

Abstract
We describe herein a two-step process for the conversion of serine to a wide array of optically pure unnatural amino acids. This method utilizes a photocatalytic cross-electrophile coupling between a bromoalkyl intermediate and a diverse set of aryl halides to produce artificial analogues of phenylalanine, tryptophan, and histidine. The reaction is tolerant of a broad range of functionalities and can be leveraged toward the scalable synthesis of valuable pharmaceutical scaffolds via flow technology.
Keywords: unnatural amino acids, photoredox catalysis, heterocycles, nickel catalysis, flow chemistry
INTRODUCTION
The amino acid motif is of paramount importance in biochemistry, constituting the basic building block of peptides and proteins and playing a crucial role in nearly every biological process.1 As such, novel analogues of the amino acids produced by nature have found broad application in chemical biology, biochemistry, and medicine.2 The versatility of these unnatural amino acids stems from their utility as chiral frameworks and their ability to confer increased potency and atypical conformations on peptide-based pharmaceuticals.3 Unnatural amino acids are an important feature of many antibody drug conjugates, as they provide bioconjugation sites orthogonal to native functional groups, thereby allowing the controlled production of homogeneous, site-specific adducts.4 In the enzymatic context, the incorporation of unnatural amino acids into novel biocatalysts can allow for tuning of a native enzyme’s activity or incorporation of unnatural metallic cofactors.5 Given the biomedical import of these motifs, there is a strong impetus to invent new methods that permit facile access to diverse collections of unnatural amino acids (Figure 1).
Figure 1.
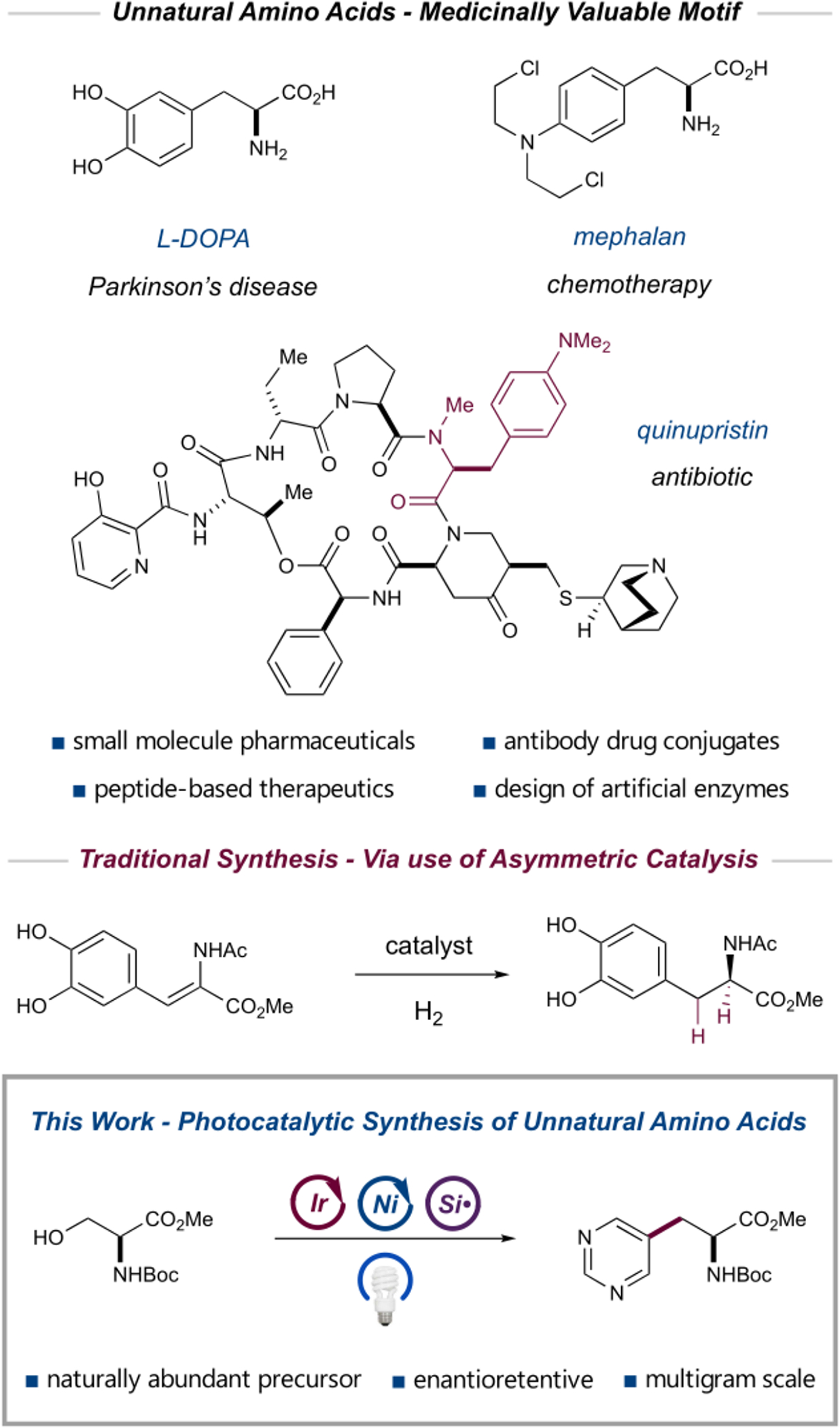
Applications and synthesis of unnatural amino acids.
The chemical synthesis of amino acids is a challenge of longstanding interest to the field of organic chemistry. Since the publication of the Strecker reaction in the mid-nineteenth century,6 a wide variety of catalytic technologies and elegant asymmetric catalyst designs have been developed for the synthesis of enantiopure unnatural amino acids.7 Notable examples include the pioneering asymmetric hydrogenation used in the synthesis of l-DOPA,8 asymmetric variants of the venerable Strecker reaction, and9 photocatalytic Michael additions of aryl radicals into dehydroalanine,10 among many others.11 Unnatural amino acids can also be accessed via derivatization of natural amino acids, although this strategy remains relatively undeveloped. Notably, in recent years, the Yu group has pioneered the use of palladium-catalyzed C–H activation to derivatize alanine into a number of unnatural analogues.12 A derivatization approach offers a clear benefit: by harnessing the resident stereocenter of the natural amino acid, it should be possible to gain access to a diverse array of structurally complex, enantiopure unnatural amino acids without the need to develop novel asymmetric catalytic platforms. The successful realization of this objective would require the development of a protocol that is sufficiently mild to avoid racemization and tolerant of a broad range of functionalities. With these considerations in mind, we sought to design a reaction platform that would allow the facile synthesis of unnatural amino acids bearing complex structural motifs of interest to the synthetic and biomedical community.
Metallaphotoredox catalysis is a powerful new platform for bond construction that involves the synergistic merger of photochemical activation with transition metal catalysis. Our laboratory and others have exploited this reactivity manifold to develop a host of novel methodologies for carbon–carbon and carbon–heteroatom bond formation.13 Notably, these photocatalytic reactions are conducted under mild conditions, with radical mechanisms proceeding at ambient temperature without the need for harsh reagents, such as strong bases or organometallic nucleophiles. We hypothesized that metallaphotoredox catalysis could be brought to bear on the challenge of synthesizing unnatural amino acids directly from natural amino acid precursors. Specifically, photochemical cross-electrophile coupling is characterized by high efficiency, mild conditions, and extraordinary substrate scope and could thus offer a means to achieve the conversion of abundant, naturally occurring amino acids to their arylated analogues.14 Herein, we report a mild and enantioretentive protocol for the conversion of serine to a range of amino acids bearing aromatic side chains via the intermediacy of a β-bromoalanine intermediate. This protocol provides ready access to a wide variety of arylated amino acid derivatives, including both carbo- and heterocyclic frameworks.
RESULTS AND DISCUSSION
Serine was selected as a starting point for amino acid derivatization due its biomass abundance and readily derivatized hydroxyl group side chain. The β-bromoalanine (6) derivative can be easily produced via an Appel reaction on scales of greater than 450 mmol. The proposed mechanism for the metallaphotoredox-catalyzed amino acid synthesis is illustrated in Scheme 1. Initial visible light excitation of the iridium(III) photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)-pyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine) (1) generates the long-lived (τ = 2.3 μs) excited-state *Ir(III) complex 2. This highly oxidizing species (E1/2red [*IrIII/IrII] = +1.21 V versus saturated calomel electrode (SCE) in CH3CN)15 can favorably undergo single-electron transfer (SET) with the bromide anion (E1/2red [Br•/Br−] = +0.80 V vs SCE in DME) to produce reduced Ir complex 3 along with an electrophilic bromine radical that can subsequently abstract a hydrogen atom from (TMS)3SiH (4, super silane) to yield silyl radical 5. Halogen atom abstraction by radical 5 from β-bromoalanine (6) affords the alkyl radical (7) and (TMS)3Si–Br as a byproduct. Concurrently, a Ni0 species 9 undergoes facile oxidative addition into an aryl bromide coupling partner (8) to generate the NiII intermediate 10. Trapping of the alkyl radical 7 by nickel species 10 affords the putative NiIII complex 11, which, upon reductive elimination, yields target amino acid 13 and a NiI intermediate (12). Finally, SET from strongly reducing iridium species 3 (E1/2red [IrIII/IrII] = −1.37 V vs SCE in CH3CN) to 12 serves to regenerate catalytically active 9 and simultaneously close the photoredox cycle.
Scheme 1.
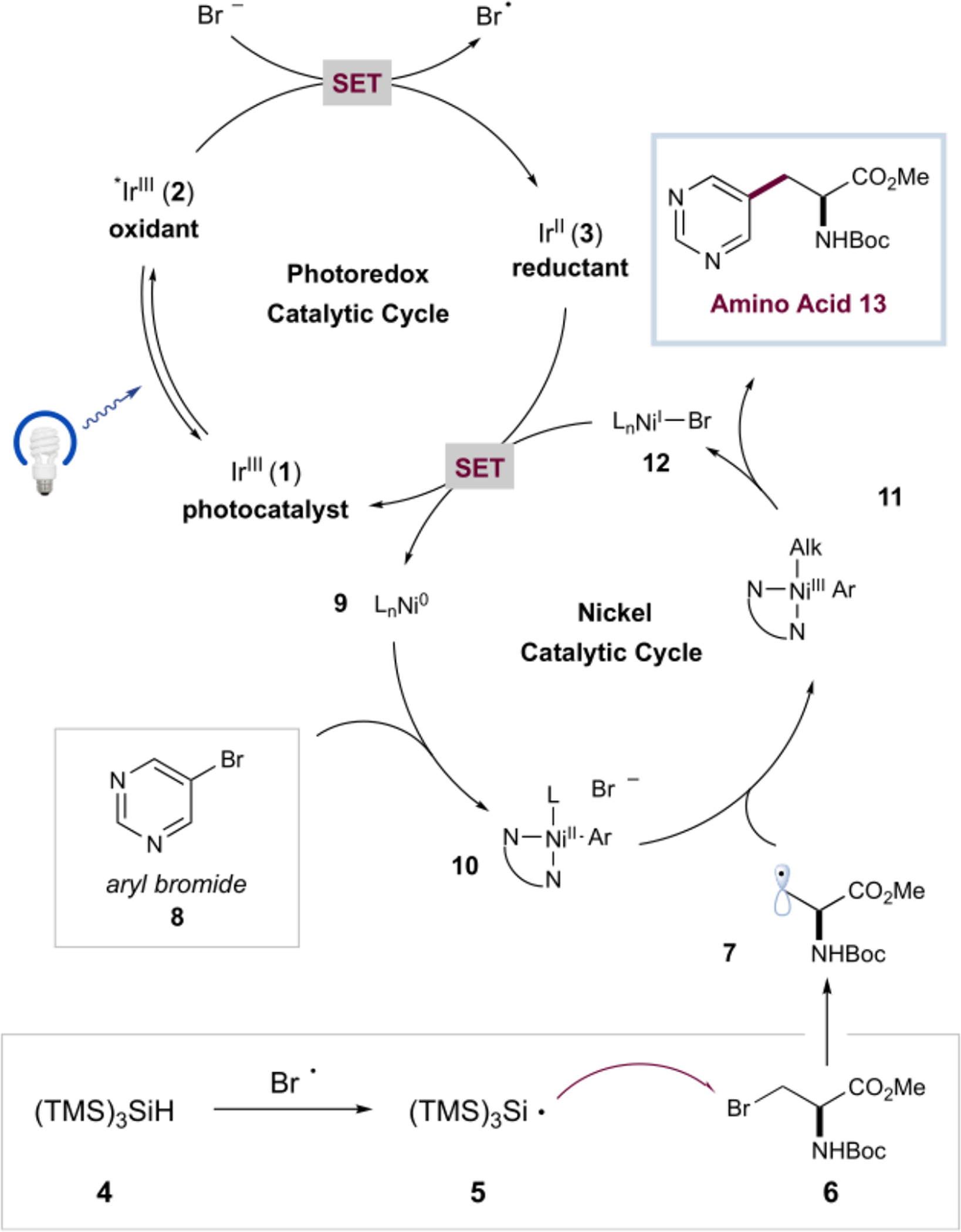
Proposed Mechanism for Cross-Electrophile Coupling
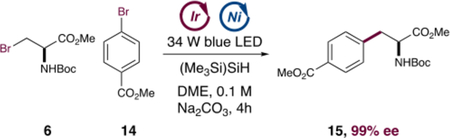
The proposed cross-electrophile coupling was explored using aryl bromide 14 as the coupling partner. As shown in Table 1, in the presence of the iridium photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)] mol %), NiCl2·dtbbpy (0.5 mol %), super silane (1 equiv), and sodium carbonate (2 equiv), the aryl bromide (14) and β-bromoalanine (6) underwent efficient cross-coupling to generate the desired amino acid 15 in 85% yield (entry 1). As anticipated, no reaction was observed when the photocatalyst, nickel catalyst, light, or silane was excluded from the reaction mixture (entries 2–5). The choice of silane proved important. As shown in entries 6 and 7, the use of silanes bearing strong Si–H bonds, compared to (TMS)3SiH, resulted in decreased reaction efficiency. These findings are consistent with those of the proposed mechanism of silyl radical formation via Si–H abstraction. Interestingly, addition of Hantzsch ester as an alternate reductant afforded 15 in 36% yield (Table 1 entry 8), indicating that a mechanism involving sequential oxidative additions into the aryl and alkyl bromides, with silane acting as a reductant, cannot be ruled out. This mechanism may be occurring in tandem with the silyl radical-mediated pathway. Finally, we examined whether the β-bromoalanine (6) could be formed in situ from a serine tosylate precursor. As shown in entry 9, in the presence of serine tosylate and tetrabutylammonium bromide, the reaction proceeded efficiently, to provide adduct 15 in 80% yield. This reaction presumably proceeds via the β-bromoalanine intermediate, as no product is observed without the addition of tetrabutylammonium bromide. Importantly, both electrophiles led to product formation in >99% ee without any detectable erosion of the enantiopurity of the starting material.
Table 1.
Cross-Electrophile Coupling Control Experimentsa
| ||
|---|---|---|
| entry | conditions | yield 15b (%) |
| 1 | as shown above | 85 |
| 2 | no photocatalyst | 0 |
| 3 | no nickel | 0 |
| 4 | no light | 0 |
| 5 | no silane | 0 |
| 6 | Ph3SiH as the silane source | 34 |
| 7 | Et3SiH as the silane source | 0 |
| 8 | Hantzsch ethyl ester instead of silane | 36 |
| 9 | serine tosylate in place of alkyl bromidec | 80 |
Performed with 1 mol % photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]-PF6, NiCl2·dtbbpy (0.5 mol %), (TMS)3SiH (1 equiv), alkyl halide (1 equiv, 0.1 mmol), and aryl halide (1.5 equiv) in DME for 4 h.
NMR yields using mesitylene as an internal standard.
Performed under identical conditions with (n-Bu)4NBr (2 equiv). No product observed without (n-Bu)4NBr added.
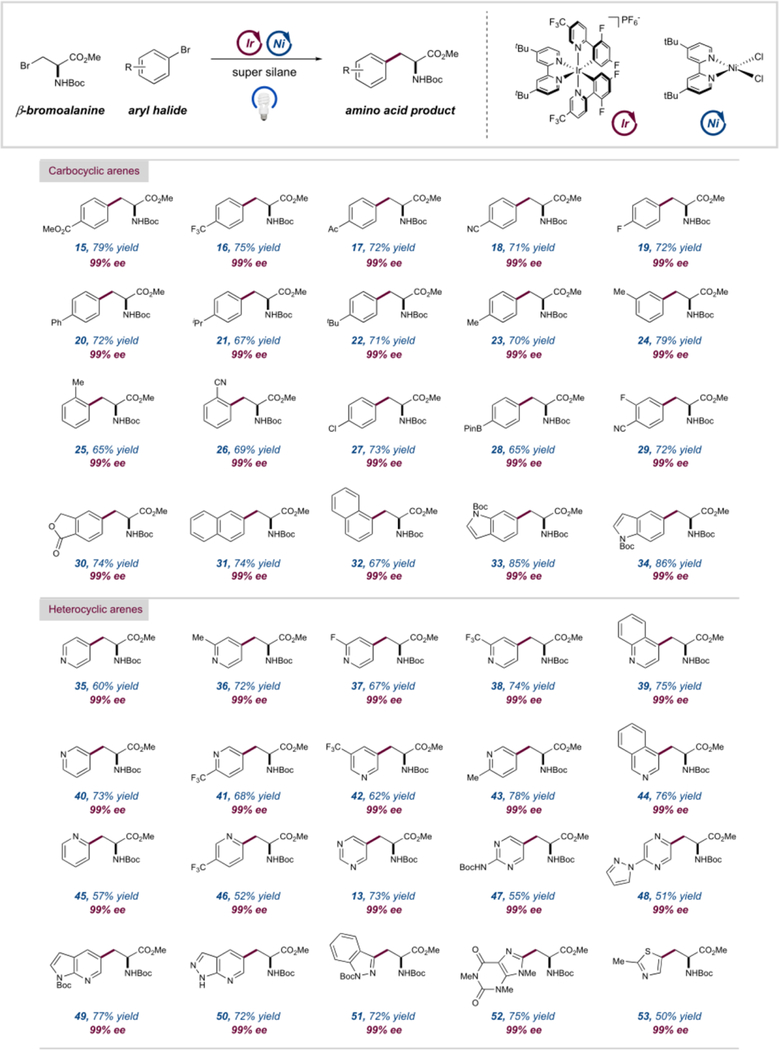
With optimized conditions in hand, we next investigated the scope of the reaction with respect to the aryl bromide coupling partner. We first turned our attention to carbocyclic aryl bromide frameworks incorporating a variety of electronic and steric motifs. As shown in Table 2, electron-deficient aryl bromides bearing ester, trifluoromethyl, acetyl, and nitrile functionalities proceed in excellent efficiency (15–18, 71–79% yields). Notable among these is p-acetophenylalanine (17), an unnatural amino acid commonly used in antibody–drug conjugates.16 Electron-rich and electron-neutral motifs, including fluoro-, aryl-, and alkyl-arenes, reacted with similar efficiency (19–25, 65–79% yields). Ring substitution at the ortho-, meta-, and para-positions was well-tolerated (23–26, 69–79% yields), although ortho-substituted aryl halides required a higher loading of the nickel catalyst (5 mol %), presumably owing to more sluggish oxidative addition. We were also able to incorporate motifs, such as chlorides and boronate esters, that allow for further modification to the arene framework of the amino acid (27 and 28, 73 and 65% yield, respectively). In these cases, exclusive functionalization was observed at the bromide-bearing position. Bifunctionalized aryl bromides reacted efficiently to give unnatural amino acids such as 29. In addition, bicyclic arenes such as naphthalenes, lactones, and indoles were readily converted to the corresponding amino acids (30–34, 67–86% yield). Perhaps most noteworthy among these are the indole-derived amino acids, 33 and 34, which correspond to unnatural 5- and 6-substituted isomers of tryptophan, respectively.
Table 2.
Scope of Unnatural Amino Acids from Serine Derived from the β-bromoalanine Starting Materiala
|
Reactions performed on the 1 mmol scale; all yields are of isolated products unless otherwise indicated. See the Supporting Information for experimental details.
Having explored the scope of carbocyclic arene coupling partners, we turned our attention to medicinally relevant heterocyclic motifs.17 Indeed, this new reaction protocol was found to be readily applied to a broad range of heterocyclic arenes. In comparison to carbocyclic aryl bromide substrates, higher loadings of the nickel catalyst (>3 mol %) were generally required to achieve good efficiency with heterocyclic substrates. Electronically varied 2-, 3-, and 4-bromopyridines were well tolerated (Table 2, 35–46, 52–78% yield). In the case of 2-pyridines, the silanol reagent (TMS)3Si–OH gave superior yields compared to the standard super silane. This could be attributed to slower generation of the alkyl radical species, which, coupled with an increase in nickel loading (5 mol %), aligns the rate of the photocatalytic and nickel cycles. Diazines, such as pyrimidines and pyrazines, were effectively converted to the aryl amino acid products (13, 47, and 48, 51–73% yields). Bicyclic heteroaromatic rings, such as quinolines, were well tolerated as were bi-heteroaryl systems, which give rise to a wide range of artificial tryptophan derivatives (39, 44, 49, and 50, 72–77% yields). Importantly, five-membered heterocyclic arenes, traditionally challenging partners in cross-coupling reactions, could be converted into unnatural tryptophan (51, 72% yield) and histidine analogues (52 and 53, 75 and 50% yield, respectively). All of these reactions proceeded with complete retention of enantiopurity of the bioderived starting material.18
We next sought to examine whether other naturally occurring amino acids might be employed in the cross-coupling reaction to provide access to distinct classes of unnatural amino acids. Along these lines, we targeted three biologically important amino acid precursors: threonine, homoserine, and β-homoserine. These structural motifs are of particular interest because derivatives of homophenylalanine and β-homophenylalanine are present in many pharmaceutical molecules and have been found to play a role in modulating the peptide function.19 As shown in Scheme 2, the bromo-derivatives of these amino acids (54–56) underwent efficient electrophilic cross-coupling to generate adducts 57–59 in good yields and with complete enantioretention. In order to be widely adopted by practitioners of synthetic chemistry, this novel protocol must be capable of providing material on a multigram scale. Flow technology is a valuable technique in process chemistry and manufacturing, enabling the scaling of reactions that cannot easily be conducted on a large scale under batch conditions.20 We thus set out to identify optimized flow conditions for electrophilic cross-coupling of β-bromoalanine and aryl bromides. Initial studies, using methyl 4-bromobenzoate as the arene component, indicated that concentrations of up to 0.5 M could be accommodated, leading to an excellent 85% yield of the desired product within a residence time of 20 min. This result represents a considerable increase in throughput compared to batch conditions. In the flow reaction, a lutidine base was used in place of sodium bicarbonate because of its enhanced solubility in DME. Under these conditions, homogeneous conditions are achieved at the outset of the reaction. Previous studies in our laboratory have demonstrated that reactions performed with high-intensity light sources are more efficient with a homogeneous base due to more rapid neutralization of the HBr byproduct.21
Scheme 2.
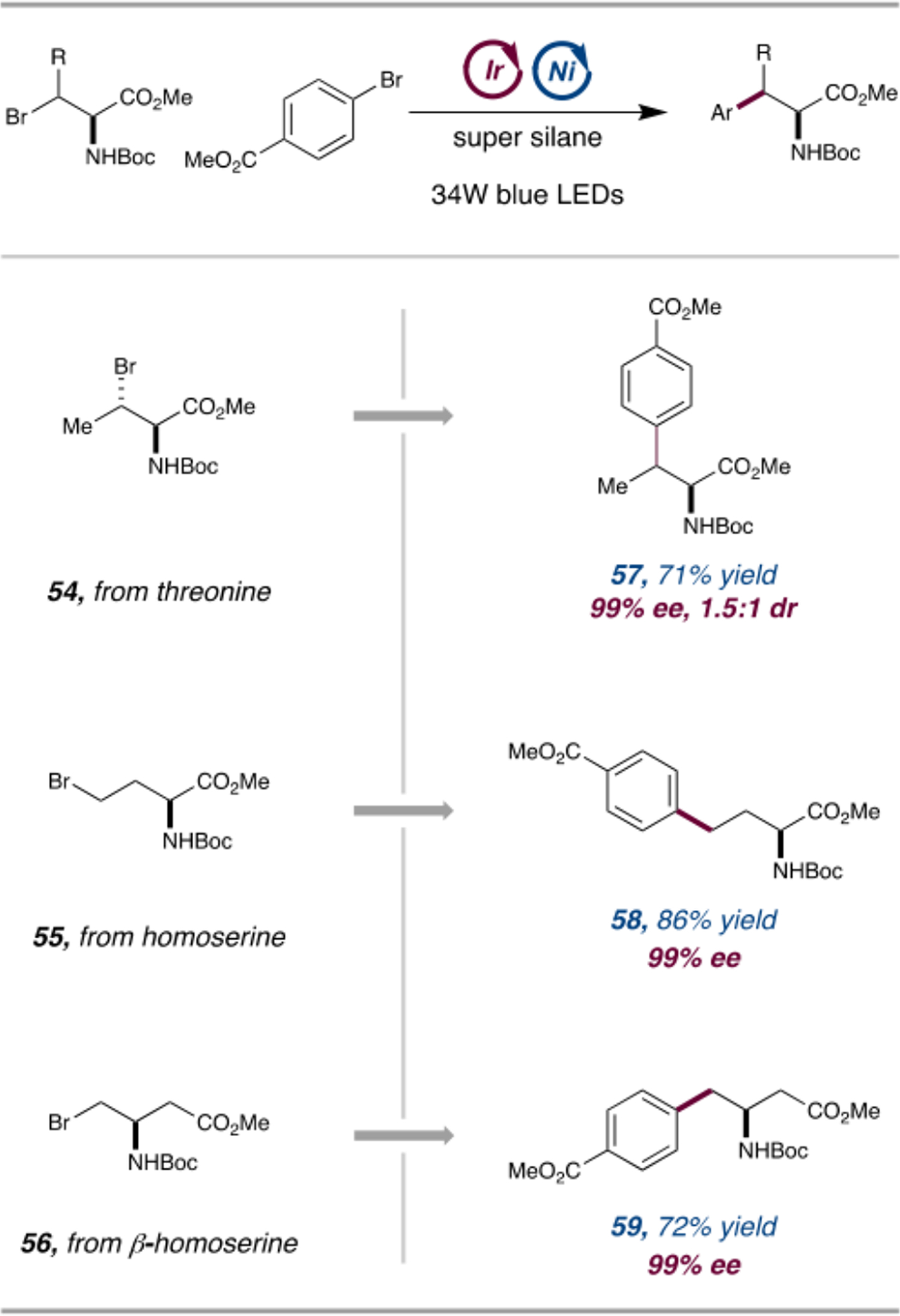
Scope of Naturally Derived Amino Acid Bromides
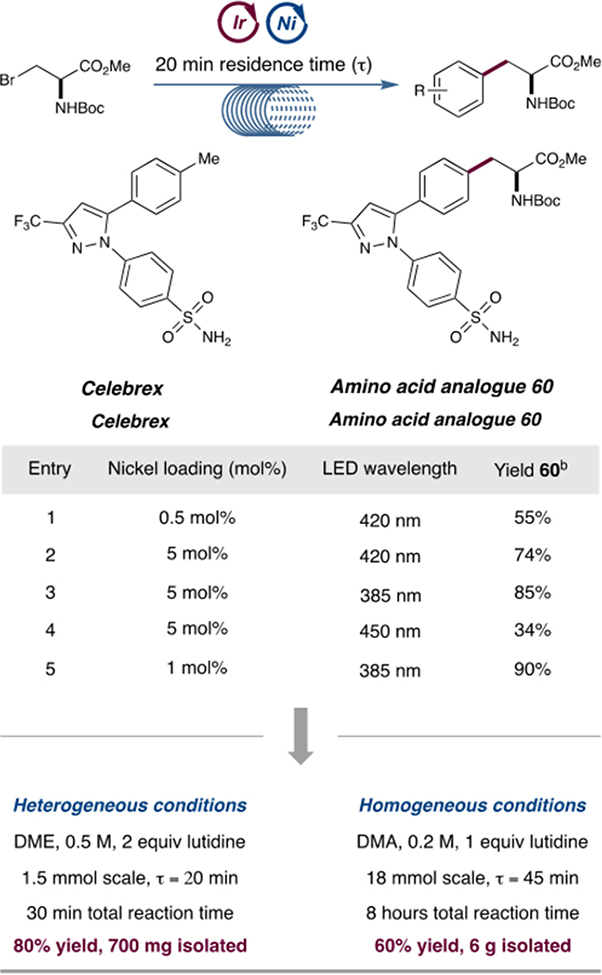
In order to demonstrate the applicability of this chemistry to valuable pharmaceutical motifs, we sought to synthesize multigram quantities of a representative, medicinally valuable scaffold. We subjected an aryl bromide analogue of Celebrex, a non-steroidal anti-inflammatory drug, to our optimized flow conditions and obtained the amino acid–Celebrex derivative 60 in 55% yield within 20 min residence time, with substantial quantities of aryl bromide remaining (Table 3, entry 1). We aimed to improve the efficiency of the reaction by increasing the loading of the nickel catalyst and modulating the LED wavelength. As shown in Table 3, a clear correlation was observed between wavelength and reaction efficiency. The lower wavelength light (385 nm), which better matches the absorption maximum of the photocatalyst, was found to substantially accelerate the reaction compared with higher wavelength sources (Table 3, entries 2–4). Under the optimal conditions shown in entry 5, the use of 1 mol % nickel catalyst and a 385 nm lamp produced 60 in 90% yield within 20 min. Using these conditions, we conducted the reaction on the 1.5 mmol scale and obtained 700 mg of product (80% yield) in a total reaction time of 30 min.
Table 3.
Development of Flow Conditions for Multigram Synthesis
|
Flow reactions performed on a Vapourtec E-series flow reactor with aryl halide (1 equiv), alkyl halide (1.5 equiv), 1 mol % photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, (TMS)3SiH (1.5 equiv), and Ni(OAc)2·4H2O; see the Supporting Information for more details.
NMR yields using mesitylene as an internal standard.
Attempts to conduct larger scale flow reactions for extended time periods were impeded by accumulation of a precipitate, lutidinium bromide, which is formed when lutidine neutralizes the HBr byproduct. In an effort to tailor the reaction conditions for homogeneity, we made a series of modifications to the original conditions: (a) DMA was used as the solvent in place of DME; (b) the reaction was run at more dilute concentrations (0.2 M); (c) the base loading was decreased to 1 equivalent; (d) the reaction temperature was increased from 30 to 40 °C; and (e) the residence time was increased to 45 min. These changes collectively served to increase the solubility of the lutidinium bromide salt, providing the desired adduct in a synthetically useful yield of 60%. We subsequently performed this reaction on scales of up to 20 mmol, obtaining 6 g of the Celebrex-derived amino acid 60 in 60% yield. We thus have developed flow conditions that allow rapid access to pharmaceutical products on a medium scale and homogeneous conditions that can afford multigram quantities of product when run continuously over extended time periods.
In summary, we have developed a mild and enantioretentive protocol for the conversion of serine to a wide range of unnatural amino acids via a β-bromoalanine intermediate. This protocol provides ready access to a vast range of arylated amino acid derivatives, including a wide variety of phenylalanine, tryptophan, and histidine derivatives. We have further demonstrated that methylphenylalanine derivatives can be prepared via arylation of threonine and that β-amino acids may be accessed via β-homoserine. The synthesis of multigram quantities of valuable amino acid derivatives of pharmaceutical agents has been demonstrated. We anticipate that this methodology will see wide adoption in industrial laboratories due to its broad scope, scalability, and ease of operation.
EXPERIMENTAL SECTION
Procedure for Cross-Electrophile Coupling Reaction.
To an oven-dried 8 mL vial was added the photocatalyst Ir[dF(CF3)ppy]2(dtbbpy) (0.6 mg, 0.5 μmol, 1 mol %), aryl bromide (0.05 mmol, 1.5 equiv), methyl 3-bromo-2-((tert-butoxycarbonyl)amino)propanoate (21 mg, 0.075 mmol, 1 equiv), and anhydrous sodium carbonate (11 mg, 0.1 mmol, 2 equiv). The vial was sealed and placed under nitrogen, and tris(trimethylsilyl)silane (23 μL, 0.075 mmol, 1.5 equiv) was added via a syringe. To a separate vial was added NiCl2·glyme (2.8 mg, 12.5 μmol, 0.25 equiv) and 4,4′-di-tert-butyl-2,2′-bipyridine (3.4 mg, 12.5 μmol, 0.25 equiv). The catalyst vial was sealed and purged with nitrogen, and then, to it was added 2.5 mL of DME. The precatalyst solution was sonicated for 10 min, after which, 0.5 mL of the solution (5 mol % catalyst, 2.5 μmol, 0.05 equiv) was syringed into the reaction vessel (0.1 M). The solution was degassed by sparging with nitrogen for 10 min before sealing with Parafilm. The reaction was stirred and irradiated with a 34 W blue LED lamp with fen cooling at 30 °C for 6 h. The reaction was quenched by exposure to air, and the solvent was removed in vacuo. Purification via flash column chromatography (C18) yielded the cross-coupled product.
Heterogeneous Flow Conditions.
A reaction solution was prepared using methyl-(R)-4-bromo-3-((tert-butoxycarbonyl)amino)butanoate (635 g, 2.25 mmol, 1.5 equiv), Ir[dF(CF3)ppy]2(dtbbpy) (17 mg, 0.015 mmol, 1 mol %), 4-(5-(4-bromophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)benzenesulfonamide (669 g, 1.5 mmol, 1 equiv), lutidine (0.35 mL, 3 mmol, 2 equiv), tris(trimethylsilyl)silane (0.7 mL, 2.25 mmol, 1.5 equiv), Ni(OAc)2·4H2O (3.7 mg, 0.015 mmol, 0.01 equiv), and 4,4′-di-tert-butyl-2,2′-bipyridine (4 mg, 0.015 mmol, 0.01 equiv). The reaction components were dissolved in DME (3 mL, 0.5 M), sonicated until homogeneous, and degassed via nitrogen sparging for 20 min. The reaction mixture was pumped through a 2 mL reactor coil (internal diameter 2.0 mm) for 30 min (0.1 mL/min, 20 min residence time) at 30 °C under irradiation from an 18 W 385 nm LED lamp. After the reaction was complete, the reaction mixture was diluted with ethyl acetate and washed with water and brine. The organic phase was dried with sodium sulfate and filtered, and the solvent was removed in vacuo. Purification via column chromatography yielded the pure product as a white solid (700 mg, 80% yield).
Homogeneous Flow Conditions.
A reaction solution was prepared using methyl-(R)-4-bromo-3-((tert-butoxycarbonyl)-amino)butanoate (7.62 g, 27 mmol, 1.5 equiv), Ir[dF(CF3)ppy]2(dtbbpy) (202 mg, 0.18 mmol, 1 mol %), 4-(5-(4-bromophenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl)-benzenesulfonamide (8 g, 18 mmol, 1 equiv), lutidine (4.2 mL, 36 mmol, 2 equiv), tris(trimethylsilyl)silane (8.33 mL, 27 mmol, 1.5 equiv), Ni(OAc)2·4H2O (45 mg, 0.18 mmol, 0.01 equiv), and 4,4′-di-tert-butyl-2,2′-bipyridine (48 mg, 0.18 mmol, 0.01 equiv). The reaction components were dissolved in DMA (90 mL, 0.2 M), sonicated until homogeneous, and degassed via nitrogen sparging for 20 min. The reaction mixture was pumped through a 10 mL reactor coil (internal diameter 2.0 mm) for 8 h (0.225 mL/min, 45 min residence time) at 40 °C under irradiation from an 18 W 385 nm LED lamp. After the reaction was complete, the reaction mixture was diluted with ethyl acetate and washed with water and brine. The organic phase was dried with sodium sulfate and filtered, and the solvent removed in vacuo. Purification via column chromatography yielded the pure product as a white solid (6 g, 60% yield).
Methyl-(S)-2-((tert-butoxycarbonyl)amino)-3-(4-(1-(4-sulfamoylphenyl)-3-(trifluoromethyl)-1H-pyrazol-5-yl)phenyl)-propanoate (60): 1H NMR (500 MHz, CDC13): δ 7.94–7.87 (m, 2H), 7.45 (d, J = 8.3 Hz, 2H), 7.15 (s, 4H), 6.76 (s, 1H), 5.03 (d, J = 8.4 Hz, 1H), 4.90 (d, J = 20.5 Hz, 2H), 4.60 (d, J = 7.3 Hz, 1H), 3.72 (d, J = 4.9 Hz, 3H), 3.10 (ddd, J = 69.8, 13.8, 6.0 Hz, 2H), 1.42 (s, 9H). 13C NMR (126 MHz, CDCl3): δ 172.17, 155.14, 144.93, 142.49, 141.65, 138.21, 130.14, 129.11, 127.65, 125.62, 106.62, 80.35, 60.57, 54.43, 52.58, 38.45, 28.42, 21.21, 14.33. HRMS (ESI-TOF) m/z calcd for C25H27F3SN4NaO6 ([M + Na]+), 591.1501; found, 591.1486. IR (film): λ(max) 2980, 1700, 1500, 1368, 1238, 1163, 976, 739 cm−1 Chiral HPLC OJ column, 20% IPA/hexanes, 1.0 mL/min, >99% ee, tR(minor) = 9.22 min, tR(major) = 24.74 min.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful for financial support provided by the National Institute of General Medical Sciences (NIGMS), the NIH (under Award R35GM134897-01), the Princeton Catalysis Initiative, and kind gifts from Eli Lilly, Merck, Janssen, BMS, Genentech, Celgene, and Pfizer. T.M.F. acknowledges Princeton University, E. Taylor, and the Taylor family for an Edward C. Taylor Fellowship. This work was supported by Eli Lilly and Company through the Lilly Research Award Program (LRAP).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.oprd.1c00208.
Experimental methods, characterization data and spectra of all isolated compounds, and optimization details of flow reactions (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.oprd.1c00208
The authors declare no competing financial interest.
Contributor Information
Tomer M. Faraggi, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States
Caroline Rouget-Virbel, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States.
Juan A. Rincón, Centro de Investigación Eli Lilly, Alcobendas-Madrid 28108, Spain;.
Mario Barberis, Centro de Investigación Eli Lilly, Alcobendas-Madrid 28108, Spain.
Carlos Mateos, Centro de Investigación Eli Lilly, Alcobendas-Madrid 28108, Spain.
Susana García-Cerrada, Centro de Investigación Eli Lilly, Alcobendas-Madrid 28108, Spain.
Javier Agejas, Centro de Investigación Eli Lilly, Alcobendas-Madrid 28108, Spain.
Oscar de Frutos, Centro de Investigación Eli Lilly, Alcobendas-Madrid 28108, Spain.
David W. C. MacMillan, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States;.
REFERENCES
- (1).Wu G Amino acids: metabolism, functions and nutrition. Amino Acids 2009, 37, 1. [DOI] [PubMed] [Google Scholar]
- (2).(a) England PM Unnatural Amino Acid Mutagenesis: A precise Tool for Probing Protein Structure and Function. Biochemistry 2004, 43, 11623. [DOI] [PubMed] [Google Scholar]; (b) Kim CH; Axup JY; Schultz PG Protein conjugation with genetically coded unnatural amino acids. Curr. Opin. Chem. Biol 2013, 17, 412. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brown W; Liu J; Deiters A Genetic Code Expansion in Animals. ACS Chem. Biol 2018, 13, 2375. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mirts EN; Bhagi Damodaran A; Lu Y Understanding and Modulating Metalloenzymes with Unnatural Amino Acids, Non-Native Metal Ions, and Non-Native Metallocofactors. Acc. Chem. Res 2019, 52, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Blaskovich MAT Unusual Amino Acids in Medicinal Chemistry. J. Med. Chem 2016, 59, 10807. [DOI] [PubMed] [Google Scholar]
- (3).(a) Fosgerau K; Hoffmann T Peptide therapeutics: current status and future directions. Drug Discovery Today 2015, 20, 122. [DOI] [PubMed] [Google Scholar]; (b) Craik DJ; Fairlie DP; Liras S; Price D The Future of Peptide-based Drugs. Chem. Biol. Drug Des 2013, 81, 136. [DOI] [PubMed] [Google Scholar]
- (4).Hallam TJ; Wold E; Wahl A; Smider VV Antibody Conjugates with Unnatural Amino Acids. Mol. Pharmaceutics 2015, 12, 1848. [DOI] [PubMed] [Google Scholar]
- (5).Yu Y; Hu C; Xia L; Wang J Artificial Metalloenzyme Design with Unnatural Amino Acids and Non-Native Cofactors. ACS Catal. 2018, 8, 1851. [Google Scholar]
- (6).Strecker A Ueber einen neuen aus Aldehyd – Ammoniak und Blausäure entstehenden Körper. Ann. Chem 1854, 91, 349. [Google Scholar]
- (7).Nájera C; Sansano JM Catalytic Asymmetrie Synthesis of α-Amino Acids. Chem. Rev 2007, 107, 4584. [DOI] [PubMed] [Google Scholar]
- (8).(a) Knowles WS Asymmetric hydrogenation. Acc. Chem. Res 1983, 16, 106. [DOI] [PubMed] [Google Scholar]; (b) Junge K; Hagemann B; Enthaler S; Spannenberg A; Michalik M; Oehme G; Monsees A; Riermeier T; Beller M Synthesis of chiral monodentate binaphthophosphepine ligands and application in asymmetric hydrogenations. Tetrahedron: Asymmetry 2004, 15, 2621. [Google Scholar]
- (9).(a) Wang J; Liu X; Feng X Asymmetric Strecker Reactions. Chem. Rev 2011, 111, 6947. [DOI] [PubMed] [Google Scholar]; (b) Akiyama T; Itoh J; Yokota K; Fuchibe K Enantioselective Mannich-Type Reaction Catalyzed by a Chiral Brønsted Acid. Angew. Chem., Int. Ed 2004, 43, 1566. [DOI] [PubMed] [Google Scholar]; (c) Uraguchi D; Terada M Chiral Brønsted Acid-Catalyzed Direct Mannich Reactions via Electrophilic Activation. J. Am. Chem. Soc 2004, 126, 5356. [DOI] [PubMed] [Google Scholar]; (d) Zuend SJ; Coughlin MP; Lalonde MP; Jacobsen EN Scalable catalytic asymmetric Strecker synthesis of unnatural α-amino acids. Nature 2009, 461, 968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Aycock RA; Vogt DB; Jui NT A practical and scalable system for heteroaryl amino acid synthesis. Chem. Sci 2017, 8, 7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Shin J-S; Kim B-G Transaminase-catalyzed asymmetric synthesis of L-2-aminobutyric acid from achiral reactants. Biotechnol. Lett 2009, 31, 1595. [DOI] [PubMed] [Google Scholar]; (b) Savile CK; Janey JM; Mundorff EC; Moore JC; Tam S; Jarvis WR; Colbeck JC; Krebber A; Fleitz FJ; Brands J; Devine PN; Huisman GW; Hughes GJ Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305. [DOI] [PubMed] [Google Scholar]; (c) Ross AJ; Lang HL; Jackson RFW Much Improved Conditions for the Negishi Cross-Coupling of Iodoalanine Derived Zinc Reagents with Aryl Halides. J. Org. Chem 2010, 75, 245. [DOI] [PubMed] [Google Scholar]; (d) Ooi T; Maruoka K Asymmetric Organocatalysis of Structurally Well-Defined Chiral Quaternary Ammonium Fluorides. Acc. Chem. Res 2004, 37, 526. [DOI] [PubMed] [Google Scholar]; (e) Ni S; Garrido-Castro AF; Merchant RR; de Gruyter JN; Schmitt DC; Mousseau JJ; Gallego GM; Yang S; Collins MR; Qiao JX; Yeung K-S; Langley DR; Poss MA; Scola PM; Qin T; Baran PS A General Amino Acid Synthesis Enabled by Innate Radical Cross-Coupling. Angew. Chem., Int. Ed 2018, 57, 14560. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Halperin SD; Kwon D; Holmes M; Regalado EL; Campeau L-C; DiRocco DA; Britton R Development of a Direct Photocatalytic C–H Fluorination for the Preparative Synthesis of Odanacatib. Org. Lett 2015, 17, 5200. [DOI] [PubMed] [Google Scholar]; (g) Rémond E; Martin C; Martinez J; Cavelier F Silicon-Containing Amino Acids: Synthetic Aspects, Conformational Studies, and Applications to Bioactive Peptides. Chem. Rev 2016, 116, 11654. [DOI] [PubMed] [Google Scholar]; (h) Leonard DJ; Ward JW; Clayden J Asymmetric α-arylation of amino acids. Nature 2018, 562, 105. [DOI] [PubMed] [Google Scholar]
- (12).He J; Li S; Deng Y; Fu H; Laforteza BN; Spangler JE; Homs A; Yu J-Q Ligand Controlled C(sp3)–H Arylation and Olefination in Synthesis of Unnatural chiral α-Amino Acids. Science 2014, 343, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Mergin photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science 2014, 345, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tellis JC; Primer DN; Molander G A Single-electron transmetalation in organoboron cross-coupling by photo-redox/nickel dual catalysis. Science 2014, 345, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DWC Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science 2016, 352, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhang P; Le CC; MacMillan DWC Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway in Cross-Electrophile Coupling. J. Am. Chem. Soc 2016, 138, 8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lowry MS; Goldsmith JI; Slinker JD; Rohl R; Pascal RA; Malliaras GG; Bernhard S Single-Layer Electroluminescent Devices and Photoinduced Hydrogen Production from an Ionic Iridium(III) Complex. Chem. Mater 2005, 17, 5712. [Google Scholar]
- (16).Axup JY; Bajjuri KM; Ritland M; Hutchins BM; Kim CH; Kazane SA; Haider R; Forsyth JS; Santidrian AF; Stafin K; Lu Y; Tran H; Seller AJ; Biroc SL; Szydlik A; Pinkstaff JK; Tian F; Sinha SC; Felding-Habermann B; Smider VV; Schultz PG Synthesis of site specific antibody drug conjugates using unnatural amino acids. Proc. Natl Acad. Sci. U.S.A 2012, 109, 16101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- (18). In all reactions, the principal byproducts were protodehalogenation of both aryl and alkyl halide components, which form most of the unaccounted-for mass balance. A small amount (<5%) of aryl halide was also coupled to the DME solvent. Approximately 5% elimination of β-bromoalanine was observed to form dehydroalanine in most reactions.
- (19).Longobardo L; Melck D; Siciliano R; Santini A; Marzo VD; Cammarota G β-Casomorphins: Substitution of phenylalanine with β-homo phenylalanine increases the β-type opioid receptor affinity. Bioorg. Med. Chem. Lett 2000, 10, 1185. [DOI] [PubMed] [Google Scholar]
- (20).(a) Webb D; Jamison TF Continuous flow multi-step organic synthesis. Chem. Sci 2010, 1, 675. [Google Scholar]; (b) Cambié D; Bottecchia C; Straathof NJW; Hessel V; Noël T Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev 2016, 116, 10276. [DOI] [PubMed] [Google Scholar]; (c) Plutschack MB; Pieber B; Gilmore K; Seeberger PH The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev 2017, 117, 11796. [DOI] [PubMed] [Google Scholar]
- (21).Le CC; Wismer MK; Shi Z-C; Zhang R; Conway DV; Li G; Vachal P; Davies IW; MacMillan DWC A General Small-Scale Reactor To Enable Standardization and Acceleration of Photocatalytic Reactions. ACS Cent. Sci 2017, 3, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.