 patients showed failed cytogenetic studies or had no cytogenetic studies done (Table 1). We compared the sensitivity and specificity of cytogenetics changes with NGS. As shown in Table S6A, the specificity of cytogenetic abnormality for MDS was 89.7%, but the sensitivity for MDS was only 61.4% (62/101). A “cytogenetic change OR mutation” criterion (cytogenetic change or mutations of VAF ≥ 1%) (Table S6A) provides 100% sensitivity and 100% NPV for MDS. This means all MDS cases have either cytogenetic changes or mutations, and we should exercise extreme caution in diagnosing MDS when there are no cytogenetic changes. and also no mutations detected by the 640-gene panel. On the other hand, a “cytogenetic change AND mutation” criterion (Table S6B) could provide 100% specificity and 100% PPV for MDS with both 640-gene and 41-gene panels, which means a diagnosis of MDS is highly likely if there are both cytogenetic changes and mutations.
4 ∣. DISCUSSION
Diagnosis by MDS is based on morphology, and is to some extent subjective due to poor inter-observer concordance in dysplasia assessment.6 Mutation profiling may provide more objective evidence for diagnosis of MDS in cytopenic patients.35,39-41 Although increasing numbers of somatic mutations have been identified in MDS, the majority of them are <5% in frequency, and none of these somatic mutations is specific for MDS.35,40 How to interpret mutation profiling results in the workup of unexplained cytopenia remains uncertain. Therefore, it is critical in clinical practice to use a comprehensive, well-designed gene panel to assess for MDS in cytopenia patients. In this study we compared the diagnostic utility of targeted NGS with a 640-gene, or a virtual 41-gene panel, focusing on clone size (VAF), number of mutations, gene function and specific genes. The comparison of the large and small panels enables optimization of panel size for optimal clinical utility.
It was noted that 21 of 52 patients in the cytopenia group show mutations ≥2% (640-gene panel) (Figure 2A), thus fulfilling the criteria for CCUS.34 Although some of the patients with AA or CCUS had somatic mutations, their mean VAF are less than 11.2%. The somatic mutation in MDS had a mean VAF at approximately 30% to 40%, indicating mutation clone size (VAF) is relevant in distinguishing the etiology of cytopenia. This is also supported by recent studies.22,42 In our study, although the 640-gene panel is more sensitive and can be useful for ruling out MDS, the “virtual” 41-gene panel based on the genes commonly mutated in MDS and AA shows similar sensitivity, but higher specificity. With the 41-gene NGS panel the presence of mutations with a VAF ≥ 20%, or two or more mutations with a VAF ≥ 10% were highly predictive of MDS (Table 2). Given the lower cost and the potential for deeper read depth, the 41-gene panel is a better fit for diagnostic workup of unexplained cytopenia than the 640-gene panel.
Cytogenetic abnormalities provide evidence of clonality, but are not sensitive for MDS diagnosis. In our study 62/115 (54%) MDS patients show cytogenetic abnormalities, and the specificity of cytogenetic abnormality for MDS is 89.7% (Table S6A). Using the 640-gene NGS panel, 2/115 of MDS patients did not carry any somatic mutations, one of whom had isolated del(5q) and the other had a complex karyotype. This suggests that a joint test combining somatic mutations and cytogenetics shows almost 100% sensitivity for MDS. Such high sensitivity is diagnostically valuable in excluding MDS as a cause of cytopenia.
Most mutated genes are not specific to either MDS or AA; however, we did observe that somatic mutations involving SRSF2, RUNX1, U2AF1, ZRSR2, CBL, IDH2, NRAS, NF1, IDH1, PHF6, ETV6, NCOR2, SMC3, FLT3, CEBPA, NPM1, KIT, GATA2, U2AF2, SETBP1, WT1, CUX1 and RIT1 were unique to the MDS group, and mutations of PIGA were unique to AA. All AA patients with PIGA mutations harbor PNH clones as shown by flow cytometry. However, given the limited cohort size, the specificity of those mutations remains to be tested. Furthermore, mutations in TP53, RUNX1 and ASXL1 occurred predominantly in high grade MDS, and spliceosome gene SF3B1 mutations occurred exclusively in low grade MDS associated with ring sideroblasts This suggests somatic mutations in TP53, RUNX1, ASXL1 and SF3B1 may distinguish low vs high grade MDS, consistent with previous studies.35,43-45 It has been shown that adding mutation data into the revised International Prognostic Scoring System (IPSS-R) improves predictive value in MDS, and adjusts MDS patients into more appropriate risk categories.44-46 Furthermore, although there are no differences in terms of the types of mutations in each group, our study suggests that the number of mutations predicts MDS, and can also be used to distinguish low- and high grade MDS, which is consistent with other studies.35,40,42
In our study, six patients were suspected of having MDS but did not meet the WHO diagnostic criteria for MDS (Table S2). Patients one, two, three and four were found to have cytopenia which persisted >6 months, mild dysplasia (present in less than 10% of all cells in one of the lineages), normal karyotype and no increase in blasts. These patients had somatic mutations with >20% VAF, including splicing factors like SRSF2 in some patients. These patients carried two or more mutations with a VAF > 20% (41 gene-NGS panel) and, based on the high PPV, we believe should be classified as MDS. On the other hand, based on mutation analysis, patients five and six could be predicted not to have a myeloid neoplasm like MDS. Follow-up of these patients will be valuable to confirm our hypotheses.
With a large cohort of cytopenia patients in a tertiary medical center, we systemically assessed the diagnostic utility of targeted NGS panel and also performed parallel comparison of large and small panels. Our study shows mutation analysis by the 41-gene NGS panel achieves similar sensitivity compared with the 640-gene panel, but has increased specificity. The presence of a mutation with ≥20% VAF, or two or more mutations with ≥10% VAF, is highly predictive of MDS (Table 2). Higher numbers of mutations also correlate with MDS (Figure S4). Furthermore, mutations of certain genes such as TP53, SF3B1, and PIGA can have specific diagnostic utility. It should be noted that diagnostic values calculated with this population of patients from our referral center may not be applicable to the general population. Another limitation of the study is a lack of long-term follow-up, especially for the six patients with no definitive diagnosis. Thus, confirmation of our findings in other populations and with a prospective longitudinal study will be of value.
Supplementary Material
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interests relevant to the manuscript.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Kwok B, Hall JM, Witte JS, et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood. 2015;126(21):2355–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malcovati L, Gallì A, Travaglino E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129(25):3371–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steensma DP. Dysplasia has a differential diagnosis: distinguishing genuine myelodysplastic syndromes (MDS) from mimics, imitators, copycats and impostors. Curr Hematol Malignancy Rep. 2012;7(4):310–320. [DOI] [PubMed] [Google Scholar]
- 4.Guralnik JM, Eisenstaedt RS, Ferrucci L, Klein HG, Woodman RC. Prevalence of anemia in persons 65 years and older in the United States: evidence for a high rate of unexplained anemia. Blood. 2004;104(8):2263–2268. [DOI] [PubMed] [Google Scholar]
- 5.Stauder R, Valent P, Theurl I. Anemia at older age: etiologies, clinical implications, and management. Blood. 2018;131(5):505–514. [DOI] [PubMed] [Google Scholar]
- 6.Parmentier S, Schetelig J, Lorenz K, et al. Assessment of dysplastic hematopoiesis: lessons from healthy bone marrow donors. Haematologica. 2012;97(5):723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Senent L, Arenillas L, Luño E, Ruiz JC, Sanz G, Florensa L. Reproducibility of the World Health Organization 2008 criteria for myelodysplastic syndromes. Haematologica. 2013;98(4):568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Font P, Loscertales J, Benavente C, et al. Inter-observer variance with the diagnosis of myelodysplastic syndromes (MDS) following the 2008 WHO classification. Ann Hematol. 2013;92(1):19–24. [DOI] [PubMed] [Google Scholar]
- 9.Schanz J, Tüchler H, Solé F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30(8):820–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sole F, Luno E, Sanzo C, et al. Identification of novel cytogenetic markers with prognostic significance in a series of 968 patients with primary myelodysplastic syndromes. Haematologica. 2005;90(9):1168–1178. [PubMed] [Google Scholar]
- 11.Haase D, Germing U, Schanz J, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110(13):4385–4395. [DOI] [PubMed] [Google Scholar]
- 12.Patel AB, Vellore NA, Deininger MW. New strategies in myeloproliferative neoplasms: the evolving genetic and therapeutic landscape. Clin Cancer Res. 2016;22(5):1037–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasine JP, Schiller GJ. Emerging strategies for high-risk and relapsed/-refractory acute myeloid leukemia: novel agents and approaches currently in clinical trials. Blood Rev. 2015;29(1):1–9. [DOI] [PubMed] [Google Scholar]
- 14.Dang L, Yen K, Attar E. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27(4):599–608. [DOI] [PubMed] [Google Scholar]
- 15.Dietrich S, Pircher A, Endris V, et al. BRAF inhibition in hairy cell leukemia with low-dose vemurafenib. Blood. 2016;127(23):2847–2855. [DOI] [PubMed] [Google Scholar]
- 16.Nazha A, Radivoyevitch T, Thota S, et al. 20 Somatic mutational model to predict response to hypomethylating agents in myelodysplastic syndromes. Leukemia Res. 2015;39:S9. [Google Scholar]
- 17.Bräuninger A, Blau W, Kunze K, et al. Targeted next-generation sequencing is a sensitive tool for differential diagnosis of myelodysplastic syndromes in bone marrow trephines. J Mol Diagn. 2018;20(3):344–354. [DOI] [PubMed] [Google Scholar]
- 18.Gnanaraj J, Parnes A, Francis CW, Go RS, Takemoto CM, Hashmi SK. Approach to pancytopenia: diagnostic algorithm for clinical hematologists. Blood Rev. 2018;32(5):361–367. [DOI] [PubMed] [Google Scholar]
- 19.Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373(1):35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langemeijer SM, Kuiper RP, Berends M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41(7):838–842. [DOI] [PubMed] [Google Scholar]
- 24.Papaemmanuil E, Gerstung M, Malcovati L, et al. Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium, Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walter MJ, Ding L, Shen D, et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia. 2011;25(7):1153–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abdel-Wahab O, Pardanani A, Patel J, et al. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia. 2011;25(7):1200–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graubert TA, Shen D, Ding L, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44(1):53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69. [DOI] [PubMed] [Google Scholar]
- 29.Reinig E, Yang F, Traer E, et al. Targeted next-generation sequencing in myelodysplastic syndrome and chronic myelomonocytic leukemia aids diagnosis in challenging cases and identifies frequent spliceosome mutations in transformed acute myeloid leukemia. Am J Clin Pathol. 2016;145(4):497–506. [DOI] [PubMed] [Google Scholar]
- 30.Pierola AA, Montalban-Bravo G, Chamseddine AN, et al. Impact of the next-generation sequencing panel on treatment choice in patients with myelodysplastic Syndrome. Am Soc Hematology. 2016;128:4340. [Google Scholar]
- 31.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. [DOI] [PubMed] [Google Scholar]
- 32.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
- 33.Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100(7):2292–2302. [DOI] [PubMed] [Google Scholar]
- 34.Valent P, Orazi A, Steensma DP, et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget. 2017;8(43):73483–73500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood. 2014;124(17):2698–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshizato T, Dumitriu B, Hosokawa K, et al. Chronological analysis of clonal evolution in acquired aplastic anemia. Am Soc Hematol. 2014;124:253. [Google Scholar]
- 39.Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2012;30(27):3376–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moyo TK, Savona MR. Molecular testing in patients with suspected myelodysplastic syndromes. Curr Hematol Malignancy Rep. 2016;11(6):441–448. [DOI] [PubMed] [Google Scholar]
- 42.Cargo CA, Rowbotham N, Evans PA, et al. Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood. 2015;126(21):2362–2365. [DOI] [PubMed] [Google Scholar]
- 43.Bejar R, Papaemmanuil E, Haferlach T, et al. Somatic mutations in MDS patients are associated with clinical features and predict prognosis independent of the IPSS-R: analysis of combined datasets from the International working group for prognosis in MDS-molecular committee. Am Soc Hematol. 2015;126:907. [Google Scholar]
- 44.Nazha A, Al-Issa K, Hamilton B, et al. Adding molecular data to prognostic models can improve predictive power in treated patients with myelodysplastic syndromes. Leukemia. 2017;31(12):2848–2850. [DOI] [PubMed] [Google Scholar]
- 45.Nazha A, Narkhede M, Radivoyevitch T, et al. Incorporation of molecular data into the revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia. 2016;30(11):2214–2220. [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Manero G, Shan J, Faderl S, et al. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008;22(3):538–543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.