

Abstract
Background and Objective
Individuals with biomarker evidence of β-amyloid (Aβ) deposition are increasingly being enrolled in clinical treatment trials but there is a need to identify markers to predict which of these individuals will also develop tau deposition. We aimed to determine whether Aβ-positive individuals can remain tau-negative for at least 5 years and identify characteristics that could distinguish between these individuals and those who develop high tau within this period.
Methods
Tau PET positivity was defined using a Gaussian mixture model with log-transformed standard uptake value ratio values from 7 temporal and medial parietal regions using all participants in the Alzheimer's Disease Neuroimaging Initiative (ADNI) with flortaucipir PET. Tau PET scans were classified as normal if the posterior probability of elevated tau was less than 1%. Aβ PET positivity was defined based on ADNI cutpoints. We identified all Aβ-positive individuals from ADNI who had normal tau PET more than 5 years after their first abnormal Aβ PET (amyloid with low tau [ALT] group) and all Aβ-positive individuals with abnormal tau PET within 5 years (biomarker AD). In a case–control design, logistic regression was used to model the odds of biomarker AD vs ALT accounting for sex, age, APOE ε4 carriership, Aβ Centiloid, and hippocampal volume.
Results
We identified 45 individuals meeting criteria for ALT and 157 meeting criteria for biomarker AD. The ALT group had a lower proportion of APOE ε4 carriers, lower Aβ Centiloid, larger hippocampal volumes, and more preserved cognition, and were less likely to develop dementia, than the biomarker AD group. APOE ε4, higher Aβ Centiloid, and hippocampal atrophy were independently associated with increased odds of abnormal tau within 5 years. A Centiloid value of 50 effectively discriminated biomarker AD and ALT with 80% sensitivity and specificity. The majority of the ALT participants did not develop dementia throughout the 5-year interval.
Discussion
Aβ-positive individuals can remain tau-negative for at least 5 years. Baseline characteristics can help identify these ALT individuals who are less likely to develop dementia. Conservative Aβ cutpoints should be utilized for clinical trials to better capture individuals with high risk of developing biomarker AD.
The National Institute on Aging–Alzheimer's Association1 and the International Working Group–22 criteria both support biomarker-defined criteria for Alzheimer disease (AD). A diagnosis of AD is supported by biomarker evidence of both β-amyloid (Aβ) and tau accumulation with the proviso that early on, Aβ, but not tau, may be detected. In such instances of Aβ without tau detection, the assumption is that tau accumulation will develop if the individual stays alive long enough; such cases are considered to have Alzheimer pathologic change.1 Given the importance of identifying and eventually treating patients as early as possible in the disease process, there has been a drive towards clinical trials enrolling cognitively normal individuals based only on positive Aβ PET status.3 While this idea is certainly reasonable, such an approach runs the risk of enrolling individuals who are many years away from developing tau or dementia. In addition, individuals with Aβ accumulation and cognitive impairment are now eligible for treatment with aducanumab.4 Yet autopsy studies have shown that some individuals with Aβ senile plaques die with minimal or absent tau neurofibrillary tangles,5,6 meeting neuropathologic criteria for low probability of AD.7,8 Hence, there is a need to identify characteristics/biomarkers that can help predict which Aβ-positive individuals have a low likelihood of developing tau deposition over a reasonable period for clinical trial study design or for exclusion criteria.
In this study, we use the Alzheimer's Disease Neuroimaging Initiative (ADNI) database, a multicenter neuroimaging study consisting of participants with the full range of cognitive status who have had molecular PET imaging to detect Aβ and paired helical filament tau in vivo. Using ADNI data allows us to investigate the relationship between Aβ and tau over time. We were specifically interested in identifying and characterizing participants with a positive Aβ status according to accepted criteria who had little evidence of tau accumulation 5 years later. We refer to such individuals who were Aβ-positive with low tau more than 5 years later as amyloid+ with low tau (ALT). We hypothesized a priori that ALT existed and set out to investigate whether there would be characteristics that could distinguish between ALT and Aβ-positive individuals who develop high tau over time.
Methods
Participants
All individuals for the study were identified from the ADNI database9 based on data downloaded on October 28, 2020. ADNI was launched in 2003 as a public–private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early AD.
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by the institutional review board at each of the participating ADNI centers and written informed consent was obtained for all participants (ClinicalTrials.gov registry numbers: ADNI GO: NCT01078636; ADNI 2: NCT0123197; ADNI 3: NCT02854033).
Defining Tau PET Positivity
We identified all individuals from ADNI (ADNI 2, ADNI 3, ADNI GO) who had a flortaucipir PET scan regardless of Aβ status. The ADNI acquisition protocol for flortaucipir is available online.9 This dataset was used as a training dataset to define tau-negative and tau-positive scans using Gaussian mixture models. Flortaucipir standardized uptake value ratios (SUVRs) from 7 regions of interest (ROIs) (entorhinal cortex, inferior temporal gyrus, parahippocampal gyrus, fusiform gyrus, middle temporal gyrus, isthmus cingulate, and precuneus) were analyzed. These ROIs were selected to capture regions that show elevated flortaucipir early in AD and preclinical AD.10-13 Each flortaucipir scan was coregistered to MRI and FreeSurfer-defined ROIs were calculated based on mean uptake over 75–105 minutes postinjection normalized by a mean inferior cerebellar gray matter uptake.14,15
Fitting a univariate Gaussian mixture model to a single biomarker is a common approach to identifying normal and abnormal subgroups with an established record in CSF biomarkers.16 More recently, a 2D Gaussian mixture model was used to cluster individuals according to both CSF Aβ42 and p-tau.17 Our approach in the current analysis is conceptually the same except instead of fitting a mixture model using 1 or 2 continuous measures, we fit a model using log-transformed SUVRs from 7 regions. This mixture model can be interpreted as describing the 7D space of log-transformed tau PET SUVRs as consisting of 2 or more 7D point clouds, with each cloud modeled as multivariate normal (i.e., each with 7 means, 7 variances, and 21 covariances).
One advantage of this parametric approach to clustering is that it provides an explicit posterior probability that a particular set of regional SUVR values is in each group.18 A classification of abnormal in a mixture model context is usually based on a posterior probability cutoff of >0.5. That is, if a value is more likely than not to be from the abnormal distribution, it is considered abnormal. For this analysis, we wanted to be much more conservative in defining a normal tau PET scan and much more liberal in defining an abnormal tau PET scan. Therefore, we classified tau PET scans as normal if the posterior probability of elevated tau was less than 1%. With this cutoff, only tau PET scans well outside the elevated tau distribution were considered normal.
We used the R language and environment for statistical computing for all analyses and the mclust package for mixture modeling.
The Bayesian information criterion (BIC) was used to determine the optimum number of clusters. As a sensitivity analysis to evaluate the influence of individual regions, we refit the mixture model 7 times, leaving 1 region out at a time. We then used kappa statistics to compare the agreement between the full model and each 6-region model in terms of classification of each individual.
Identifying ALT Individuals
We identified all individuals in ADNI GO, ADNI2, or ADNI3 who were Aβ-positive on PET (either with florbetapir [18F-AV-45] or [18F]florbetaben). The ADNI acquisition protocols for florbetapir and florbetaben are available online.9 Global composite SUVRs were downloaded from the ADNI dataset and Aβ PET values were converted to the Centiloid scale using methods described on the ADNI website. The ADNI processing pipeline includes coregistering the Aβ PET scans to the structural MRI, FreeSurfer (V5.3.0) was used to generate regional data, and the global composite SUVRs were calculated by referencing cortical uptake to the whole cerebellum.15,19 Aβ PET positivity was defined based on the ADNI cutpoints for florbetapir (>1.11) and florbetaben (>1.08),9 which map onto a Centiloid cutpoint of 23.
We then identified all individuals who had undergone flortaucipir PET. Because of the relatively recent introduction of tau PET, extensive prospective follow-up is limited. Still, because many ADNI participants with Aβ PET have extensive follow-up, we were able to determine tau status at the 5-year mark for a subset of individuals. Setting time zero as an ADNI participant's first abnormal Aβ PET, individuals who had no abnormal tau PET scan within 5 years and a normal tau PET scan after 5 years were classified as ALT. As a comparison group, we identified all individuals who had an abnormal tau PET scan—that is, a posterior probability ≥1%—within 5 years (biomarker AD group). Note that individuals with an indeterminate tau status at 5 years are not included in our analysis. For example, someone having an abnormal tau PET at 7 years cannot be assumed to have qualified as biomarker AD at 5 years.
The classification of individuals is depicted graphically in Figure 1 for 3 ALT scenarios, 3 biomarker AD scenarios, and 3 indeterminate scenarios. In the ALT scenarios, a normal tau PET scan at least 5 years after the first abnormal Aβ PET implies normal tau PET levels at the 5-year mark. For the biomarker AD scenarios, an abnormal tau PET scan within 5 years of the first abnormal Aβ PET implies elevated tau PET at or before the 5-year mark. The indeterminate scenarios result from either insufficient follow-up or from ambiguity as to the individual's status at the 5-year mark.
Figure 1. Graphical Depiction of Scenarios Informing the ALT Group, the Biomarker AD Group, and the Indeterminate Group.
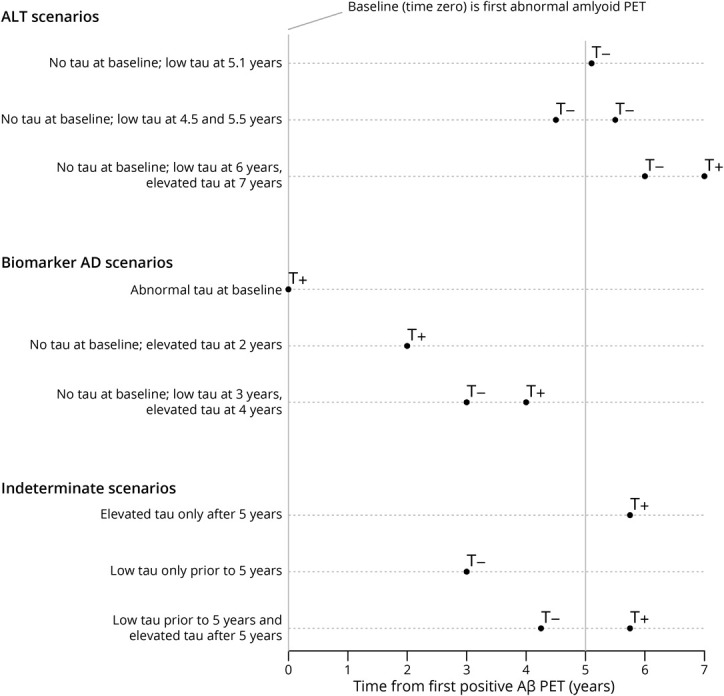
Each row in the plot represents a prototypical scenario. The origin on the horizontal axis is the time of the individual's first abnormal amyloid PET scan. The symbol T− denotes a normal tau PET scan while T+ denotes an abnormal tau PET scan. The amyloid+ with low tau (ALT) and biomarker Alzheimer disease (AD) scenarios show how the individual's tau status at the 5-year mark can be inferred. The indeterminant scenarios show how the data do not allow the individual's status at 5 years to be inferred. Aβ = β-amyloid.
Statistical Analysis
We used χ2 and Wilcoxon rank-sum tests to compare the ALT and biomarker AD groups at the time of their first abnormal Aβ PET scan in univariate analysis. We used logistic regression to model the odds of biomarker AD vs ALT accounting for sex, age, APOE ε4 carriership, amyloid Centiloid, FreeSurfer-based hippocampal volume, and total intracranial volume. Age, Centiloid, and volume measurements were from the time of the first abnormal Aβ PET. To understand the relationship between age, amyloid Centiloid, and ALT vs biomarker AD, we used quantile regression. This allowed us to model Centiloid values by age in a way that was robust to a few high Centiloid values. We performed an area under the receiver operating characteristic curve (AUC) analysis of Centiloids to identify a cutpoint that maximized sensitivity and specificity. We were also interested in further understanding the ALT group and compared individuals with higher (>36) vs lower (≤36) Centiloid values split at the median Centiloid of 36.
Data Availability
Qualified researchers may obtain access to all de-identified ADNI data from the ADNI website.9
Results
Defining Tau Positivity in the Training Dataset
The training dataset consisted of 774 individuals who had undergone flortaucipir PET in ADNI (Figure 2). The characteristics of these individuals are shown in eTable 1 (links.lww.com/WNL/B897). The BIC from the univariate Gaussian mixture model improved when increasing from 1 cluster to 2 clusters (one cluster, −13,812; 2 clusters, −15,940), but did not improve for additional clusters (3 clusters, −15,899; 4 clusters, −15,748; 5 clusters −15,607), and so 2 clusters was found to be optimal. In the sensitivity analysis, kappa values ranged from κ = 0.88 with a model omitting the fusiform to κ = 0.97 with a model omitting the parahippocampal gyrus, indicating a robustness of the clustering and that no region was particularly influential. Figure 3 shows the distribution of posterior probabilities of abnormal tau PET among 774 individuals in the training dataset. The large number of individuals with posterior probabilities <1% or >99% indicates very good separation between the clusters.
Figure 2. Flowchart Illustrating the Selection of the Training Dataset and the ALT and Biomarker AD Groups.

Aβ = β-amyloid; AD = Alzheimer disease; ADNI = Alzheimer's Disease Neuroimaging Initiative; ALT = amyloid+ with low tau.
Figure 3. Histogram of Posterior Probability of Abnormal Tau PET in the Training Dataset.
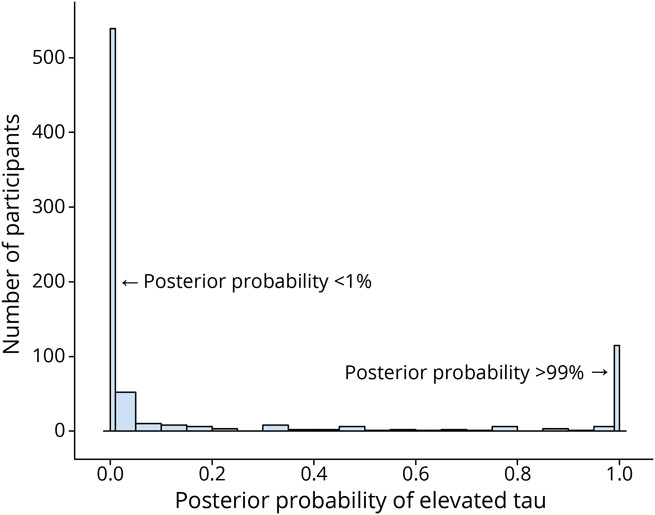
The distribution of the posterior probability of abnormal tau PET in the tau PET training dataset (n = 774) is summarized with a histogram. The first bin in the histogram tabulates cases with a posterior probability <0.01. The last bin tabulates cases with a posterior probability >0.99.
Baseline Characteristics of the ALT and Biomarker AD Cases
A flowchart depicting the identification of the ALT and biomarker AD groups is shown in Figure 2. From a total of 857 individuals with positive Aβ PET, 361 had undergone flortaucipir PET. From these, 45 individuals were identified who met criteria for the ALT group, while 157 met the criteria for biomarker AD by 5 years. The median time from first positive Aβ PET to tau PET in the ALT group was 7.1 years (range 5.0–8.6 years). The median Centiloid at the time of first positive Aβ PET was significantly lower in the ALT group (36; interquartile range [IQR] 29–44) compared to the biomarker AD group (83; IQR 58–109) (Figure 4 and Table 1), and Centiloid tended to increase with age in the ALT group but decrease with age in the biomarker AD group (Figure 4). The ALT group had a lower proportion of APOE ε4 carriers compared to biomarker AD (31% vs 67%; p < 0.001) and was significantly less likely to have a dementia diagnosis at first Aβ PET scan compared to the biomarker AD group (2% vs 22%; p < 0.001). The majority (58%) of individuals in the ALT group were cognitively normal at the time of first positive Aβ PET, with 40% having a diagnosis of MCI. In keeping with the spectrum of clinical diagnoses, the ALT group also performed better on the Mini-Mental State Examination (MMSE) and Alzheimer's Disease Assessment Scale–cognitive subscale (ADAS-Cog) tests and had more preserved hippocampal volumes, compared with biomarker AD (Table 1). There were no clear differences identified between the ALT and biomarker AD groups in age at baseline, sex, education, or vascular measures (Table 1).
Figure 4. Centiloid at First Abnormal Amyloid PET Scan Vs Age.
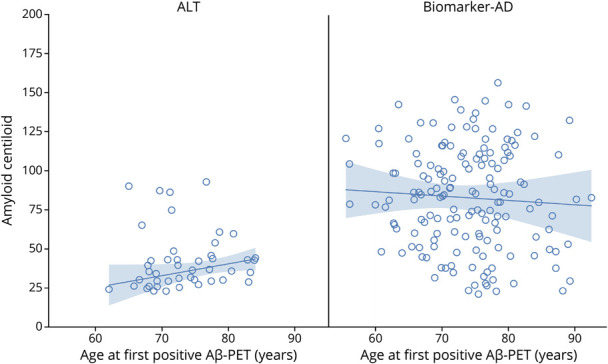
The trend line and 95% CI represent the estimated median Centiloid by age based on quantile regression. This model is used because the Centiloid values in the amyloid+ with low tau (ALT) group are positively skewed. Aβ = β-amyloid; AD = Alzheimer disease.
Table 1.
Demographic, Imaging, and Clinical Features of the ALT and Biomarker AD Groups Overall and Among the Subset That Was Cognitively Normal at Baseline

Among the subset of 72 individuals who were cognitively normal at baseline, 36% were in the ALT group and 64% in the biomarker AD group (Table 1). As with the full sample, the ALT group had a lower proportion of APOE ε4 carriers (27% vs 56%; p = 0.04), lower amyloid levels (median Centiloid 36 vs 72; p < 0.001), and better cognitive performance on the ADAS-Cog (median 5 vs 7; p = 0.006).
The logistic regression model showed that APOE ε4 status was associated with a 3-fold increase in the odds (odds ratio [OR] 3.0, 95% CI 1.2–7.5; p = 0.02) of being biomarker AD (i.e., having elevated tau within 5 years from baseline Aβ PET) (Table 2). A 10-unit higher Centiloid was associated with a 70% increase in the odds of biomarker AD (OR 1.7, 95% CI 1.4–2.1; p < 0.001) and a 0.5 mL smaller hippocampus was independently associated with a 50% increase in the odds of biomarker AD (OR 1.5, 95% CI 1.2–2.1; p = 0.002). Amyloid Centiloid discriminated between the 2 groups with an AUC of 0.85 (95% CI 0.77–0.90) and a Centiloid value of 50 effectively discriminates between biomarker AD and ALT with both 80% sensitivity and 80% specificity. Among the subset of participants who were cognitively normal at baseline, OR effect sizes appeared similar but with wider CIs. For example, a 10-unit higher level of Centiloid was associated with an OR of 2.7 (95% CI 1.6–5.5; p = 0.001) and APOE ε4 carriers also appeared to have an elevated odds of biomarker AD (OR 2.5, 95% CI 0.7–9.2; p = 0.14).
Table 2.
Results From Logistic Regression Model Showing Odds of Elevated Tau Within 5 Years From Baseline β-Amyloid PET Overall and Among the Subset That Was Cognitively Normal at Baseline

Dividing the ALT group according to the median Centiloid value of 36, those above 36 (n = 23) tended to be older (median 75 vs 71 years) and have a much higher proportion of APOE ε4 carriers (43% vs 18%). The distribution of clinical diagnoses, sex, and education was similar between the 2 groups.
Longitudinal Characteristics of the ALT Individuals
Figure 5 displays all available longitudinal clinical diagnosis information and tau PET for each ALT individual. The clinical diagnosis recorded at the first Aβ PET remained stable or reverted to a more normal state (i.e., MCI reverting to cognitively normal) in the majority of cases (34/45 [76%]). Of the remaining 11 cases, 7 converted to a dementia diagnosis over time and one obtained a dementia diagnosis before reverting to MCI. Of the 9 ALT individuals who received a dementia diagnosis during follow-up, 8 were considered to have dementia due to AD and 1 was determined to have encephalitis. Only 1 individual who obtained a diagnosis of dementia was an APOE ε4 carrier. There were 23 individuals (51%) in the ALT group with more than 1 tau PET scan after 5 years (Figure 5). With only one exception, these individuals continued to have a very low posterior probability of elevated tau.
Figure 5. Follow-up Information for ALT Cases.

(A) Clinical diagnosis over time for 45 amyloid+ with low tau (ALT) cases numbered sequentially. (B) Posterior probability of elevated tau over time for a subset of 23 individuals identified by ALT case number who had more than 1 tau PET scan at least 5 years after the first abnormal amyloid scan. For both panels, zero on the X-axis corresponds to the time of the individual's first abnormal β-amyloid (Aβ) scan. CN = cognitively normal; MCI = mild cognitive impairment.
Discussion
Our findings demonstrate that a relatively large group of individuals who are Aβ-positive based on generally accepted criteria have subthreshold/very low levels of tau on tau PET for at least 5 years after their first Aβ-positive PET scan. Previous studies have reported on the existence of Aβ-positive, tau-negative individuals,20,21 but this study examines individuals who remain tau PET-negative for at least 5 years. We show that these ALT individuals are less likely to have AD-related clinical, neuroimaging, and genetic features compared to biomarker AD individuals and that the majority do not develop dementia over at least 5 years of follow-up.
The ALT individuals were less likely to have dementia at the baseline Aβ PET compared with Aβ-positive individuals who became tau-positive within 5 years (biomarker AD) and performed better on tests of general cognitive function. Their hippocampal volumes were also more preserved. These results are perhaps unsurprising given the observed close relationship among tau deposition, neurodegeneration, and subsequent clinical decline in AD,22-26 and the lack of tau deposition in this cohort. These findings concord with previous studies that have found greater neurodegeneration in Aβ- and tau-positive individuals compared with Aβ-positive but tau-negative individuals.23,27 However, another striking feature of the ALT group was the low frequency of the APOE ε4 allele, which was only observed in 31% of these individuals. This is significantly lower than the APOE ε4 frequency observed in the biomarker AD group, is lower than the frequency typically observed in autopsy-confirmed AD,26 and is more in line with the frequency that has been observed in cognitively normal cohorts.28 This relatively low frequency of APOE ε4 suggests that these individuals do not have a high risk of developing AD. In fact, APOE ε4 status was associated with a 3-fold increase in the odds of developing tau deposition within 5 years from first positive Aβ PET. Previous studies have observed APOE ε4 frequencies of 49%20 and 43%21 in Aβ-positive, tau-negative cohorts, but those cohorts were not followed longitudinally and it is likely that many individuals did develop high tau within 5 years. APOE ε4 allele has been associated with higher rates of tau accumulation in cognitively impaired, but not cognitively unimpaired, cohorts.28
The ALT group had lower median Centiloid compared with the biomarker AD group, with a 10-unit increase in Centiloid associated with a 1.7-fold increase in the odds of developing tau deposition over 5 years from first positive Aβ PET. This fits with a previous study that found that higher baseline Aβ PET was associated with a higher rate of subsequent tau accumulation in cognitively normal and abnormal cohorts.28 Within the ALT group, some individuals started with a higher than median Centiloid of more than 36 units and these individuals had a higher APOE ε4 frequency of 43%, suggesting that the ALT cohort is heterogeneous in regards to the risk of developing AD but that the risk will be higher in those who start off with higher Aβ PET burden.
Sex differences in brain structure and function as well as a higher prevalence of dementia in women make sex an increasingly important consideration in AD research.29 We did not observe clear sex differences between ALT and biomarker AD but larger samples will be needed to clarify the association if effect sizes are moderate or if sex modifies the effect of other factors such as APOE.30
In assessment of the longitudinal trajectory of the ALT individuals, it was clear that the risk of developing dementia remained low over the 5 years after the first Aβ PET scan and even up to 8 years after in some individuals. The tau PET SUVR in 1 ALT individual did eventually increase, climbing to a 30% posterior probability of elevated tau more than 7 years after the first Aβ-positive PET scan. This person remained cognitively normal. One of the ALT individuals had dementia at the baseline Aβ-positive PET and a further 7 individuals developed dementia during the course of follow-up. However, none of these individuals showed an elevated tau PET profile and so the biological underpinnings of their dementia are unclear. There was no particular relationship between those who developed dementia and age, and only one of them was an APOE ε4 carrier. The median Centiloid at the first Aβ-positive scan was 43 vs 35 for those who did not obtain a dementia diagnosis. MCI was observed in 40% of the ALT cohort at first positive Aβ PET, and MCI developed in a handful of individuals who were cognitively normal at baseline. However, we also had individuals who reverted from a diagnosis of MCI back to cognitively normal, and even one from a diagnosis of dementia to normal, a phenomenon that has been previously reported.31 It is possible that the individuals with dementia or MCI have another non-Aβ neurodegenerative disease, such as diffuse Lewy body disease or TDP-43 proteinopathy; a nonpaired helical filament tauopathy, such as diffuse argyrophilic grain disease; or a non-neurodegenerative disease, such as vascular disease.32-37 However, we cannot rule out the fact that these individuals may have low levels of paired helical filament tau in the brain despite normal tau PET levels, as it is clear that tau PET is not sensitive enough to detect low levels of neurofibrillary tangle pathology.38-41
It remains unclear whether and how many of the individuals in the ALT group will go on to develop high tau uptake and eventually meet criteria for biomarker-confirmed AD. Follow-up in this cohort was relatively long, up to 8 years in many cases, but the development of tau deposition could take longer in some cases. One study that assessed longitudinal Aβ PET suggested that it could take 14 years from the first detection of Aβ on PET to the onset of MCI,42 and another study suggested it could be up to 19 years between the first detection of Aβ and the first detection of tau on PET.43 However, the individuals in our study may have already been Aβ-positive for many years, perhaps even for 20 years before entering ADNI. These findings have important implications for studies and clinical treatment trials that define preclinical AD based on Aβ PET positivity. Clinical treatment trials that aim to enrich their cohort with individuals who are likely to develop tau deposition and hence AD within 5 years of trial enrollment should select individuals with higher Centiloid values and smaller hippocampal volumes; this would likely also enrich for APOE ε4 carriers. Our findings also beg the question of whether the current Aβ PET cutpoints need to be refined, and increased, to truly capture preclinical AD. A previous study came to a similar conclusion and suggested that recruiting individuals into trials with a Centiloid of 68 or higher would be appropriate to enrich for people who will likely show tau accumulation over time.44 In our analysis, a Centiloid of 50 was sensitive and specific for distinguishing between biomarker AD and ALT. The choice of cutpoint should, however, depend on the context, with higher cutpoints most appropriate for clinical trials targeting tau and lower cutpoints most appropriate when more inclusiveness is required. The inclusion criteria for our study of 23 Centiloid units is already higher than the cutpoint range of between 15 and 18.5 that has been proposed as optimal to predict future Aβ accumulation and cognitive decline.45 Whereas these results from ADNI, a convenience sample, may not generalize to the entire population,46 the study protocols in ADNI were designed to mirror recruitment mechanisms in clinical treatment trials.
The findings of our study are strengthened by the fact that we required ALT individuals to have at least 5 years of follow-up since the Aβ PET, and many individuals had follow-up for 8 years. Further follow-up will be necessary to determine the fate of these individuals, specifically which participants may still go on to develop biomarker AD and dementia. Our definition of tau positivity was based on a clustering approach that examined tau uptake in 7 regions with no one region particularly important in the classification. We used an extremely conservative cutpoint to define tau-negative or low individuals by requiring that individuals have less than 1% posterior probability of being in the high tau cluster, which was based on 7 of the most sensitive regions for tau deposition. The ALT and biomarker AD individuals were included in the training dataset as it was not possible to exclude them up front without a means to determine whether the tau PET was positive or negative.
Our definition of biomarker AD was designed to be extremely sensitive and prevent false-negatives whereby an individual with some indications of abnormal tau PET was called normal. A consequence of this definition is that the biomarker AD group includes individuals with a low posterior probability of abnormal tau, that is, any value >1%. If we compare ALT to a biomarker AD group consisting only of individuals with >99% posterior probability of abnormal tau—essentially contrasting the 2 extremes—the differences in terms of APOE, Centiloid, and hippocampal volume increase. In particular, in this case APOE ε4+ is associated with more than a 9-fold increase in the odds of biomarker AD. However, we do not think it is appropriate to omit potentially equivocal cases from our analysis. Although our study could be considered limited by the lack of autopsy confirmation, in reality, biomarker defined AD is based on PET and CSF, not pathologic confirmation. Whereas in this study we focused on PET, it will be interesting for future studies to examine whether a similar ALT group is identified with CSF or blood biomarkers because although they have good concordance with PET47,48 it is possible they could yield different results. The relatively limited tau PET follow-up currently available in ADNI, and hence the small number of transitions occurring between normal and abnormal tau levels, meant a survival analysis or time-to-event approach was not feasible but this approach could prove powerful in the future.
We have demonstrated that a significant proportion of Aβ−positive individuals have absent or low tau burden at least 5 years out with characteristics that differ from those who have biomarker-define AD. The findings have important consequences to the entire AD field ranging from diagnosis to clinical trials.
Acknowledgment
The authors thank Nha Trang Thu Pham (Mayo Clinic, Rochester, MN) for running the FreeSurfer pipeline to generate hippocampal volumes.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADAS-Cog
Alzheimer's Disease Assessment Scale–cognitive subscale
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- ALT
amyloid+ with low tau
- AUC
area under the receiver operating characteristic curve
- BIC
Bayesian information criterion
- IQR
interquartile range
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- OR
odds ratio
- ROI
region of interest
- SUVR
standardized uptake value ratio
Appendix. Authors

Study Funding
K.A.J. and J.W. are funded by NIH grants R01-AG50603, R01-AG37491, R01-DC12519, R01-DC14942, R01-NS89757, and RF1-NS112153. Data collection and sharing for this project was funded by ADNI (NIH grant U01 AG024904) and Department of Defense ADNI (award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and contributions from the following: AbbVie; Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol Myers Squibb; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health. The grantee organization is the Northern California Institute for Research and Education and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The sponsor played no role in study design; collection, analysis, or interpretation of data; writing of the report; or the decision to submit the article for publication.
Disclosure
K.A. Josephs receives funding from the NIH. S.D. Weigand reports no disclosures relevant to the manuscript. J.L. Whitwell receives funding from the NIH. Go to Neurology.org/N for full disclosures.
References
- 1.Jack CR Jr., Bennett DA, Blennow K, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. 2014;13(6):614-629. [DOI] [PubMed] [Google Scholar]
- 3.Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6:228fs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunn B, Stein P, Cavazzoni P. Approval of aducanumab for Alzheimer disease: the FDA's perspective. JAMA Intern Med. 2021;181(10):1276-1278. [DOI] [PubMed] [Google Scholar]
- 5.Knopman DS, Parisi JE, Salviati A, et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol. 2003;62(11):1087-1095. [DOI] [PubMed] [Google Scholar]
- 6.Wennberg AM, Whitwell JL, Tosakulwong N, et al. The influence of tau, amyloid, alpha-synuclein, TDP-43, and vascular pathology in clinically normal elderly individuals. Neurobiol Aging. 2019;77:26-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8(1):1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alzheimer's Disease Neuroimaging Initiative. Alzheimer's Disease Neuroimaging Initiative website. adni.loni.usc.edu/.
- 10.Mishra S, Gordon BA, Su Y, et al. AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: defining a summary measure. Neuroimage. 2017;161:171-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lockhart SN, Schöll M, Baker SL, et al. Amyloid and tau PET demonstrate region-specific associations in normal older people. Neuroimage. 2017;150:191-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Josephs KA, Tosakulwong N, Graff-Radford J, et al. MRI and flortaucipir relationships in Alzheimer's phenotypes are heterogeneous. Ann Clin Transl Neurol. 2020;7(5):707-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowe VJ, Bruinsma TJ, Min HK, et al. Elevated medial temporal lobe and pervasive brain tau PET signal in normal participants. Alzheimers Dement. 2018;10:210-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maass A, Landau S, Baker SL, et al. Comparison of multiple tau PET measures as biomarkers in aging and Alzheimer's disease. Neuroimage. 2017;157:448-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jagust WJ, Landau SM, Koeppe RA, et al. The Alzheimer's disease neuroimaging initiative 2 PET core: 2015. Alzheimers Dement. 2015;11(7):757-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Meyer G, Shapiro F, Vanderstichele H, et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol. 2010;67:949-956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Harten AC, Wiste HJ, Weigand SD, et al. CSF biomarkers in Olmsted County: evidence of 2 subclasses and associations with demographics. Neurology. 2020;95:e256–e267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLachlan G, Peel D. Finite Mixture Models. John Wiley & Sons, Inc.; 2000. [Google Scholar]
- 19.Landau SM, Fero A, Baker SL, et al. Measurement of longitudinal beta-amyloid change with 18F-florbetapir PET and standardized uptake value ratios. J Nucl Med. 2015;56:567-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo T, Korman D, Baker SL, Landau SM, Jagust WJ. Alzheimer's Disease Neuroimaging Initiative. Longitudinal cognitive and biomarker measurements support a unidirectional pathway in Alzheimer's disease pathophysiology. Biol Psychiatry. 2021;89(8):786-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jack CR Jr., Wiste HJ, Therneau TM, et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA. 2019;321:2316-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 2019;76(8):915-924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrison TM, Du R, Klencklen G, Baker SL, Jagust WJ. Distinct effects of beta-amyloid and tau on cortical thickness in cognitively healthy older adults. Alzheimers Dement. 2021;17(7):1085-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitwell JL, Josephs KA, Murray ME, et al. MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology. 2008;71(10):743-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Josephs KA, Martin PR, Weigand SD, et al. Protein contributions to brain atrophy acceleration in Alzheimer's disease and primary age-related tauopathy. Brain. 2020;143(11):3463-3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitwell JL, Dickson DW, Murray ME, et al. Neuroimaging correlates of pathologically defined subtypes of Alzheimer's disease: a case-control study. Lancet Neurol. 2012;11(10):868-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jack CR Jr, Wiste HJ, Weigand SD, et al. Age-specific and sex-specific prevalence of cerebral beta-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50-95 years: a cross-sectional study. Lancet Neurol. 2017;16:435-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jack CR, Wiste HJ, Weigand SD, et al. Predicting future rates of tau accumulation on PET. Brain. 2020;143(10):3136-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer's disease: assessing sex and gender differences. Clin Epidemiol. 2014;6:37-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altmann A, Tian L, Henderson VW, Greicius MD, Alzheimer's Disease Neuroimaging Initiative. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol. 2014;75(4):563-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koepsell TD, Monsell SE. Reversion from mild cognitive impairment to normal or near-normal cognition: risk factors and prognosis. Neurology. 2012;79(15):1591-1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Botha H, Mantyh WG, Graff-Radford J, et al. Tau-negative amnestic dementia masquerading as Alzheimer disease dementia. Neurology. 2018;90(11):e940–e946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol. 2018;83(1):74-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buciuc M, Whitwell JL, Tosakulwong N, et al. Association between transactive response DNA-binding protein of 43 kDa type and cognitive resilience to Alzheimer's disease: a case-control study. Neurobiol Aging. 2020;92:92-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chui HC, Zarow C, Mack WJ, et al. Cognitive impact of subcortical vascular and Alzheimer's disease pathology. Ann Neurol. 2006;60(6):677-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Josephs KA, Whitwell JL, Weigand SD, et al. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol. 2014;127(6):811-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63(5):665-672. [DOI] [PubMed] [Google Scholar]
- 38.Ghirelli A, Tosakulwong N, Weigand SD, et al. Sensitivity-specificity of tau and amyloid beta positron emission tomography in frontotemporal lobar degeneration. Ann Neurol. 2020;88:1009-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lowe VJ, Lundt ES, Albertson SM, et al. Tau-positron emission tomography correlates with neuropathology findings. Alzheimers Dement. 2020;16(3):561-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fleisher AS, Pontecorvo MJ, Devous MD Sr., et al. Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol. 2020;77:829-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soleimani-Meigooni DN, Iaccarino L, La Joie R, et al. 18F-flortaucipir PET to autopsy comparisons in Alzheimer's disease and other neurodegenerative diseases. Brain. 2020;143:3477-3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jagust WJ, Landau SM, Alzheimer's Disease Neuroimaging Initiative. Temporal dynamics of beta-amyloid accumulation in aging and Alzheimer's disease. Neurology. 2021;96:e1347–e1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barthélemy NR, Li Y, Joseph-Mathurin N, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer's disease. Nat Med. 2020;26(3):398-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knopman DS, Lundt ES, Therneau TM, et al. Association of initial beta-amyloid levels with subsequent flortaucipir positron emission tomography changes in persons without cognitive impairment. JAMA Neurol. 2021;78:217-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farrell ME, Jiang S, Schultz AP, et al. Defining the lowest threshold for amyloid PET to predict future cognitive decline and amyloid accumulation. Neurology. 2021;96(4):e619–e631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitwell JL, Wiste HJ, Weigand SD, et al. Comparison of imaging biomarkers in the Alzheimer disease neuroimaging initiative and the mayo clinic study of aging. Arch Neurol. 2012;69(5):614-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid-beta PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14:1470-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative Accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2020;324(8):772-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Qualified researchers may obtain access to all de-identified ADNI data from the ADNI website.9