

Abstract
The merging of photoredox and nickel catalysis has revolutionized the field of C-C cross-coupling. However, in comparison to the development of synthetic methods, detailed mechanistic investigations of these catalytic systems are lagging. To improve the mechanistic understanding, computational tools have emerged as powerful tools to elucidate the factors controlling reactivity and selectivity in these complex catalytic transformations. Based on the reported computational studies, it appears that the mechanistic picture of catalytic systems is not generally applicable, but is rather dependent on the specific choice of substrate, ligands, photocatalysts, etc. Given the complexity of these systems, the need for more accurate computational methods, readily available and user-friendly dynamics simulation tools, and data-driven approaches is clear in order to understand at the molecular level the mechanisms of these transformations. In particular, we anticipate that such improvement of theoretical methods will become crucial to advance the understanding of excited-state properties and dynamics of key species, as well as to enable faster and unbiased exploration of reaction pathways. Further, with greater collaboration between computational, experimental, and spectroscopic communities, the mechanistic investigation of photoredox/Ni dual-catalytic reactions is expected to thrive quickly, facilitating the design of novel catalytic systems and promoting our understanding of the reaction selectivity.
Keywords: Photoredox/nickel dual catalysis, metallaphotoredox catalysis, C-C cross-coupling, olefin difunctionalization, density functional theory, molecular dynamics, reaction selectivity
Graphical Abstract

1. INTRODUCTION
Transition-metal-catalyzed C(sp2)-C(sp2) cross-coupling reactions have revolutionized the field of carbon-carbon bond formation in organic synthesis and are widely used in the pharmaceutical industry. However, the broader application of these synthetic strategies to the formation of C(sp2)-C(sp3) and C(sp3)-C(sp3) bonds remains a challenge due to, inter alia, unwanted side reactions from beta-hydride elimination and the requirement of harsh conditions (e.g., high temperature, strong bases, etc.).1 To overcome these challenges, modifications to the catalytic systems have focused on ligand design, manipulation of oxidation states, and electronic excitation.2 In this vein, the merging of nickel and photoredox catalysis has become an attractive method to construct carbon-carbon bonds. Compared to traditional cross-coupling chemistry, they not only require milder conditions but also expand the chemical space by allowing the incorporation of sp3-hybridized carbons into bioactive chemicals and drug candidates.2–9 Moreover, photoredox-mediated radical generation has acted as an effective strategy to expand the toolbox of cross-coupling partners that are rarely utilized in transition metal-catalyzed protocols (Figure 1A). In this context, a broad variety of radical precursors have found applications in photoredox and nickel dual-catalyzed cross-coupling reactions, including alkyl halides,10 carboxylic acids,4 silicates,11,12 4-alkyl-1,4-dihydropyridines (DHPs),13–15 trifluoroborates,3,16 and oxalates17 (Figure 1B). In addition, sp3-hybridized C-H bonds are also eligible as latent radical precursors in photoredox/Ni dual-catalyzed C-C cross-coupling reactions via the combination of photo-assisted hydrogen-atom-transfer (HAT) and nickel catalysts (Figure 1C).18,19 The HAT process can be realized by the utilization of an external agent (e.g., quinuclidine), electrophile (heteroatom), or an excited photocatalyst (biphenylketone, decatungstate, etc.)
FIGURE 1.
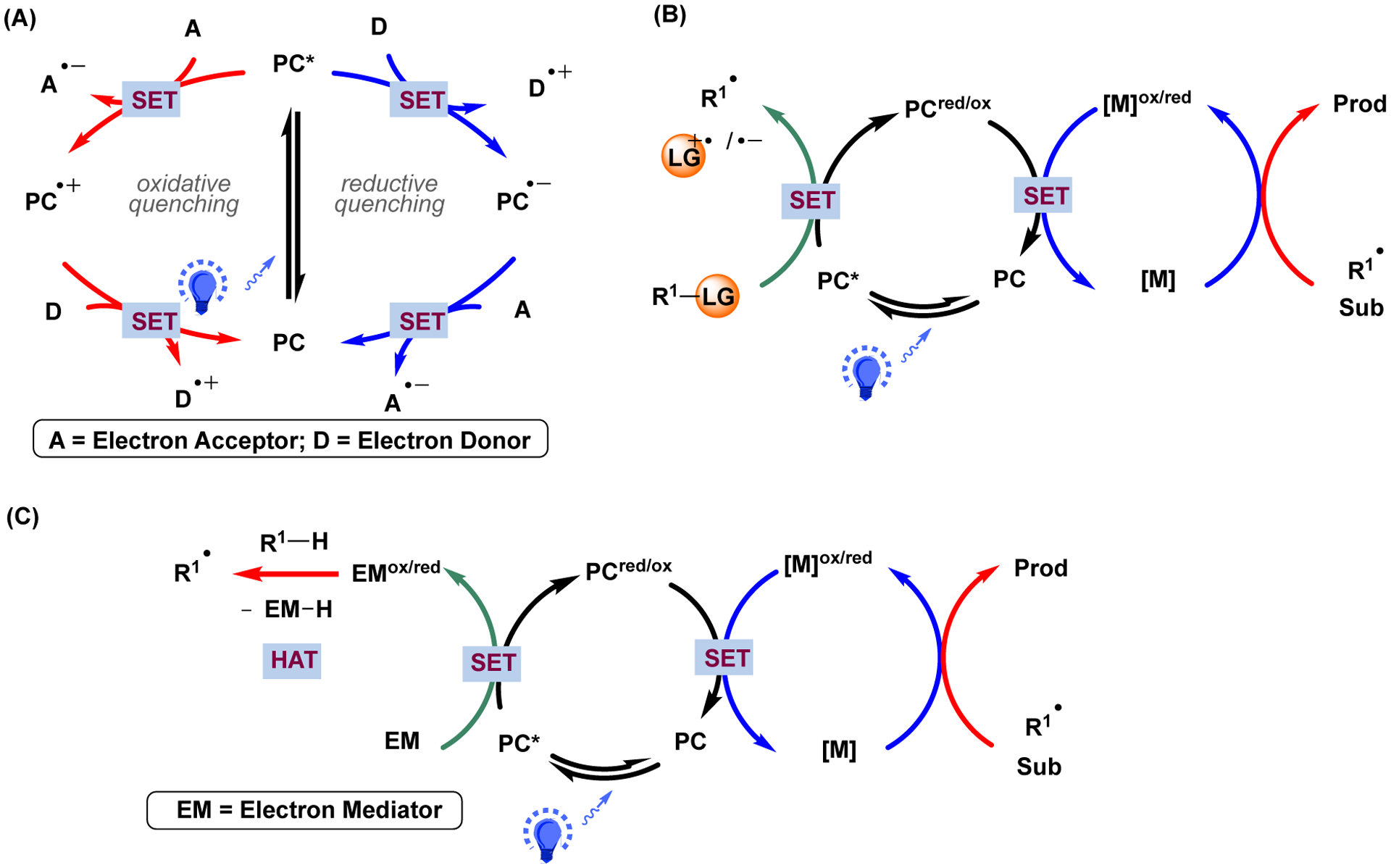
Commonly proposed mechanisms of photoredox/metal dual-catalyzed reactions. (A) Electron transfer events involved in light-irradiated conditions between the electron acceptor/donor and the excited photocatalyst (i.e., via oxidative or reductive quenching, respectively). (B) Proposed mechanism of metallaphotoredox-catalyzed carbon-carbon cross-coupling reactions, where radicals are generated via single-electron transfer between radical precursors and the excited photocatalyst. Sub = substrates; Prod = products. (C) Proposed mechanism of metallaphotoredox-catalyzed carbon-carbon cross-coupling reactions via H-atom abstraction in the presence of electron mediator.
Overall, in stark contrast to the rapid growth of synthetic methods in this area, the knowledge about the molecular-level interactions in many of these catalytic reactions is rudimentary. On the contrary, the operating mechanisms in traditional transition-metal-catalyzed cross-couplings reactions (e.g. Pd-catalyzed cross-coupling reactions) have been successfully characterized by both experimental and computational methods and, in turn, one can argue that this information has led to more rational reaction design.20,21 In all, palladium chemistry is found to mainly undergo electron transfer in pairs and change in oxidation states by two units, i.e., Pd0/PdII and PdII/PdIV catalytic cycles. However, Ni is more accessible to multiple oxidation states compared to Pd, involving Ni0, NiI, NiII, NiIII, and high-valent species NiIV, thus is more prone to undergo SET processes in the presence of suitable reagents.1 In these dual catalytic systems, both polar and radical mechanisms might operate, and multiple species could undergo single electron transfers in the reaction system. Moreover, Ni complexes could also undergo electronic excitation in light-irradiated conditions, either via energy transfer (EnT) in the presence of an excited photocatalyst or via direct excitation by light, which is usually difficult to distinguish using traditional mechanistic tools.22–29 As a consequence of the inherent complexity in these dual nickel/photoredox catalytic systems, information about the factors that control chemo-, regio- and stereoselectivity for many of these transformations remains virtually unknown.
To understand and design new photo-induced transition-metal-catalyzed reactions, novel aspects of mechanistic investigation need to be considered compared to traditional transition metal-catalyzed reactions. For example, multiple single-electron transfer events, different spin states of metal, excited-state properties of photosensitizers, and dynamical behaviors of high-energy photoexcited electronic states.30 In this context, the computational study of these highly reactive species and complex reaction mechanisms can provide a blueprint for the rational design of catalyst and reaction. In this short review, we first focus on commonly used methods (both experimental and computational) to investigate the mechanisms of these transformations. Next, we briefly examine the encountered challenges in mechanistic exploration and the potential opportunities expected to enhance the understanding of photoredox/Ni dual-catalyzed reactions and facilitate the design of new catalysts. Then, we will highlight several computational studies on the area of C(sp2)-C(sp3) bond-forming reactions mainly from our laboratory. Since we intend to provide an overview of our recent work on the computational study of the mechanisms of photoredox/Ni dual-catalyzed C(sp2)-C(sp3) cross-coupling reactions, other reviews and representative computation studies are recommended for extended interest, including the mechanism of C-heteroatom cross-coupling reactions (C-N,31–35 C-O,29,36–41 and C-S bond-forming systems42) and the excited-state properties of Ni complexes22,24,29,32,38,40,43–47. Notably, compared to C(sp2)-C(sp3) cross-coupling reactions, C-heteroatom cross-coupling reactions likely proceed via significantly different catalytic mechanisms. For example, Maseras and coworkers showed that in a C-N cross-coupling reaction, the photocatalyst was involved in the change of oxidation state of Ni via single electron transfer prior to reductive elimination, and the iodide ion was not attached to the Ni center when aryl iodide was applied as electrophile.33
Nonetheless, similar to photoredox/Ni dual catalysis, nickel-catalyzed reductive coupling and nickel-catalyzed electrochemical reactions also show access to multiple oxidation states of Ni and the involvement of single-electron transfer events.8 Therefore, mechanistic study of photoredox/Ni dual catalysis is expected to provide insight into the fundamental steps applicable to the other two fields.
2. EXPERIMENTAL AND COMPUTATIONAL METHODS TO INVESTIGATE THE MECHANISMS OF PHOTOREDOX/NICKEL DUAL-CATALYTIC REACTIONS AND EMERGING OPPORTUNITIES
2.1. Commonly used experimental methods
Photoredox/Ni dual-catalyzed reactions often rely on the ability of a light-induced excited photoredox catalyst to undergo single-electron transfer with the quencher (substrate, metal catalyst, external reagent, etc.; Figure 1A). Therefore, mechanistic studies in this field have benefited from methods developed to study radical chemistry48–50and photochemistry51–54.
2.1.1. Study of ground-state and excited-state thermodynamics
Experimental methods to study the properties of ground-state species include UV-Visible (UV-vis) absorption spectroscopy55 and cyclic voltammetry (CV)56,57. With these methods, the concentration of chromophores, and the redox potential of electron donors and electron acceptors can be measured, respectively. Since many photocatalytic transformations are realized by the transfer of electrons (e.g., single-electron transfer),58 the evaluation of the redox properties is critical to analyze the thermodynamic feasibility of such a process. Compared to the ground-state species, the excited-state entities are often significantly different (both structurally and electronically) from their ground-state counterparts.59 Nonetheless, a range of spectroscopic methods59–61 and photo-modulated voltammetry62,63 can be used to study the properties of excited-state species and understand their reactivity.
Since the open-shell reactivity of transient species lies at the center of many photo-induced transformations, the mechanistic investigation of photocatalytic processes often relies on experimental methods that are typically used in the field of radical chemistry.7,49,64 For example, radical trapping agents (e.g., phenyl N-tert-butylnitrone, PNB; 2,2,6,6-tetramethylpiperidine 1-oxyl, TEMPO)65 and radical clock experiments66,67 are used to probe the generation and lifetime of radical species. In particular, the latter method can provide kinetic information of a given radical pathway by incorporating the moieties that can undergo rapid unimolecular radical rearrangement pathways (e.g., ring-opening of cyclopropylmethyl radical and radical cyclization of 5-hexenyl radical).
However, due to the fleeting nature of short-lived species in photo-induced reactions, their isolation or trapping with organic species is often challenging. In these cases, spectroscopic methods are often applied to investigate the properties of these reactive open-shell species. In particular, electron paramagnetic resonance (EPR) spectroscopy is commonly used to detect and characterize paramagnetic organic and organometallic species.68–70 In addition, NMR spectroscopy can be applied to detect the presence of radicals via Chemical Induced Dynamic Nuclear Polarization (CDNIP) spectroscopy that requires in situ illumination within the NMR spectrometer.71 Further, the application of more sensitive time-resolved spectroscopic methods like laser flash photolysis (LFP)72,73 allows for the observation of species with lifespans in the order of pico/femtoseconds (i.e., singlet/triplet excited-state molecules and transient ionic radicals).74
2.1.2. Kinetic study of photochemical processes and radical reactivity
Although the thermodynamic preference of bimolecular radical processes can be assessed by determining the redox potentials, the feasibility of a redox event is also dependent on the kinetic parameters.54 Common experimental methods used to investigate the excited-state kinetics in these transformations involve time-resolved spectroscopic methods like Stern-Volmer analysis51,75 and LFP analysis72, where the kinetics of bimolecular events of emissive excited-state molecules can be obtained (e.g., rate constant of dynamic quenching processes). Apart from these methods, the kinetics of the overall photochemical processes can be studied by the method of initial-rate measurements.76
2.2. Computational methods
The identification of the complex mechanistic scenarios using only with experimental methods is extremely challenging due to the short lifetime of relevant reactive intermediates, the competing side pathways of excited-state species, and the difficulty of assessing kinetic parameters of individual steps. As such, in addition to experimental methods, computational tools have become more common, practical, and powerful to study the molecular-level interactions of reactive species (i.e., excited-state species and radical intermediates)21,30,78–81 involved in photoredox/Ni dual catalysis.31,32,77
2.2.1. Density functional theory
Advances in hardware, software, and algorithms in the past decades have led to wider application of quantum mechanical calculations to understand the operating mechanism and facilitate the design of novel catalysts and substrates in photoredox/Ni dual catalysis.30,82,83 In particular, density functional theory (DFT) is commonly used in locating intermediates and transition state structures due to its great balance between computational cost and chemical accuracy.84–93 Moreover, recent efforts aiming to tackle some of the well-known drawbacks of DFT have led to increasing applications in catalysis. For example, DFT is well known for its poor performance in treating noncovalent interactions, which results from the failure of local and semilocal functions to correctly examine the dynamic electron correlation.30 To overcome this issue, developments have been made in terms of optimized semilocal functions, effective one-electron potentials, density functionals with a nonlocal kernel, and the addition of (semi)empirical pairwise corrections for describing noncovalent interactions.94–98 Notably, Grimme’s D3 correction97 with the Becke-Johnson damping function99,100 has been widely used to treat noncovalent interactions in DFT calculations to mitigate the aforementioned issues.
Oftentimes, the choice of functional, basis set, and solvent for DFT calculations is based on prior literature, experience, and the intuition of computational chemists. However, methods appropriate for the study of organic molecules and closed-shell transition metals systems might not be suitable for investigating the electronic, structural properties, thermodynamics, and kinetics of open-shell 3d transition metal complexes.30 As such, benchmarking and calibration with available experimental data are always recommended to study these systems.101,102 To verify the accuracy of the computed structural properties and electronic configuration (e.g., spin states), the expectation value of the <S2> operator and frequency analysis of open-shell and closed-shell configurations is recommended.30 The inspection of these parameters is important as multiple spin states are readily accessible for late 3d transition metal systems.103–106 In sum, the development of a physically accurate, broadly applicable, and practical computational method remains one of the holy grails in computational chemistry.107Therefore, caution is advised when studying the mechanisms using purely computational tools, especially when related to nickel/photoredox catalysis, as the absolute energetics for each step in the catalytic cycles are likely to be dependent on the choice of DFT functional and basis set.21,30,108,109
2.2.2. Coupled-Cluster theory
Beyond DFT, coupled-cluster singles-and-doubles with perturbative triples (CCSD(T)) theory has been considered as the “golden standard” of quantum chemistry. However, this method remains virtually impractical for the analysis of (large) complex organic and organometallic systems (such as dual nickel/photoredox cross-coupling catalysis) due to its high computational cost.110 To address this issue, Neese and coworkers have developed DLPNO-CCSD(T) method that involves domain-based local pair approximation.111 Such a method can emulate CCSD(T) accuracy with the use of a suitable basis set with a low computational cost comparable to DFT methods and thus likely leads to wider application to studying nickel/photoredox dual catalysis.112–116 In this vein, our group has applied this method to calculate the single-point energies of the full catalytic cycle of a challenging single-electron Tsuji-Trost reaction117 and a three-component C-H activation/alkene dicarbofunctionalization reaction118 in photoredox/Ni dual catalytic conditions (vide infra).
2.2.3. Multireference and multiconfigurational theories
Under photoredox/Ni dual catalytic conditions, nickel complexes can access different spin states and, as a consequence, single-reference methods like DFT could lead to erroneous conclusions. In these scenarios, the application of multireference or multiconfigurational methods including configuration interaction (CI) and multiconfigurational self-consistent-field (MCSCF) are essential to validate the computational method.79 For example, the complete-active-space self-consistent-field (CASCCF) method builds its success on its proper description of the electronic structure and excited-state properties at the nonequilibrium geometries of mono- and polynuclear transition metal complexes.119 In addition, multireference methods including CASPT2, NEVPT2, and MRCI can also be used to intrinsically to analyze the conical intersections between various electronic states.30,79 An alternative method, DFT/MRCI method, uses Kohn-Sham orbitals to construct CSFs and to modify an MRCI-type Hamiltonian.120,121 However, the applicability of these methods is limited by the dependence on the definition and size of active space. Thus, due to the generally greater computational cost of multireference methods, time-dependent DFT (TD-DFT) has been widely used in the study of electronic spectroscopy and photochemistry of transition metal complexes.85,122,123 However, the main error of TD-DFT arises from the erroneous long-range behavior of most density functionals, the lack of double and higher excitations, and the limited accuracy of higher spin states.124 Regarding these issues, the computed results of TD-DFT are dependent on the choice of exchange-correlation functional, and the accuracy can be greatly promoted when it is combined with range-separated functionals or functionals with 100% HF exchange.125
2.3. Emerging opportunities in the computational analysis of mechanisms
2.3.1. More accurate and affordable computational methods for the description of excited-state and multiconfiguration properties
To facilitate the computational study of photoredox and Ni systems, multireference (MR) methods are needed to map the excited-state potential energy surface.79 However, compared to single reference methods, application of MR methods usually require (or benefits from) more advanced knowledge (or assumptions) of the structural and electronic properties of the species under investigation.79,126,127 Therefore, the development of more accurate, faster multireference methods is needed to map the excited-state PES at the cost of similar computational efficiency to single-reference methods. In addition to the excited-state nature of electronic configurations under photoexcited conditions, the multiconfigurational effect of Ni along with its ability to undergo spin crossover in reaction conditions necessitates the application of MR methods. Notably, under light irradiation, the properties of Ni intermediates in high spin states (i.e., triplet spin state) have been studied but the photophysics and dynamical properties of such intermediates are still not well-understood.29,38,45 In all, increasingly accurate and efficient electronic structure calculation methods are needed for broader applicability to larger, more realistic (rather than highly truncated) catalytic systems.30
2.3.2. Study of ground-state and excited-state dynamical behavior
Quantum mechanical calculations are useful to examine the properties of static points on the potential energy surface (PES), including local minima (reactants, intermediates, and products) and saddle points (transition states). After obtaining the structures of stationary points, reactivity and selectivity of the reaction are often rationalized based on the calculated kinetics (i.e., barriers using transition state theory) and thermodynamics.83,108 However, sometimes transition state theory (TST) fails to rationalize or predict product selectivity.128–131 In such scenarios, Born-Oppenheimer molecular dynamics (BOMD) calculations on the QM-calculated PES can be used to generate a statistically representative number of trajectories from the transition states, as well as to monitor the dynamics of reactive events of relevant metal species.132 In addition, quasi-classical molecular dynamics has been applied with great success in both organic reactions with bifurcation points and homogeneous transition metal catalysis.133–135 For smaller systems, ab initio molecular dynamics (AIMD) can also be utilized.136–139 In addition, since excited-state PES is accessed under light irradiation, the commonly applied molecular dynamics simulation tools need to be modified to take into consideration the behavior of highly energetic species.140 For example, the nonadiabatic molecular-dynamics (NAMD) simulation method was developed to describe the nonadiabatic coupling effects between nuclear and electronic motions.141 Recently, the Lopez group reported a study of the photoinduced denitrogenation of bicyclo azoalkanes with multireference calculations and non-adiabatic molecular dynamics (NAMD) simulations.142 With simulations starting at the S1 excited state of cyclic azoalkanes, its photophysics and reactivity were studied, revealing the stereoselectivity of such transformation. In all, with these dynamics simulation tools, opportunities to obtain more detailed information of the dynamical bond-forming and breaking processes of reactants and intermediates will likely lead to greater understanding of these transformations and rational design of dynamically-controlled systems.
2.3.3. Unbiased mapping of potential energy surface and exploration of potential reaction pathways
Apart from the challenges in describing the reactivity of excited-state species, determining speciation and conformation of solvated reactive transition metal species, the dynamical behavior of intermediates, and potential non-productive pathways remain difficult to study.83 Further, to accelerate the study of potential reaction mechanisms, the initial inputs are often biased by chemical intuition and previous knowledge of related systems.143 In this vein, automated reaction mapping methods are developed to realize the unbiased exploration of PES and exhaustively explore plausible reaction pathways.143–147 However, the computation of excited-state PES becomes more complicated beyond the Franck-Condon region due to the strong mixing between different electronic configurations.79 This situation becomes even more complicated with the consideration of possible interaction between excited-state species and transition metals.30 Due to such challenges, improvements of automated mapping approaches of excited-state PES are of great potential with the addition of multireference calculations and the appropriate description of the entire excited-state surfaces. With the development of unbiased analysis of reaction networks, the computational mechanistic study can be employed to describe the non-productive pathways accounting for the loss of reaction selectivity as well as the deactivation of catalysts.
2.3.4. Data-driven approaches for catalyst design and reaction optimization
Recently, data-driven approaches, benefiting from the large sets of chemical data from online and industrial databases and the improvement of applicable algorithms, have been developed and applied in the field of chemistry.148,149 One of the advantages of data-driven approaches is that an in-depth mechanistic understanding of the catalytic system is not required, thus facilitates the study of complicated systems where multiple reaction pathways might exist. Common data-driven approaches applied in the field of chemistry include classical statistical analysis and supervised machine learning algorithms.150 For example, multilinear regression analysis, employed by Sigman and coworkers in several studies, correlates a set of chemical descriptors (obtained through preliminary calculations) to reaction efficiency and, in turn, allows for the prediction of the performance of different substrates and catalysts.151–153
Apart from preliminary calculations, data applied for further analysis can also come from high-throughput experimentation (HTE) capable of rapidly generating enormous amounts of data.154–156 As an example, Doyle and coworkers, in collaboration with Merck, applied a set of QM descriptors obtained from 4608 C-N cross-coupling reactions to random forest algorithm for supervised machine learning. As a result, mechanistic findings and prediction of reaction performance were achieved, which were also verified by experiments.157 However, it raised a dispute on the algorithm’s validity and the variety of dataset used to develop the algorithm by Keiser and Chuang.158 In response, the authors argued that their model is valid to predict out-of-sample reactions.159 Nonetheless, the application of data-analyzing approaches to chemistry need to be done with care, which requires a broad collaboration between the communities of data scientists, synthetic chemists and computational chemists.160
2.3.5. Automated systems combined with data-driven approaches
The application of automated experimentation systems has revolutionized the reaction discovery process in synthetic chemistry.161,162 To this end, the design of new experiments can be improved with adaption to new reaction outcomes in autonomous experimentation with the involvement of decision-making algorithms.163,164 As an example of application to the field of visible-light photoredox catalysis, Noël and coworkers developed a Python-automated flow platform. Incorporating iniline UV/Vis spectroscopy and Stern-Volmer analysis, this platform not only identified the promising combinations of photocatalyst and quencher but also measured the quenching rate constants.165
Apart from automated experimentation, automated computation has also been developed based on data-driven approaches. As an example, Moitessier, Norrby, and coworkers developed the VIRTUAL CHEMIST program that helps bench chemists predict the outcomes of a series of asymmetric chemical reactions. Gratifyingly, it could achieve accuracies within ~1 kcal/mol and provided rapid analysis and a user-friendly working environment. Overall, this technology is expected to accelerate catalyst and reaction discovery.166
Moreover, the rapid development of data-driven approaches and related algorithms have facilitated computational chemists with the mechanistic investigation. Commonly, mechanistic exploration is performed based on quantum mechanical calculations to map out the potential energy surface and main reaction pathways based on the experimental findings. With the help of data-driven machine learning (ML) methods, qualitative reaction prediction models can be generated based on the given data as training sets.160 For cases where insight-dependent approaches and quantum mechanical calculations are challenging or time-consuming, data-driven approaches are powerful complements. Therefore, quantum mechanical calculations combined with machine learning methods can greatly expand the toolbox of computational methods for the mechanistic exploration of photoredox/Ni dual-catalytic systems.
3. EXAMPLES OF MECHANISTIC STUDIES OF PHOTOREDOX AND NICKEL DUAL-CATALYZED CROSS-COUPLING REACTIONS
Herein, we will discuss several representative cases of computationally studied mechanisms of photoredox/nickel dual-catalyzed C-C cross-coupling reactions primarily from our lab. Such comparison illustrates the complexity of intrinsic mechanistic scenarios of different systems and showcases the difficulty of analyzing the mechanisms of metallaphotoredox-catalyzed reactions. In light of these efforts, we strongly recommend examining the mechanism of each system based on the specific reaction condition and catalytic system rather than making generalizations of the mechanistic paradigms.
3.1. Two-component direct cross-coupling
Initially, Kozlowski, Molander, and Gutierrez explored the mechanism of two-component photoredox/Ni dual catalytic cross-coupling reactions between alkyl potassium trifluoroborates and aryl bromides (Figure 2A).3 Prior work determined that the photocatalyst is responsible for generating the alkyl radical 4 presumably via the single-electron oxidation between potassium benzyltrifluoroborate reagent and the excited Ir photocatalyst 1 (Figure 2B).77 In parallel, oxidative addition of aryl halides to Ni(0) catalyst 5 can occur to afford Ni(II)-aryl-bromo complex 7 (downhill by 25.6 kcal/mol), which can capture the generated alkyl radical 4. Alternatively, the alkyl radical 4 can be directly captured by the Ni(0) species 5 and give Ni(I)-alkyl complex 6 (downhill by 10.2 kcal/mol), which can, in turn, undergo oxidative addition with aryl halide (barrier of 9.3 kcal/mol from 6). Notably, independent of the order of radical capture and oxidative addition, both of these two pathways converge at the same Ni(III)-alkyl-aryl-bromo complex 8, which can undergo reductive elimination (barrier of 8.7 kcal/mol) to give the cross-coupling product with concomitant formation of Ni(I)-bromo species 9. Finally, the Ni(I) species can undergo single-electron reduction with the reduced Ir photocatalyst 2 to generate the starting Ni(0) catalyst 5, closing both catalytic cycles.
FIGURE 2.
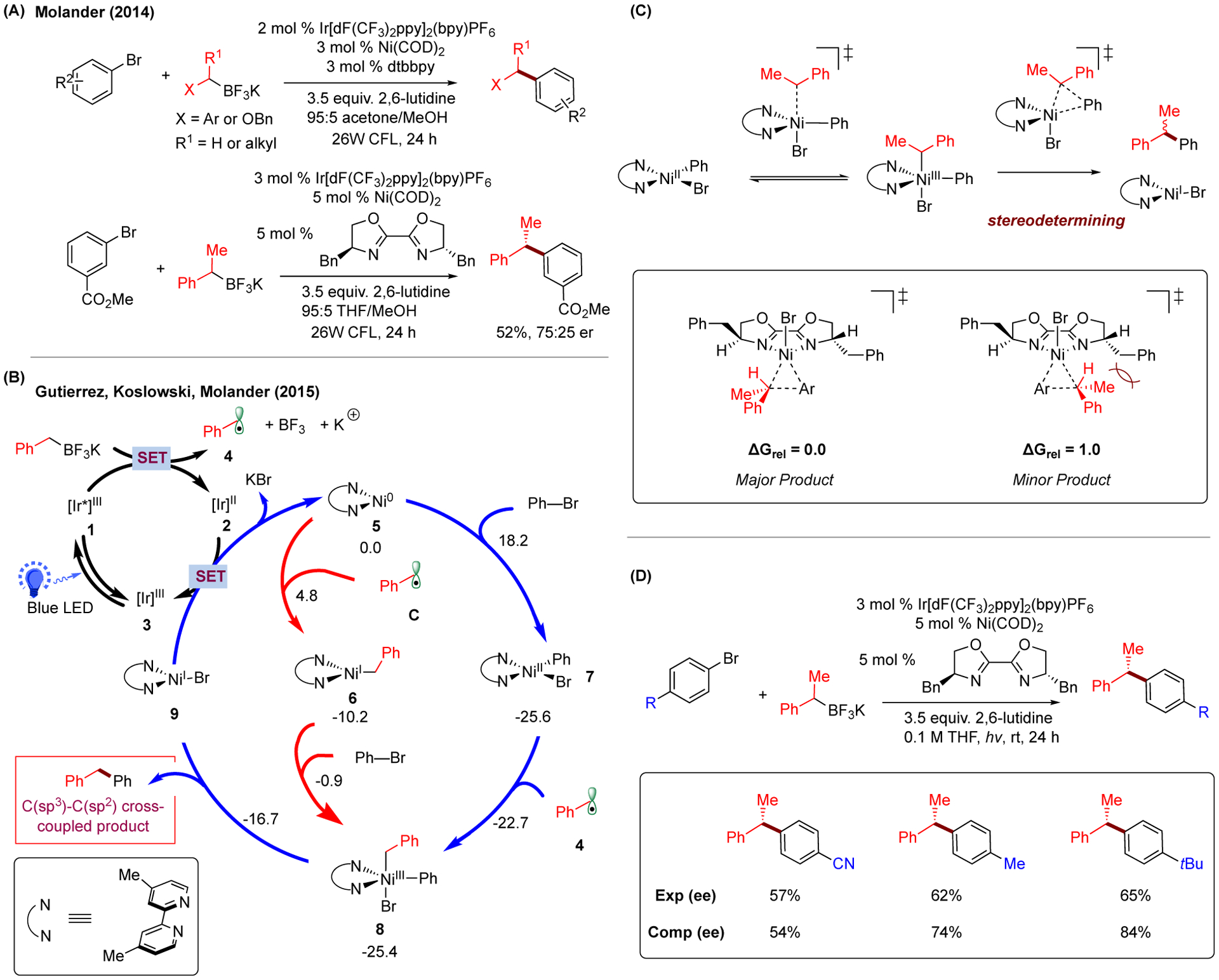
Proposed mechanism of photoredox/Ni dual-catalyzed C(sp2)-C(sp3) bond-forming reaction. (A) Photoredox/Ni dual-catalyzed C(sp2)-C(sp3) cross-coupling between aryl bromides and alkyltrifluoroborate salts, including the construction of chiral centers via asymmetric catalysis. (B) Proposed catalytic cycles with calculated energetic values. Photocatalyst [Ir]III = Ir[dF(CF3)2ppy]2(bpy)PF6. Relative Gibbs free energy values (kcal/mol;298 K) were calculated with (U)M06/6–311+G(d,p)-SMD(water)//UB3LYP/6–31G(d) level of theory. Both Ni(I)/Ni(II)/Ni(III)/Ni(I) and Ni(I)/Ni(III)/Ni(I) pathways are feasible. (C) Identification of reductive elimination as the stereodetermining step and the origin of stereoselectivity. (D) Comparison of experimental and calculated enantiomeric excess (ee) values for different aryl systems based on the proposed stereocontrol model.
Importantly, the dissociation of the alkyl radical from Ni(III) complex 8 was found reversible and occurred more rapidly than the reductive elimination from the same intermediate (−22.7 vs −16.7 kcal/mol). This finding opens new opportunities to study the mechanistic details of asymmetric photoredox/Ni dual catalytic cross-coupling reactions. Considering that the homolytic equilibration of Ni(III)/Ni(II) pair is faster than reductive elimination, the addition of alkyl radical to Ni(II) center operates under Curtin-Hammett conditions and the enantioselectivity is determined by the energy difference between the diastereomeric reductive elimination transitions states (Figure 2C). Specifically, the diastereomeric Ni(III) complexes (generated from radical addition and/or oxidative addition) could rapidly interconvert via alkyl radical dissociation/association prior to undergoing enantioselective reductive elimination. The calculation of the reductive elimination transition states leading to the corresponding products supports this finding (Figure 2C), where the calculated enantiomeric ratio (85:15) is in general consistent with the experimental value (75:25). This proposed stereochemical model was further validated via experiments (Figure 2D) that showed improved enantioselectivity by varying the para-substituents of the aryl ring.
Apart from our investigation of catalytic cycles involving primary and secondary benzyl radicals, the reactivity of tertiary aminooxetanyl radicals was also studied in the photoredox/Ni dual-catalyzed oxetanylation of aryl halides by Terrett, Huestis, and coworkers (Figure 3).167 Similar to the previous case, both Ni(0)/Ni(II)/Ni(III) and Ni(0)/Ni(I)/Ni(III) pathways are feasible, merging in the formation of Ni(III) intermediate 14 prior to reductive elimination to afford the C(sp3)-C(sp2) cross-coupling product. Interestingly, in this system Ni(0)/Ni(II)/Ni(III) pathway was found to be dominant, with the oxidative addition of aryl halides to Ni(0) 11 as the turnover-limiting step. At the same time, the Ni(0)/Ni(I)/Ni(III) pathway was less favorable due to the slightly higher barrier for the radical addition of tertiary alkyl radical 10 to Ni(0) 11 (13.9 kcal/mol vs. 11.8 kcal/mol for oxidative addition) and the high barrier for oxidative addition from Ni(I) 12. Despite such differences, the computational results suggested that the radical addition of aminooxetanyl radical 10 to Ni(II)-aryl-bromo intermediate 13 was reversible (barrier of only 6.2 kcal/mol), which is consistent with the previous findings with benzyl radicals. Overall, this study suggests that the operative mechanism in radical cross-coupling reactions is likely substrate-dependent and emphasizes the necessity of conducting a case-by-case mechanistic investigation for photoredox/Ni dual-catalytic systems.
FIGURE 3.
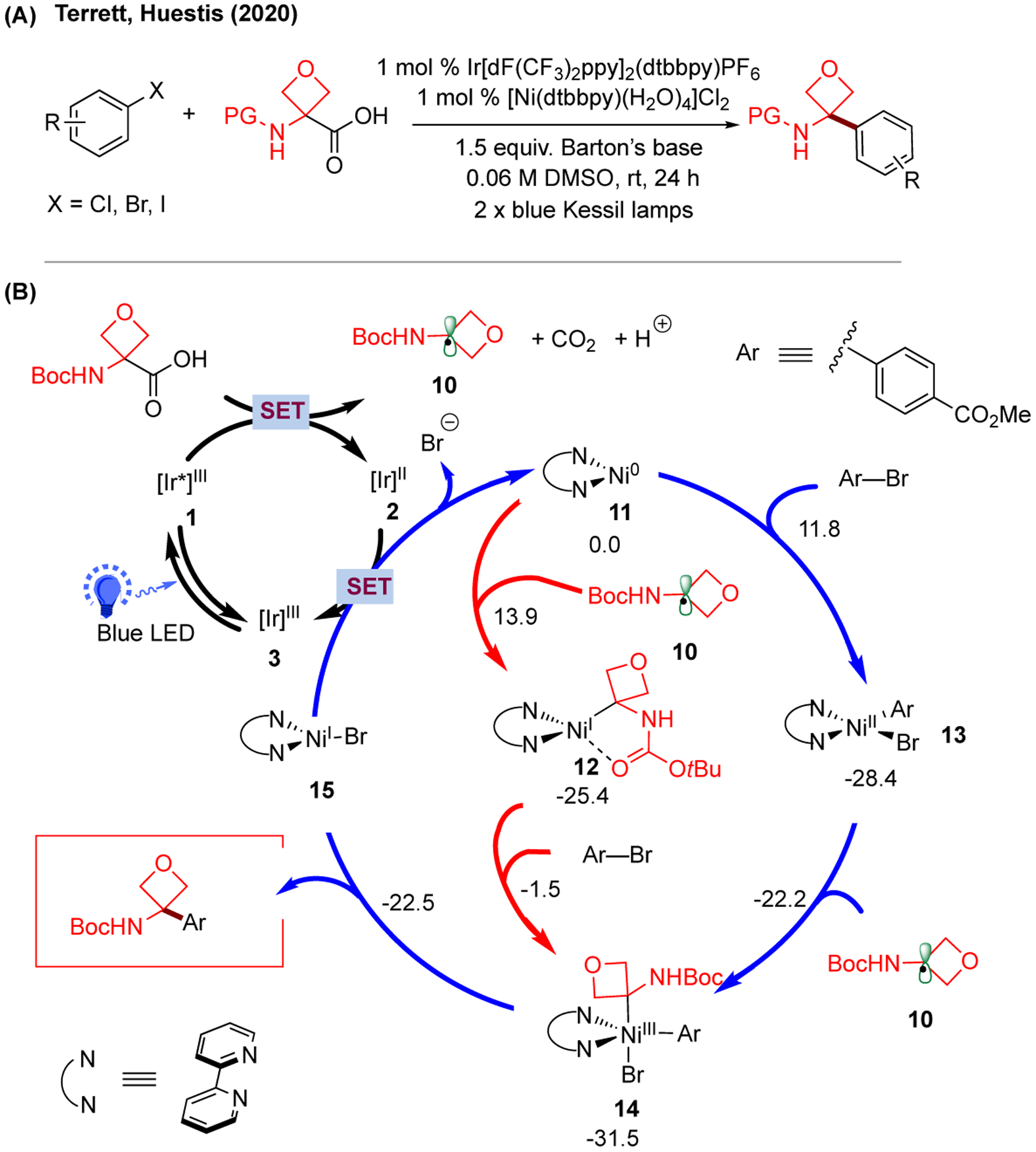
Proposed mechanism of photoredox/Ni dual-catalyzed oxetanylation of aryl halides. (A) Experimental condition of photoredox/Ni dual-catalyzed construction of aryl aminooxetanes. (B) Proposed catalytic cycles with calculated energetics. Relative Gibbs free energy values (kcal/mol;298 K) were calculated with M06/6-311+G(d,p)-SDD(Ni)-SMD(DMSO)//B3LYP/6-31G(d)-LANL2DZ(Ni) level of theory.
More recently, in collaboration with Molander and coworkers, our group studied the mechanism of a photoredox/Ni dual-catalyzed single-electron Tsuji-Trost reaction between trifluoroborate salts and vinyl epoxides using both DFT and DLPNO-CCSD(T) calculation (Figure 4A).117 Compared to the alkylation of aryl bromide system (Figure 2), a Ni(0)/Ni(II) oxidative addition with a vinyl epoxide to afford 22 is favorable (via 21-TS-22, barrier of only 3.1 kcal/mol from Ni(0)-vinyl epoxide complex 21), followed by radical addition of benzyl radical 16 (blue; Figure 4B). In this case, the Ni(0) to Ni(I) radical addition transition state 18-TS-19 from Ni(0)-benzyl complex 18 (1.5 kcal/mol) is higher in energy than the Ni(0)/Ni(II) oxidative addition transition state 21-TS-22 (−5.8 kcal/mol). Similar to the previous systems, both pathways converged at the formation of Ni(III) intermediate 20 prior to inner-sphere reductive elimination (via 20-TS-23) to forge a C-C bond and generate Ni(I) species 23. Finally, the Ni(I) species can undergo SET with the reduced photocatalyst 2 to retrieve Ni(0) catalyst and give the desired product after protonation (inset; not computed).
FIGURE 4.
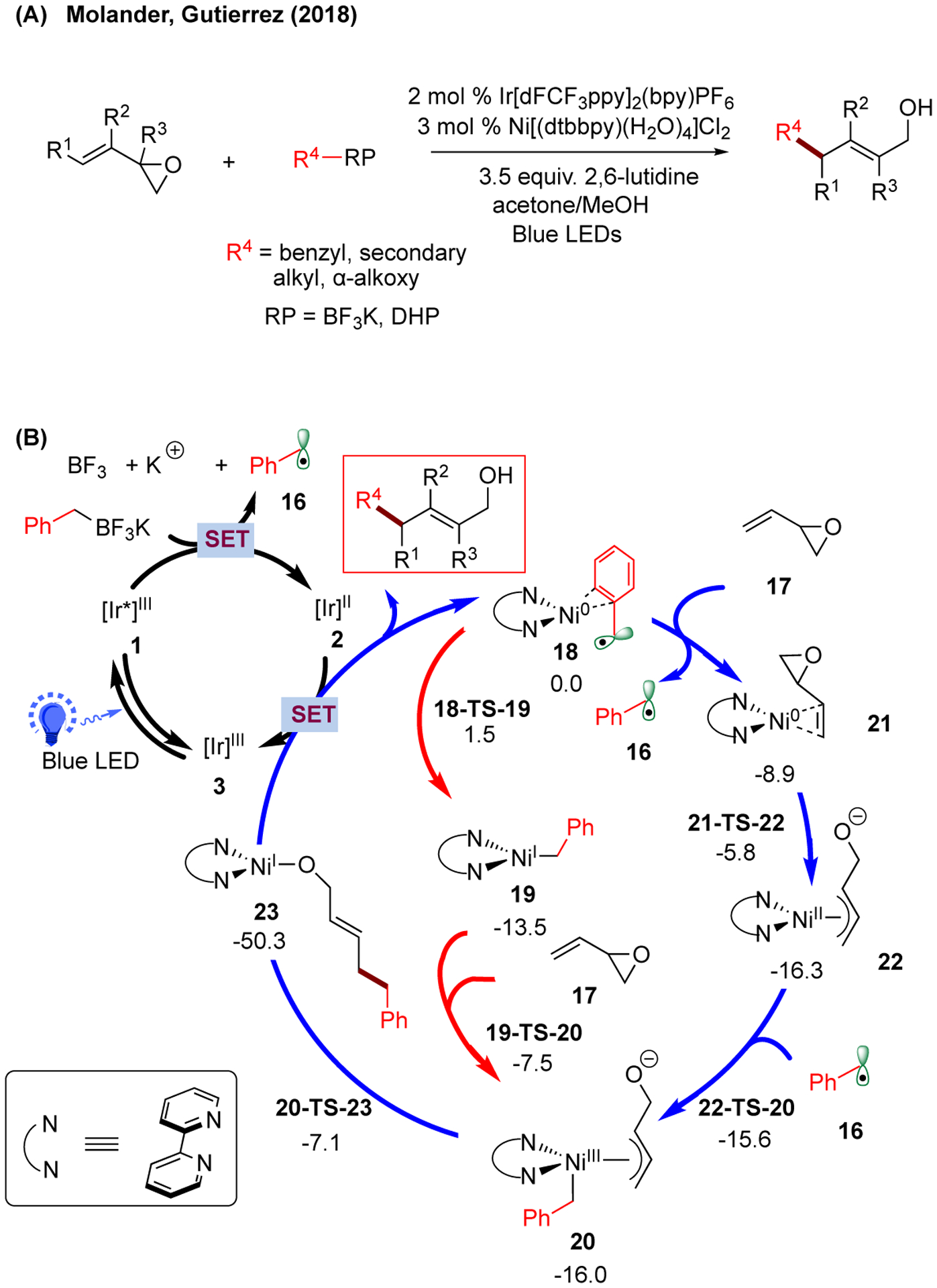
(A) Photoredox/Ni dual-catalyzed single-electron Tsuji-Trost reaction (DHP = 1,4-dihydropyridine). (B) Proposed catalytic cycles with calculated energetics of the allylation of benzyl radical precursor with vinyl epoxide. Relative Gibbs free energy values (kcal/mol;298 K) were calculated with UM06/6-311+G(d,p)-SMD(water)//UB3LYP/6-31G(d) level of theory.
Apart from vinyl epoxides, oxa- and azabenzonorbornadienes can also be used as electrophilic partners in photoredox/Ni dual catalysis, allowing for the formation of 2-alkyl-1,2-dihydronaphthalenols (Figure 5A).168 In this case, a non-metal-based photocatalyst 2,4,5,6-tetra-9H-carbazol-9-yl-1,3-benzenedicarbonitrile (4-CzIPN) was used in collaboration with radical precursor 4-alkyl-1,4-dihydropyridines (DHPs). With diimine ligand L1 employed, this system exhibited a similar mechanism as the single-electron Tsuji-Trost reaction (Figure 5B). Specifically, the syn-complexed Ni(0)-epoxynaphthalene 28 can undergo direct Ni(0)/Ni(II) oxidative addition (via 28-TS-29, barrier of 24.1 kcal/mol) to forge Ni(II)-O-π-allyl complex 29 (exothermic by 7.1 kcal/mol). After facile π-σ rearrangement to generate 1,4-Ni(II)-O-alkyl complex 30 (via 29-TS-30), benzyl radical 27 (generated from oxidative cleavage of alkyl-DHP via SET with excited photocatalyst) can add to the Ni center to afford Ni(III) intermediate 31. Starting from 31, it can undergo direct reductive elimination to deliver the syn-1,4 product (green; via 31-TS-32, barrier of 26.1 kcal/mol with respect to 30), or undergo facile σ-π-σ isomerization (via 31-TS-33) followed by reductive elimination from Ni(III) 33 to generate the syn-1,2 product (blue; via 33-TS-34). Comparing these two pathways, the syn-1,2 pathway is more favorable due to its lower barrier of reductive elimination (via 33-TS-34, ~ 11 kcal/mol from 33) compared to the syn-1,4 pathway (via 31-TS-32, ~ 26 kcal/mol from 30). Overall, the equilibrium between 30, 31, and 33 (via rapid σ-π-σ isomerization and benzyl addition) results in a Curtin-Hammett scenario, where the exclusive 1,2-regioselectivity was determined by the regio-determining reductive elimination transition states.
FIGURE 5.
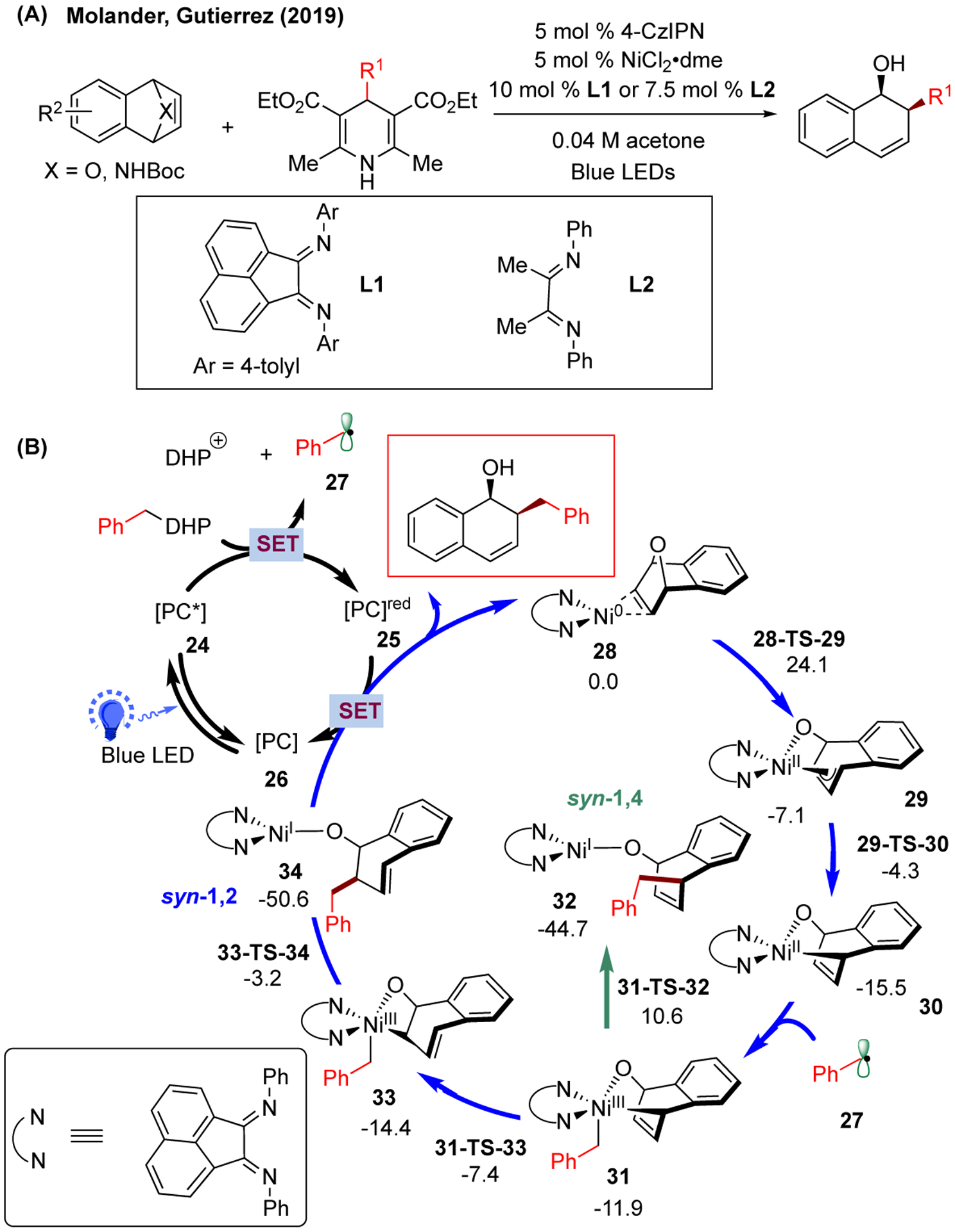
(A) Photoredox/Ni dual-catalyzed alkylation of oxa- and azabenzonorbornadienes (4-CzIPN = 2,4,5,6-tetra-9H-carbazol-9-yl-1,3-benzenedicarbonitrile). (B) Proposed catalytic cycles with calculated energetics of the alkylation of oxabenzonorbornadiene with benzyl radical precursor. Relative Gibbs free energy values (kcal/mol;298 K) were calculated with UM06/6-311+G(d,p)-CPCM(acetone)//UB3LYP-D3/6-31G(d)-CPCM(acetone) level of theory.
More recently, we used DFT calculations to shed light on the divergent reactivity reported by Molander and coworkers in the nickel/photoredox-catalyzed cross-coupling of tertiary alkyl radicals (Figure 6A).169 In particular, we were interested in understanding the effect of ligand (neutral versus anionic) on the efficiency of cross-couplings.170 As shown in Figure 6A, the neutral bipyridine-based ligand (4,4’-di-tert-butyl-2,2’-bipyridine; dtbbpy) was found effective with cyclic tertiary alkyltrifluoroborate reagents while ineffective with the acyclic compounds (e.g., tert-butyltrifluoroborate). On the contrary, the anionic diketonoate-based ligands (tetramethylheptanedione; TMHD) became eligible in forging the C-C bonds between tert-butyltrifluoroborate and aryl bromides.
FIGURE 6.
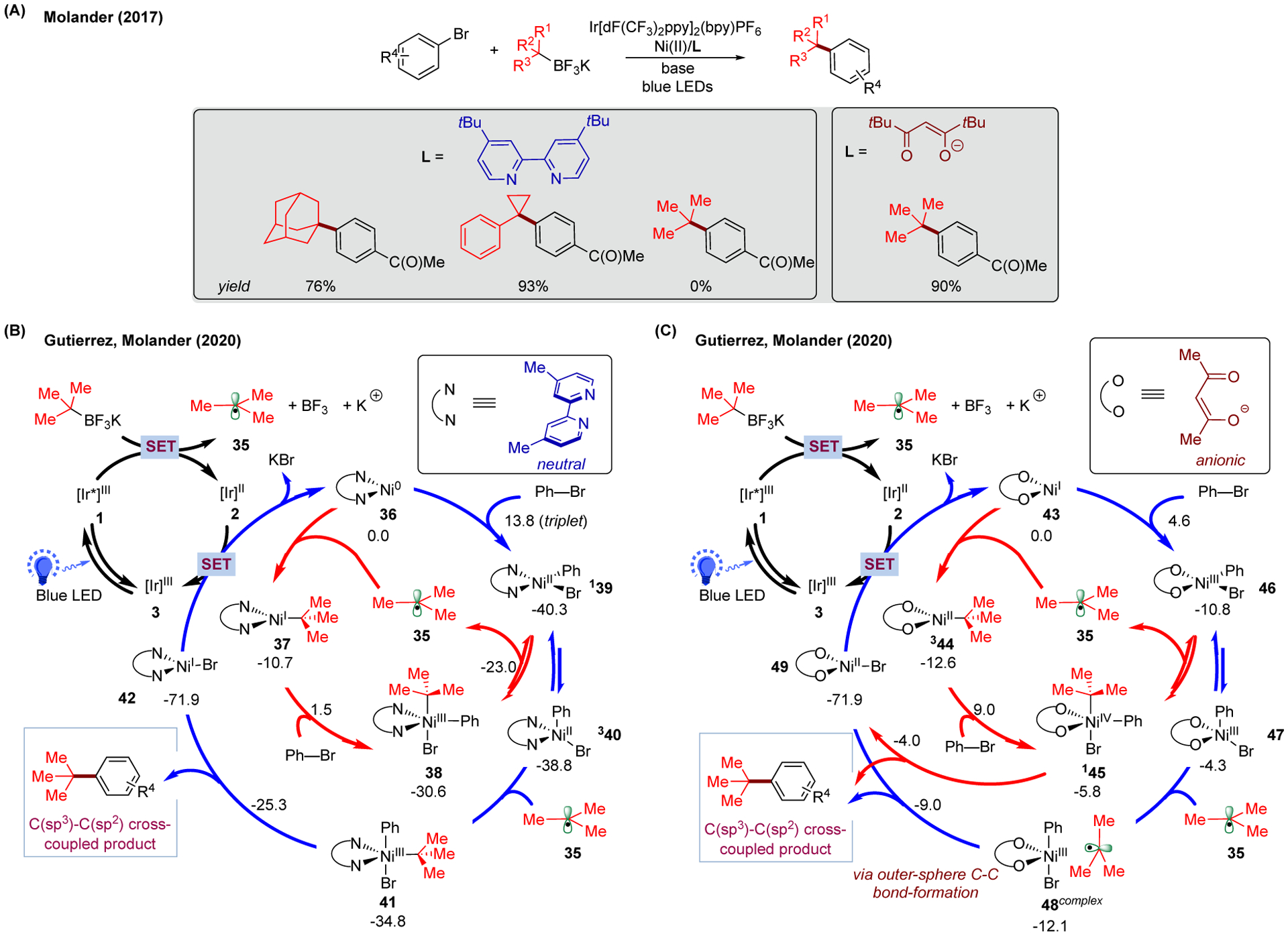
Proposed ligand-dependent mechanisms of photoredox/Ni dual-catalyzed C(sp2)-C(sp3) bond-forming reaction between tertiary alkyltrifluoroborates and aryl bromides. (A) Reported experimental results on the ligand-dependent performance of cross-coupling reactions of tertiary alkyl radicals in bipyridine (blue) and diketonoate (red) systems. (B) Proposed catalytic cycles of neutral bipyridine system. Superscripts indicate the spin state (2S+1) of the corresponding Ni complex. Relative Gibbs free energy values (kcal/mol;298 K) were calculated with UB3LYP-D3/def2-TZVPP-CPCM(THF)//UB3LYP-D3/def2-SVP-CPCM(THF) level of theory. (C) Proposed catalytic cycles of anionic diketonoate system. Relative Gibbs free energy values (kcal/mol;298 K) were calculated with UB3LYP-D3/def2-TZVPP-CPCM(DMA)//UB3LYP-D3/def2-SVP-CPCM(THF) level of theory.
The computation of both systems started by structural optimizations using open-shell, dispersion-corrected DFT in THF with implicit CPCM solvent model [noted as UB3LYP-D3/def2-SVP-CPCM(THF)], followed by single-point energy calculations using other functionals and larger basis sets to compare the energetics. For the neutral bipyridine system (Figure 6B), similar to the previous results, both Ni(0)/Ni(II)/Ni(III) and Ni(0)/Ni(I)/Ni(III) pathways were found feasible. However, the Ni(III) intermediate 38 generated from oxidative addition of Ni(I)-alkyl species 37 (barrier of 12.2 kcal/mol) was found to undergo reversible radical dissociation to afford the singlet-spin, square planar Ni(II)-phenyl-bromo 139 rather than undergoing reductive elimination to give the C-C cross-coupled product (i.e., a novel Ni(0)/Ni(I)/Ni(III)/Ni(II) pathway). Notably, all attempts to locate the transition state of reductive elimination from such conformation failed.
To further probe the reactivity of Ni(III) species 38 in the system, we conducted, a first for nickel-catalyzed cross-couplings, preliminary quasi-classical dynamics simulations from the transition state of oxidative addition from Ni(I) species 37 (with the truncated model due to computational cost). The simulated trajectories propagated towards the generation of Ni(III) showed that 60% of the trajectories retained the formation of Ni(III) intermediate, while 40% formed the Ni(III) intermediate followed by radical dissociation of tBu radical to afford square planar Ni(II)-phenyl-bromo 139. Consistent with the DFT results, no cross-coupling product was observed in all simulated trajectories. We anticipate that the fate of the purported Ni(III) intermediate is highly dependent on its conformation with profound implications for its reactivity and selectivity in photoredox/Ni dual catalysis and, more broadly, nickel-catalyzed cross-couplings.
Apart from the radical dissociation from Ni(III) species 38, Ni(II)-phenyl-bromo 139 can be accessed directly by oxidative addition from Ni(0) 36. After, this singlet-spin, square planar Ni(II) can isomerize to the triplet-spin, tetrahedral Ni(II) 340 under photoinduced conditions. Such singlet-triplet isomerization was also reported by the Doyle group in the study of (bpy)Ni(II)-(o)Tol-chloro species.29,38 In addition, Ni(III) intermediate 41 (in different conformation from 38) can be obtained from the radical addition of tert-butyl radical via the “equatorial” trajectory of triplet-spin, tetrahedral Ni(II) species 340. Importantly, this process was found endothermic by 5.5 kcal/mol with respect to the singlet-spin, square planar Ni(II)-phenyl-bromo 139.
In stark contrast, with the anionic acac ligand, the mechanism of the C-C bond-formation is significantly different. Specifically, the common inner-sphere low-spin (doublet spin state) reductive elimination pathway is less favorable (Figure 6C; red), while a novel outer-sphere high-spin (triplet spin state) C(sp3)-C(sp2) bond-forming mechanism is preferred by such a system (Figure 6C; blue). Notably, in the inner-sphere C-C bond-forming pathway (red), the generation of high-valent Ni(IV)-alkyl-phenyl-bromo intermediate 145 from Ni(II)-alkyl 344 is endergonic by 6.8 kcal/mol, and the barrier for oxidative addition in this process is as high as 21.6 kcal/mol. On the contrary, the starting Ni species 43 can undergo facile oxidative addition (barrier of only 4.6 kcal/mol) of phenyl bromide to generate Ni(III)-phenyl-bromo intermediate 46, followed by the formation of complexed species 48complex in the presence of tBu radical. After, the outer-sphere C-C bond-forming pathway is facile (barrier of 3.1 kcal/mol from the complexes species 48complex), affording the cross-coupled product and Ni(II)-bromo 49. Such outer-sphere C-C bond-forming mechanism was also observed in other studies of transition-metal-catalyzed cross-coupling reactions.171–174 Notably, novel reactivity of high-spin Ni species was reported in both neutral and anionic ligand systems. Such spin-controlled reactivity was not unprecedented, as two-state reactivity has been observed in many transition metal systems.104,175 Regarding the need to study the properties of different spin states, the transformation between singlet and triplet spin states can be studied in more detail with the use of minimum energy crossing point (MECP) calculation.105,176
In addition to investigating the ligand effect on the mechanism, we also explored the origin of the distinct reactivity shown by various secondary and tertiary alkyl radicals in the neutral bipyridine system (Figure 7).169 We hypothesize that the reversible radical addition to Ni(II)-phenyl-bromo species to form the Ni(III) intermediate in addition to subsequent reductive elimination are likely to be crucial for reactivity in these transformations (Figure 7A). Among the investigated secondary (for a similar system, see Figure 2) and tertiary alkyl radicals (i.e., tert-butyl, 1-phenylcyclopropyl, isopropyl, and 1-adamantyl radicals), the generation of Ni(III) intermediate with tert-butyl radical is endergonic, while for other radicals the radical addition is exergonic (Figure 6B). Moreover, the barrier for reductive elimination transition state with tert-butyl radical with respect to Ni(II) is much higher than that with other radicals (15.0 kcal/mol). Taken together, the thermodynamic drive to form the Ni(III) intermediate in addition to the subsequent barrier for reductive elimination determines the reactivity of alkyl radicals in this C-C cross-coupling system.
FIGURE 7.
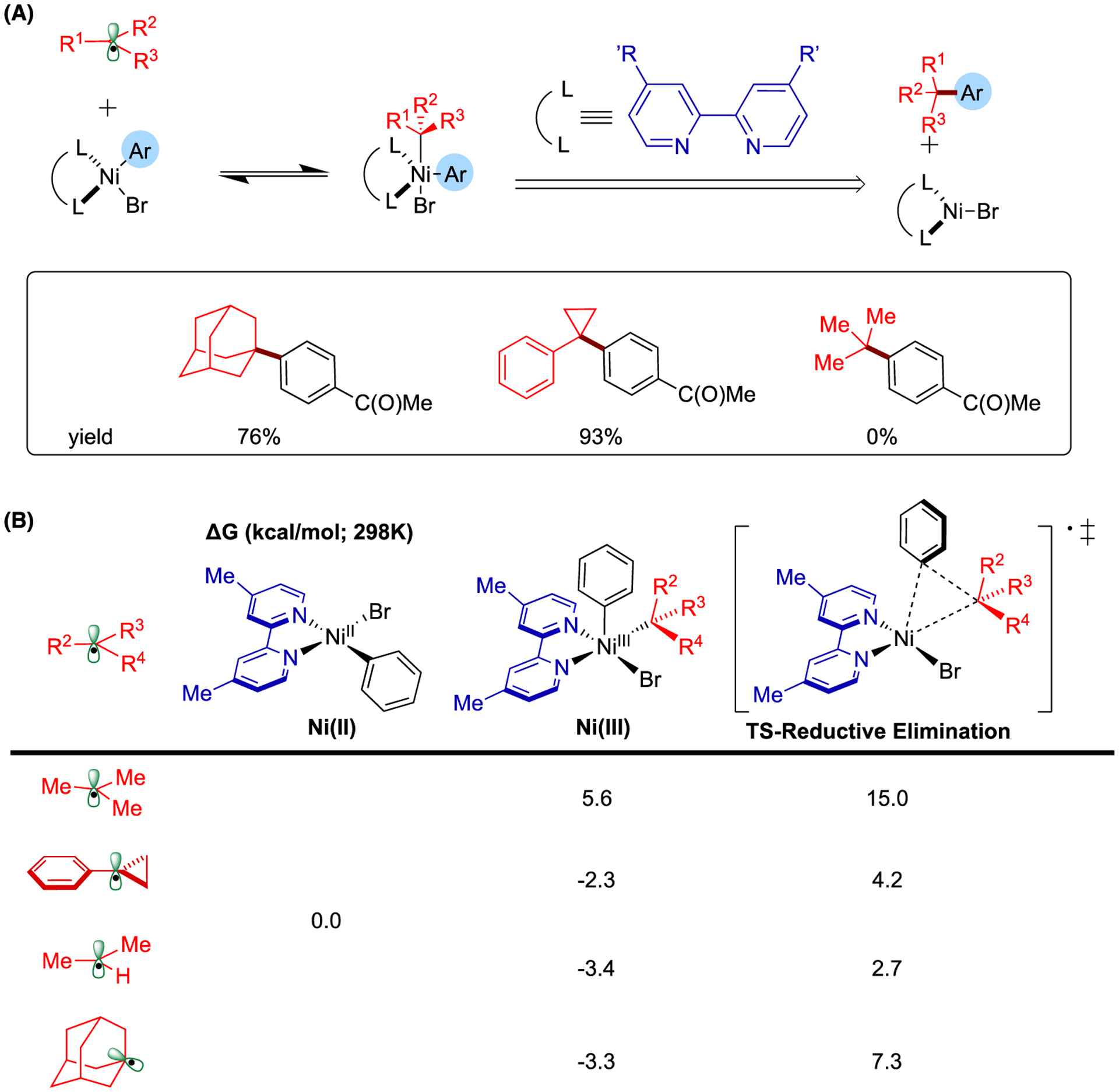
Study of the different reactivity of various secondary and tertiary alkyl radicals. (A) The key pathway determining the reactivity of alkyl radicals and the relevant experimental results.170 (B) Relative Gibbs free energy values (kcal/mol; 298 K) of the generation of Ni(III) intermediate and the following reductive elimination step with respect to the square planar Ni(II)-phenyl-bromo intermediate. Relative Gibbs free energy values (kcal/mol;298 K) were calculated with respect to the sum of separated Ni(II) species and alkyl radical with UB3LYP-D3/def2-TZVPP-CPCM(THF)//UB3LYP-D3/def2-SVP-CPCM(THF) level of theory.
Such finding on the origin of different reactivity of alkyl radicals was supported by the mechanistic studies of another photoredox/Ni dual-catalytic system that involved fluorinated radicals (Figure 8).177 In this system, we observed that the alkyl radical was generated from carboxylic acid via decarboxylation consisting of deprotonation and single-electron oxidation with the excited Ir photocatalyst. Interestingly, the fluorinated benzyl carboxylic acid was found less reactive than the non-fluorinated compound despite the small difference of oxidation potential between the corresponding carboxylates (+1.20 V and +1.27 V, correspondingly; vs. SCE in MeCN). Based on the previous finding (vide supra), the overall pathways for these two alkyl radicals were calculated (Figure 8), and the performance of these two alkyl radicals was found dependent on the reversible radical addition to Ni(II) and the subsequent reductive elimination steps. Based on the computed energetics, the weaker reactivity of fluorinated radical was attributed to the endergonic nature of radical addition (uphill by 4.0 kcal/mol) and the higher energetics of reductive elimination transition state (13.7 vs 6.9 kcal/mol).
FIGURE 8.
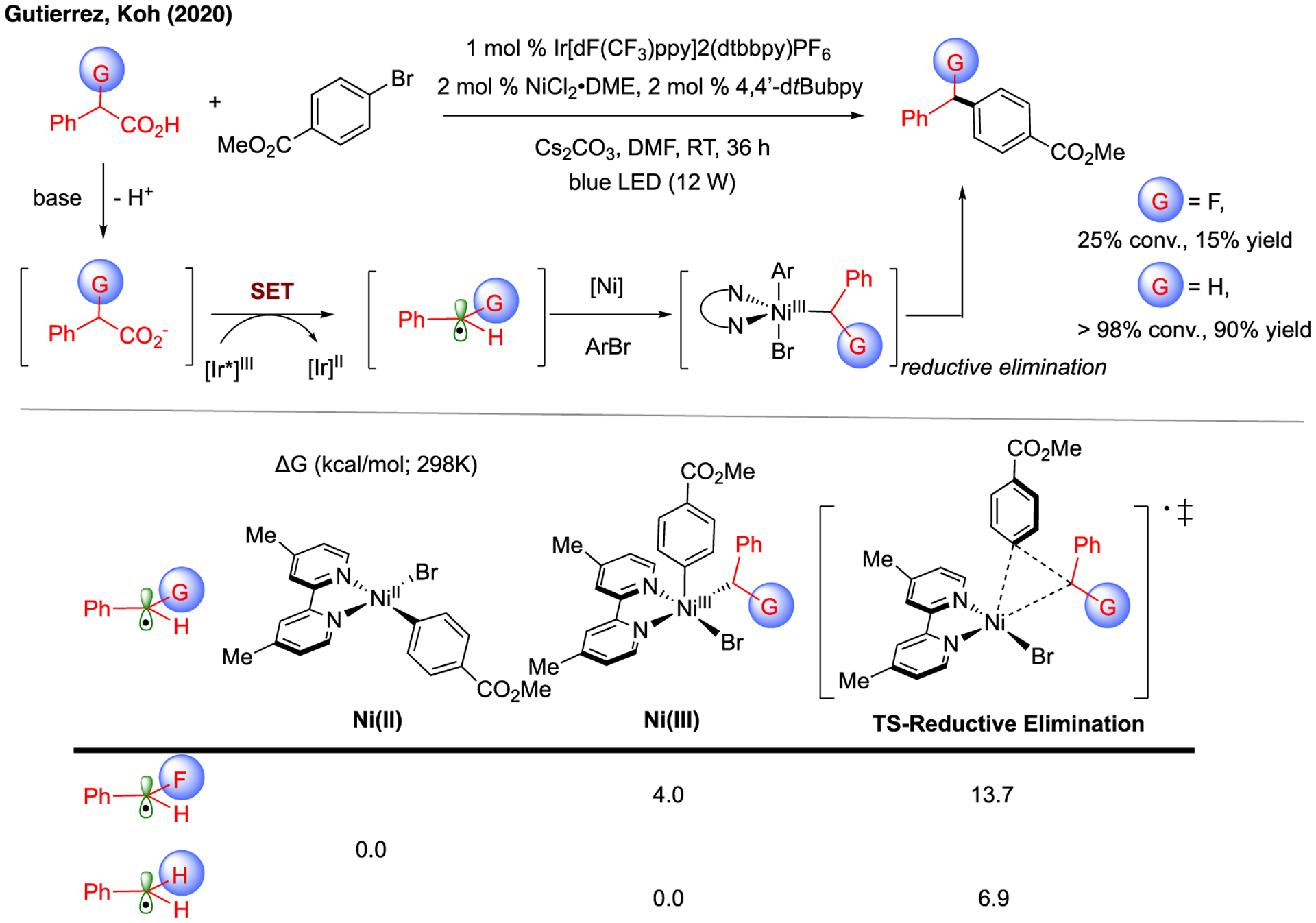
Proposed origin of different radical reactivity in the photoredox/Ni dual-catalyzed decarboxylative C(sp2)-C(sp3) bond-forming reaction. Relative Gibbs free energy values (kcal/mol;298 K) were calculated with respect to the sum of separated Ni(II) species and alkyl radical with UB3LYP-D3/def2-TZVPP-CPCM(DMA)//UB3LYP-D3/def2-SVP-CPCM(DMA) level of theory.
Despite the great achievement of photoredox/Ni dual catalysis in C-C cross-coupling reactions, a prefunctionalized alkyl precursor is often required for the success of these transformations. In this context, the well-established radical C-H activation chemistry can be combined into the photoredox-Ni catalytic system to utilize a broader scope of substrates via activation of C(sp3)-H bonds. As one of the pioneering studies, MacMillan and coworkers realized the activation of α-amino C(sp3)-H bond by the use of quinuclidine as a hydrogen-atom-transfer reagent in its oxidized form.18,19,178 Utilizing this photoredox/Ni/HAT triple catalytic system, both C(sp2)-C(sp3) and C(sp3)-C(sp3) bond-formation can be achieved, depending on the applied electrophile (Figure 9A). However, compared to the dual catalytic systems (vide supra), this triple nickel, photoredox, and H-atom abstraction catalytic system makes the mechanistic exploration more complicated. Targeting this complex system, Rueping, Cavallo, and coworkers explored the mechanistic picture using a combined computational and spectroscopic approach (Figure 9B) with a focus on the interplay between different cycles (Ni, photoredox, HAT).179 Due to the multiple possibilities of SET events in the system, the authors computed the thermodynamics as well as the kinetics of single-electron transfer processes between different electron donors and acceptors (e.g., Ni catalyst, excited Ir photocatalyst, quinuclidine, etc.). As a result, after considering the concentration of quinuclidine, the first step was proposed to be the single-electron reduction of excited Ir(III) photocatalyst 1 by quinuclidine catalyst 50 to afford the oxidized quinuclidine 51 and corresponding Ir(II) species 2. Next, alkyl radical 54 can be generated by HAT of oxidized quinuclidine 51 from α-amino C(sp3)-H, which can then be captured by Ni(I)-bromo species 55 to generate Ni(II)-alkyl-bromo intermediate 56. Finally, SET between Ni(II) 56 and the reduced Ir photocatalyst 2 forms the corresponding Ni(I)-alkyl intermediate 57. This intermediate can then undergo oxidative addition with R-Br (R = alkyl/aryl) to give Ni(III) intermediate 58 that is prone for reductive elimination to generate the corresponding C-C cross-coupling products (i.e., Ni(I)/Ni(II)/Ni(I)/Ni(III)).
FIGURE 9.
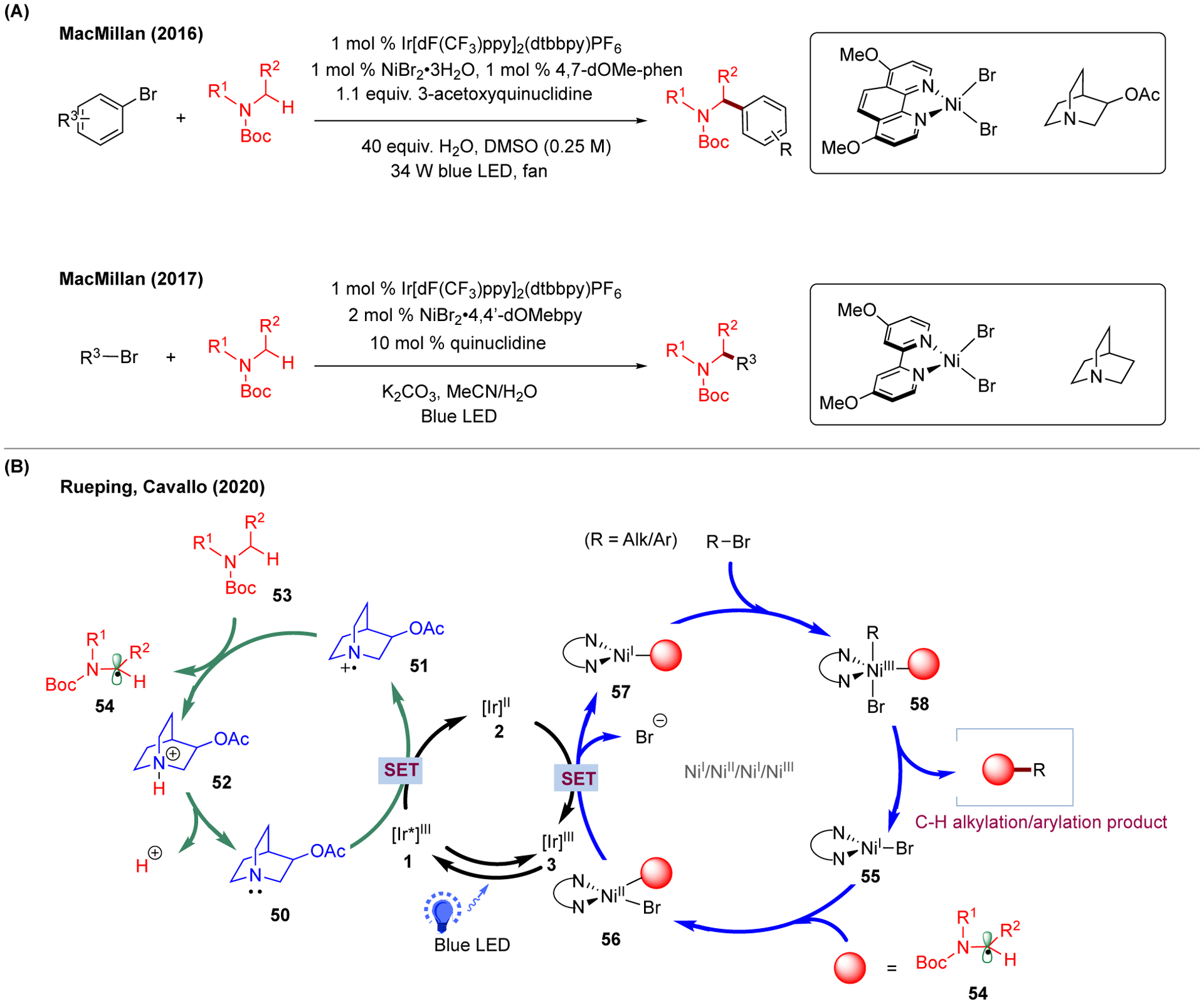
C-C cross-coupling enabled by C-H activation with the photoredox-Ni-HAT triple catalytic system. (A) Experimental condition and catalytic system (inset) of C(sp2)-C(sp3) and C(sp3)-C(sp3) bond-formation reactions. (B) Proposed catalytic cycles of the photoredox-Ni-HAT triple catalytic system.
3.2. Three-component C-C cross-coupling reactions
Apart from the cross-coupling reactions between two components, nickel-catalyzed 1,2-difunctionalizations of alkenes and alkynes have attracted more and more attention since they allow for the building of two sequential chemical bonds from readily available and abundant starting materials. Compared to conventional two-electron-based 1,2-difunctionalization strategies, the cooperative use of photoredox and nickel catalysts circumvents the requirement of organometallic reagent and high temperature.180–185 Thus, the application of photoredox/Ni dual catalysis in the field of 1,2-dicarbofunctionalization (DCF) has grown rapidly to install multiple molecular entities across the C-C π bonds.186–189 However, the broader development of these strategies is limited by inter alia the competing two-component coupling reactions, facile β-hydride elimination, and dimerization of radicals.187 Further, the mechanistic understanding of the origin of regio-, chemo- and stereoselectivity in these transformations remains limited, hindering the design of catalytic systems with increased selectivity. Therefore, a detailed mechanistic exploration (both experimental and computational) can no doubt provide insight into the molecular-level understanding of the key interactions and factors that determine the selectivity, thus opening opportunities to overcome the current obstacles faced by photoredox/Ni dual-catalyzed 1,2-difunctionalization reactions.
As an example, our group looked into the operating mechanism of a photoredox/Ni dual-catalyzed asymmetric alkylarylation of activated alkenes that generate enantioenriched α-aryl carbonyls reported by the groups of ours and Chu’s (Figure 10A).190 This strategy provides an efficient way to forge high-value stereocenters from readily available starting materials considering the prevalence of chiral α-aryl carbonyls in many bioactive compounds, pharmaceuticals, and agricultural chemicals.191–193 Moreover, due to the rare reports of asymmetric 1,2-difunctionalization of alkenes via photoredox/Ni dual catalysis, this system serves as an ideal platform to investigate the origin of the exhibited chemo- and stereoselectivity. Specifically, for this system, we were interested in the following questions: (1) why is the three-component alkylarylation product favored over the two-component direct cross-coupling between aryl bromide and alkyltrifluoroborate reagent, and (2) what is the stereodetermining step and the determining factors of the enantioselectivity.
FIGURE 10.
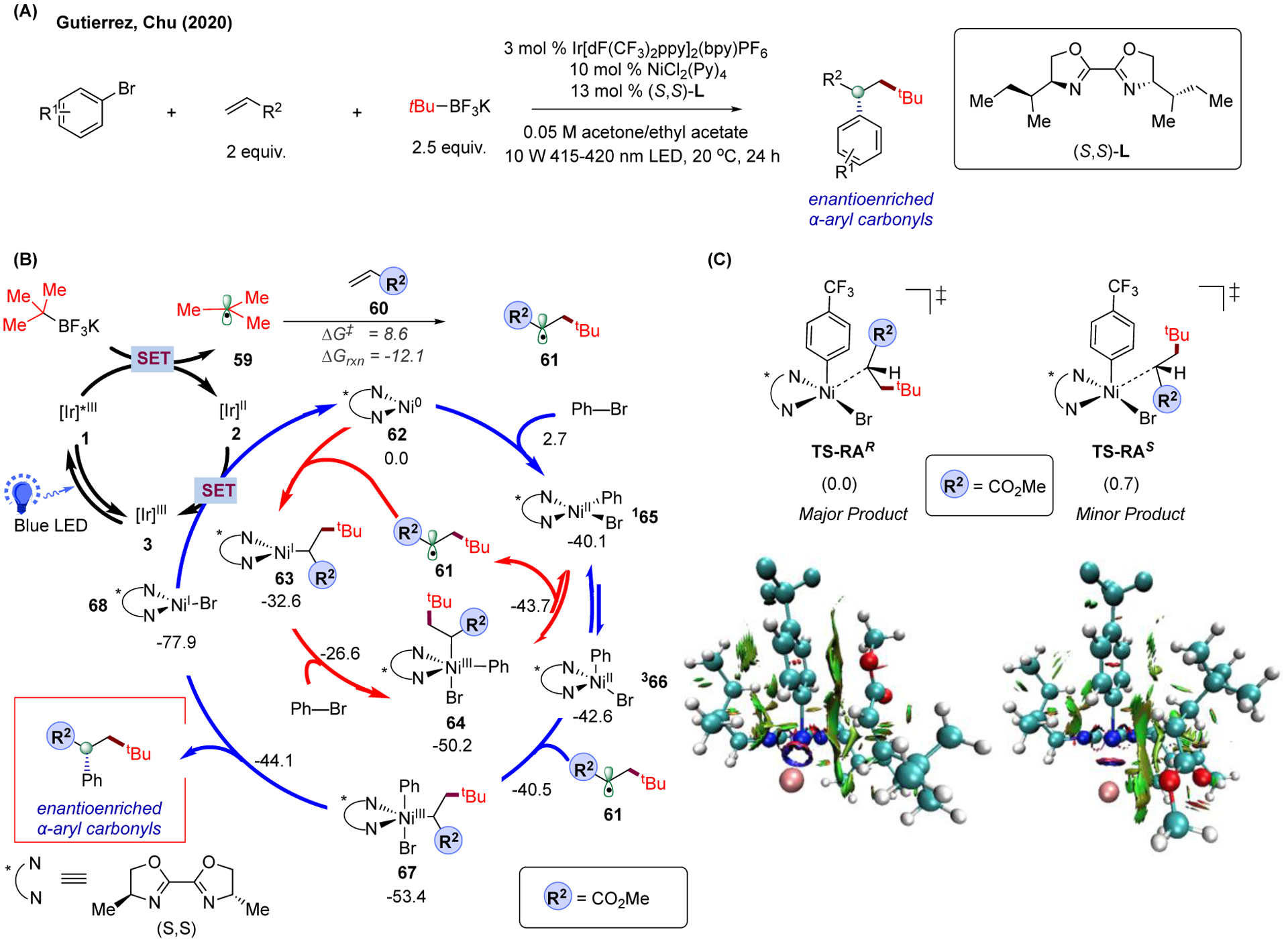
Enantioselective photoredox/Ni dual-catalyzed dicarbofunctionalization of alkenes. (A) Experimental condition of the catalytic strategy. (B) Proposed catalytic cycles of the operating mechanism. Relative Gibbs free energy values (kcal/mol;298 K) were calculated with UB3LYP-D3(BJ)/def2-SVP-CPCM(THF) level of theory. Superscript indicates the spin state (2S+1) of Ni. (C) Origin of the enantioselectivity of the transformation. Color-filled reduced density gradient surface was created by the VMD program.197 Relative electronic energy values given were calculated at the same level of theory as structural optimization.
The mechanistic investigation began with the exploration of the catalytic cycles (Figure 10B) using truncated ligand (i.e., the isopropyl group on the ligand chain was truncated to methyl) and the structural optimization was performed with open-shell, dispersion-corrected DFT (with Becke-Johnson damping function) in THF solvent with implicit CPCM solvent model [noted as UB3LYP-D3(BJ)/def2-SVP-CPCM(THF)]. Under photocatalytic conditions, the radical precursor tert-butyltrifluoroborate salt can be converted to tBu radical 59 via single-electron oxidation with the excited Ir photocatalyst 1. In turn, this alkyl radical can then undergo fast Giese addition (barrier of 8.6 kcal/mol) to the acrylate 60 to form, irreversibly, the corresponding α-carbonyl radical 61 (exergonic by 12.1 kcal/mol). Consistent with the previous findings, the newly formed α-carbonyl radical can then add to the Ni catalyst in an enantioselective manner. Specifically, the addition of 61 to tetrahedral Ni(II) species 366 (barrier of 2.1 kcal/mol) is predicted to be facile and irreversible to afford the corresponding Ni(III) intermediate 67 (downhill by 11.8 kcal/mol from 366 to 67). Finally, subsequent reductive elimination (barrier of only 9.3 kcal/mol from 67) will deliver the final chiral α-arylated carbonyl product. Since the radical addition transition state (−40.5 kcal/mol) is higher in energy level than the reductive elimination step (−44.1 kcal/mol), the former step is likely to be the stereodetermining step in this transformation.
Based on this finding, further analysis of the stereodetermining step (i.e., radical addition to tetrahedral Ni(II) species) was conducted, using the full ligand (S,S)-sec-Bu-BiOx and p-CF3-PhBr as model substrates (Figure 10C). Overall, numerous rotamers of the alkyl chains on the ligand were considered and a Boltzmann distribution analysis was conducted on the stereodetermining radical addition step (Note: only the two of the lowest-energy radical addition transition states are shown in Figure 10C). Consistent with the experiment, the computational results predicted the same enantiomeric product albeit, not surprisingly given the implicit models used to account for implicit solvent and complex reaction medium, these calculations underestimate somewhat the enantiomeric ratio (~80:20 vs ~95:5 in the experiment). In addition, to gain insights into the origin of enantioselectivity, we performed the noncovalent interaction (NCI) analysis194 with Multiwfn program195 on the two lowest-energy competing diastereomeric radical addition transition states (Figure 10C). Based on the obtained results, the attractive noncovalent interaction (i.e., van der Waals interaction) was found between the aryl ring and the alkyl chain as well as the ester group, which can be attributed to the C-H•••π and π-π interactions, correspondingly. Overall, these noncovalent interactions play an important role in determining the stereochemistry of this enantioselective olefin dicarbofunctionalization protocol. Similarly, the important contribution of the C-H•••π interaction between the alkyl chain of chiral BiOx ligand and the aryl ring to the stereoselectivity was reported in the reductive asymmetric Ni catalysis by Doyle and Sigman via multivariate analysis.196
Despite the advantages of olefin dicarbofunctionalization strategies in constructing complex molecules, their broader application in organic synthesis is limited by the need for prefunctionalized starting materials. In this vein, the combination of radical-based C-H functionalization chemistry and multicomponent cross-couplings could expand the reaction scope and enable the utilization of simpler, more abundant reagents as starting materials (e.g., alcohols and ethers). Recently, in collaboration with Molander and coworkers, we reported a photoredox/Ni dual-catalytic system realizing the multicomponent C-H activation/cross-coupling, where diaryl ketone was employed as the photoactive, hydrogen-atom-transfer (HAT) catalyst (Figure 11A).118 In contrast to other photo-induced HAT catalysis utilizing intermolecular HAT catalysts (e.g., tertiary amines or tungsten-based salts) activated by the excited photocatalyst,18,19,178,198–201 the photoactive diaryl ketone catalyst employed in this transformation overcomes the inherent challenges of these HAT agents as it is chemoselective and avoids the use of external organometallic photocatalysts.44,202–204
FIGURE 11.
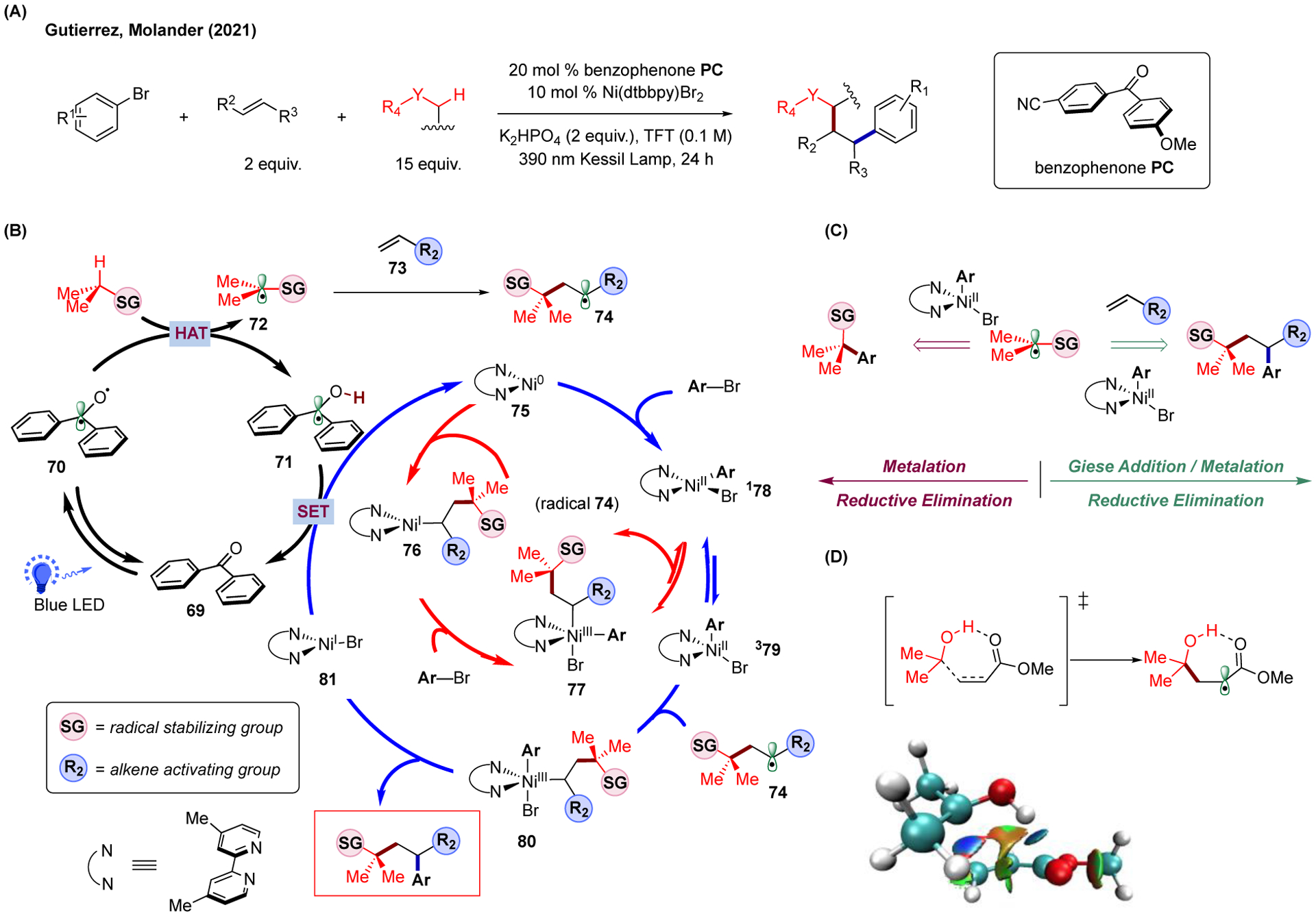
Photoredox/Ni dual-catalyzed dicarbofunctionalization of alkenes by photoinduced C-H activation via diaryl ketone hydrogen-atom transfer (HAT) catalysis. (A) Experimental condition of the catalytic strategy. (B) Proposed catalytic cycles of the operating mechanism. (C) Divergent reactivity of alkyl radical is determined by the competition between the metalation of the alkyl radical and the Giese addition processes. (D) Effect of hydrogen bonding in assisting the Giese addition step. Color-filled reduced density gradient surface was obtained via NCI analysis and created by the VMD program.
Based on the previous mechanistic studies of photo-induced diaryl-ketone-catalyzed HAT catalysis and multicomponent olefin dicarbofunctionalization (DCF) reactions,118,202 this novel catalytic strategy is proposed to proceed via the mechanism as outlined in Figure 11B. After excitation by light, the excited diaryl ketone 70 can undergo HAT with starting substrate to deliver alkyl radical 72, which is prone to undergo Giese addition in the presence of activated alkene 73. After, the newly formed Giese adduct 74 could be captured by the catalytically active Ni0 species 75 to give Ni(I)-alkyl intermediate 76. In turn, oxidative addition with aryl bromide to afford Ni(III) intermediate 77 followed by reversible radical dissociation will afford the Ni(II)-aryl-bromo species 178. Alternatively, the Ni0 species could undergo direct oxidative addition with electron-deficient aryl bromide to generate Ni(II)-aryl-bromo species 178, which was expected to undergo singlet-triplet isomerization to give tetrahedral Ni(II) intermediate 379 under light irradiation. This active tetrahedral Ni(II) intermediate could capture the Giese adduct 74 to give Ni(III) intermediate 80, followed by reductive elimination and form the desired difunctionalization product.
Notably, the competition between the metalation and the Giese addition of alkyl radical was found to determine the chemoselectivity of this catalytic strategy. In particular, the former step leads to the formation of the arylated product while the latter step generates Giese adduct that could undergo metalation with Ni(II) species and reductive elimination to deliver the dicarbofunctionalization product (Figure 11C). Importantly, we found evidence for hydrogen bonding as a crucial element to promote the Giese addition between alkyl radical and activated alkene, resulting in the selective formation of dicarbofunctionalization product over the direct arylation of the alkyl radical (Figure 11D). As an example, when iPrOH is used as an alkyl precursor in the system, the use of acrylate was found to increase the ratio of DCF product versus arylation product compared to acrylonitrile, where H-bonding played a key role in promoting the Giese addition.
4. CONCLUSION
Compared to traditional transition-metal-catalyzed cross-couplings, the merger of photoredox and Ni catalysis expands the chemical space, allowing the incorporation of C(sp3) centers that are previously inaccessible due to side reaction pathways. Moreover, using light as energy input, dual photoredox/Ni catalysis has enabled mild reaction conditions and shows great tolerance of functional groups. Despite the rapid advancements in the field of photoredox/Ni dual catalysis and the growing number of mechanistic studies, the lack of in-depth mechanistic investigation limits the wider application and discovery of novel catalytic systems of this field. Based on the mechanistic studies of photoredox/Ni dual-catalyzed C(sp2)-C(sp3) cross-coupling reactions, the mechanistic picture is dependent on the substrates and catalytic systems employed. Therefore, overgeneralization of mechanism should be avoided, and the analysis of selectivity of reaction needs to be performed based on the specific systems.
To promote the understanding of the complicated mechanistic pictures of developed systems, the use of computational methodologies is of great significance. In particular, properties and molecular-level interactions of the high-energy, short-lived intermediates can be learned with computational approaches. However, improvements need to be made to broaden the application of computational investigation and to facilitate the experimental chemists to take advantage of the studied results. Regarding this issue, the accuracy and reliability of computational methods can be advanced to deepen the study of properties and dynamical behavior of the key species. Moreover, wider collaboration among computational, experimental, and spectroscopic groups is expected to accelerate reaction discovery, especially for asymmetric transformations. We expect that with the rapid development of many emerging computational tools and data-driven approaches, the mechanistic exploration of photoredox/Ni dual catalytic systems can advance and facilitate the design of novel catalytic systems.
Funding Information
We acknowledge the NIGMS NIH (R35GM137797) for funding.
Research Resources
We are thankful for the UMD Deepthought2, MARCC/BlueCrab HPC clusters, and XSEDE (CHE160082 and CHE160053) for computational resources.
Contributor Information
Mingbin Yuan, University of Maryland, College Park.
Osvaldo Gutierrez, University of Maryland, College Park; Texas A&M University.
References
- 1.Tasker SZ, Standley EA, Jamison TF. Recent advances in homogeneous nickel catalysis. Nature. 2014;509(7500):299–309. 10.1038/nature13274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Twilton J, Zhang P, Shaw MH, Evans RW, MacMillan DW. The merger of transition metal and photocatalysis. Nat Rev Chem. 2017;1(7):1–19. 10.1038/s41570-017-0052 [DOI] [Google Scholar]
- 3.Tellis JC, Primer DN, Molander GA. Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science. 2014;345(6195):433–6. 10.1126/science.1253647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DW. Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science. 2014;345(6195):437–40. 10.1126/science.1255525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tellis JC, Kelly CB, Primer DN, Jouffroy M, Patel NR, Molander GA. Single-electron transmetalation via photoredox/nickel dual catalysis: unlocking a new paradigm for sp3–sp2 cross-coupling. Acc Chem Res. 2016;49(7):1429–39. 10.1021/acs.accounts.6b00214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cavalcanti LN, Molander GA. Photoredox catalysis in nickel-catalyzed cross-coupling. Ni-and Fe-Based Cross-Coupling Reactions. Springer, Cham; 2017. 10.1007/978-3-319-49784-6_2 [DOI] [PubMed] [Google Scholar]
- 7.Matsui JK, Lang SB, Heitz DR, Molander GA. Photoredox-mediated routes to radicals: the value of catalytic radical generation in synthetic methods development. ACS Catal. 2017;7(4):2563–75. 10.1021/acscatal.7b00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diccianni JB, Diao T. Mechanisms of nickel-catalyzed cross-coupling reactions. Trends Chem. 2019;1(9):830–44. 10.1016/j.trechm.2019.08.004 [DOI] [Google Scholar]
- 9.Milligan JA, Phelan JP, Badir SO, Molander GA. Alkyl carbon–carbon bond formation by nickel/photoredox cross-coupling. Angew Chem Int Ed. 2019;58(19):6152–63. 10.1002/anie.201809431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang P, Le CC, MacMillan DW. Silyl radical activation of alkyl halides in metallaphotoredox catalysis: a unique pathway for cross-electrophile coupling. J Am Chem Soc. 2016;138(26):8084–7. 10.1021/jacs.6b04818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jouffroy M, Primer DN, Molander GA. Base-free photoredox/nickel dual-catalytic cross-coupling of ammonium alkylsilicates. J Am Chem Soc. 2016;138(2):475–8. 10.1021/jacs.5b10963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corcé V, Chamoreau LM, Derat E, Goddard JP, Ollivier C, Fensterbank L. Silicates as latent alkyl radical precursors: visible‐light photocatalytic oxidation of hypervalent bis‐catecholato silicon compounds. Angew Chem Int Ed. 2015;54(39):11414–8. 10.1002/anie.201504963 [DOI] [PubMed] [Google Scholar]
- 13.Nakajima K, Nojima S, Sakata K, Nishibayashi Y. Visible‐light‐mediated aromatic substitution reactions of cyanoarenes with 4‐Alkyl‐1, 4‐dihydropyridines through double carbon–carbon bond cleavage. ChemCatChem. 2016;8(6):1028–32. 10.1002/cctc.201600037 [DOI] [Google Scholar]
- 14.Gutiérrez-Bonet Al, Tellis JC, Matsui JK, Vara BA, Molander GA. 1, 4-Dihydropyridines as alkyl radical precursors: introducing the aldehyde feedstock to nickel/photoredox dual catalysis. ACS Catal. 2016;6(12):8004–8. 10.1021/acscatal.6b02786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakajima K, Nojima S, Nishibayashi Y. Nickel‐and photoredox‐catalyzed cross‐coupling reactions of aryl halides with 4‐Alkyl‐1, 4‐dihydropyridines as formal nucleophilic alkylation reagents. Angew Chem Int Ed. 2016;55(45):14106–10. 10.1002/anie.201606513 [DOI] [PubMed] [Google Scholar]
- 16.Karakaya I, Primer DN, Molander GA. Photoredox cross-coupling: Ir/Ni dual catalysis for the synthesis of benzylic ethers. Org Lett. 2015;17(13):3294–7. 10.1021/acs.orglett.5b01463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo L, Tu H-Y, Zhu S, Chu L. Selective, intermolecular alkylarylation of alkenes via photoredox/nickel dual catalysis. Org Lett. 2019;21(12):4771–6. 10.1021/acs.orglett.9b01658 [DOI] [PubMed] [Google Scholar]
- 18.Jeffrey JL, Terrett JA, MacMillan DW. O–H hydrogen bonding promotes H-atom transfer from α C–H bonds for C-alkylation of alcohols. Science. 2015;349(6255):1532–6. 10.1126/science.aac8555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DW. Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science. 2016;352(6291):1304–8. 10.1126/science.aaf6635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.García-Melchor M, Braga AA, Lledós A, Ujaque G, Maseras F. Computational perspective on Pd-catalyzed C–C cross-coupling reaction mechanisms. Acc Chem Res. 2013;46(11):2626–34. 10.1021/ar400080r [DOI] [PubMed] [Google Scholar]
- 21.Sperger T, Sanhueza IA, Kalvet I, Schoenebeck F. Computational studies of synthetically relevant homogeneous organometallic catalysis involving Ni, Pd, Ir, and Rh: an overview of commonly employed DFT methods and mechanistic insights. Chem Rev. 2015;115(17):9532–86. 10.1021/acs.chemrev.5b00163 [DOI] [PubMed] [Google Scholar]
- 22.Hwang SJ, Powers DC, Maher AG, Anderson BL, Hadt RG, Zheng S-L, et al. Trap-free halogen photoelimination from mononuclear Ni (III) complexes. J Am Chem Soc. 2015;137(20):6472–5. 10.1021/jacs.5b03192 [DOI] [PubMed] [Google Scholar]
- 23.Hwang SJ, Anderson BL, Powers DC, Maher AG, Hadt RG, Nocera DG. Halogen photoelimination from monomeric nickel (iii) complexes enabled by the secondary coordination sphere. Organometallics. 2015;34(19):4766–74. 10.1021/acs.organomet.5b00568 [DOI] [Google Scholar]
- 24.Heitz DR, Tellis JC, Molander GA. Photochemical nickel-catalyzed C–H arylation: synthetic scope and mechanistic investigations. J Am Chem Soc. 2016;138(39):12715–8. 10.1021/jacs.6b04789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shields BJ, Doyle AG. Direct C (sp3)–H cross coupling enabled by catalytic generation of chlorine radicals. J Am Chem Soc. 2016;138(39):12719–22. 10.1021/jacs.6b08397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen MK, Shields BJ, Liu J, Williams MJ, Zacuto MJ, Doyle AG. Mild, redox‐neutral formylation of aryl chlorides through the photocatalytic generation of chlorine radicals. Angew Chem Int Ed. 2017;56(25):7191–4. 10.1002/anie.201702079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welin ER, Le C, Arias-Rotondo DM, McCusker JK, MacMillan DW. Photosensitized, energy transfer-mediated organometallic catalysis through electronically excited nickel (II). Science. 2017;355(6323):380–5. 10.1126/science.aal2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ackerman LK, Martinez Alvarado JI, Doyle AG. Direct C–C bond formation from alkanes using Ni-photoredox catalysis. J Am Chem Soc. 2018;140(43):14059–63. 10.1021/jacs.8b09191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ting SI, Garakyaraghi S, Taliaferro CM, Shields BJ, Scholes GD, Castellano FN, et al. 3d-d Excited states of Ni (II) complexes relevant to photoredox catalysis: spectroscopic identification and mechanistic implications. J Am Chem Soc. 2020;142(12):5800–10. 10.1021/jacs.0c00781 [DOI] [PubMed] [Google Scholar]
- 30.Vogiatzis KD, Polynski MV, Kirkland JK, Townsend J, Hashemi A, Liu C, et al. Computational approach to molecular catalysis by 3d transition metals: Challenges and opportunities. Chem Rev. 2018;119(4):2453–523. 10.1021/acs.chemrev.8b00361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qi Z-H, Ma J. Dual role of a photocatalyst: generation of Ni (0) catalyst and promotion of catalytic C–N bond formation. ACS Catal. 2018;8(2):1456–63. 10.1021/acscatal.7b03024 [DOI] [Google Scholar]
- 32.Lim C-H, Kudisch M, Liu B, Miyake GM. C–N Cross-coupling via photoexcitation of nickel–amine complexes. J Am Chem Soc. 2018;140(24):7667–73. 10.1021/jacs.8b03744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Aguirre A, Funes‐Ardoiz I, Maseras F. Four oxidation states in a single photoredox nickel‐based catalytic cycle: A computational study. Angew Chem Int Ed. 2019;58(12):3898–902. 10.1002/anie.201814233 [DOI] [PubMed] [Google Scholar]
- 34.Li R-H, Zhu B, Wang S, Geng Y, Yan L-K, Su Z-M, et al. Theoretical mechanistic study of metallaphotoredox catalysis: C–N cross-coupling via Ni (II)-mediated σ-bond metathesis. Org Chem Front. 2020;7(16):2168–78. 10.1039/D0QO00458H [DOI] [Google Scholar]
- 35.Till NA, Tian L, Dong Z, Scholes GD, MacMillan DW. Mechanistic analysis of metallaphotoredox C–N coupling: Photocatalysis initiates and perpetuates Ni (I)/Ni (III) coupling activity. J Am Chem Soc. 2020;142(37):15830–41. 10.1021/jacs.0c05901 [DOI] [PubMed] [Google Scholar]
- 36.Chu C-q, Dang L. Esterification of aryl and alkyl amides enabled by tailor-made and proposed nickel catalyst: Insights from theoretical investigation. J Org Chem. 2018;83(9):5009–18. 10.1021/acs.joc.8b00160 [DOI] [PubMed] [Google Scholar]
- 37.Zhu B, Yan L-K, Geng Y, Ren H, Guan W, Su Z-M. Ir III/Ni II-Metallaphotoredox catalysis: the oxidation state modulation mechanism versus the radical mechanism. Chem Commun. 2018;54(47):5968–71. 10.1039/C8CC03550D [DOI] [PubMed] [Google Scholar]
- 38.Shields BJ, Kudisch B, Scholes GD, Doyle AG. Long-lived charge-transfer states of nickel (II) aryl halide complexes facilitate bimolecular photoinduced electron transfer. J Am Chem Soc. 2018;140(8):3035–9. 10.1021/jacs.7b13281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun R, Qin Y, Ruccolo S, Schnedermann C, Costentin C, Nocera DG. Elucidation of a redox-mediated reaction cycle for nickel-catalyzed cross coupling. J Am Chem Soc. 2018;141(1):89–93. 10.1021/jacs.8b11262 [DOI] [PubMed] [Google Scholar]
- 40.Ma P, Wang S, Chen H. Reactivity of transition-metal complexes in excited states: C–O bond coupling reductive elimination of a Ni (II) complex is elicited by the metal-to-ligand charge transfer state. ACS Catal. 2019;10(1):1–6. 10.1021/acscatal.9b03827 [DOI] [Google Scholar]
- 41.Tian L, Till NA, Kudisch B, MacMillan DW, Scholes GD. Transient absorption spectroscopy offers mechanistic insights for an iridium/nickel-catalyzed C–O coupling. J Am Chem Soc. 2020;142(10):4555–9. 10.1021/jacs.9b12835 [DOI] [PubMed] [Google Scholar]
- 42.Ren H, Li G-F, Zhu B, Lv X-D, Yao L-S, Wang X-L, et al. How does iridium (III) photocatalyst regulate nickel (II) catalyst in metallaphotoredox-catalyzed C–S cross-coupling? Theoretical and experimental insights. ACS Catal. 2019;9(5):3858–65. 10.1021/acscatal.9b00375 [DOI] [Google Scholar]
- 43.Abdiaj I, Fontana A, Gomez MV, de la Hoz A, Alcázar J. Visible‐Light‐Induced nickel‐catalyzed negishi cross‐couplings by exogenous‐photosensitizer‐free photocatalysis. Angew Chem Int Ed. 2018;57(28):8473–7. 10.1002/anie.201802656 [DOI] [PubMed] [Google Scholar]
- 44.Dewanji A, Krach PE, Rueping M. The dual role of benzophenone in visible‐Light/nickel photoredox‐catalyzed C− H arylations: Hydrogen‐atom transfer and energy transfer. Angew Chem Int Ed. 2019;58(11):3566–70. 10.1002/anie.201901327 [DOI] [PubMed] [Google Scholar]
- 45.Cagan DA, Stroscio GD, Cusumano AQ, Hadt RG. Multireference description of nickel–aryl homolytic bond dissociation processes in photoredox catalysis. J Phys Chem A. 2020;124(48):9915–22. 10.1021/acs.jpca.0c08646 [DOI] [PubMed] [Google Scholar]
- 46.Kariofillis SK, Doyle AG. Synthetic and mechanistic implications of chlorine photoelimination in nickel/photoredox c (sp3)–h cross-coupling. Acc Chem Res. 2021;54(4):988–1000. 10.1021/acs.accounts.0c00694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wenger OS. Photoactive nickel complexes in cross‐coupling catalysis. Chem Eur J. 2020;27(7):2270–8. 10.1002/chem.202003974 [DOI] [PubMed] [Google Scholar]
- 48.Studer A, Curran DP. Catalysis of radical reactions: a radical chemistry perspective. Angew Chem Int Ed. 2016;55(1):58–102. 10.1002/anie.201505090 [DOI] [PubMed] [Google Scholar]
- 49.Staveness D, Bosque I, Stephenson CR. Free radical chemistry enabled by visible light-induced electron transfer. Acc Chem Res. 2016;49(10):2295–306. 10.1021/acs.accounts.6b00270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan M, Lo JC, Edwards JT, Baran PS. Radicals: reactive intermediates with translational potential. J Am Chem Soc. 2016;138(39):12692–714. 10.1021/jacs.6b08856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pitre SP, McTiernan CD, Scaiano JC. Understanding the kinetics and spectroscopy of photoredox catalysis and transition-metal-free alternatives. Acc Chem Res. 2016;49(6):1320–30. 10.1021/acs.accounts.6b00012 [DOI] [PubMed] [Google Scholar]
- 52.Marchini M, Bergamini G, Cozzi PG, Ceroni P, Balzani V. Photoredox catalysis: The need to elucidate the photochemical mechanism. Angew Chem Int Ed. 2017;56(42):12820–1. 10.1002/anie.201706217 [DOI] [PubMed] [Google Scholar]
- 53.Ghosh I, Bardagi JI, König B. Reply to “Photoredox catalysis: the need to elucidate the photochemical mechanism”. Angew Chem Int Ed. 2017;56(42):12822–4. 10.1002/anie.201707594 [DOI] [PubMed] [Google Scholar]
- 54.Buzzetti L, Crisenza GE, Melchiorre P. Mechanistic studies in photocatalysis. Angew Chem Int Ed. 2019;58(12):3730–47. 10.1002/anie.201809984 [DOI] [PubMed] [Google Scholar]
- 55.Perkampus H-H. UV-VIS Spectroscopy and its Applications. Springer Science & Business Media; 2013. [Google Scholar]
- 56.Mabbott GA. An introduction to cyclic voltammetry. J Chem Educ. 1983;60(9):697. 10.1021/ed060p697 [DOI] [Google Scholar]
- 57.Roth H, Romero N, Nicewicz D. Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry. Synlett. 2016;27(05) :714–23. 10.1055/s-0035-1561297 [DOI] [Google Scholar]
- 58.Kavarnos GJ. Fundamentals of photoinduced electron transfer. Vol. 1. New York: VCH; 1993. [Google Scholar]
- 59.Balzani V, Ceroni P, Juris A. Photochemistry and photophysics: concepts, research, applications. John Wiley & Sons; 2014. [Google Scholar]
- 60.Farid S, Dinnocenzo JP, Merkel PB, Young RH, Shukla D, Guirado G. Reexamination of the Rehm–Weller data set reveals electron transfer quenching that follows a Sandros–Boltzmann dependence on free energy. J Am Chem Soc. 2011;133(30):11580–7. 10.1021/ja2024367 [DOI] [PubMed] [Google Scholar]
- 61.Rosspeintner A, Angulo G, Vauthey E. Bimolecular photoinduced electron transfer beyond the diffusion limit: The Rehm–Weller experiment revisited with femtosecond time resolution. J Am Chem Soc. 2014;136(5):2026–32. 10.1021/ja4118279 [DOI] [PubMed] [Google Scholar]
- 62.Jones WE Jr, Fox MA. Determination of excited-state redox potentials by phase-modulated voltammetry. J Phys Chem. 1994;98(19):5095–9. 10.1021/j100070a025 [DOI] [Google Scholar]
- 63.ODA N TSUJI K, ICHIMURA A, editors. Voltammetric measurements of redox potentials of photo-excited species. Analytical Sciences/Supplements Proceedings of IUPAC International Congress on Analytical Sciences 2001 (ICAS 2001). The Japan Society for Analytical Chemistry; 2002. [Google Scholar]
- 64.Goddard J-P, Ollivier C, Fensterbank L. Photoredox catalysis for the generation of carbon centered radicals. Acc Chem Res. 2016;49(9):1924–36. 10.1021/acs.accounts.6b00288 [DOI] [PubMed] [Google Scholar]
- 65.Ingold KU, Pratt DA. Advances in radical-trapping antioxidant chemistry in the 21st century: a kinetics and mechanisms perspective. Chem Rev. 2014;114(18):9022–46. 10.1021/cr500226n [DOI] [PubMed] [Google Scholar]
- 66.Griller D, Ingold KU. Free-radical clocks. Acc Chem Res. 1980;13(9):317–23. 10.1021/ar50153a004 [DOI] [Google Scholar]
- 67.Newcomb M Radical kinetics and clocks. Encyclopedia of Radicals in Chemistry, Biology and Materials. 2012. [Google Scholar]
- 68.Weil JA, Bolton JR. Electron paramagnetic resonance: elementary theory and practical applications. John Wiley & Sons; 2007. [Google Scholar]
- 69.Abragam A, Bleaney B. Electron paramagnetic resonance of transition ions. OUP Oxford; 2012. [Google Scholar]
- 70.Al’Tshuler SA, Kozyrev BM. Electron paramagnetic resonance. Academic Press; 2013. [Google Scholar]
- 71.Goez M Photo-CIDNP spectroscopy. Annu Rep NMR Spectrosc. 2009;66:77–147. 10.1016/S0066-4103(08)00403-1 [DOI] [Google Scholar]
- 72.Scaiano J Nanosecond laser flash photolysis: a tool for physical organic chemistry. Reactive Intermediate Chemistry. Wiley Online Library; 2004. p. 797–845. 10.1002/0471721492.ch18 [DOI] [Google Scholar]
- 73.Farr EP, Quintana JC, Reynoso V, Ruberry JD, Shin WR, Swartz KR. Introduction to time-resolved spectroscopy: Nanosecond transient absorption and time-resolved fluorescence of eosin b. J Chem Educ. 2018;95(5):864–71. 10.1021/acs.jchemed.7b00941 [DOI] [Google Scholar]
- 74.Stolow A, Bragg AE, Neumark DM. Femtosecond time-resolved photoelectron spectroscopy. Chem Rev. 2004;104(4):1719–58. 10.1021/cr020683w [DOI] [PubMed] [Google Scholar]
- 75.Lakowicz JR. Principles of fluorescence spectroscopy. Springer science & business media; 2013. [Google Scholar]
- 76.Bahamonde A, Murphy JJ, Savarese M, Brémond Er, Cavalli A, Melchiorre P. Studies on the enantioselective iminium ion trapping of radicals triggered by an electron-relay mechanism. J Am Chem Soc. 2017;139(12):4559–67. 10.1021/jacs.7b01446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC. Nickel-catalyzed cross-coupling of photoredox-generated radicals: uncovering a general manifold for stereoconvergence in nickel-catalyzed cross-couplings. J Am Chem Soc. 2015;137(15):4896–9. 10.1021/ja513079r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ghosh S, Verma P, Cramer CJ, Gagliardi L, Truhlar DG. Combining wave function methods with density functional theory for excited states. Chem Rev. 2018;118(15):7249–92. 10.1021/acs.chemrev.8b00193 [DOI] [PubMed] [Google Scholar]
- 79.Lischka H, Nachtigallova D, Aquino AJ, Szalay PG, Plasser F, Machado FB, et al. Multireference approaches for excited states of molecules. Chem Rev. 2018;118(15):7293–361. 10.1021/acs.chemrev.8b00244 [DOI] [PubMed] [Google Scholar]
- 80.Garavelli M Computational organic photochemistry: strategy, achievements and perspectives. Theor Chem Acc. 2006;116(1):87–105. 10.1007/s00214-005-0030-z [DOI] [Google Scholar]
- 81.Mai S, González L. Molecular photochemistry: Recent developments in theory. Angew Chem Int Ed. 2020;59(39):16832–46. 10.1002/anie.201916381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Demissie TB, Ruud K, Hansen JH. DFT as a Powerful Predictive Tool in Photoredox Catalysis: Redox Potentials and Mechanistic Analysis. Organometallics. 2015;34(17):4218–28. 10.1021/acs.organomet.5b00582 [DOI] [Google Scholar]
- 83.Funes-Ardoiz I, Schoenebeck F. Established and Emerging Computational Tools to Study Homogeneous Catalysis—From Quantum Mechanics to Machine Learning. Chem. 2020;6(8):1904–13. 10.1016/j.chempr.2020.07.008 [DOI] [Google Scholar]
- 84.Cohen AJ, Mori-Sánchez P, Yang W. Insights into current limitations of density functional theory. Science. 2008;321(5890):792–4. 10.1126/science.1158722 [DOI] [PubMed] [Google Scholar]
- 85.Cramer CJ, Truhlar DG. Density functional theory for transition metals and transition metal chemistry. Phys Chem Chem Phys. 2009;11(46):10757–816. 10.1039/B907148B [DOI] [PubMed] [Google Scholar]
- 86.Simón L, Goodman JM. How reliable are DFT transition structures? Comparison of GGA, hybrid-meta-GGA and meta-GGA functionals. Org Biomol Chem. 2011;9(3):689–700. 10.1039/C0OB00477D [DOI] [PubMed] [Google Scholar]
- 87.Minenkov Y, Singstad Å, Occhipinti G, Jensen VR. The accuracy of DFT-optimized geometries of functional transition metal compounds: a validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012;41(18):5526–41. 10.1039/C2DT12232D [DOI] [PubMed] [Google Scholar]
- 88.Cohen AJ, Mori-Sánchez P, Yang W. Challenges for density functional theory. Chem Rev. 2012;112(1):289–320. 10.1021/cr200107z [DOI] [PubMed] [Google Scholar]
- 89.Burke K Perspective on density functional theory. J Chem Phys. 2012;136(15):150901. 10.1063/1.4704546 [DOI] [PubMed] [Google Scholar]
- 90.Becke AD. Perspective: Fifty years of density-functional theory in chemical physics. J Chem Phys. 2014;140(18):18A301. 10.1063/1.4869598 [DOI] [PubMed] [Google Scholar]
- 91.Jones RO. Density functional theory: Its origins, rise to prominence, and future. Rev Mod Phys. 2015;87(3):897. 10.1103/RevModPhys.87.897 [DOI] [Google Scholar]
- 92.Pribram-Jones A, Gross DA, Burke K. Dft: A theory full of holes? Annu Rev Phys Chem. 2015;66:283–304. 10.1146/annurev-physchem-040214-121420 [DOI] [PubMed] [Google Scholar]
- 93.Yu HS, Li SL, Truhlar DG. Perspective: Kohn-Sham density functional theory descending a staircase. J Chem Phys. 2016;145(13):130901. 10.1063/1.4963168 [DOI] [PubMed] [Google Scholar]
- 94.Grimme S Semiempirical hybrid density functional with perturbative second-order correlation. J Chem Phys. 2006;124(3):034108. 10.1063/1.2148954 [DOI] [PubMed] [Google Scholar]
- 95.Wu Q, Yang W. Empirical correction to density functional theory for van der Waals interactions. J Chem Phys. 2002;116(2):515–24. 10.1063/1.1424928 [DOI] [Google Scholar]
- 96.Grimme S Accurate description of van der Waals complexes by density functional theory including empirical corrections. J Comput Chem. 2004;25(12):1463–73. 10.1002/jcc.20078 [DOI] [PubMed] [Google Scholar]
- 97.Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132(15):154104. 10.1063/1.3382344 [DOI] [PubMed] [Google Scholar]
- 98.de la Roza AO, DiLabio GA. Non-covalent Interactions in Quantum Chemistry and Physics: Theory and Applications. Elsevier; 2017. [Google Scholar]
- 99.Johnson ER, Becke AD. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections. J Chem Phys. 2006;124(17):174104. 10.1063/1.2190220 [DOI] [PubMed] [Google Scholar]
- 100.Grimme S, Ehrlich S, Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J Comput Chem. 2011;32(7):1456–65. 10.1002/jcc.21759 [DOI] [PubMed] [Google Scholar]
- 101.Mardirossian N, Head-Gordon M. ωB97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation. J Chem Phys. 2016;144(21):214110. 10.1063/1.4952647 [DOI] [PubMed] [Google Scholar]
- 102.Haoyu SY, He X, Li SL, Truhlar DG. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem Sci. 2016;7(8):5032–51. 10.1039/C6SC00705H [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Poli R Open-shell organometallics as a bridge between Werner-type and low-valent organometallic complexes. The effect of the spin state on the stability, reactivity, and structure. Chem Rev. 1996;96(6):2135–204. 10.1021/cr9500343 [DOI] [PubMed] [Google Scholar]
- 104.Schröder D, Shaik S, Schwarz H. Two-state reactivity as a new concept in organometallic chemistry. Acc Chem Res. 2000;33(3):139–45. 10.1021/ar990028j [DOI] [PubMed] [Google Scholar]
- 105.Harvey JN, Poli R, Smith KM. Understanding the reactivity of transition metal complexes involving multiple spin states. Coord Chem Rev. 2003;238:347–61. 10.1016/S0010-8545(02)00283-7 [DOI] [Google Scholar]
- 106.Holland PL. Distinctive reaction pathways at base metals in high-spin organometallic catalysts. Acc Chem Res. 2015;48(6):1696–702. 10.1021/acs.accounts.5b00036 [DOI] [PubMed] [Google Scholar]
- 107.Houk K, Liu F. Holy grails for computational organic chemistry and biochemistry. Acc Chem Res. 2017;50(3):539–43. 10.1021/acs.accounts.6b00532 [DOI] [PubMed] [Google Scholar]
- 108.Sperger T, Sanhueza IA, Schoenebeck F. Computation and experiment: a powerful combination to understand and predict reactivities. Acc Chem Res. 2016;49(6):1311–9. 10.1021/acs.accounts.6b00068 [DOI] [PubMed] [Google Scholar]
- 109.Verma P, Truhlar DG. Status and challenges of density functional theory. Trends Chem. 2020;2(4):302–18. 10.1016/j.trechm.2020.02.005 [DOI] [Google Scholar]
- 110.Raghavachari K, Trucks GW, Pople JA, Head-Gordon M. A fifth-order perturbation comparison of electron correlation theories. Chem Phys Lett. 1989;157(6):479–83. 10.1016/S0009-2614(89)87395-6 [DOI] [Google Scholar]
- 111.Riplinger C, Sandhoefer B, Hansen A, Neese F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J Chem Phys. 2013;139(13):134101. 10.1063/1.4821834 [DOI] [PubMed] [Google Scholar]
- 112.Liakos DG, Neese F. Is it possible to obtain coupled cluster quality energies at near density functional theory cost? Domain-based local pair natural orbital coupled cluster vs modern density functional theory. J Chem Theory Comput. 2015;11(9):4054–63. 10.1021/acs.jctc.5b00359 [DOI] [PubMed] [Google Scholar]
- 113.Liakos DG, Sparta M, Kesharwani MK, Martin JM, Neese F. Exploring the accuracy limits of local pair natural orbital coupled-cluster theory. J Chem Theory Comput. 2015;11(4):1525–39. 10.1021/ct501129s [DOI] [PubMed] [Google Scholar]
- 114.Guo Y, Riplinger C, Becker U, Liakos DG, Minenkov Y, Cavallo L, et al. Communication: An improved linear scaling perturbative triples correction for the domain based local pair-natural orbital based singles and doubles coupled cluster method [DLPNO-CCSD (T)]. J Chem Phys. 2018;148(1):011101. 10.1063/1.5011798 [DOI] [PubMed] [Google Scholar]
- 115.Liakos DG, Guo Y, Neese F. Comprehensive benchmark results for the domain based local pair natural orbital coupled cluster method (DLPNO-CCSD (T)) for closed-and open-shell systems. J Chem Phys A. 2019;124(1):90–100. 10.1021/acs.jpca.9b05734 [DOI] [PubMed] [Google Scholar]
- 116.Guo Y, Riplinger C, Liakos DG, Becker U, Saitow M, Neese F. Linear scaling perturbative triples correction approximations for open-shell domain-based local pair natural orbital coupled cluster singles and doubles theory [DLPNO-CCSD (T0/T)]. J Chem Phys. 2020;152(2):024116. 10.1063/1.5127550 [DOI] [PubMed] [Google Scholar]
- 117.Matsui JK, Gutiérrez‐Bonet Á, Rotella M, Alam R, Gutierrez O, Molander GA. Photoredox/Nickel‐Catalyzed Single‐Electron Tsuji–Trost Reaction: Development and Mechanistic Insights. Angew Chem Int Ed. 2018;57(48):15847–51. 10.1002/anie.201809919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Campbell MW, Yuan M, Polites VC, Gutierrez O, Molander GA. Photochemical C–H Activation Enables Nickel-Catalyzed Olefin Dicarbofunctionalization. J Am Chem Soc. 2021;143(10):3901–10. 10.1021/jacs.0c13077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Szalay PG, Muller T, Gidofalvi G, Lischka H, Shepard R. Multiconfiguration self-consistent field and multireference configuration interaction methods and applications. Chem Rev. 2012;112(1):108–81. 10.1021/cr200137a [DOI] [PubMed] [Google Scholar]
- 120.Grimme S, Waletzke M. A combination of Kohn–Sham density functional theory and multi-reference configuration interaction methods. J Chem Phys. 1999;111(13):5645–55. 10.1063/1.479866 [DOI] [Google Scholar]
- 121.Escudero D, Thiel W. Assessing the density functional theory-based multireference configuration interaction (DFT/MRCI) method for transition metal complexes. J Chem Phys. 2014;140(19):194105. 10.1063/1.4875810 [DOI] [PubMed] [Google Scholar]
- 122.Daniel C Electronic spectroscopy and photoreactivity in transition metal complexes. Coord Chem Rev. 2003;238:143–66. 10.1016/S0010-8545(02)00295-3 [DOI] [Google Scholar]
- 123.Neese F, Petrenko T, Ganyushin D, Olbrich G. Advanced aspects of ab initio theoretical optical spectroscopy of transition metal complexes: Multiplets, spin-orbit coupling and resonance Raman intensities. Coord Chem Rev. 2007;251(3–4):288–327. 10.1016/j.ccr.2006.05.019 [DOI] [Google Scholar]
- 124.Ipatov A, Cordova F, Doriol LJ, Casida ME. Excited-state spin-contamination in time-dependent density-functional theory for molecules with open-shell ground states. J Mol Struct: THEOCHEM. 2009;914(1–3):60–73. 10.1016/j.theochem.2009.07.036 [DOI] [Google Scholar]
- 125.Adamo C, Jacquemin D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem Soc Rev. 2013;42(3):845–56. 10.1039/C2CS35394F [DOI] [PubMed] [Google Scholar]
- 126.Domcke W, Yarkony D, Köppel H. Conical intersections: electronic structure, dynamics & spectroscopy. World Scientific; 2004. [Google Scholar]
- 127.Domcke W, Yarkony DR, Köppel H. Conical intersections: theory, computation and experiment. World Scientific; 2011. [Google Scholar]
- 128.Ess DH, Wheeler SE, Iafe RG, Xu L, Celebi‐Oelcuem N, Houk KN. Bifurcations on potential energy surfaces of organic reactions. Angew Chem Int Ed. 2008;47(40):7592–601. 10.1002/anie.200800918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hare SR, Tantillo DJ. Post-transition state bifurcations gain momentum–current state of the field. Pure Appl Chem. 2017;89(6):679–98. 10.1515/pac-2017-0104 [DOI] [Google Scholar]
- 130.Yang Z, Houk K. The dynamics of chemical reactions: atomistic visualizations of organic reactions, and homage to van’t Hoff. Chem Eur J. 2018;24(16):3916–24. 10.1002/chem.201706032 [DOI] [PubMed] [Google Scholar]
- 131.Yang Z, Jamieson CS, Xue X-S, Garcia-Borràs M, Benton T, Dong X, et al. Mechanisms and dynamics of reactions involving entropic intermediates. Trends Chem. 2019;1(1):22–34. 10.1016/j.trechm.2019.01.009 [DOI] [Google Scholar]
- 132.Pu M, Sanhueza IA, Senol E, Schoenebeck F. Divergent reactivity of stannane and silane in the trifluoromethylation of PdII: cyclic transition state versus difluorocarbene release. Angew Chem Int Ed. 2018;57(46):15081–5. 10.1002/anie.201808229 [DOI] [PubMed] [Google Scholar]
- 133.Wang Z, Hirschi JS, Singleton DA. Recrossing and dynamic matching effects on selectivity in a Diels-Alder reaction. Angew Chem Int Ed. 2009;48(48):9156–9. 10.1002/anie.200903293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hare SR, Tantillo DJ. Cryptic post-transition state bifurcations that reduce the efficiency of lactone-forming Rh-carbenoid C–H insertions. Chem Sci. 2017;8(2):1442–9. 10.1039/C6SC03745C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mandal N, Datta A. Gold (I)-catalyzed intramolecular diels–alder reaction: evolution of trappable intermediates via asynchronous transition states. J Org Chem. 2018;83(18):11167–77. 10.1021/acs.joc.8b01752 [DOI] [PubMed] [Google Scholar]
- 136.Car R, Parrinello M. Unified approach for molecular dynamics and density-functional theory. Phys Rev Lett. 1985;55(22):2471. 10.1103/PhysRevLett.55.2471 [DOI] [PubMed] [Google Scholar]
- 137.Remler DK, Madden PA. Molecular dynamics without effective potentials via the Car-Parrinello approach. Mol Phys. 1990;70(6):921–66. 10.1080/00268979000101451 [DOI] [Google Scholar]
- 138.Payne MC, Teter MP, Allan DC, Arias T, Joannopoulos JD. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev Mod Phys. 1992;64(4):1045. 10.1103/RevModPhys.64.1045 [DOI] [Google Scholar]
- 139.Tse JS. Ab initio molecular dynamics with density functional theory. Annu Rev Phys Chem. 2002;53(1):249–90. 10.1146/annurev.physchem.53.090401.105737 [DOI] [PubMed] [Google Scholar]
- 140.Worth GA, Cederbaum LS. Beyond Born-Oppenheimer: molecular dynamics through a conical intersection. Annu Rev Phys Chem. 2004;55:127–58. 10.1146/annurev.physchem.55.091602.094335 [DOI] [PubMed] [Google Scholar]
- 141.Agostini F, Curchod BF. Different flavors of nonadiabatic molecular dynamics. Wiley Interdiscip Rev Comput Mol Sci. 2019;9(5):e1417. 10.1002/wcms.1417 [DOI] [Google Scholar]
- 142.Li J, Stein R, Lopez SA. A Theoretical Stereoselectivity Model of Photochemical Denitrogenations of Diazoalkanes Toward Strained 1, 3-Dihalogenated Bicyclobutanes. J Org Chem. 2021;86(5):4061–70. 10.1021/acs.joc.0c02905 [DOI] [PubMed] [Google Scholar]
- 143.Foscato M, Jensen VR. Automated in silico design of homogeneous catalysts. ACS Catal. 2020;10(3):2354–77. 10.1021/acscatal.9b04952 [DOI] [Google Scholar]
- 144.Maeda S, Morokuma K. Finding reaction pathways of type A+ B→ X: toward systematic prediction of reaction mechanisms. J Chem Theory Comput. 2011;7(8):2335–45. 10.1021/ct200290m [DOI] [PubMed] [Google Scholar]
- 145.Maeda S, Morokuma K. Toward predicting full catalytic cycle using automatic reaction path search method: a case study on HCo (CO) 3-catalyzed hydroformylation. J Chem Theory Comput. 2012;8(2):380–5. 10.1021/ct200829p [DOI] [PubMed] [Google Scholar]
- 146.Varela Carrete JÁ, Vázquez Rodríguez SA, Martínez Núñez E. An automated method to find reaction mechanisms and solve the kinetics in organometallic catalysis. Chem Sci. 2017;8(5):3843–51. 10.1039/C7SC00549K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Guan Y, Ingman VM, Rooks BJ, Wheeler SE. AARON: an automated reaction optimizer for new catalysts. J Chem Theory Comput. 2018;14(10):5249–61. 10.1021/acs.jctc.8b00578 [DOI] [PubMed] [Google Scholar]
- 148.Chen WL. Chemoinformatics: past, present, and future. J Chem Inf Model. 2006;46(6):2230–55. 10.1021/ci060016u [DOI] [PubMed] [Google Scholar]
- 149.Haghighatlari M, Li J, Heidar-Zadeh F, Liu Y, Guan X, Head-Gordon T. Learning to Make chemical predictions: the Interplay of feature representation, data, and machine learning methods. Chem. 2020;6(7):1527–42. 10.1016/j.chempr.2020.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Toyao T, Maeno Z, Takakusagi S, Kamachi T, Takigawa I, Shimizu K-i. Machine learning for catalysis informatics: recent applications and prospects. ACS Catal. 2019;10(3):2260–97. 10.1021/acscatal.9b04186 [DOI] [Google Scholar]
- 151.Harper KC, Bess EN, Sigman MS. Multidimensional steric parameters in the analysis of asymmetric catalytic reactions. Nat Chem. 2012;4(5):366–74. 10.1038/nchem.1297 [DOI] [PubMed] [Google Scholar]
- 152.Santiago CB, Guo J-Y, Sigman MS. Predictive and mechanistic multivariate linear regression models for reaction development. Chem Sci. 2018;9(9):2398–412. 10.1039/C7SC04679K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Reid JP, Sigman MS. Comparing quantitative prediction methods for the discovery of small-molecule chiral catalysts. Nat Rev Chem. 2018;2(10):290–305. 10.1038/s41570-018-0040-8 [DOI] [Google Scholar]
- 154.Williams T, McCullough K, Lauterbach JA. Enabling catalyst discovery through machine learning and high-throughput experimentation. Chem Mater. 2020;32(1):157–65. 10.1021/acs.chemmater.9b03043 [DOI] [Google Scholar]
- 155.McCullough K, Williams T, Mingle K, Jamshidi P, Lauterbach J. High-throughput experimentation meets artificial intelligence: a new pathway to catalyst discovery. Phys Chem Chem Phys. 2020;22(20):11174–96. [DOI] [PubMed] [Google Scholar]
- 156.Eyke NS, Koscher BA, Jensen KF. Toward machine learning-enhanced high-throughput experimentation. Trends Chem. 2021;3(2):120–32. 10.1016/j.trechm.2020.12.001 [DOI] [Google Scholar]
- 157.Ahneman DT, Estrada JG, Lin S, Dreher SD, Doyle AG. Predicting reaction performance in C–N cross-coupling using machine learning. Science. 2018;360(6385):186–90. 10.1126/science.aar5169 [DOI] [PubMed] [Google Scholar]
- 158.Chuang KV, Keiser MJ. Comment on “Predicting reaction performance in C–N cross-coupling using machine learning”. Science. 2018;362(6416):eaat8603. 10.1126/science.aat8603 [DOI] [PubMed] [Google Scholar]
- 159.Estrada JG, Ahneman DT, Sheridan RP, Dreher SD, Doyle AG. Response to Comment on “Predicting reaction performance in C–N cross-coupling using machine learning”. Science. 2018;362(6416):eaat8763. 10.1126/science.aat8763 [DOI] [PubMed] [Google Scholar]
- 160.Jorner K, Tomberg A, Bauer C, Sköld C, Norrby P-O. Organic reactivity from mechanism to machine learning. Nat Rev Chem. 2021;5(4):240–55. 10.1038/s41570-021-00260-x [DOI] [PubMed] [Google Scholar]
- 161.Bibette J Gaining confidence in high-throughput screening. Proc Natl Acad Sci. 2012;109(3):649–50. 10.1073/pnas.1119350109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Breen CP, Nambiar AM, Jamison TF, Jensen KF. Ready, set, flow! Automated continuous synthesis and optimization. Trends Chem. 2021;3(5):373–386. 10.1016/j.trechm.2021.02.005 [DOI] [Google Scholar]
- 163.Coley CW, Eyke NS, Jensen KF. Autonomous discovery in the chemical sciences part I: Progress. Angew Chem Int Ed. 2020;59(51):22858–93. 10.1002/anie.201909987 [DOI] [PubMed] [Google Scholar]
- 164.Coley CW, Eyke NS, Jensen KF. Autonomous discovery in the chemical sciences part II: outlook. Angew Chem Int Ed. 2020;59(52):23414–36. 10.1002/anie.201909989 [DOI] [PubMed] [Google Scholar]
- 165.Kuijpers KP, Bottecchia C, Cambié D, Drummen K, König NJ, Noël T. A fully automated continuous‐flow platform for fluorescence quenching studies and Stern–Volmer analysis. Angew Chem Int Ed. 2018;57(35):11278–82. 10.1002/anie.201805632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Patrascu MB, Pottel J, Pinus S, Bezanson M, Norrby P-O, Moitessier N. From desktop to benchtop with automated computational workflows for computer-aided design in asymmetric catalysis. Nat Catal. 2020;3(7):574–84. 10.1038/s41929-020-0468-3 [DOI] [Google Scholar]
- 167.Kolahdouzan K, Khalaf R, Grandner JM, Chen Y, Terrett JA, Huestis MP. Dual photoredox/nickel-catalyzed conversion of aryl halides to aryl aminooxetanes: Computational evidence for a substrate-dependent switch in mechanism. ACS Catal. 2019;10(1):405–11. 10.1021/acscatal.9b03596 [DOI] [Google Scholar]
- 168.Luo Y, Gutiérrez-Bonet Al, Matsui JK, Rotella ME, Dykstra R, Gutierrez O, et al. Oxa-and azabenzonorbornadienes as electrophilic partners under photoredox/nickel dual catalysis. ACS Catal. 2019;9(9):8835–42. 10.1021/acscatal.9b02458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yuan M, Song Z, Badir SO, Molander GA, Gutierrez O. On the nature of C(sp3)–C(sp2) bond formation in nickel-catalyzed tertiary radical cross-couplings: A case study of ni/photoredox catalytic cross-coupling of alkyl radicals and aryl halides. J Am Chem Soc. 2020;142(15):7225–34. 10.1021/jacs.0c02355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Primer DN, Molander GA. Enabling the cross-coupling of tertiary organoboron nucleophiles through radical-mediated alkyl transfer. J Am Chem Soc. 2017;139(29):9847–50. 10.1021/jacs.7b06288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Trost BM, Xu J, Schmidt T. Palladium-catalyzed decarboxylative asymmetric allylic alkylation of enol carbonates. J Am Chem Soc. 2009;131(51):18343–57. 10.1021/ja9053948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Keith JA, Behenna DC, Sherden N, Mohr JT, Ma S, Marinescu SC, et al. The reaction mechanism of the enantioselective Tsuji allylation: inner-sphere and outer-sphere pathways, internal rearrangements, and asymmetric C–C bond formation. J Am Chem Soc. 2012;134(46):19050–60. 10.1021/ja306860n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Cusumano AQ, Stoltz BM, Goddard III WA. Reaction mechanism, origins of enantioselectivity, and reactivity trends in asymmetric allylic alkylation: A comprehensive quantum mechanics investigation of a C(sp3)–C(sp3) cross-coupling. J Am Chem Soc. 2020;142(32):13917–33. 10.1021/jacs.0c06243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lee W, Zhou J, Gutierrez O. Mechanism of Nakamura’s Bisphosphine-Iron-Catalyzed Asymmetric C(sp(2))-C(sp(3)) Cross-Coupling Reaction: The Role of Spin in Controlling Arylation Pathways. J Am Chem Soc. 2017;139(45):16126–33. 10.1021/jacs.7b06377 [DOI] [PubMed] [Google Scholar]
- 175.Sun Y, Tang H, Chen K, Hu L, Yao J, Shaik S, et al. Two-state reactivity in low-valent iron-mediated C–H activation and the implications for other first-row transition metals. J Am Chem Soc. 2016;138(11):3715–30. 10.1021/jacs.5b12150 [DOI] [PubMed] [Google Scholar]
- 176.Harvey JN, Aschi M, Schwarz H, Koch W. The singlet and triplet states of phenyl cation. A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces. Theor Chem Acc. 1998;99(2):95–9. 10.1007/s002140050309 [DOI] [Google Scholar]
- 177.Wang H, Liu C-F, Song Z, Yuan M, Ho YA, Gutierrez O, et al. Engaging α-fluorocarboxylic acids directly in decarboxylative C–C bond formation. ACS Catal. 2020;10(7):4451–9. 10.1021/acscatal.0c00789 [DOI] [Google Scholar]
- 178.Le C, Liang Y, Evans RW, Li X, MacMillan DW. Selective sp 3 C–H alkylation via polarity-match-based cross-coupling. Nature. 2017;547(7661):79–83. 10.1038/nature22813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Maity B, Zhu C, Yue H, Huang L, Harb M, Minenkov Y, et al. Mechanistic insight into the photoredox-nickel-HAT triple catalyzed arylation and alkylation of α-amino Csp3–H bonds. J Am Chem Soc. 2020;142(40):16942–52. 10.1021/jacs.0c05010 [DOI] [PubMed] [Google Scholar]
- 180.Giri R, Kc S. Strategies toward dicarbofunctionalization of unactivated olefins by combined heck carbometalation and cross-coupling. J Org Chem. 2018;83(6):3013–22. 10.1021/acs.joc.7b03128 [DOI] [PubMed] [Google Scholar]
- 181.Saini V, Stokes BJ, Sigman MS. Transition‐Metal‐Catalyzed laboratory‐scale carbon–carbon bond‐forming reactions of ethylene. Angew Chem Int Ed. 2013;52(43):11206–20. 10.1002/anie.201303916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Coombs JR, Morken JP. Catalytic enantioselective functionalization of unactivated terminal alkenes. Angew Chem Int Ed. 2016;55(8):2636–49. 10.1002/anie.201507151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Dhungana RK, Kc S, Basnet P, Giri R. Transition metal‐catalyzed dicarbofunctionalization of unactivated olefins. Chem Rec. 2018;18(9):1314–40. 10.1002/tcr.201700098 [DOI] [PubMed] [Google Scholar]
- 184.Derosa J, Apolinar O, Kang T, Tran VT, Engle KM. Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes. Chem Sci. 2020;11(17):4287–96. 10.1039/C9SC06006E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tu H-Y, Zhu S, Qing F-L, Chu L. Recent advances in nickel-catalyzed three-component difunctionalization of unactivated alkenes. Synthesis. 2020;52(09):1346–56. 10.1055/s-0039-1690842 [DOI] [Google Scholar]
- 186.Zhu C, Yue H, Chu L, Rueping M. Recent advances in photoredox and nickel dual-catalyzed cascade reactions: pushing the boundaries of complexity. Chem Sci. 2020;11(16):4051–64. 10.1039/D0SC00712A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Badir SO, Molander GA. Developments in photoredox/nickel dual-catalyzed 1, 2-difunctionalizations. Chem. 2020;6(6):1327–39. 10.1016/j.chempr.2020.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Diccianni J, Lin Q, Diao T. Mechanisms of nickel-catalyzed coupling reactions and applications in alkene functionalization. Acc Chem Res. 2020;53(4):906–19. 10.1021/acs.accounts.0c00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Qi X, Diao T. Nickel-catalyzed dicarbofunctionalization of alkenes. ACS Catal. 2020;10(15):8542–56. 10.1021/acscatal.0c02115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Guo L, Yuan M, Zhang Y, Wang F, Zhu S, Gutierrez O, et al. General method for enantioselective three-component carboarylation of alkenes enabled by visible-light dual photoredox/nickel catalysis. J Am Chem Soc. 2020;142(48):20390–9. 10.1021/jacs.0c08823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Harrington PJ, Lodewijk E. Twenty years of naproxen technology. Org Process Res Dev. 1997;1(1):72–6. 10.1021/op960009e [DOI] [Google Scholar]
- 192.Venkatesan H, Davis MC, Altas Y, Snyder JP, Liotta DC. Total synthesis of SR 121463 A, a highly potent and selective vasopressin V2 receptor antagonist. J Org Chem. 2001;66(11):3653–61. 10.1021/jo0004658 [DOI] [PubMed] [Google Scholar]
- 193.Sarabu R, Bizzarro FT, Corbett WL, Dvorozniak MT, Geng W, Grippo JF, et al. Discovery of piragliatin-first glucokinase activator studied in type 2 diabetic patients. J Med Chem. 2012;55(16):7021–36. 10.1021/jm3008689 [DOI] [PubMed] [Google Scholar]
- 194.Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang W. Revealing noncovalent interactions. J Am Chem Soc. 2010;132(18):6498–506. 10.1021/ja100936w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lu T, Chen F. Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem. 2012;33(5):580–92. 10.1002/jcc.22885 [DOI] [PubMed] [Google Scholar]
- 196.Woods BP, Orlandi M, Huang C-Y, Sigman MS, Doyle AG. Nickel-catalyzed enantioselective reductive cross-coupling of styrenyl aziridines. J Am Chem Soc. 2017;139(16):5688–91. 10.1021/jacs.7b03448 [DOI] [PubMed] [Google Scholar]
- 197.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–8. 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 198.Capaldo L, Ravelli D. Hydrogen Atom Transfer (HAT): A versatile strategy for substrate activation in photocatalyzed organic synthesis. European J Org Chem. 2017;2017(15):2056–71. 10.1002/ejoc.201601485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Perry IB, Brewer TF, Sarver PJ, Schultz DM, DiRocco DA, MacMillan DWC. Direct arylation of strong aliphatic C-H bonds. Nature. 2018;560(7716):70–5. 10.1038/s41586-018-0366-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sarver PJ, Bacauanu V, Schultz DM, DiRocco DA, Lam YH, Sherer EC, et al. The merger of decatungstate and copper catalysis to enable aliphatic C(sp3)-H trifluoromethylation. Nat Chem. 2020;12(5):459–67. 10.1038/s41557-020-0436-1 [DOI] [PubMed] [Google Scholar]
- 201.Xu S, Chen H, Zhou Z, Kong W. Three‐Component alkene difunctionalization by direct and selective activation of aliphatic C− H bonds. Angew Chem Int Ed. 2021;60(13):7405–11. 10.1002/anie.202014632 [DOI] [PubMed] [Google Scholar]
- 202.Shen Y, Gu Y, Martin R. sp(3) C-H Arylation and alkylation enabled by the synergy of triplet excited ketones and nickel catalysts. J Am Chem Soc. 2018;140(38):12200–9. 10.1021/jacs.8b07405 [DOI] [PubMed] [Google Scholar]
- 203.Abadie B, Jardel D, Pozzi G, Toullec P, Vincent JM. Dual benzophenone/copper-photocatalyzed Giese-type alkylation of C(sp3)-H bonds. Chem Eur J. 2019;25(70):16120–7. 10.1002/chem.201904111 [DOI] [PubMed] [Google Scholar]
- 204.Zhu D-L, Young DJ, Li H-X. Carbonyl-Photoredox/Metal dual catalysis: Applications in organic synthesis. Synthesis. 2020;52(23):3493–510. 10.1055/s-0040-1707183 [DOI] [Google Scholar]