

Abstract
Background:
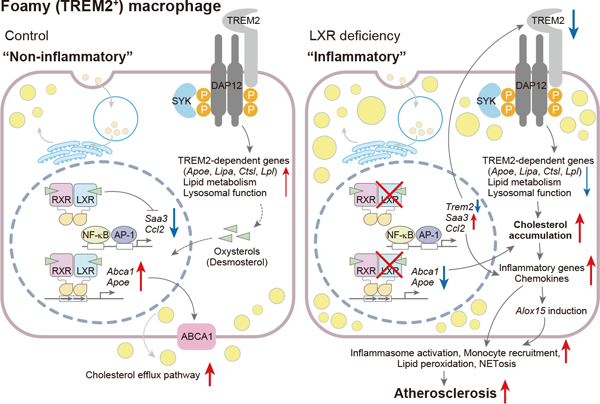
Cholesterol loaded macrophage foam cells are a prominent feature of atherosclerotic plaques. Single cell RNA sequencing has identified foam cells as triggering receptor expressed on myeloid cells 2 (TREM2) positive populations with low expression of inflammatory genes, resembling the TREM2 positive microglia of neurodegenerative diseases. Cholesterol loading of macrophages in vitro results in activation of liver X receptor (LXR) transcription factors and suppression of inflammatory genes.
Methods:
To test the hypothesis that LXRs mediate anti-inflammatory effects in Trem2 expressing atherosclerotic plaque foam cells, we carried out RNA profiling on plaque cells from hypercholesterolemic mice with myeloid LXR deficiency.
Results:
Myeloid LXR deficiency led to a dramatic increase in atherosclerosis with increased monocyte entry, foam cell formation and plaque inflammation. Bulk cell-RNA profiling of plaque myeloid cells showed prominent upregulation of inflammatory mediators including oxidative, chemokine and chemotactic genes. Single cell-RNA sequencing revealed increased numbers of foamy TREM2-expressing macrophages; however, these cells had reduced expression of the Trem2 gene expression module, including phagocytic and cholesterol efflux genes, and had switched to a pro-inflammatory and proliferative phenotype. Expression of Trem2 was suppressed by inflammatory signals but not directly affected by LXR activation in bone marrow-derived macrophages.
Conclusions:
Our current studies reveal the key role of macrophage LXRs in promoting the Trem2 gene expression program and in suppressing inflammation in foam cells of atherosclerotic plaques.
Keywords: liver X receptor, atherosclerosis, macrophage, form cell, triggering receptor expressed on myeloid cells 2
Subject terms: Basic Science Research
Graphical Abstract
Introduction
Macrophages play essential roles in the progression of atherosclerosis in both early and advanced stages. In the traditional view, these cells take up modified low-density lipoprotein (LDL), induce cholesterol efflux pathways, and promote inflammatory signaling1. Macrophages are also involved in the regression of atherosclerosis, promoting apoptotic cell clearance and inflammation resolution2. Recent studies using single-cell RNA sequencing (scRNA-seq) have provided novel insights into the complexity and potential functions of different macrophage populations accumulating in damaged tissues in atherosclerosis and Alzheimer’s disease. In atherosclerosis, these populations include resident macrophages, inflammatory macrophages, interferon-inducible macrophages and triggering receptor expressed on myeloid cells 2 (TREM2)-positive foamy macrophages, each with a distinctive transcriptome, suggesting important functional distinctions3. Paradoxically, foamy TREM2-positive macrophages are noninflammatory, whereas nonfoamy macrophages have elevated expression of inflammatory genes4.
The TREM2 receptor is expressed on the surface of phagocytic macrophages in the brain, adipose tissue and liver. Under conditions of tissue damage and lipid accumulation, TREM2 is activated by lipids and damage-associated molecular patterns instituting a signaling program that activates a gene expression module that promotes phagocytic, endocytic and lipid metabolic functions5. TREM2-positive macrophages appear to have beneficial roles in Alzheimer’s disease and obesity6. However, the function and regulation of TREM2-positive macrophages during the progression of atherosclerosis remains poorly understood.
The LXR nuclear receptors (LXR)-α and -β are highly expressed in macrophages. LXRs are activated by oxysterols and regulate the expression of genes related to lipid metabolism and immunity. LXR activators have beneficial effects in atherosclerosis, inflammatory diseases, Alzheimer’s disease and cancer7. In macrophages, LXRs induce the expression of ATP-binding cassette transporter A1 (ABCA1), ABCG1 and apolipoprotein E (Apo E), and promote reverse cholesterol transport8–10. In lipopolysaccharide (LPS) stimulated-cultured macrophages, LXRs suppress expression of inflammatory genes by multiple mechanisms including cis-repression11, trans-repression12 and ABC transporter and cholesterol efflux-mediated regulation13, 14. Foam cells formed in the peritoneal cavity of Western-type diet (WTD)-fed low-density lipoprotein receptor deficient (Ldlr−/−) mice accumulate desmosterol, an LXR activator, suggesting that LXRs could suppress inflammatory genes in foam cells of atherosclerotic plaques15, 16. Similarly, LXR activation represses inflammatory genes and up-regulates cholesterol efflux genes in myelin damage-associated microglia17. LXR activation in TREM2-deficient mice upregulates genes associated with phagocytic processes and cholesterol efflux and reverses cholesterol and cholesteryl ester accumulation in demyelination associated phagocytic microglia, suggesting that LXR may act collaboratively with TREM2 to activate cholesterol clearance genes18. Additionally, Trem2 expression could be regulated by LXR binding cooperatively with activating transcription factor 3 in Kupffer cells during nonalcoholic liver disease progression19.
Together, these findings suggest that LXRs could have a key role in cholesterol clearance and inflammatory gene repression in TREM2 expressing macrophages in atherosclerosis. However, this important concept has not been directly tested in vivo. To explore this hypothesis, we developed a new mouse model with myeloid cell LXR deficiency and performed detailed RNA profiling and immunocytochemical characterization of their atherosclerotic lesions. These studies reveal a central role of LXRs in inducing TREM2 and an associated anti-atherogenic gene expression program, including up-regulation of endocytic and lipid metabolic genes, and suppression of macrophage inflammation and proliferation.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some of the methods were shown in Supplemental Materials.
Animals and treatments
Nr1h3floxed/floxedNr1h2floxed/floxed (Lxrαfl/flLxrβfl/fl (floxed control)) mice was reported previously20 and crossed with B6.129P2-Lyz2tm1(cre)Ifo/J mice (The Jackson Laboratory, stock No: 004781) to generate myeloid cell-selective LXRα/β-deficient (Lxrαfl/flLxrβfl/fl-LysM-Cre (Myl-Lxrdko)) mice. Mice were maintained under controlled temperature at 71–73 F (21.7–22.8 C) and humidity (45–55%) with a 12-h light, 12-h dark cycles, and with free access to water and normal laboratory diet (Purina Mills diet 5053). For atherosclerosis study, 7–8 weeks old male B6.129S7-Ldlrtm1Her/J mice (Ldlr−/−, The Jackson Laboratory, stock No: 002207) were lethally irradiated twice at 6.5 Gy with an interval of 4 hours and were injected intravenously with bone marrow cells (1 × 107 cells per 20 g of body weight) isolated from floxed control (male, 7–8 weeks old) or Myl-Lxrdko (male, 7–8 weeks old) mice. To compare with previous studies of an atherosclerosis model using LXR-deficient male mice21–23, the same sex mice were utilized for all experiments in this study. After 5 weeks of bone marrow transplantation, mice were fed with Western-type diet (WTD, Harlan Teklad, TD88137) for the indicated time in each experiment. All protocols were approved by the Institutional Animal Care and Use Committee of Columbia University (protocol No. AAAY-8464).
Blood analysis
Plasma total cholesterol content was measured with Cholesterol E (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). Plasma IL-18 levels was measured with enzyme-linked immunosorbent assay kits (MBL, Woborn, MA).
Bone marrow-derived macrophages (BMDMs)
Bone marrow cells were obtained by femurs and tibias of mice and incubated in Dulbecco’s Modified Eagle Medium containing 10 % fetal bovine serum, 1 % penicillin/streptomycin and 20 % L929-conditioned medium for 7 days.
Quantitative polymerase chain reaction (qPCR) analysis
Total RNA was extracted by TRIzol Reagent (Thermo Fisher Scientific, Waltham, MA) or RNeasy Micro Kit (Qiagen, Venlo, Netherlands) and cDNA was synthesized with First Strand cDNA Synthesis Kit for RT-qPCR (Thermo Fisher Scientific). Quantitative real-time polymerase chain reaction was performed with the StepOnePlus Real-time PCR system (Thermo Fisher Scientific) using Fast Power SYBR Green PCR Master Mix reagent (Thermo Fisher Scientific). Intron-spanning mouse primer sequences were described previously11, 15, 24 or listed in Table S1. Each mRNA expression level was normalized with the expression of 36b4 or Ppib.
Atherosclerosis lesion analysis
For the lesion analysis, we adhere to the guidelines for experimental atherosclerosis studies described in the AHA Statement25. Heart tissues were isolated from Ldlr−/− mice after WTD feeding and fixed with 10 % formalin solution for 72 hours. Fixed heart tissues were dehydrated and embedded with paraffin, and then were cut into 6 μm width, de-paraffinized, and stained with hematoxylin and eosin solution. Lesion area and necrotic core area were calculated with the average of 6 sections for individual sample using ImageJ software by an independent observer in a blind manner. Analyses of immunofluorescent staining, immunohistochemistry, monocyte tracking and intracellular lipid staining were described in Supplemental Materials.
Isolation of immune cells from whole aorta
Whole aortae were perfused twice with PBS, and isolated from mice, and then cut into small pieces with scissors after removal of adipose tissues under microscopy. The tissues were incubated with digestive solution including Liberase TM (Roche Diagnostics, Mannheim, Germany, 2.5 mg/mL), Hyaluronidase (Sigma-Aldrich, 120 U/mL), and DNase I (Sigma-Aldrich, 80 U/mL) in DMEM for 40 minutes at 37 degrees. Cell sorting was performed by FACSAria cell sorter (BD Biosciences) and the detail was described in Supplemental Materials.
Bulk-cell RNA sequencing in aortic CD11b+ cells
The CD11b+ cells were isolated from digested whole aorta using magnetic beads (Miltenyi Biotec), and 1 ng of total RNA (combined two mice samples) for each group was utilized for cDNA synthesis using SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (TaKaRa Bio. Inc., Otsu, Japan). The libraries were prepared with Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA) and subjected to NextSeq 550 sequencing system (Illumina). Reads were aligned, reads counts per gene for RefSeq genes were computed, and counts were normalized as reported previously with false discovery rate 10 %11. GO analysis was performed using the PANTHER database.
Single cell RNA-sequencing
The 31,193 CD45+ cells isolated from floxed control-BMT Ldlr−/− group (cell viability: 78 %) or 33,575 cells from Myl-Lxrdko-BMT Ldlr−/− group (cell viability: 78 %) were subjected to Chromium library preparation system (10x Genomics, Pleasanton, CA), respectively.
For the analysis, Cell Ranger v3.1.0 was used to process FASTQ files. The raw sequencing reads were aligned to the mm10 mouse reference genome build and Ensembl 93 was applied to annotate mouse transcriptome. Unique molecular identifier (UMI) counts were obtained for each of floxed control- and Myl-Lxrdko-BMT Ldlr−/−, and Cell Ranger was also employed for calling cell barcodes. Single-cell RNA-seq analysis was performed using Seurat v3.2.226 in R software v.3.6. Pre-processing was performed using Seurat functions. Genes expressed in less than 10 cells were excluded in downstream analysis. The input data were also filtered to remove cells that have gene counts less than 200 and have over 10% mitochondrial counts. Preliminary processing was also applied to the maximum number of genes and the maximum number of UMIs for each cell, based on sequencing depth per sample. Detailed filtering procedure was provided in Fig. S5B.
The scRNA-seq integration methodology was implemented to integrate the two pre-processed datasets and to perform downstream analysis. Seurat was used to identify the top 2,000 genes with highly variable expression for each sample and to select the top 2,000 highly variable genes across the two datasets for integration. In order to specify the anchors for integration, the first 20 dimensions from canonical correlation analysis were selected. The first 20 principal components (PC), a liberal estimate by the elbow plot from Seurat, were used as input features for the t-distributed Stochastic Neighbor Embedding (tSNE) dimensional reduction as well as for the Shared Nearest-neighbor graph with 20 nearest neighbors. Resolution parameter of 0.7 was applied to identify the heterogeneous clusters based on Louvain clustering. Wilcoxon Rank Sum test was performed to identify differentially expressed genes by comparing each cluster to all remaining clusters based on the normalized and log-transformed gene expressions. For each cluster, genes that were expressed in more than 25% cells with at least 0.182 natural log-fold (0.5-fold) difference were considered differentially expressed.
Statistical analysis
All data are represented as mean ± SD and the dots for each data indicate values from individual samples. The statistical analysis was performed by Prism 9.3 (GraphPad Software, La Jolla, CA). The normality were analyzed by Shapiro-Wilk test and the variance were by F test for two groups or Bartlett’s test for more than two groups. For data that passed tests for normality and variance, we performed parametric tests including unpaired Student’s t test to compare two groups or one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons to analyze data of more than two groups. For nonparametric tests, we performed Mann-Whitney’s test to compare two groups or Kruskal-Wallis test followed by Dunn’s multiple comparisons to analyze more than two groups. For the experiments in Fig. 5, we performed parametric tests without analyzing normality and variance because each sample was prepared from an individual mouse.
Results
Myeloid cell deficiency of LXRα/LXRβ.
The anti-atherogenic effect of LXRs has been shown by administration of small molecule LXR activators or by transplantation of LXR-deficient bone marrow in Ldlr−/− or Apoe−/− mice21–23. These reports suggest that LXRs in hematopoietic cells play important roles in the atheroprotective effect of LXRs. However, since bone marrow-derived cells include many types of immune cells, these studies do not address the in vivo function of LXR in myeloid cells. To establish myeloid cell-selective LXR-knockout mice, we generated Lxrαfloxed/floxedLxrβfloxed/floxed (Lxrαfl/flLxrβfl/fl)-LysM-Cre (Myl-Lxrdko) mice by crossing Lxrαfl/flLxrβfl/fl mice with LysM-Cre transgenic mice. Tissue weight, blood cell counts and plasma cholesterol levels were similar to littermate control mice (Fig. S1A–C). To confirm cell specific knockout of LXR genes, we evaluated expression of Nr1h3 (the gene encoding LXRα) and Nr1h2 (the gene encoding LXRβ) in sorted lymphocytes and myeloid cells from spleen or blood. Expression of Nr1h3 and Nr1h2 was significantly reduced in neutrophils, monocytes and macrophages, but not in B cells and T cells in Myl-Lxrdko mice. (Fig. S1D). Expression of these genes was also reduced in bone marrow-derived macrophages (BMDMs) but not in the liver (Fig. S1E). Ligand-dependent expression of known LXR targets such as Abca1, Abcg1 and Scd2 was abolished in BMDMs derived from Myl-Lxrdko mice (Fig. S1F). These results show that LXRα and LXRβ are selectively and functionally deficient in myeloid cells in Myl-Lxrdko mice.
Accelerated inflammatory atherosclerosis in Myl-Lxrdko-BMT Ldlr−/− mice.
To examine the effects of myeloid LXRα/β deficiency on the progression of atherosclerosis, Ldlr−/− mice were lethally irradiated and injected intravenously with bone marrow cells isolated from floxed control or Myl-Lxrdko mice. After 5 weeks of recovery, mice were fed the WTD for 10 weeks. Hematoxylin and eosin staining of aortic root showed that Myl-Lxrdko-bone marrow transplanted (BMT) Ldlr−/−mice have much larger lesions with increased necrotic cores compared to controls (Fig. 1A, G, H). Body weight, liver weight, spleen weight, blood cell counts and plasma cholesterol levels were not different between floxed control- and Myl-Lxrdko-BMT Ldlr−/− mice (Fig. S2).
Fig. 1. Increased atherosclerosis, myeloid cell infiltration and activation in Myl-Lxrdko-BMT Ldlr−/− mice.
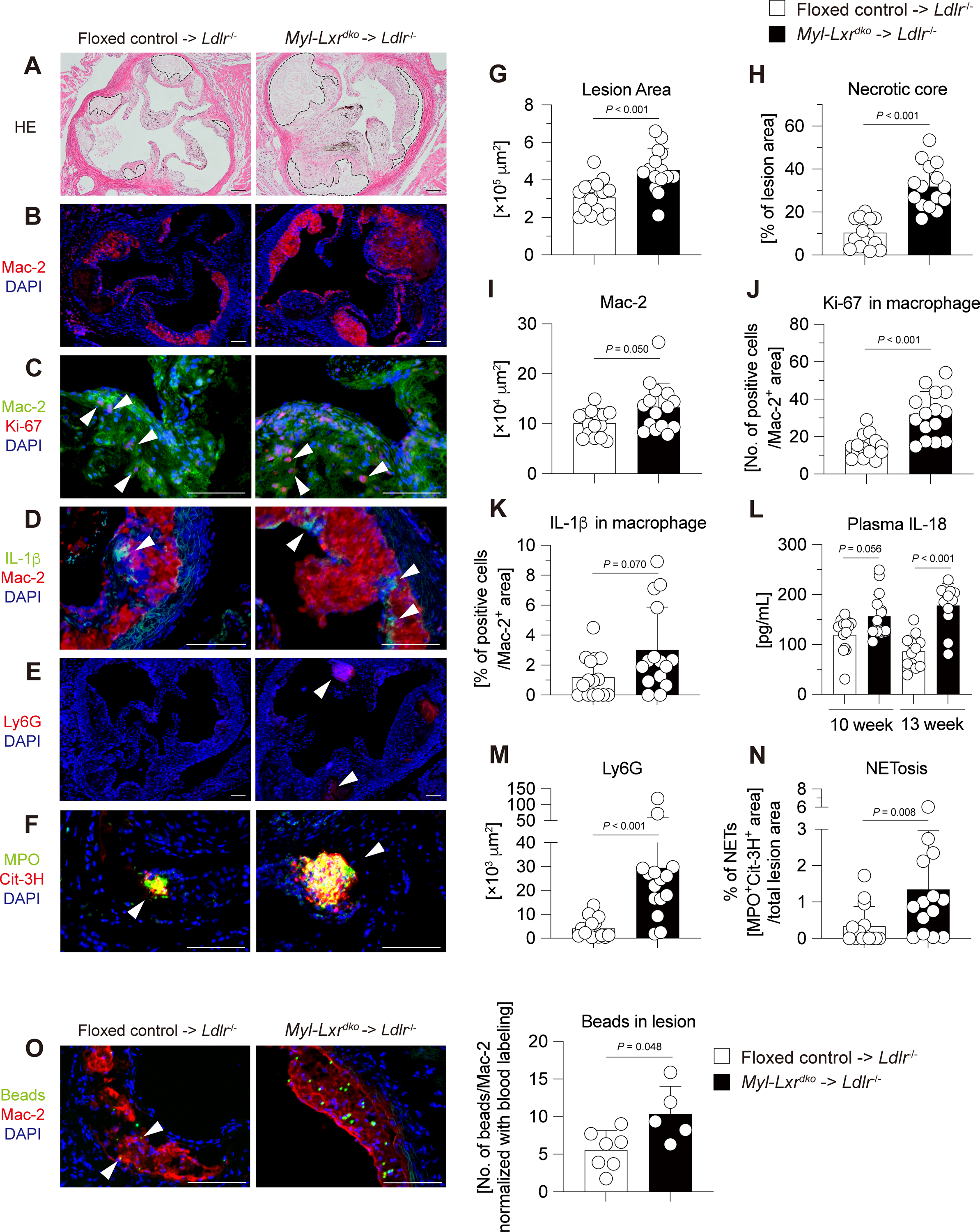
Ldlr−/− male mice were transplanted with bone marrow cells from floxed control or Myl-Lxrdko mice and fed with WTD for 10 weeks (n = 15 for each group). (A, G, H) Hematoxylin and eosin staining of atherosclerotic plaques in aortic roots. The surrounding area with dot line indicates necrotic core. Quantification of total lesion size (G) and necrotic core area (H) in aortic root. Immunofluorescent staining and its quantification in atherosclerosis lesion in 10-week WTD-fed mice for macrophages (Mac-2 (red) and DAPI (blue)) (B, I), proliferative macrophages (Mac-2 (green), Ki-67 (red) and DAPI (blue)) (C, J), inflammasome activation in macrophages (IL-1β (green), Mac-2 (red) and DAPI (blue)) (D, K), neutrophils (Ly6G (red) and DAPI (blue)) (E, M), and NETosis (MPO (green), Cit-3H (red) and DAPI (blue)) (F, N). (n = 15 for each group). (L) Plasma IL-18 levels in 10- or 13-week WTD-fed mice. (O) Recruitment of fluorescence beads-labeled monocytes into the atherosclerotic lesion. The number of beads in macrophages was normalized to the frequency of blood Gr-1hi monocyte (CD115+CD11b+Gr-1hi cells) labeling after 24 hours of beads injection. (n = 5–7 for each group). Scare bar = 100 μm. All data are shown as mean ± SD and white dots indicate individual samples. Statistical analysis was performed by Mann-Whitney U test.
Mac-2, a macrophage marker, was significantly increased in the Myl-Lxrdko-BMT Ldlr−/− group (Fig. 1B, I). Ki-67-staining was increased in Mac-2-positive regions of plaque, indicating increased macrophage proliferation as reported in mice with disruption of LXRα phosphorylation27 (Fig. 1C, J). The staining for interleukin (IL)-1β a potential product of inflammasome activation, was increased in Mac-2-positive regions (Fig. 1D, K). An increase of Caspase-1 cleavage was also observed in splenic CD11b+ cells in Myl-Lxrdko-BMT Ldlr−/− mice (Fig. S3). Additionally, plasma IL-18 levels were significantly elevated in both 10 and 13 weeks WTD-fed mice, consistent with inflammasome activation in Myl-Lxrdko-BMT Ldlr−/− mice (Fig. 1L). Since inflammasome activation in macrophages can induce neutrophil extracellular traps (NETs) formation28, we stained with markers for neutrophils (Ly6G) and NETosis (myeloperoxidase, (MPO) and citrullinated histone H3 (Cit-3H)). The Ly6G-positive area (Fig. 1E, M), and the colocalization of MPO and Cit-3H (Fig. 1F, N) were both markedly increased in the Myl-Lxrdko-BMT Ldlr−/− lesions. Recent studies have indicated up-regulation of integrins in LXR-deficient leukocytes suggesting the potential for increased recruitment into lesions11. To examine monocyte tracking into lesions, we injected clodronate liposome, and then fluorescent beads into BMT-Ldlr−/− mice to label Gr-1hi classical monocytes (CD115+CD11b+-gated Gr-1hi cells) selectively29. After 5 days an increased number of beads was detected in the Mac-2-positive lesion in Myl-Lxrdko-BMT Ldlr−/− group (Fig. 1O). These observations demonstrate a marked monocyte-macrophage mediated inflammatory process in the atherosclerotic lesions of Myl-Lxrdko-BMT Ldlr−/− mice.
Elevated expression of inflammatory genes in aortic CD11b+ cells in Myl-LXRdko-BMT Ldlr−/− mice.
To more directly address the hypothesis that LXRs suppress the expression of inflammatory genes in plaque myeloid cells, we carried out bulk-cell RNA-sequencing (RNA-seq) using aortic CD11b+ cells, comprising macrophages, monocytes and neutrophils, isolated from BMT-Ldlr−/− mice fed with WTD for 13 weeks. 112 genes were significantly induced and 84 genes were repressed by LXR deficiency in myeloid cells (Fig. 2A). The most highly induced genes were Alox15 (the gene encoding arachidonate 15-lipoxygenase (15-LOX)), Saa3 (the gene encoding serum amyloid-3), scavenger receptor Marco, and chemokine (C-X-C motif) ligand family Cxcl13 (Fig. 2B). Although these genes are involved in inflammatory responses, they have not been previously identified as LXR repressed genes in prior studies that employed LPS-treated peritoneal macrophages15. This highlights the distinctive environment of the atherosclerotic plaque that may not be precisely recapitulated in LPS-treated cultured macrophages. Gene ontology (GO) analysis showed that the genes induced by Myl-Lxrdko were highly enriched in the pathways mediating inflammatory signaling such as the cellular response to interferon-β, chemokine-mediated signaling pathways and leukocyte chemotaxis (Fig. 2C). The increase in expression of these genes was confirmed by qPCR analysis using gene-specific primers (Fig. 2D). We also confirmed reduced expression of LXRs (Nr1h3 and Nr1h2), and their target genes (Abca1 and Abcg1) in Myl-Lxrdko-BMT Ldlr−/− plaque myeloid cells.
Fig. 2. Increased expression of inflammatory genes in aortic CD11b+ cells in Myl-Lxrdko-BMT Ldlr−/− mice.
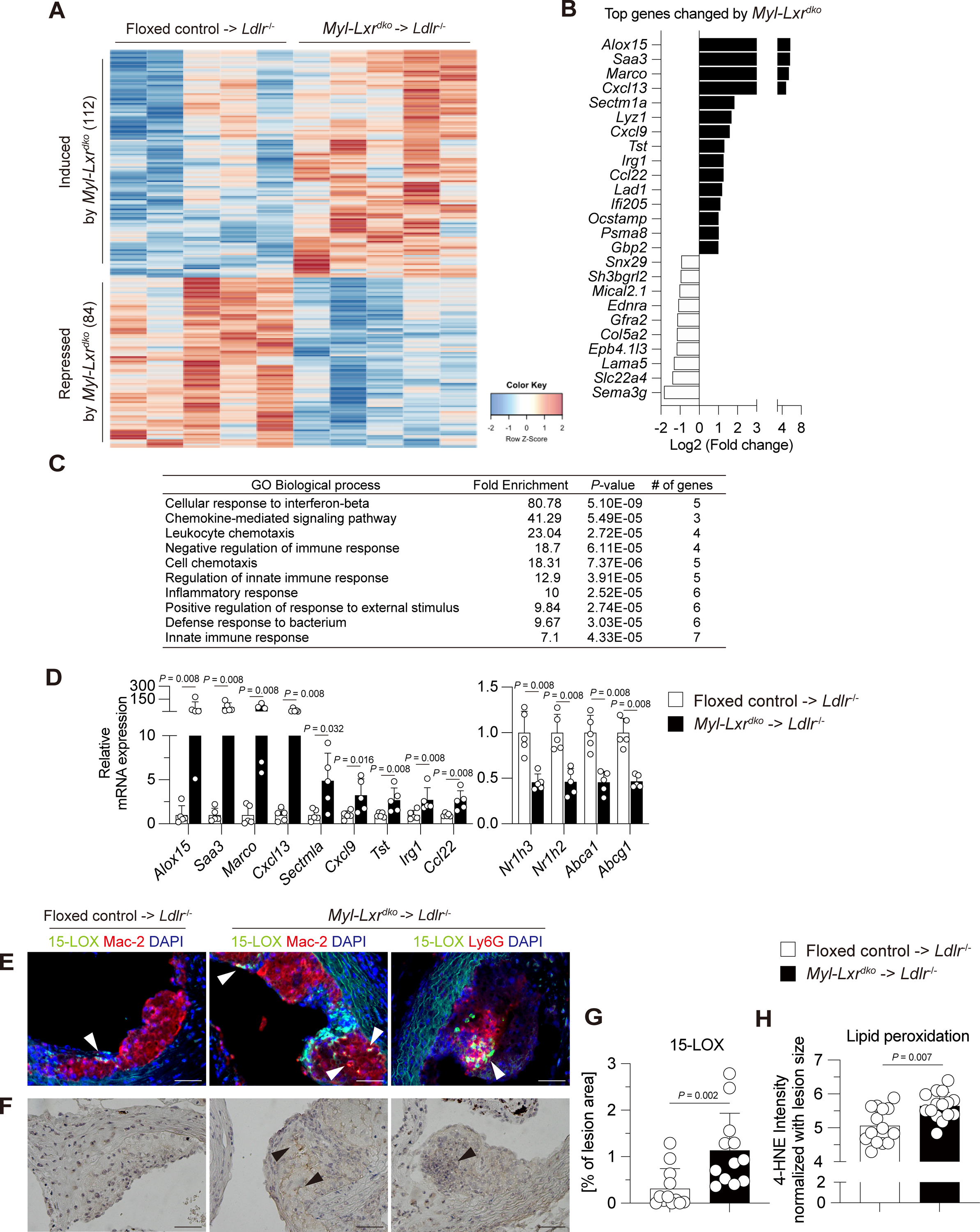
(A) Heatmap of RNA-sequencing analysis in aortic CD11b+ cells isolated from Ldlr−/− mice transplanted with bone marrow cells from floxed control- or Myl-Lxrdko-BMT Ldlr−/− mice fed with WTD for 13 weeks. The 112 genes were induced and 84 genes were repressed by absence of myeloid LXRα/β. Gene ontology (GO) analysis (> 1.5-fold, Bonferroni-adjusted P value < 0.05) (B) and top genes (C) altered by myeloid LXRα/β deficiency (n = 5 for each group). (D) Expression of increased genes by LXR deficiency, LXRs (Nr1h3 and Nr1h2 encoding LXRα and LXRβ, respectively) and their target genes (Abca1 and Abcg1) were evaluated by qPCR. (E, G) Expression and distribution of 15-LOX in atherosclerotic lesion in floxed control- or Myl-Lxrdko-BMT Ldlr−/− mice. Aortic root sections were co-stained with antibodies for 15-LOX (green), macrophage marker (Mac-2) or neutrophil marker (Ly6G) (red), and DAPI (blue). (F, H) Immunohistochemistry of lipid peroxidation in the aortic plaques. Aortic root sections were stained with anti-4-HNE antibody. (n = 15 for each group) Scare bar = 50 μm. All data are shown as mean ± SD and white dots represent individual samples. Statistical analysis was performed by Mann-Whitney U test.
The most highly upregulated gene Alox15 has been shown to promote progression of atherosclerosis in mice fed high-fat, high-cholesterol diets30–33. 15-LOX catalyzes oxidation of polyunsaturated fatty acids, and promotes formation of oxidized LDL and oxidized phospholipids34, 35. We confirmed increased levels of 15-LOX by immunofluorescence staining in Myl-Lxrdko-BMT Ldlr−/− lesions and showed co-localization with Mac-2 (Fig. 2E, G). Interestingly, 15-LOX staining was also prominent in Ly6G-positive areas, especially in regions containing NETs, indicating 15-LOX is induced not only in macrophages but also in activated neutrophils. Consistent with the oxidative role of 15-LOX, lipid peroxidation as assessed by 4-hydroxynonenal (HNE) staining was increased in Myl-Lxrdko-BMT Ldlr−/− lesions (Fig. 2F, H).
scRNA-seq of cells from atherosclerotic lesions.
To more clearly define the immune cell profile in atherosclerotic lesions of Myl-Lxrdko-BMT Ldlr−/− mice, we carried out scRNA-seq analysis of aortic CD45+ cells isolated from floxed control- and Myl-Lxrdko-BMT Ldlr−/− fed with WTD for 12 weeks, and compared gene expression profiles in each cell cluster. The t-distributed stochastic neighbor embedding (tSNE) plot revealed a total of 21 clusters (Fig. S6A). In addition, all of these clusters were detected in both floxed control and Myl-Lxrdko-BMT Ldlr−/− (Fig. S6B). Based on the cluster-specific transcriptome profile3, we identified 8 clusters of T cells (cluster 0–4, 9, 10 and 13) 3 clusters of macrophages (cluster 5, 11 and 17), 4 clusters of monocytes (cluster 7, 8, 14 and 16), 3 clusters of dendritic cells, and a single cluster of neutrophils (cluster 6) as well as natural killer cells (cluster 12) and B cells (cluster 15) (Fig. S6C, S7A, B). To evaluate our hypothesis, we focused on the clusters of monocytes and macrophages including resident-like TREM2+ macrophages (cluster 5), monocytes (cluster 7 and 8), TREM2+ macrophages (cluster 11), classical monocytes (cluster 16) and cavity macrophages (cluster 17) (Fig. 3A). Clusters 5 and 11 macrophages had relatively high expression of Trem2, as well as some markers of resident macrophages such as Folr2 and Pf4 (Fig. S6D). The percentage of resident-like TREM2+ macrophages (cluster 5) was increased from 23.7 % to 39.0 % in Myl-Lxrdko-BMT Ldlr−/− plaques while TREM2+ macrophages (cluster 11) were increased from 11.7 % to 20.8 % (Fig. 3B). Consistent with increased monocyte entry into lesions (Fig. 1O), classical monocytes (cluster 16) were increased in Myl-Lxrdko-BMT Ldlr−/− mice. However, the main monocyte populations (clusters 7,8) were substantially decreased possibly indicating rapid conversion of monocytes into macrophages in Myl-Lxrdko-BMT Ldlr−/− mice. Violin plots of gene expression showed that Il1b and Ccl2 were increased in the cluster 5 TREM2+ macrophages of Myl-Lxrdko-BMT Ldlr−/− (Fig. 3C). Several TREM2-downstream genes such as Apoe and Lpl were reduced in this cluster (Fig. 3D). The expression of Apoe was also decreased in cluster 11 and 16. The violin plots from floxed control showed that Nr1h3 is highly expressed in the cluster 5, 8 and 11 similar to Trem2 whereas Nr1h2 is observed in all myeloid cell clusters (Fig. S6E). Since Nr1h3 undergoes positive autoregulation owing to conserved LXR response elements in its promoter36, this could indicate either a specific role of LXRα in upregulating Trem2 or that the Trem2 phagocytic program provides cholesterol that upregulated LXR target genes including Nr1h3. GO analysis in the main TREM2+ resident macrophage population (cluster 5) showed that genes that were increased in Myl-Lxrdko-BMT Ldlr−/− macrophages compared to floxed controls were enriched in pathways mediating inflammatory responses such as “lymphocyte chemotaxis”, “neutrophil chemotaxis”, “cellular response to interleukin-1”, and “inflammatory response” (Fig. 3E). Interestingly, there was also enrichment of cell cycle genes, including “microtubule cytoskeleton organization involved in mitosis”, “mitotic nuclear division” and “cell division”, suggesting that some of these cells had a proliferative phenotype. In addition to Ccl2, inflammatory genes including Ccl8, Ccl6, Cebpb and Mapk3 were increased in cluster 5 (Fig. 3F). In contrast, genes involved in lipid metabolism such as Fabp5, Mylip and Abcg1 were reduced together with Apoe and Lpl. In addition to the TREM+ macrophages, GO analysis in neutrophils (cluster 6) of Myl-Lxrdko-BMT Ldlr−/− mice compared to controls were showed enrichment in pathways involved in inflammatory responses such as “positive regulation of reactive oxygen species metabolic process”, “neutrophil chemotaxis”, “positive regulation of tumor necrosis factor production” and “positive regulation of inflammatory response” (Fig. S7C). Together with increased accumulation of neutrophils and its NETosis in the plaques of Myl-Lxrdko-BMT Ldlr−/− mice (Fig. 1E, F), scRNA-seq analysis showed that not only macrophages but also lesional neutrophils were activated by LXR deficiency and contributed to the inflammatory plaque phenotype.
Fig. 3. Distinct composition and gene expression in monocytes/macrophages in Myl-Lxrdko-BMT Ldlr−/− mice.
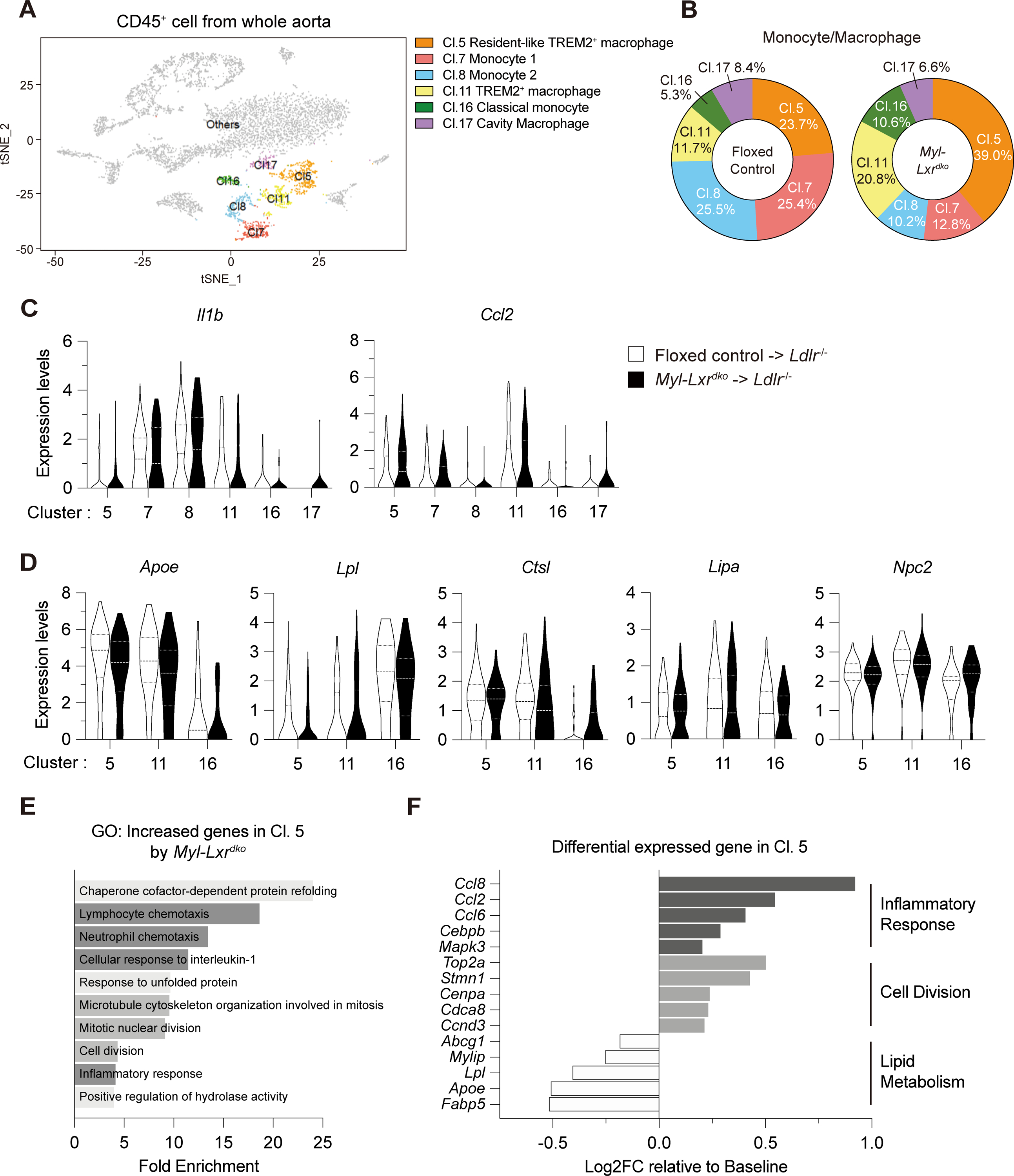
(A) Representative t-distributed stochastic neighbor embedding plot of monocytes and macrophages (Cl. 5, 7, 8, 11, 16 and 17) in total CD45+ cells. (B) Percentage of each cluster for monocytes and macrophages in floxed control- or Myl-Lxrdko-BMT Ldlr−/− mice. Each percentage was normalized with total cell number. (C) Violin plots for expression of Il1b and Ccl2 in the cluster for monocytes and macrophages. (D) Violin plots for the expression of TREM2-downstream genes (Apoe, Lpl, Ctsl, Lipa and Npc2) in cluster 5, 11 and 16. (E) GO analysis in increased genes in cluster 5 by myeloid LXRα/β deficiency (> 1.2-fold, Bonferroni-adjusted P value < 0.05). The 145 genes were induced and 40 genes were repressed by myeloid LXRα/β deficiency. (F) The levels of differential expressed genes by myeloid LXRα/β deficiency compared to baseline (control) involved in inflammatory response, cell division and lipid metabolism.
Myl-LXRdko-BMT Ldlr−/− decreases expression of TREM2 and downstream genes in foamy macrophages
We used orthogonal approaches to substantiate the findings suggested by the bulk cell-RNA-seq and scRNA-seq data. We confirmed that there was an increased BODIPY-positive area that corresponded to increased Oil Red O staining and partly co-localized with Mac-2 in Myl-Lxrdko-BMT Ldlr−/− mouse lesions, indicating an increase in macrophage foam cells (Fig. 4A). A significant increase of BODIPY intensity was observed in the lesion of Myl-Lxrdko-BMT Ldlr−/− mice (Fig. 4B). In addition, TREM2 staining was clearly seen within the BODIPY-positive region in controls. Notably there was markedly reduced TREM2 staining in Myl-Lxrdko-BMT Ldlr−/− mice. We also used flow cytometry to isolate foamy macrophages (CD64+CD11b+BODIPYhiSSChi cells) and nonfoamy macrophages (CD64+CD11b+BODIPYloSSClo cells) as well as a mixed fraction of monocytes and neutrophils (CD64−CD11b+ cells) in CD45+ aortic cells as reported previously4 (Fig. 4C, Fig. S8A). We confirmed that the expression of Nr1h3, Nr1h2 and Abca1 was significantly reduced in all cell types including foam cells from Myl-Lxrdko-BMT Ldlr−/− mice (Fig. S8B). Trem2 mRNA levels were decreased in foamy macrophages isolated from plaques together with its downstream metabolic and endocytic genes including Apoe, Lpl, Ctsl, and Lipa (Fig. 4D). Even though not detected in the scRNA-seq data (probably because there were too few transcripts), we found that Alox15 expression was selectively increased in foamy macrophages (Fig. 4E). The expression of Saa3 was also increased in foamy cells consistent with bulk RNA-seq data. However, Saa3 was also increased in nonfoamy cells and some other inflammatory genes such as Il1b were more prominently increased in nonfoamy cells and monocyte/neutrophil fraction rather than in foam cells. These results demonstrate a major reduction in expression of TREM2 and its downstream genes as well as an increase in inflammatory genes in foamy macrophages isolated from Myl-Lxrdko-BMT Ldlr−/− mice.
Fig. 4. Increased inflammatory gene and impaired TREM2-downstream gene expression in foamy macrophages in Myl-Lxrdko-BMT Ldlr−/− mice.
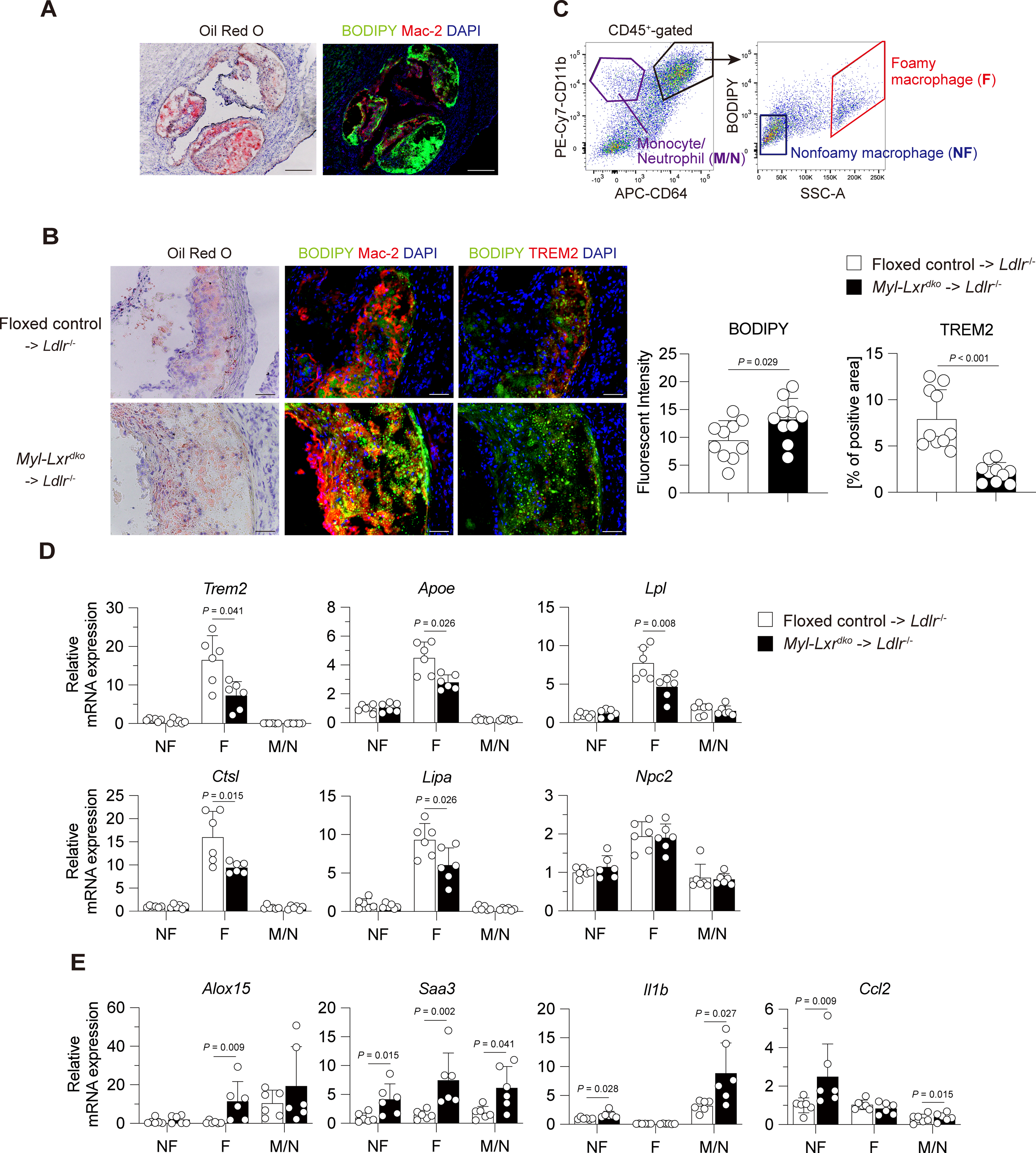
(A) Lipid staining in aortic roots using Oil red O or BODIPY. Scare bar = 200 μm. (B) Staining and quantification of neutral lipid and TREM2+ foamy macrophages in atherosclerotic lesion. (n = 10 for each group) Frozen section was stained with Oil red O (left panel), co-stained with BODIPY (green), Mac-2 (red) and DAPI (blue) (middle panel) or BODIPY (green), or TREM2 (red) and DAPI (blue) (middle or right panel). Scare bar = 50 μm. (C) Gating strategy for sorting foamy macrophages (indicated with F, CD64+CD11b+-gated BODIPYhiSSChi cells), nonfoamy macrophages (NF, CD64+CD11b+-gated BODIPYloSSClo cells) and monocyte/neutrophils (M/N, CD64−CD11b+cells). Gene expression of Trem2 and its downstream genes (Apoe, Lpl, Ctsl, Lipa and Npc2) (D), Inflammatory genes (E) in sorted foamy macrophages, nonfoamy macrophages and monocyte/neutrophil mixed fraction. (n = 6 for each group) All data are shown as mean ± SD and white dots indicate individual samples. Statistical analysis was performed by Mann-Whitney U test.
Effects of inflammatory signals and LXR activation on the expression of Trem2 in BMDMs
To assess the mechanism of regulation of Trem2 expression by LXRs in macrophages, WT BMDMs we treated with toll-like receptor ligands.Trem2 expression levels were decreased by the stimulation with LPS or CpG-DNA in a time-dependent manner (Fig. 5A) as reported previously in alveolar macrophages37. Stimulation with recombinant IL-1β for 6 hours also reduced Trem2 expression (Fig. 5B), suggesting that inflammasome pathways also suppress Trem2. To assess the effect of LXR activation on the expression of Trem2 gene, BMDMs were treated with LXR agonists, T0901317 and 22R-hydroxycholesterol, and then stimulated with LPS for 6 hours. Unexpectedly, the expression of Trem2 was not affected by direct LXR activation in LPS-untreated group (Fig. 5C). Additionally, LXR activation did not reverse the suppression of Trem2 expression by LPS treatment. The Il1b expression was significantly repressed and Abca1 was induced by T0901317 and 22R-hydroxycholesterol, indicating that LXR was effectively activated by its ligands. These results demonstrate that Trem2 is repressed by inflammatory signals and not directly affected by LXR activation in BMDMs and suggest that macrophage Trem2 is decreased in LXR deficiency as a result of increased levels of IL-1β and other inflammatory factors.
Fig. 5. Effects of inflammatory signals and LXR activation on the expression of Trem2 in BMDMs.
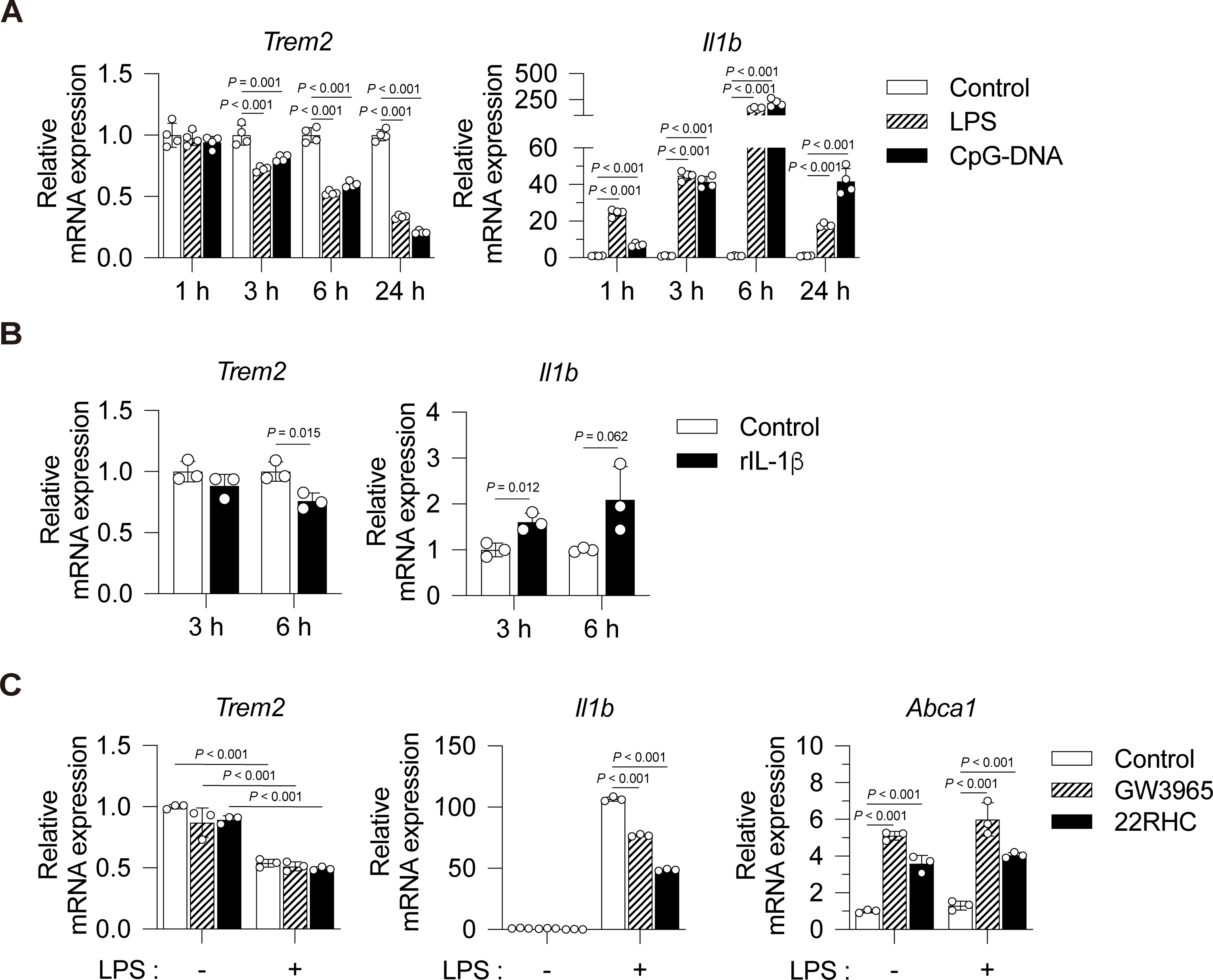
(A) WT BMDMs were stimulated with vehicle control (PBS), lipopolysaccharide (LPS, 1 ng/mL) or CpG-DNA (1 μg/mL) for 1, 3, 6 and 24 hours. Expressions of Trem2 and Il1b were evaluated by qPCR (n = 4). (B) Effect of IL-1β stimulation on the expression of Trem2. WT BMDMs were treated with vehicle control (PBS) or recombinant mouse IL-1β (rIL-1β, 100 ng/mL) for 3 or 6 hours (n = 3). (C) Effect of LXR activation on suppression of Trem2 expression by inflammatory signaling. BMDMs were treated with DMSO (control), GW3965 (1 μM) or 22R-hydroxycholesterol (22RHC, 10 μM) for 16 hours and then stimulated with vehicle control (PBS) or LPS (1 ng/mL) for 6 hours (n = 3). All data are shown as mean ± SD. Statistical analyses were performed by one-way ANOVA followed by Tukey’s multiple comparisons (A, C) or unpaired Student t-tests (B).
Discussion
Our findings in myeloid LXR-deficient mice provide direct evidence for an anti-inflammatory, anti-atherogenic role of macrophage LXRs in atherosclerotic plaques (Fig. 1, 2) as previously suspected from bone marrow transplantation studies22, 23. High resolution RNA profiling and immunohistochemistry reveals that in myeloid LXR deficiency there is increased accumulation of foamy, TREM2+ macrophages in atherosclerotic lesions. However, as shown by isolation of foam cells from plaques and by immunostaining, LXR-deficient foamy macrophages have relatively low TREM2 expression and a diminished downstream program of expression of endocytic and lipid metabolism genes alongside prominent upregulation of inflammatory and proliferative genes (Fig. 4). Our studies provide new insights into the mechanisms underlying the potent anti-atherogenic effects of myeloid cell LXRs, including their key role in monocyte recruitment, maintenance of the Trem2 gene expression module in foam cells and suppression of inflammatory gene expression in both foamy and nonfoamy myeloid cells.
Previous studies have suggested that TREM2+ macrophages have a beneficial effect on disease outcomes in Alzheimer disease and obesity. A loss-of-function variant of TREM2 has been associated with early onset dementia and late onset Alzheimer’s disease38, 39. In mouse models of Alzheimer’s disease and obesity, the TREM2 receptor has been identified as a major pathology-induced immune signaling hub that senses tissue damage and activates a robust immune remodeling response40. TREM2 also activates a program to decrease cholesterol and cholesteryl ester accumulation via induction of Apoe, Lpl, Ctsl, Lipa and Npc2 in demyelination associated phagocytic microglia, the same genes that showed reduced expression in TREM2+ foamy macrophages on the present study18. Together, these findings suggest that LXRs in TREM2+ macrophages have a similar favorable homeostatic role in atherosclerosis, helping to clear lipid debris derived from circulating lipoproteins and necrotic cells while tamping down inflammation.
Our studies suggest an increased number of foamy, TREM2+ macrophages that have reduced Trem2 expression levels. While this may seem paradoxical the TREM2+ macrophage classification in the scRNA-seq data is based on a gene expression signature which includes genes involved in lipid metabolism, cholesterol efflux and efferocytosis, not just the level of Trem2 expression. Since a number of TREM2 downstream genes are LXR targets such as Lpl, Apoe, Abca1, Abcg1 and Mertk, LXR may act collaboratively with TREM2 to induce LXR target genes in lesional macrophages. For example, by inducing the phagocytic function of macrophages, TREM2 may induce cholesterol accumulation that in turn leads to desmosterol accumulation and activation of LXR15, 16. Indeed, oxidized cholesterol derivatives and desmosterol that are formed as a result of tissue damage and cholesterol uptake may provide an important signal to activate the TREM2 program. Unexpectedly, we found that LXR activators did not upregulate Trem2 expression in BMDMs. Rather TREM2 expression was repressed by LPS and IL-1β (Fig. 5). Thus, increased inflammatory effects including inflammasome activation likely explain the reduced TREM2 expression in LXR deficient macrophages in the plaque. This may also explain why Nr1h3 has higher expression in Trem2 expressing cells since Nr1h3 undergoes positive autoregulation36. While our studies confirmed that LXRs suppress inflammatory genes in TREM2+ foamy macrophages, they also indicate anti-inflammatory effects in nonfoamy macrophages and monocytes (Fig. 4). These findings suggest that LXRs may suppress expression of some inflammatory genes independent of cholesterol loading, consistent with the finding that LXRs bind to many macrophage enhancer elements in regions of chromatin closure independent of LXR activation11.
Bulk-cell RNA-seq and qPCR analysis with foamy or nonfoamy macrophages demonstrated that the expression of 15-LOX is highly induced in foamy macrophages by LXR deficiency (Fig. 2). It has been reported that 15-LOX is increased by LXR activation in human alternatively activated macrophages41. IL-4 or IL-13 signals are needed for ALOX15 induction, indicating that 15-LOX is not a direct LXR target gene. The expression of 15-LOX is also stimulated by prostaglandin E2 (PGE2) through cAMP-PKA-CREB pathway in human neutrophils42, and by IL-1β in mouse microglia43. PGE2 synthesis is mediated by IL-1β receptor signaling during Mycobacterium tuberculosis infection44, and LPS-induced PGE2 synthase expression is suppressed by LXR activation45. These findings suggest that elevated expression of IL-1β by inflammasome activation triggers PGE2 synthesis and leads to upregulation of Alox15 in Myl-Lxrdko.
In addition to macrophages, our study also reveals a major anti-atherogenic role of LXRs in monocytes. We recently showed that LXRs repress expression of monocyte cell surface integrins that are known to mediate binding and entry of monocyte into atherosclerotic plaques11. Accordingly, we showed a marked increase in monocyte entry into plaque in Myl-Lxrdko-BMT Ldlr−/− mice (Fig. 2N). However, the scRNA-seq data indicated depletion of the main monocyte populations in plaques of these mice (Fig. 3B). We speculate that this could reflect a rapid conversion of monocytes into resident-like TREM2+ foamy macrophages as a result of defective cholesterol efflux genes and the heightened inflammatory milieu of the plaque in Myl-Lxrdko-BMT Ldlr−/− mice.
In conclusion, our study shows that LXRs play important beneficial roles in TREM2+ foamy macrophages during atherogenesis, and reveal major anti-inflammatory effects of LXRs in plaques in both foamy and non-foamy myeloid cells. The ultimate failure of the LXR/TREM2 program in progressive atherosclerosis may reflect an excessive burden of atherogenic lipoprotein uptake and cell death in plaques, suboptimal levels of LXR activation in plaque cells or bottlenecks in downstream pathways such as ABC transporter and HDL-mediated cholesterol efflux. The clinical development of LXR activators was halted owing to adverse effects in the liver leading to steatosis and increased LDL levels46. Our findings underline the therapeutic potential of selective activation of monocyte/macrophage LXRs and the macrophage TREM2 program in a wide variety of disease processes including Alzheimer disease, obesity and atherosclerosis.
Supplementary Material
Highlights.
LXR deficiency in myeloid cells accelerates the progression of atherosclerosis by increasing monocyte entry, foam cell formation and plaque inflammation.
Bulk cell-RNA profiling of plaque myeloid cells showed an upregulation of inflammatory mediators including oxidative, chemokine and chemotactic genes.
Single cell-RNA sequencing of plaque leukocytes revealed increased numbers of foamy TREM2-expressing macrophages.
TREM2-expressing cells had reduced expression of the Trem2 gene expression module, including phagocytic and cholesterol efflux genes, and had switched to a pro-inflammatory and proliferative phenotype.
Acknowledgements
We thank members of the Tall laboratory, Hongxue Shi (Columbia University), Michael Kissner (Columbia Stem Cell Initiative Flow Cytometry Core Facility) and Erin Bush (Columbia Genome Center) for technical assistance and helpful comments.
Sources of Funding
This work was supported by National Institutes of Health (NIH) grant HL107653 (to A.R.T.), the Swedish Research Council and the Robert A. Welch Foundation (to J-Å.G.). K.E-U. was supported by Nihon University Overseas Research Fellowships. This research was also funded in part through the NIH/NCI Cancer Center Support Grant P30CA013696 and used the Genomics and High Throughput Screening Shared Resource. This publication was supported by the National Center for Advancing Translational Sciences, NIH, through Grant Number UL1TR001873. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations
- LXR
liver X receptor
- TREM2
triggering receptor expressed on myeloid cells 2
- scRNA-seq
single-cell RNA sequencing
- ABCA1
ATP-binding cassette transporter A1
- Apo E
apolipoprotein E
- LPS
lipopolysaccharide
- WTD
Western-type diet
- LDLR
low-density lipoprotein receptor
- BMDM
bone marrow-derived macrophage
- BMT
bone marrow transplanted
- IL
interleukin
- MPO
myeloperoxidase
- 15-LOX
15-lipoxygenase
- tSNE
t-distributed stochastic neighbor embedding
Footnotes
Disclosures
None.
References
- 1.Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol 2015;15:104–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bäck M, Yurdagul A Jr., Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat Rev Cardiol 2019;16:389–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zernecke A, Winkels H, Cochain C et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res 2020;127:402–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim K, Shim D, Lee JS et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res 2018;123:1127–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaitin DA, Adlung L, Thaiss CA et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell 2019;178:686–698.e614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deczkowska A, Weiner A, Amit I. The physiology, pathology, and potential therapeutic applications of the trem2 signaling pathway. Cell 2020;181:1207–1217 [DOI] [PubMed] [Google Scholar]
- 7.Wang B, Tontonoz P. Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol 2018;14:452–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid x receptor. J Biol Chem 2000;275:28240–28245 [DOI] [PubMed] [Google Scholar]
- 9.Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 2000;289:1524–1529 [DOI] [PubMed] [Google Scholar]
- 10.Naik SU, Wang X, Da Silva JS, Jaye M, Macphee CH, Reilly MP, Billheimer JT, Rothblat GH, Rader DJ. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation 2006;113:90–97 [DOI] [PubMed] [Google Scholar]
- 11.Thomas DG, Doran AC, Fotakis P, Westerterp M, Antonson P, Jiang H, Jiang XC, Gustafsson J, Tabas I, Tall AR. LXR suppresses inflammatory gene expression and neutrophil migration through cis-repression and cholesterol efflux. Cell Rep 2018;25:3774–3785.e3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. Parallel sumoylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell 2007;25:57–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: Free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 2008;118:1837–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, Gratton E, Parks J, Tontonoz P. LXRs link metabolism to inflammation through ABCA1-dependent regulation of membrane composition and TLR signaling. Elife 2015;4:e08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spann NJ, Garmire LX, McDonald JG et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 2012;151:138–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, McDonald JG, Aryal B et al. Desmosterol suppresses macrophage inflammasome activation and protects against vascular inflammation and atherosclerosis. Proc Natl Acad Sci U S A 2021;118. [DOI] [PMC free article] [PubMed]
- 17.Berghoff SA, Spieth L, Sun T et al. Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis. Nat Neurosci 2021;24:47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nugent AA, Lin K, van Lengerich B et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 2020;105:837–854.e839 [DOI] [PubMed] [Google Scholar]
- 19.Seidman JS, Troutman TD, Sakai M et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity 2020;52:1057–1074.e1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan CT, Fenn AM, Harder NK et al. Liver X receptors are required for thymic resilience and T cell output. J Exp Med 2020;217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joseph SB, McKilligin E, Pei L et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A 2002;99:7604–7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tangirala RK, Bischoff ED, Joseph SB, Wagner BL, Walczak R, Laffitte BA, Daige CL, Thomas D, Heyman RA, Mangelsdorf DJ, Wang X, Lusis AJ, Tontonoz P, Schulman IG. Identification of macrophage liver X receptors as inhibitors of atherosclerosis. Proc Natl Acad Sci U S A 2002;99:11896–11901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levin N, Bischoff ED, Daige CL, Thomas D, Vu CT, Heyman RA, Tangirala RK, Schulman IG. Macrophage liver X receptor is required for antiatherogenic activity of LXR agonists. Arterioscler Thromb Vasc Biol 2005;25:135–142 [DOI] [PubMed] [Google Scholar]
- 24.Fotakis P, Kothari V, Thomas DG, Westerterp M, Molusky MM, Altin E, Abramowicz S, Wang N, He Y, Heinecke JW, Bornfeldt KE, Tall AR. Anti-inflammatory effects of HDL (high-density lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques. Arterioscler Thromb Vasc Biol 2019;39:e253–e272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, García-Cardeña G, Lusis AJ, Owens AP 3rd, Rosenfeld ME, Virmani R. Recommendation on design, execution, and reporting of animal atherosclerosis studies: A scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol 2017;37:e131–e157 [DOI] [PubMed] [Google Scholar]
- 26.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, 3rd, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive integration of single-cell data. Cell 2019;177:1888–1902.e1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gage MC, Bécares N, Louie R, Waddington KE, Zhang Y, Tittanegro TH, Rodríguez-Lorenzo S, Jathanna A, Pourcet B, Pello OM, De la Rosa JV, Castrillo A, Pineda-Torra I. Disrupting LXRα phosphorylation promotes FOXM1 expression and modulates atherosclerosis by inducing macrophage proliferation. Proc Natl Acad Sci U S A 2018;115:E6556–e6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Westerterp M, Fotakis P, Ouimet M et al. Cholesterol efflux pathways suppress inflammasome activation, netosis, and atherogenesis. Circulation 2018;138:898–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest 2007;117:185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF, Funk CD. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in Apo E-deficient mice. J Clin Invest 1999;103:1597–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.George J, Afek A, Shaish A, Levkovitz H, Bloom N, Cyrus T, Zhao L, Funk CD, Sigal E, Harats D. 12/15-lipoxygenase gene disruption attenuates atherogenesis in LDL receptor-deficient mice. Circulation 2001;104:1646–1650 [DOI] [PubMed] [Google Scholar]
- 32.Huo Y, Zhao L, Hyman MC et al. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation 2004;110:2024–2031 [DOI] [PubMed] [Google Scholar]
- 33.Kotla S, Singh NK, Heckle MR, Tigyi GJ, Rao GN. The transcription factor CREB enhances interleukin-17a production and inflammation in a mouse model of atherosclerosis. Sci Signal 2013;6:ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Donnell VB, Aldrovandi M, Murphy RC, Krönke G. Enzymatically oxidized phospholipids assume center stage as essential regulators of innate immunity and cell death. Sci Signal 2019;12. [DOI] [PubMed] [Google Scholar]
- 35.Singh NK, Rao GN. Emerging role of 12/15-lipoxygenase (Alox15) in human pathologies. Prog Lipid Res 2019;73:28–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ulven SM, Dalen KT, Gustafsson JA, Nebb HI. Tissue-specific autoregulation of the LXRalpha gene facilitates induction of apoe in mouse adipose tissue. J Lipid Res 2004;45:2052–2062 [DOI] [PubMed] [Google Scholar]
- 37.Gao X, Dong Y, Liu Z, Niu B. Silencing of triggering receptor expressed on myeloid cells-2 enhances the inflammatory responses of alveolar macrophages to lipopolysaccharide. Mol Med Rep 2013;7:921–926 [DOI] [PubMed] [Google Scholar]
- 38.Jonsson T, Stefansson H, Steinberg S et al. Variant of TREM2 associated with the risk of alzheimer’s disease. N Engl J Med 2013;368:107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guerreiro R, Wojtas A, Bras J et al. TREM2 variants in alzheimer’s disease. N Engl J Med 2013;368:117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017;169:1276–1290.e1217 [DOI] [PubMed] [Google Scholar]
- 41.Snodgrass RG, Benatzy Y, Schmid T, Namgaladze D, Mainka M, Schebb NH, Lütjohann D, Brüne B. Efferocytosis potentiates the expression of arachidonate 15-lipoxygenase (ALOX15) in alternatively activated human macrophages through LXR activation. Cell Death Differ 2021;28:1301–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat Immunol 2001;2:612–619 [DOI] [PubMed] [Google Scholar]
- 43.Rivera-Escalera F, Pinney JJ, Owlett L, Ahmed H, Thakar J, Olschowka JA, Elliott MR, O’Banion MK. Il-1β-driven amyloid plaque clearance is associated with an expansion of transcriptionally reprogrammed microglia. J Neuroinflammation 2019;16:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayer-Barber KD, Andrade BB, Oland SD et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014;511:99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ninomiya Y, Yasuda T, Kawamoto M, Yuge O, Okazaki Y. Liver X receptor ligands inhibit the lipopolysaccharide-induced expression of microsomal prostaglandin E synthase-1 and diminish prostaglandin E2 production in murine peritoneal macrophages. J Steroid Biochem Mol Biol 2007;103:44–50 [DOI] [PubMed] [Google Scholar]
- 46.Kirchgessner TG, Sleph P, Ostrowski J et al. Beneficial and adverse effects of an LXR agonist on human lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab 2016;24:223–233 [DOI] [PubMed] [Google Scholar]
- 47.Fidler TP, Xue C, Yalcinkaya M et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021;592:296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang W, Liu W, Fidler T et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in JAK2 (V617F) mice. Circ Res 2018;123:e35–e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.