

Abstract
Background and Aims:
Immunotherapy with PD-1/PD-L1 blockade has shown low response rates in liver cancer patients, with the underlying mechanisms unclear. To decipher a specific impact of the liver microenvironment, we compared the effects of anti-PD-L1 antibody (αPD-L1) blockade on the same tumor grown subcutaneously or in the liver.
Approach and Results:
We generated subcutaneous tumors in mice by inoculating MC38 colorectal cancer (CRC) cells under the skin and metastatic liver tumors by portal vein or splenic injection of the CRC cells. Tumor-bearing mice were treated by intraperitoneal injection of αPD-L1, polyinosinic:polycytidylic acid (polyIC) or both. αPD-L1 monotherapy significantly suppressed subcutaneous tumor growth but showed no effect on metastatic liver tumors. However, combination of αPD-L1 with polyIC, an innate immunity-stimulating reagent, robustly inhibited tumor progression in the liver. The combination therapy effectively down-regulated myeloid-derived suppressor cells (MDSC) but upregulated the ratios of M1/M2 macrophages, CD8/CD4 and CD8/Treg cells infiltrated into liver tumors and the whole liver. A group of long-lasting T-bet+Eomes−PD-1− cytotoxic T cells was maintained in the Combo-treated liver, leading to resistance to tumor recurrence. Depleting macrophages or blocking type I interferon signaling abrogated the synergistic anti-tumor effect of αPD-L1 and polyIC, indicating a requirement of boosting innate immunity for optimized activation of cytotoxic T cells by PD-1/PD-L1 blockade.
Conclusion:
The poor response of liver cancers to αPD-L1 therapy is largely due to a unique hepatic immunotolerant microenvironment, independent of tumor origins or types. The success of a combinatorial immunotherapy relies on coordinated inhibition or activation of various innate and adaptive immune cell activities.
Keywords: metastatic liver cancer, anti-PD-L1 blockade, polyIC, type I interferon
Graphical Abstract
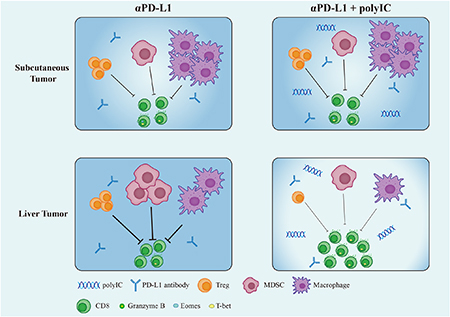
INTRODUCTION
Therapy with immune checkpoint inhibitors (ICIs) has shown great promise in a variety of cancers.(1, 2) Of note, the proof of principle for this transformative oncological treatment was initially obtained in mouse tumor models. In 1996, Allison’s group reported that intraperitoneal injection of a specific antibody against cytotoxic T lymphocyte antigen-4 (CTLA-4, CD152) suppressed tumor progression in mice inoculated subcutaneously with colon carcinoma 51BLim10 cells or Sa1N fibrosarcoma cells.(3) Honjo and colleagues showed that a specific antibody to programmed death-ligand 1 (PD-L1, CD274) inhibited subcutaneous tumor growth in mice with myeloma cells injected under the skin.(4) These groundbreaking studies in animal models were quickly translated into clinical trials in cancer patients, with a phase I result reported in 2012, on a humanized anti-PD-1 antibody therapy in non-small-cell lung cancer, melanoma and renal cell carcinoma.(5) Despite the great success of immunotherapy in a broad range of cancers, even some life-saving miracles in a few patients at the advanced stages of melanoma or kidney tumor,(6) the vast majority of cancer patients showed poor response or developed resistance to the checkpoint blockers.(5, 7)
Primary liver cancer, mainly hepatocellular carcinoma (HCC), is now a leading cause of cancer-associated death.(8) Following the initial encouraging outcomes in melanoma and lung cancer, phase I and II trials with ICIs started immediately in advanced liver cancer patients.(9) However, approximately 17% HCC patients responded to monotherapy of pembrolizumab(10) and 20% responded to nivolumab.(11) A combination of nivolumab and ipilimumab increased the response rate to 30% in HCC.(12) Simultaneous blockade of PD-L1 and VEGF signaling using atezolizumab and bevacizumab achieved better overall and progression-free survival than sorafenib in HCC (IMbrave 150).(13) As a major organ of metabolism and detoxification, liver develops an intricate immunotolerant environment,(9, 14, 15) which may restrict the effectiveness of immunotherapy.(16) Metastatic liver tumors are often detected in association with colorectal cancer (CRC), pancreatic cancer, and neuroendocrine tumor.(17) As many as 30–50% of CRC patients develop liver metastasis,(18, 19) with only 15–20% of them eligible for surgical resection.(20) Although ICIs have demonstrated efficacy in CRCs with mismatch-repair deficiency or high microsatellite instability, the majority of CRCs that are mismatch repair proficient or microsatellite stable are refractory to immunotherapy.(21) Patients with colorectal liver metastasis have poor prognosis, and various combinatorial strategies are being evaluated in clinical trials.(22)
Our previous data showed that monotherapy with anti-PD-L1 antibody (ɑPD-L1) exhibited no inhibitory effect in primary liver cancer driven by classical oncogenes in mice.(23) Given a robust induction of PD-L1 expression by polyinosinic:polycytidylic acid (polyIC) in the liver, as revealed by RNA-sequencing,(24) we reasoned that the synthetic dsRNA could sensitize hepatic response to αPD-L1 treatment. Indeed, combined treatment of polyIC and αPD-L1 showed markedly improved efficacy in mouse HCC models.(23) polyIC is a TLR3 ligand that stimulates production of interferons and other cytokines, and has been used as an immune adjuvant.(25) However, it is not understood how polyIC regulates communications between innate and adaptive immune cells and their activation status in the liver tumor microenvironment.
In this study, we compared the efficacy of these reagents between subcutaneous and metastasized liver tumors derived from the same CRC cells, to identify liver-specific factors responsible for the poor response to immunotherapy. Despite potent suppression on tumor growth under the skin, αPD-L1 alone had no effect on the liver-grafted tumor progression. A combinatorial therapy of αPD-L1 and polyIC exhibited additive effect in subcutaneous tumors but an intriguing synergistic effect in liver tumors. We interrogated the compositions and changes of immune cell subtypes infiltrated into the tumors grown subcutaneously or in the liver, as well as the whole liver of tumor-bearing mice. This comparative analysis provides a fresh view on the unique immune ecosystem of liver tumors, and also prompts us propose a new thought on how to design effective liver cancer immunotherapy.
MATERIALS AND METHODS
Animals.
Wild-type C57BL/6J and type I IFN knockout mouse line B6(Cg)-Ifnar1tm1.2Ees/J (Ifnar1−, stock# 028288) were obtained from the Jackson Laboratory, and male mice at age of 8–12 weeks were used for the experiments. The animal protocols (S09108) were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California San Diego, following National Institutes of Health guidelines.
Cell culture.
MC38 tumor cells were a gift from Karin lab at UCSD. Cells were cultured in antibiotic-free DMEM containing 10% fetal bovine serum and tested negative for mycoplasma using a universal mycoplasma detection kit (30-1012K, ATCC, Manassas, Virginia).
Tumor cell inoculation.
Subcutaneous inoculation, portal vein injection or intrasplenic transplantation of tumor cells was performed as described previously.(26) Mice were inoculated with 1×105 cells per 100 μl under anesthesia via an anesthesia machine by continuous inhalation of 3% isoflurane gas for 5–20 min. Subcutaneous tumor volumes were measured twice weekly using digital calipers. Tumor volume = (length × width × high)/2. The tumors were measured on day 7, 10, 12, 14, 17, and 21.
Drug delivery.
PolyIC (GE Healthcare, Chicago, Illinois) was injected intraperitoneally (i.p.) at 4 mg/kg every other day for five doses as indicated in the figures. Anti-mouse PD-L1 antibody (αPD-L1) (BE0101, Bioxcell, Upper Valley, New Hampshire) and anti-mouse IFNαR1 (BE0241, Bioxcell) were injected i.p. at 200 μg as indicated. Macrophages were depleted by i.p. injection of 200 μL of clodronate liposome, or 200 μL of PBS control liposome (CP-005–005, Liposoma BV, Amsterdam, Netherlands).
Histopathology.
The livers were fixed in Z-fix for 24 hrs, dehydrated, cleared, and embedded in paraffin. Then, the tissues were stained using hematoxylin and eosin (H&E) stains. The H&E slides were scanned by nanozoomer 2.0-HT slide scanner and screened by NDP view 2.7.25 (Hamamatsu Photonics, Hamamatsu, Japan).
Antibody for flow cytometry.
FITC-Granzyme B (515403), FITC-Ly6C (128006), PE-PD-L1 (124308), BV421-F4/80 (123124), APC-NK1.1 (108710), APC-CD11c (117310), APC-CD44 (103012), APC-Cy7-CD19 (115530), APC-Cy7-I-A/I-E (107628), PE-Cy7-CD8a (100722), PE-Cy7-CD206 (141720), BV605-CD4 (100548), BV605-CD11b (101237), BV711-CD69 (104537), BV711-CD45R/B220 (103255), BV711-Tbet (644819), Percp-Cy5.5-CD45 (147706) were purchased from BioLegend (San Diego, California). PE-CD62L (12-0621-82), PE-Foxp3 (12-5773-82), Pacific Blue-Ki67 (48-5698-82), APC-eFluor780-PD1 (47-9985-80), PE-Cy5-CD3 (15-0031-81), PE-Cy5-Eomes (15-4875-80), anti-CD16/32 (14-0161-85) were purchased from Thermo Fisher (Waltham, Massachusetts).
Cell preparation for flow cytometry.
Subcutaneous tumors or liver-grafted tumors (short for liver tumors and liv T) were separated from skin or surrounding liver tissues by a sterile forceps, the weights of collected tumors were measured before crushing with perfusion buffer and filtrated from 100 μm (Celltreat, Pepperell, Massachusetts) and 35 μm (352235, Falcon polystyrene test tube) cell strainers. The immune cells from the whole liver were collected as described previously.(23) Cells were stained for 30 mins using a LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (L34957, Thermo Fisher), following staining of an antibody cocktail for 15 mins. The intracellular staining followed a protocol of Foxp3/transcription factor staining buffer set (00-5523, Thermo Fisher). Flow cytometry analysis was done on BD LSRFortessa X-20 Cell Analyzer at UCSD Human Embryonic Stem Cell Core facility at Sanford Consortium for Regenerative Medicine. Data was then analyzed using FlowJo 10.6.2 (Becton, Dickinson & Company, Franklin Lakes, New Jersey), the gating strategy is indicated in Supporting Figure 7.
Statistical analysis.
Experimental results were expressed as means ± SEM. One-way ANOVA with Holm-Sidak’s multiple comparisons test or Kruskal-Wallis test with Dunn’s multiple comparisons test were performed to compare differences. Statistical analyses were performed using Prism 8.02 (GraphPad Software, La Jolla, CA).
RESULTS
Monotherapy of anti-PD-L1 antibody effectively suppresses tumors grown subcutaneously but not in the liver
In previous experiments, we demonstrated that primary HCCs induced by pairs of oncogenes, such as Ras and Myc or MET and b-catenin, were poorly responsive to therapy with anti-PD-L1 antibody (αPD-L1).(23) However, combined treatment of αPD-L1 with polyIC exhibited an improved tumor-inhibiting effect,(23) with the underlying mechanisms to be elucidated. To pinpoint exclusively a hepatic environmental influence, we compared the anti-tumor effects of αPD-L1 blockade on the same tumor grown subcutaneously or in the liver. We inoculated mouse MC38 CRC cells under the skin in the left and right abdomen, and tumor nodules were visible 6 days after implantation. The tumor-bearing mice were then treated by intraperitoneal (i.p.) injection of αPD-L1 at day 7, 9 and 11, polyIC at day 6, 8 and 10, or both αPD-L1 and polyIC (Combo) at these time points (Fig. 1A). Tumor sizes were measured during the course, and tumor tissues were collected at day 21 for histopathological, cell and molecular analyses. Monotherapy with either αPD-L1 or polyIC showed significant suppression of subcutaneous tumor growth; the mean tumor sizes decreased from 291.5 mm3 for the control group to 159.6 mm3 in the αPD-L1 group and 159.2 mm3 in the polyIC group, respectively (Fig. 1B–D). The Combo treatment showed an additive tumor-suppressive effect, with the mean tumor size at 104.2 mm3 (Fig. 1B–D).
Fig. 1. Differential therapeutic effects of αPD-L1 and/or polyIC on subcutaneous and liver tumors.
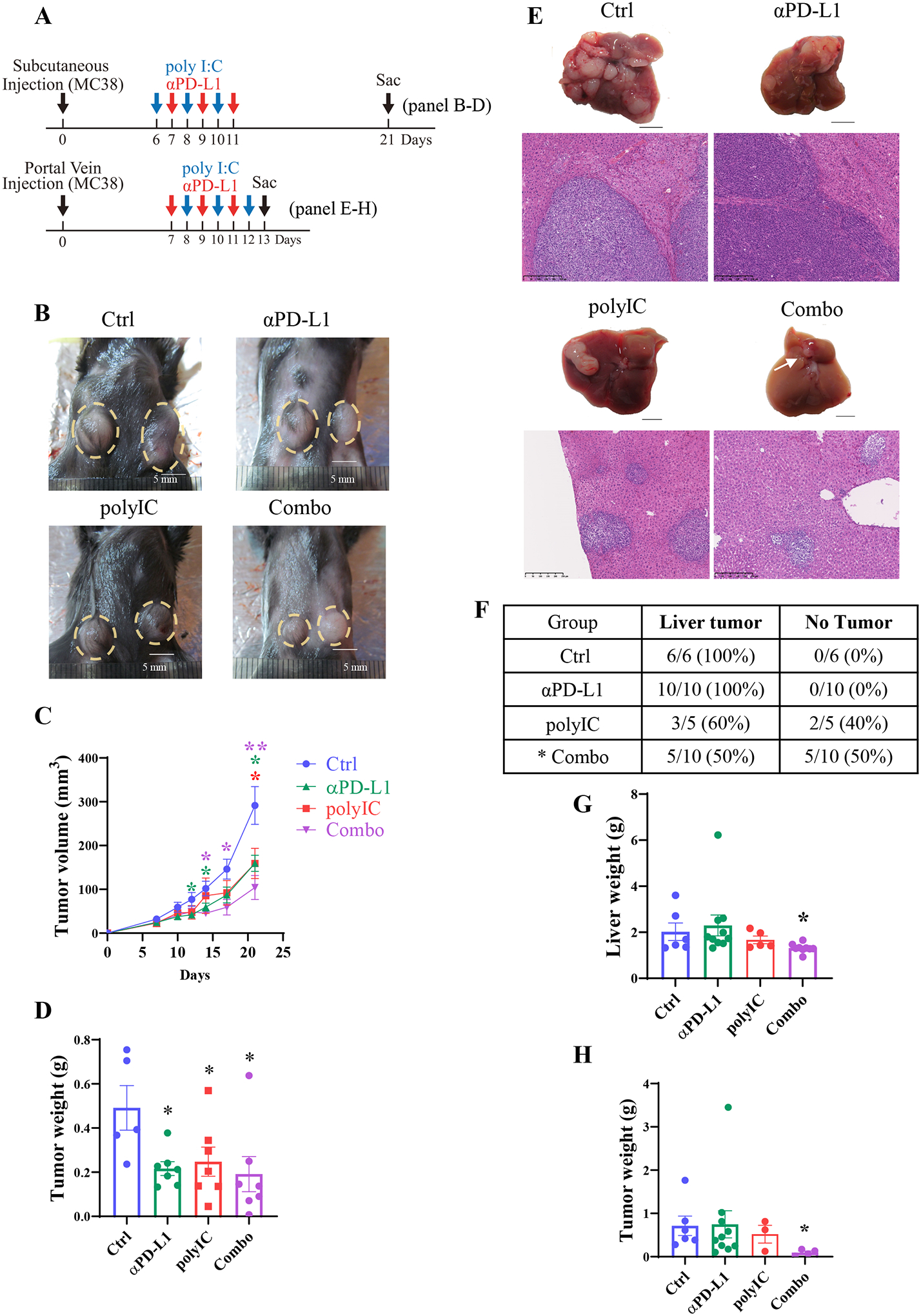
A). Upper panel: the experimental scheme of the subcutaneous tumor model with relevant data in 1B-D. After MC38 cell inoculation, polyIC was injected intraperitoneally (i.p.) at day 6, 8 and 10, αPD-L1 injected at day 7, 9, and 11. The Combo group received both at these time points. Lower panel: the scheme of the liver-grafted tumor model with data shown in 1E-H. After portal vein injection of MC38 cells, polyIC was injected at day 8, 10, and 12, αPD-L1 injected at day 7, 9, and 11, and the Combo group received both.
B). Macroscopic views of representative subcutaneous tumors of untreated (Ctrl), treated with polyIC, αPD-L1, or the combination (Combo). A yellow dotted circle indicates the tumors.
C). The subcutaneous tumor volumes were measured at various time points, n = 5–8.
D). Subcutaneous tumor weights of each group. Samples were collected at day 21 after tumor cell inoculation, n = 5–7.
E). Macroscopic views of representative tumors, and H&E staining of liver sections for each group on day 13.
F). The liver tumor formation rates of each group. Statistical analysis: Chi-square test, * p < 0.05.
G). Liver weights of each group, n = 5–10.
H). Tumor weights of each group, n = 4–10.
Statistical analysis: One-way ANOVA or Kruskal-Wallis, compared with Ctrl. * p<0.05, **p<0.01.
Meanwhile, we induced the same tumor growth in the liver following portal vein injection of MC38 CRC cells, and treated the mice similarly with αPD-L1, polyIC or both (Fig. 1A). Most of the mice died around 15 days after MC38 cell inoculation, due to aggressive tumor progression in the liver. Of note, monotherapy with αPD-L1 showed no inhibition on tumor growth in the liver, with variable effects observed for polyIC (Fig. 1E–F). However, a robust tumor repression was observed in the Combo group, with half of the mice (5/10) having no visible or microscopic tumor nodules and another half having only small tumor foci (Fig. 1E–F). The tumor burdens in the Combo group were significantly lower than the control, αPD-L1 and polyIC groups, when comparing the liver or tumor weights (Fig. 1G–H). Together, these results unveil a unique liver influence on the outcome of immunotherapy: a) despite significant suppression of subcutaneous tumors, αPD-L1 monotherapy had no effect on the same tumor growth in the liver; b) combination of αPD-L1 with polyIC showed an additive tumor-inhibitory effect under the skin but a synergistic effect in the liver.
polyIC promotes liver response to αPD-L1 by suppressing Treg cells and boosting cytotoxic CD8 T cells
To interrogate the differential tumor-inhibitory effects, we compared the compositions of immune cells infiltrated into the tumors in these two sites. The ratios of T cells (CD3+) versus total leukocytes (CD45+) were similar in subcutaneous tumors without or with treatment, but a significant increase of CD3+ T cell infiltration was detected in liver-grafted tumors (liver tumors or liv T) following the Combo treatment (Fig. 2A, Supporting Fig. 1A). The ratios of CD8 to CD4 T cells were elevated in subcutaneous tumors, following mono- or Combo treatments, but a drastically increased CD8/CD4 T cell ratio was observed in liver tumors only after the Combo therapy (Fig. 2B). In particular, the Combo induced significantly increased CD8 but decreased CD4 T cell infiltration into the liver tumors (Supporting Fig. 1B, 1C).
Fig. 2. The immune cell profiles in subcutaneous and liver tumors.
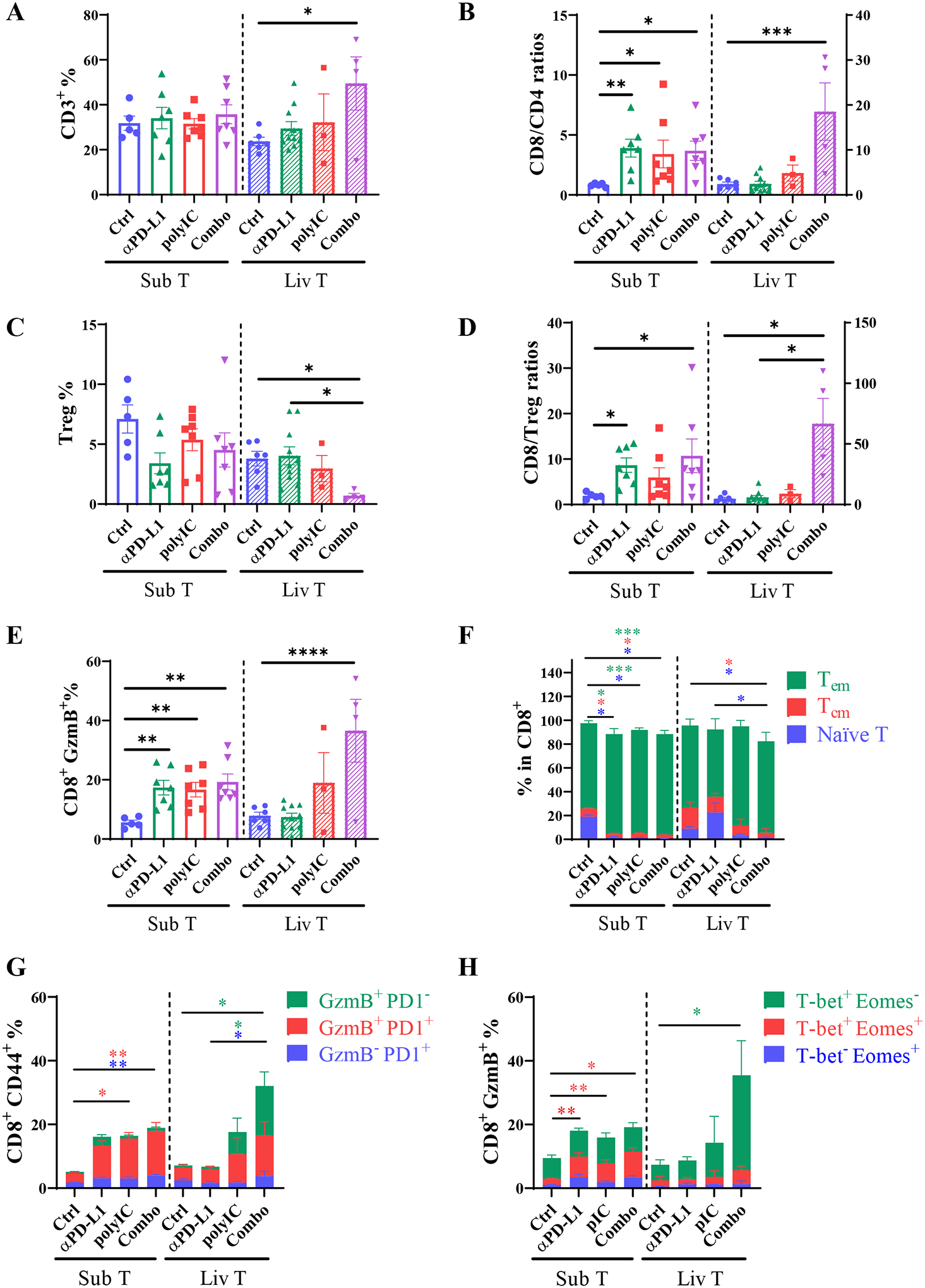
A). The percentages (%) of CD3+ cells (CD45+ L/D− CD3+ NK1.1−) in CD45+ cells, in subcutaneous tumors (Sub T, left to dotted line) or metastasized liver tumors (Liv T, right to dotted line). The numbers of total CD45+ cells in each gram of tumor tissues were first determined and the relative percentages or ratios of various cell types were calculated.
B). The CD8 versus CD4 T cell ratios.
C). The percentages of Treg cells (CD3+ NK1.1+ CD4+ Foxp3+) in CD45+ cells.
D). The CD8 to Treg cell ratios.
E). The percentages of CD8+ GzmB+ T cells (CD3+ NK1.1+ CD8+ Granzyme B+) in CD45 cells.
F). The percentages of indicated cell subsets in CD8 T cells. Green bar: effector memory T cell (Tem, CD62L−CD44+); blue bar: naïve T cells (CD62L+CD44−); red bar, central memory T cells (Tcm, CD62L+CD44+). Asterisks in each color represent the p values.
G). The percentages of Tem cells of Granzyme B or PD1 expression in total CD45 cells. Blue bar: exhausted Tem (GzmB− PD1+); red bar, Tem cells that are GzmB+ and PD1+; green Bar: functional Tem cells (GzmB+PD1−) without the feature of exhaustion. Asterisks in each color represent the p values.
H). The percentages of CD8+GzmB+ T cells distinguished by T-bet and Eomes expression in total CD45 cells. Green bar: effector CD8 (T-bet+Eomes−); red bar: intermediate state of activated CD8 cells (T-bet+Eomes+); blue bar: memory CD8 cells (T-bet−Eomes+); Asterisks in each color represent the p values. n = 4–10.
Tumor samples of Sub T and Liv T were collected from 3 individual experiments. Statistical analysis: one-way ANOVA or Kruskal-Wallis. * p<0.05, **p<0.01.
Meanwhile, the Combo treatment induced a most severe decrease of the regulatory T (Treg) cell subpopulation only in liver tumors (Fig. 2C, Supporting Fig. 1D). The potential of anti-tumor immune response, as evaluated by the CD8/Treg cell ratio, was enhanced in all 3 treatment groups of subcutaneous tumors but was not detected in liver tumors following monotherapies (Fig. 2D). Of note, the CD8/Treg ratio was dramatically elevated in the Combo-treated group of liver tumors (Fig. 2D). These observations were further substantiated by a similar change of functional cytotoxic T cells marked by granzyme B (GzmB) (Fig. 2E). The increase of CD8 T cells was accompanied by a decrease of B cells in all three treated groups of subcutaneous tumors, and αPD-L1 induced a higher population of B cells in liver tumors as compared to the other 3 groups (Supporting Fig. S1E). We did not find dramatic changes of NK cells either in subcutaneous or liver tumors following various treatments (Supporting Fig. S1F). Together, these results suggest that αPD-L1 monotherapy effectively boosted the CD8+/Treg cell ratios in subcutaneous tumors but failed to do so in liver tumors, leading to defective tumor regression. Nonetheless, its combination with polyIC showed more robust regression and even eradication of liver tumors, relative to subcutaneous tumors, by suppressing Treg cells and activating more GzmB+ CD8 T cells.
The activated cytotoxic T cells in liver tumors are sustained and less exhausted
We further examined the sub-types of cytotoxic T cells infiltrated into subcutaneous or liver tumors. Consistent to the tumor-inhibitory effect, the ratios of effector memory T cells (Tem, CD62L−CD44+) increased in subcutaneous tumors following the three types of treatment, accompanied by decrease of naïve T cells (CD62L+CD44−) and central memory (Tcm, CD62L+CD44+) T cells (Fig. 2F). However, in liver tumors, only the Combo group showed a drastic reduction of naïve T and Tcm subtypes (Fig. 2F). We further analyzed the subsets in Tem cells (Fig. 2G), a rise of GzmB+PD1+ Tem subset was detected in subcutaneous tumors following mono- and Combo treatment. Of note, the Combo-treated liver tumors displayed a dramatic upregulation of GzmB+PD1− subset, in addition to increased GzmB+PD1+ Tem cells (Fig. 2G, green bar). Further, we interrogated the status of activated T cells by two transcription factors T-bet and Eomes (Fig. 2H). An increase of CD8+Eomes+CD69+ subset was viewed as a sign of response to PD-1/PD-L1 blockade in melanoma patients.(27) We detected expansion of T-bet+Eomes+ cells in all 3 treated groups of subcutaneous tumors. However, no significant change was detected in this population in liver tumors. Instead, a markedly expanded group of GzmB+T-bet+Eomes− was observed in liver tumors following the Combo treatment (Fig. 2H). These results suggest that the liver tumor-infiltrated cytotoxic T cells activated by polyIC and αPD-L1 were persistently functional, rather than going through an exhaustion state.
Hepatic innate immune cells contribute to the low efficacy of αPD-L1 in liver tumors
We also examined myeloid-derived suppressor cells (MDSC, CD11b+Ly6C+F4/80−) in subcutaneous and liver tumors. The ratios of myeloid cells in subcutaneous tumors remained similar in the control and treated groups, although the liver tumors showed a lower level of CD11b+ cells following the Combo treatment (Fig. 3A). In subcutaneous tumors, MDSC infiltration was relatively low in the control group and remained low following the mono- or Combo treatments (Fig. 3B–C), accompanied by relatively higher numbers of F4/80+ macrophages (Fig. 3D), consistent with a previous report.(28) Compared to subcutaneous tumors, liver tumors contained much more MDSCs in the control group, and the abundance of MDSCs did not change significantly after monotherapy with αPD-L1 or polyIC but markedly decreased in the Combo-treated group (Fig. 3C). In consequence, the Combo treatment induced a significant increase in the CD8 to MDSC ratios, as compared to all other groups in subcutaneous or liver tumors (Fig. 3D). Liver tumors contained fewer macrophages than subcutaneous tumors in general. Again, the Combo treatment, but neither of the monotherapy, significantly decreased the abundance of tumor-infiltrating macrophages (Fig. 3B, 3E).
Fig. 3. The profiles of myeloid cells infiltrated into subcutaneous or liver tumors.
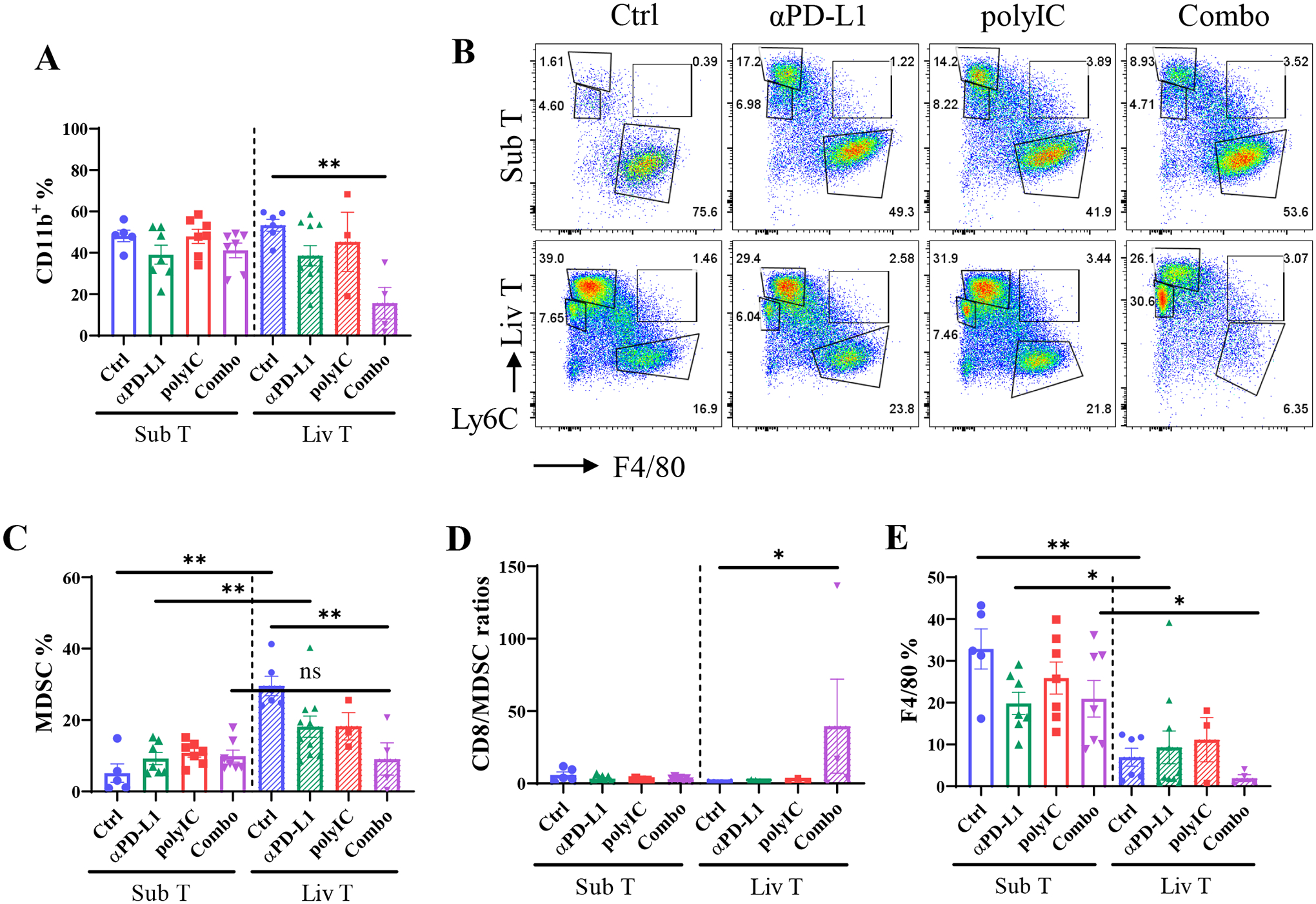
A). The percentages of CD11b+ cells in CD45+ cells in each group.
B). The graph of CD11b+ myeloid cells that were stained with Ly6C and F4/80 to determine MDSC (Ly6C+F4/80−), MoMF (Ly6C+F4/80+), and Kupffer (Ly6C−F4/80+).
C-E). Statistical analysis of MDSC (C), CD8 to MDSC ratios (D) and macrophage (E) populations in subcutaneous or liver-grafted tumors. MDSC: CD45+CD11b+Ly6C+F4/80−; macrophage: CD45+CD11b+F4/80+. n = 4–10.
Metastasized liver tumors are suppressed by combined αPD-L1 and polyIC treatment
As described above, the comparative analysis between subcutaneous and liver tumors revealed a liver-specific immunosuppressive environment that lowered the efficacy of αPD-L1 monotherapy. To extend this observation, we set up another liver tumor metastasis model by intrasplenic transplantation (IST) of CRC cells (Fig. 4A). The tumor-bearing mice were then treated by intraperitoneal injection with αPD-L1, polyIC or the Combo (Fig. 4A). In the control group, visible tumor nodules were detected as early as 13 days following tumor cell injection (Supporting Fig. S2). After 17 days, multiple tumor nodules were observed in the livers of control and αPD-L1 groups. As expected, αPD-L1 monotherapy failed to induce any significant changes in the ratios of liver/body or spleen/body weights (Fig. 4B–D). polyIC monotherapy reduced the tumor numbers and the liver/body or spleen/body weight ratios. Strikingly, the Combo treatment displayed the most robust tumor-inhibitory effect, as demonstrated by the lack of tumor nodules on liver surface, H&E staining of liver sections, and significant changes in the ratios of liver/body and spleen/body weights (Fig. 4B–D). Tumors were also visible in the spleen of 4/5 control mice, 3/5 in the polyIC group, but no tumor was detected in the spleen of the Combo group (Fig. 4B; Supporting Fig. S3A). αPD-L1 treatment did not show any survival benefit with the median survival time at 15 days, even shorter than the control group at 19 days (Fig. 4E). Half of the mice (5/10) in the polyIC group died in 35–50 days, but only 2/10 (20%) mice in the Combo group died during the time period (Fig. 4E). Together, these results indicate no therapeutic benefit for αPD-L1 monotherapy, but its combination with polyIC effectively inhibited or even eliminated metastatic liver tumors.
Fig. 4. The dynamic changes of immune cell populations in the whole liver.
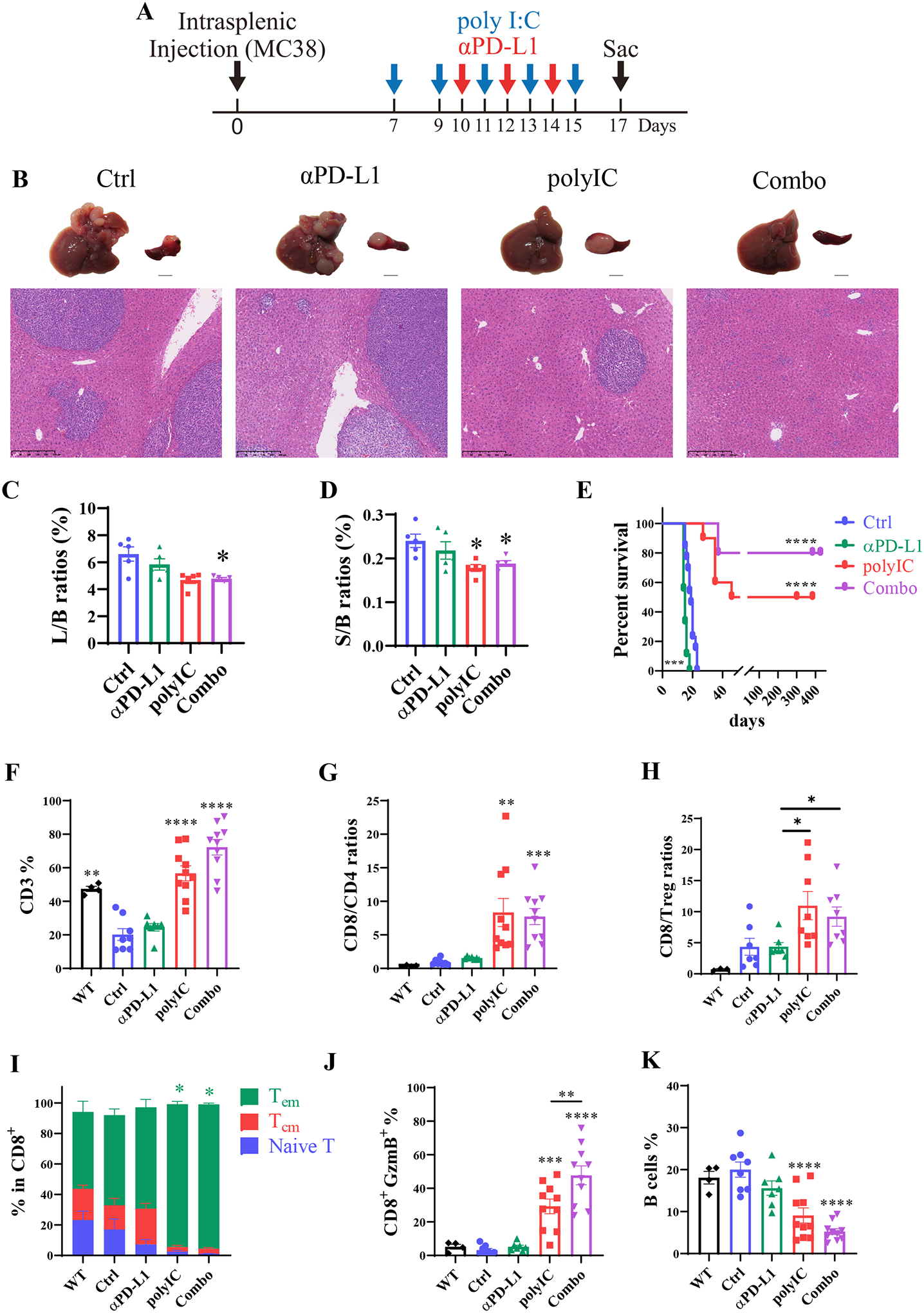
A). The scheme of the metastasized liver tumor model via intrasplenic transplantation (IST). polyIC was injected at day 7, 9, 11,13, and 15, αPD-L1 injected at day 10, 12, and 14, and the Combo group received both at these time points. Mice were sacrificed on day 17 for analyses.
B). Representative macroscopic and microscopic views (H&E staining) of the tumor phenotypes.
C-D). The liver to body (L/B) weight (C) and spleen to body (S/B) weight (D) ratios, n = 5. One-way ANOVA or Kruskal-Wallis compared with Ctrl. * p<0.05.
E). The survival curves of mice in each group. n = 9–13, Statistical analysis: Log-rank (Mantel-Cox) test.
F). The percentages of CD3 T cells in total CD45 cells. In 4F-4K, healthy livers (WT) that received no tumor cells and no treatment were also included in comparative analysis with the other four groups, same as in Fig.4A–4E.
G-H). The ratios of CD8 to CD4 (G) and CD8 to Treg cells (H) in each group, n = 3–8.
I). The sub-types of CD8 T cells as indicated. Green bar: effector memory T cell (Tem, CD62L−CD44+); red bar: central memory T cell (Tcm, CD62L+CD44+); blue bar: naïve T cells (CD62L+CD44−); Asterisks in each color represent the p values.
J). The CD8+ Granzyme B+ population in total CD45 cells.
K). The percentages of B cells, n = 4–10. Statistical analysis: One-way ANOVA or Kruskal-Wallis. * p<0.05, **p<0.01, ***p<0.005, ****p<0.001.
We examined the infiltrated immune cells in the whole liver in untreated (Ctrl), mono- or Combo-treated mice, following splenic tumor cell injection. Also included in this analysis was healthy liver of age- and gender-matched WT mice that received no tumor cell injection and no treatment, to evaluate the impact of tumor growth on hepatic immune cell infiltration. Relative to the healthy WT liver, CD3+ T cells decreased in tumor-bearing untreated (Ctrl) mice and similarly in αPD-L1-treated mice (Fig. 4F). However, the CD3+ T cell deficiency in the liver was reversed by polyIC treatment and more so in the Combo group. CD4+ T cells and Treg cells did not change significantly between the untreated (Ctrl) and the treated groups (Supporting Fig. S3B–C). However, CD8+ T cells were expanded in the polyIC group and further increased in the Combo group, with no change in Ctrl and αPD-L1 groups (Supporting Fig. S3D). Accordingly, the ratios of hepatic CD8/CD4 and CD8/Treg cells increased significantly in polyIC and Combo groups, relative to Ctrl and αPD-L1 groups (Fig. 4G–H). Furthermore, the CD8+ effector memory (Tem) cell subtype in the liver was expanded in the polyIC and Combo groups, accompanied by the decrease of Tcm and naïve T cell subtypes (Fig. 4I). More importantly, polyIC treatment induced expansion of the functional GzmB+ CD8 T cell population, which was further boosted in the Combo group (Fig. 4J). The ratios of B cells were similar in the WT, Ctrl and αPD-L1 groups, but decreased in the polyIC and Combo groups (Fig. 4K). Together, these results suggest that the combinatorial treatment with αPD-L1 and polyIC effectively suppresses metastatic liver tumor growth, by boosting infiltration and activation of GzmB+ CD8 T cells into the liver.
Liver tumor recurrence is prevented following the Combo treatment
Given that a few mice in the Combo-treated group exhibited long time tumor-free survival, we asked if the therapy could prevent tumor relapse. As the lifespan of mice with partial remission was approximately 45 days (Fig. 4E), we performed second IST of CRC cells into the tumor-free mice that survived at least 60 days after the first tumor cell injection (Fig. 5A). After 17 days of tumor cell re-injection, we did not observe visible tumor nodules in the liver (Fig. 5B; Supporting Fig. S4). The CD8/CD4 ratio was elevated in the 2nd IST group relative to the control group, similar to the 1st IST group (Fig. 5C). Both the 1st and 2nd IST groups showed higher ratios of CD8/Treg cells (Fig. 5D), and higher numbers of CD8+ Tem cells (Fig. 5E), indicating long-term residency of the CD8+ T cell sub-population in mice with complete tumor remission. Indeed, the GzmB+ cytotoxic T cells remained high in the 1st and 2nd IST groups (Fig. 5F). Within the CD8 T cell subsets, the T-bet+Eomes− and T-bet+PD1− T cell subtypes were expanded in the 2nd IST group, demonstrating persistently boosting activated CD8+ cells by the Combo treatment (Fig. 5G–H). The proliferation rate of CD8+ T cells decreased in the 2nd IST group, as evaluated by Ki67 staining (Fig. 5I). Interestingly, the ratios of CD11b− cDCs significantly increased in the 1st and 2nd IST groups, accompanied by a decrease of CD11b+ cDCs (Fig. 5J–5K). Of note, the subset of CD11b+ cDCs was considered to promote tumor metastasis by inhibiting CD8+ T cell-mediated tumor immunity in a pancreatic liver metastatic model, while CD11b− cDCs support Th1 response.(29) Therefore, the Combo treatment established a sustained immune surveillance environment in the liver, which can prevent tumor recurrence, due to persistent residency of cytotoxic CD8+ T cells.
Fig. 5. The preventive effect of liver tumor recurrence by the Combo treatment.
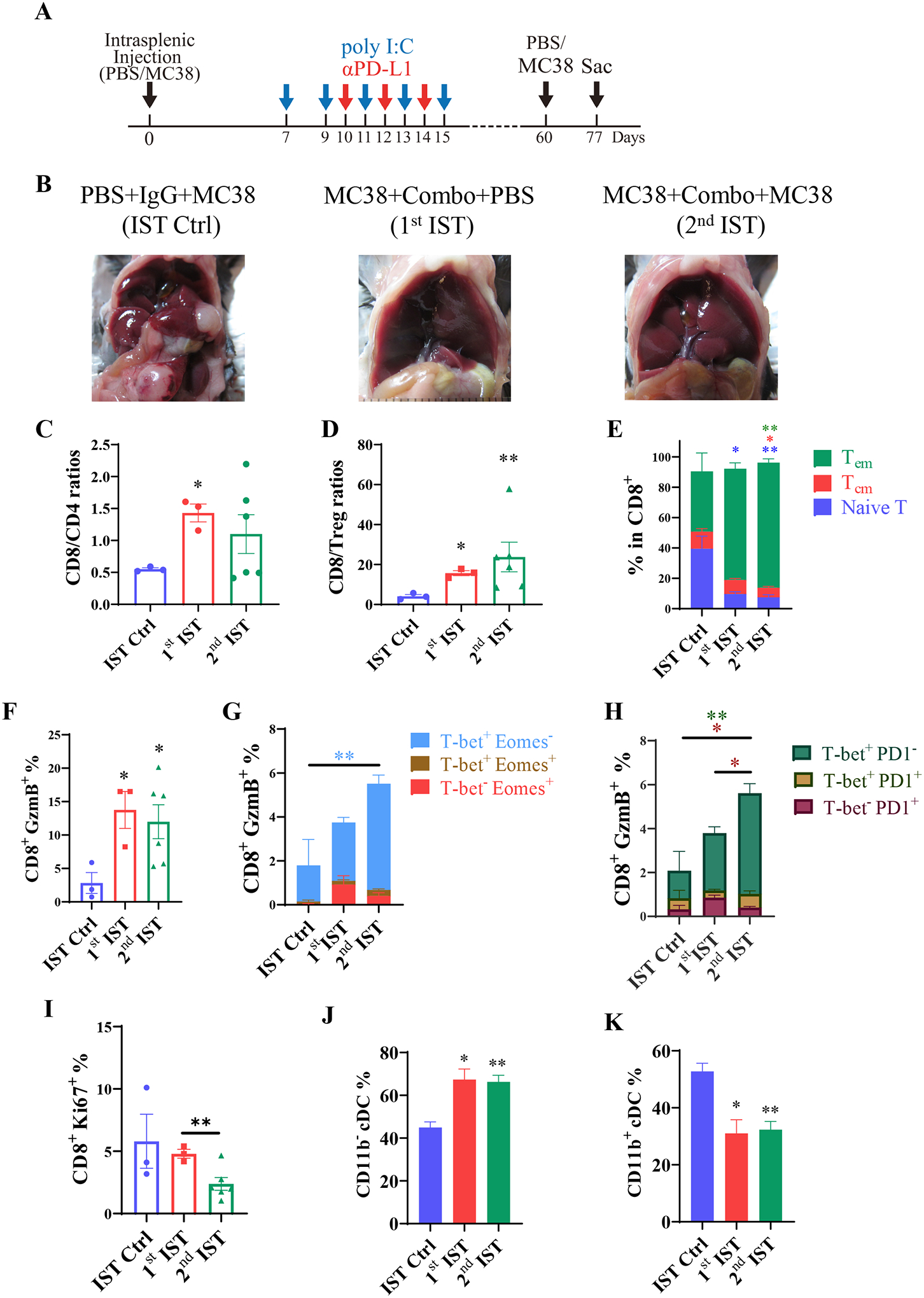
A). The scheme of the tumor recurrence model. Mice received the first intrasplenic injection of MC38 cells and were treated with polyIC and αPD-L1 as indicated. At 60 days after the 1st tumor cell inoculation (1st IST), tumor-free mice received the 2nd injection of MC38 cells (2nd IST). A control group of mice (IST Ctrl) received PBS at day 0, and were injected with tumor cells at 60 days. All mice were sacrificed at day 77 for analysis.
B). Representative macroscopic view of the livers in each group on day 77.
C-D). The ratios of CD8 to CD4 (C) and CD8 to Treg cells (D).
E). The subsets of CD8+ T cells on day 77. Green bar: effector memory T cells (Tem, CD62L−CD44+); red bar: central memory T cells (Tcm, CD62L+CD44+); blue bar: naïve T cells (CD62L+ CD44−).
F). The percentages of CD8+ GzmB+ T cells in total CD45 cells.
G). The expression of T-bet and Eomes in the CD8+ GzmB+ subset of the indicated groups. Water blue bar: effector CD8 (T-bet+ Eomes−); brown bar: intermediate state of activated CD8 cells (T-bet+Eomes+); red bar: memory CD8 cells (T-bet−Eomes+). Asterisks in colors represent the p values.
H). The expression of T-bet and PD1 in the CD8+ GzmB+ T cells. Green bar: effector CD8 (T-bet+ PD1−); yellow bar: the effector cells with exhausted feature (T-bet+PD1+); red bar: exhausted CD8 T cells (T-bet−PD1+).
I). The proliferation rates of CD8+ T cells based on Ki67 staining.
J-K). The relative percentages of CD11b− cDCs (J) and CD11b+ cDCs (K).
IST Ctrl: n = 3, 1st IST: n = 3, 2nd IST: n = 6. Statistical analysis: One-way ANOVA or Kruskal-Wallis, compared with Ctrl (asterisk only) or the line of the compared groups indicated on the top of the graph. * p<0.05, **p<0.01.
M1 macrophages promote anti-tumor effect induced by αPD-L1 and polyIC
We further examined the innate immune cell profiles in the healthy WT liver and livers of tumor-bearing mice that were untreated (Ctrl) or treated with polyIC and/or the αPD-L1, following intrasplenic tumor cell injection (Fig. 6A–B). Of note, the basal level of MDSCs was very low in the tumor-free healthy liver (Fig. 6A, black), but tumor growth induced a dramatic recruitment of MDSCs into the liver (Fig. 6A, blue versus black). The abundance of MDSCs in tumor-bearing livers was modestly downregulated by αPD-L1 or polyIC and was further suppressed by the Combo treatment. As expected, the basal level of macrophages was very high in the tumor-free healthy liver, which was lowered by tumor growth (Fig. 6A, Ctrl versus Healthy). Monotherapy with αPD-L1, but not polyIC or the Combo, upregulated the macrophage numbers in the liver (Fig. 6A). Consistent with the previous observation,(24) we detected polyIC-induced recruitment of monocyte-derived macrophages (MoMF, Ly6C+F4/80+) into the liver (Fig. 6B). The basal level of Kupffer cells (Ly6C−, F4/80+) was very high in the liver, as expected, and remained relatively high in αPD-L1-treated liver, but was robustly suppressed by the Combo treatment (Fig. 6B). The anti-tumor M1 type macrophages significantly increased in the Combo-treated liver (Fig. 6C), accompanied by a dramatic suppression of the pro-tumor M2 macrophages (Fig. 6D). The opposite changes in M1 and M2 likely generated an tumor-suppressive niche, by enhancing the activation of adaptive immune response in the Combo-treated liver. Of note, the ratio of tumor-promoting M2 macrophages was higher in the αPD-L1 than the untreated control group (Fig. 6D). Together with the decreased CD8/Treg ratio (Fig. 2D), these data suggest that the low efficacy of αPD-L1 monotherapy is due to higher infiltration of MDSCs and pro-tumor macrophages in the liver, resulting in defective activation of cytotoxic CD8 T cells in the liver.(30)
Fig. 6. The effects of macrophage depletion in the liver.
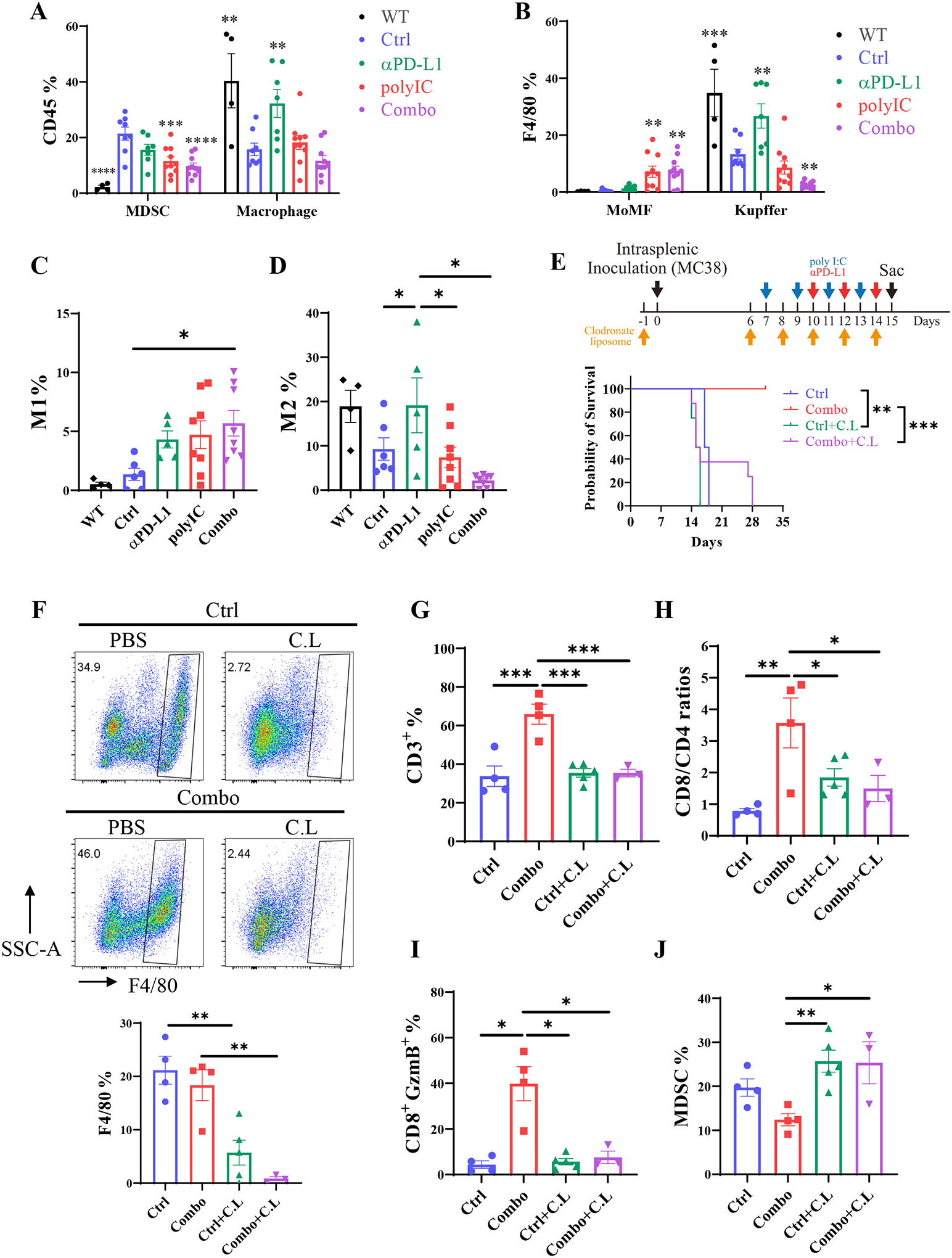
A). The percentages of MDSC and macrophages in CD45+ cells in the whole liver, analyzed as in Fig.4F–K, following intrasplenic injection of MC38 cells. WT mice received no tumor cells and no treatment. Asterisk indicates a comparison of the group with the Ctrl group.
B). The percentages of monocyte-derived macrophages (MoMF) and Kupffer cells in total CD45+ cells in the whole liver, n = 4–10. Asterisk indicates a comparison of the group with the Ctrl.
C-D). The polarization of M1 (C) and M2 (D) macrophages in total CD45 cells. M1: F4/80+CD11c+CD206−; M2: F4/80+CD11c− CD206+, n = 4–8. Asterisk indicates a comparison of the group with the Ctrl.
E). The survival curves of mice in each group. Macrophages were depleted in untreated (Ctrl) and Combo-treated mice by clodronate liposome (C.L.) injection at day −1, 6, 10, 12, and 14, before or after tumor cell inoculation. polyIC and αPD-L1 were injected as described in Fig.4A. n = 4–8.
F). The efficiency of macrophage depletion. Upper: the graphs of Ly6C and F4/80 in CD45+ CD11b+ subset. Lower: statistical analysis of the results in total CD45 cells. n = 3–5.
G). The percentages of CD3 T cells in CD45 cells, CD3: CD45+ CD3+ NK1.1−.
H). The CD8 to CD4 T cell ratios.
I). The percentages of functional GzmB+ CD8 T cell subpopulation.
J). The percentages of MDSCs in each group.
Statistical analysis: One-way ANOVA or Kruskal-Wallis, compared with Ctrl (asterisk only) or the line of the compared groups indicated on the top of the graph. * p<0.05, **p<0.01, ***p<0.005, ****p<0.001.
To define a putative role of macrophages in mediating the synergistic effect of the Combo treatment, we depleted macrophages by clodronate liposome (C.L.) injection (Fig. 6E). Removal of macrophages had no remarkable impact on tumor progression and mouse survival in the control group. However, depleting macrophages abolished the anti-tumor effect of the Combo treatment in the liver (Fig. 6E; Supporting Fig. S5A). After C.L. and the Combo treatment, 5 mice died around 14–16 days, and the other 3 mice died between 27–29 days after the last C.L. injection at day 16 (Fig. 6F). Injection of C.L effectively depleted macrophages, but was not effective in depleting monocytes, which can generate monocyte-derived macrophages (MoMF). In mice bearing small or no tumors, we found the population of macrophages was not completely depleted by C.L treatment (Fig. 6G and Supporting Fig. S5B, Combo+C.L-2). The expansion and activation of CD3, CD8/CD4 ratios, and CD8+GzmB+ cytotoxic CD8+ T cells were significantly suppressed following depletion of macrophages (Fig. 6H–J). In contrast, the ratio of MDSCs, which was suppressed by the Combo treatment, was elevated significantly in clodronate-treated livers (Fig. 6K). Together, these results suggest a critical role of macrophages in mediating communications between immune cell types, especially in activation of cytotoxic CD8 T cells, to achieve durable anti-tumor effect of polyIC and αPD-L1 in this liver tumor metastasis model.
Interferon signaling is required for the tumor-inhibitory effect of polyIC and αPD-L1
The synthetic dsRNA polyIC is known to induce a milieu of cytokines, in particular interferons (IFNs). We took two different approaches to determine a putative role of type I IFNs in mediating the polyIC effect in sensitizing liver response to αPD-L1 therapy (Fig. 7A). First, we injected antibodies against type I IFN receptor (αIFNαR1) and examined tumor progression and mouse survival (Fig. 7A, Supporting Fig. S6A–E). Blocking the basal level of interferon signaling had no effect on either tumor progression or lifespan of the tumor-bearing mice, as compared to the controls (Supporting Fig. 6B–C). However, injecting the αIFNαR1 antibody almost abolished the anti-tumor effect of the Combo therapy (Fig. 7B–D). All mice in the αIFNαR1+Combo group died around day 20, similar to the control group, while the mice in the Combo group lived much longer (Fig. 7E). We further examined the efficacy of the Combo treatment in type I interferon receptor knockout (Ifnar1−) mice (Fig. 7F). Similar to αIFNαR1 antibody, genetic ablation of the IFN receptor removed the therapeutic benefit of polyIC+αPD-L1 Combo (Fig. 7G). These data indicate an essential role of interferon signaling in priming the liver to fight against metastatic liver tumors by αPD-L1 blockade.
Fig. 7. Disruption of type I interferon signaling abrogates the synergistic effect of polyIC and αPD-L1 in the liver.
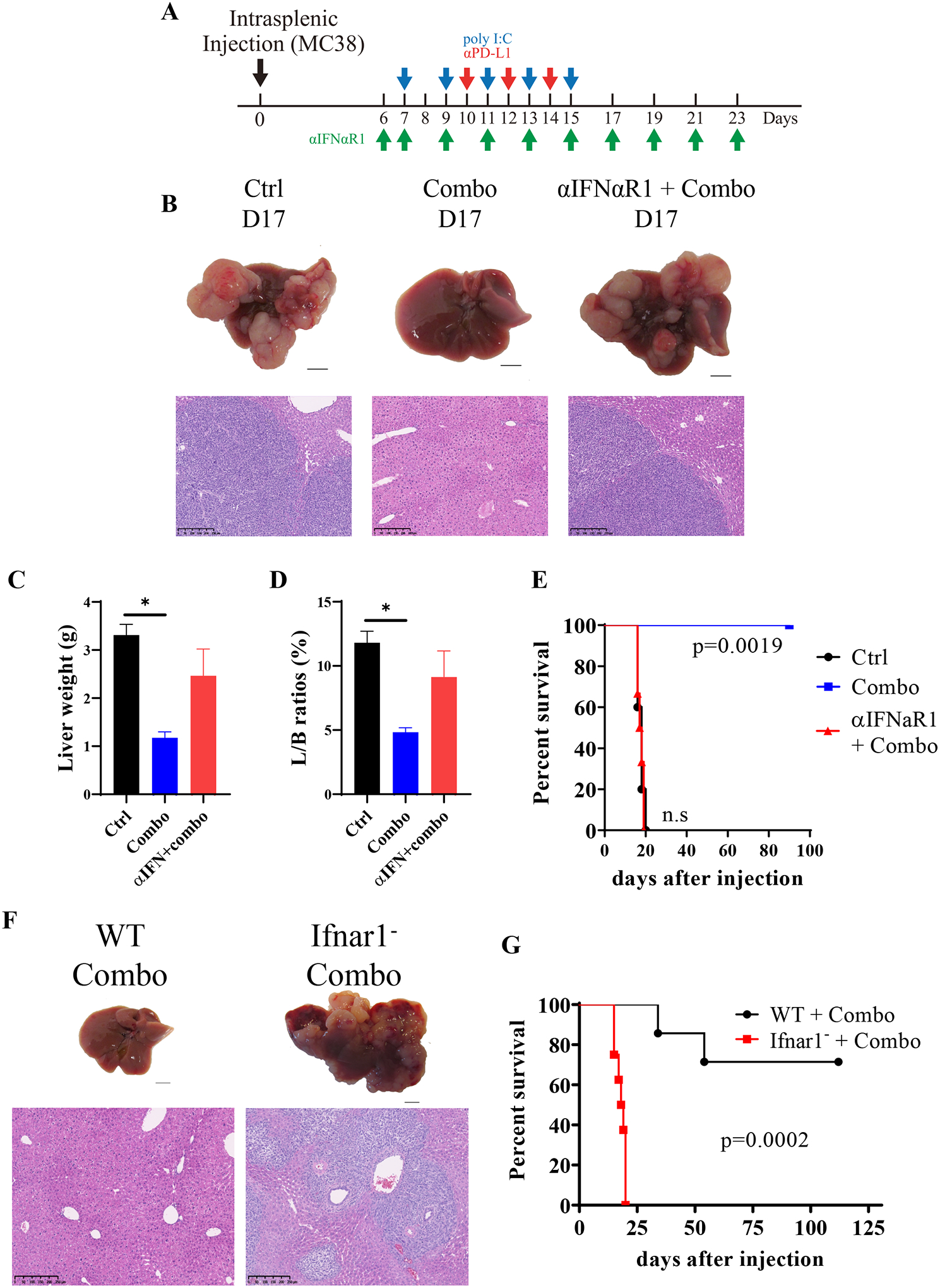
A). The scheme of Combo treatment with or without antibody to type I interferon receptor (αIFNαR1).
B). Representative macroscopic view and H&E staining of liver sections with samples collected on day 17.
C-D). The liver weights (C) and liver to body (L/B) weight ratios (D), n = 5. Statistical analysis: unpaired t-test with Welch’s correction or Mann-Whitney test, compared with Ctrl.
E). The survival curves of mice, n = 5–6. Log-Rank (Mantel-Cox) test compared with Ctrl.
F). The macroscopic view of livers and H&E staining of liver sections. WT: the C57BL/6J mice; Ifnar1−: type I interferon receptor knockout mice.
G). The survival curves of mice, n = 7–8. Log-Rank (Mantel-Cox) test.
DISCUSSION
In this study, we have deciphered a liver-specific environmental impact on αPD-L1 immunotherapy, by comparing the responses of identical tumors growing under the skin or in the liver. Of note, the pioneering work of cancer immunotherapy was initially performed in mice with inoculated subcutaneous tumors,(3, 4) which remains to be a most widely used animal tumor model for oncological treatment. Consistent with a body of literature, we demonstrated a robust inhibition of αPD-L1 blockade on subcutaneous tumor progression. However, this antibody injection had no impact on the same tumor metastasized into the liver, following portal vein or intrasplenic tumor cell injection. Our previous experiments also showed that αPD-L1 alone had minimal efficacy on primary liver tumors in mice induced by oncogenes that are frequently detected in HCC patients.(23) Together, these results unveil a unique negative impact of the hepatic environment on PD-1/PD-L1 blockade, independent of tumor origins or types.
Our comparative analysis of the tumor-infiltrated immune cells revealed several important aspects of mechanisms underlying the contrasting therapeutic effects between the two sites. αPD-L1 significantly upregulated the ratios of CD8/CD4 and CD8/Treg cells, and increased the numbers of GzmB+ cytotoxic CD8 T cells in subcutaneous tumors, leading to remarkable tumor regression. However, these anti-tumor immune responses were not boosted in αPD-L1-treated liver tumors. By examining the immune cell populations in the whole liver, we also found that αPD-L1 treatment failed to augment the CD3 T cell numbers and the CD8/CD4 or CD8/Treg cell ratios. MDSCs were reported to suppress cytotoxic T cell activity by inducing Treg cells;(31) αPD-L1 blockade was reported to restrain MDSC infiltration into subcutaneous tumors,(32) and to downregulate MDSC functions, leading to enhanced T cell activation in subcutaneous tumor models.(33) However, we detected relatively low numbers of MDSCs in subcutaneous tumors in the untreated control group, which was not significantly influenced by αPD-L1 blockade. In contrast, much higher numbers of MDSCs were infiltrated into liver tumors (Fig. 3), and the tumor growth also recruited more MDSCs in the whole liver, relative to the WT mouse (Fig. 6). αPD-L1 alone did not significantly reduce MDSCs in either liver tumor or the whole liver of tumor-bearing mice. In addition, αPD-L1 monotherapy failed to change the M1/M2 macrophage ratios, as both M1 and M2 macrophages increased in the liver. Therefore, despite its significant inhibition of subcutaneous tumors, αPD-L1 monotherapy failed to inhibit the same tumor growth in the liver, because of a failure in boosting the tumor-suppressive innate and adaptive immune functions.
Strikingly, the combination of αPD-L1 with polyIC showed synergistic effect in suppressing the metastasized liver tumor progression, which may be contributed by several mechanisms. First, the Combo therapy significantly reduced infiltration of MDSCs in the liver tumor and the whole liver, resulting in a drastic upregulation of CD8/MDSC ratios. Second, M1 macrophages increased, accompanied by decreased M2 macrophages, leading to an elevated M1/M2 ratio in the Combo group. Third, CD3 cell numbers increased with much higher ratios of CD8/CD4 and CD8/Treg cells. Fourth, the Combo regimen drastically enhanced GzmB+ CD8 T cells in the liver tumor and the whole liver. Therefore, the high efficacy of the Combo therapy is due to orchestrated and optimized activation of multiple innate and adaptive immune cells in the liver tumor microenvironment, as compared to subcutaneous tumor. A strong support to this notion was rendered by the data showing that depleting macrophages or blocking type I IFN signaling abrogated polyIC-mediated synergistic effect with αPD-L1.
Although αPD-L1 alone had a significant inhibitory effect in the subcutaneous tumor model, the Combo treatment showed only an additive effect, causing only more tumor regression but no tumor elimination. In this regard, it is of great interest that liver tumors were even eradicated in half of the mice by the Combo therapy, despite the inefficiency of αPD-L1 monotherapy. Based on these results, we argue here that as long as the hepatic immunotolerant mechanism is disrupted by another assisting regimen, PD-1/PD-L1 blockade can even be more beneficial for liver cancer patients that are otherwise poorly responsive to the ICIs. Furthermore, the fully cured tumor-free mice effectively resisted a second tumor cell challenge, indicating a preventive effect of tumor relapse. We believe this is due to upregulation of a long-lived subset of T-bet+Eomes−PD1− effector CD8+ T cells in the Combo-treated liver. Further elucidation of the underlying molecular and cellular mechanisms is warranted.
Due to the disappointedly low response rate of HCC patients and liver-metastatic CRCs to monotherapy with various ICIs, many different combinations are being tested in clinical trials without clear rationale. Further in-depth mechanistic analyses of the dynamic communications of both primary and metastasized liver tumors with the hepatic microenvironment are necessary to guide design of more efficacious combinatorial therapy.(9) In this regard, it is extremely important to explore new therapeutic strategies for liver cancer using proper animal tumor models. Despite being most widely used, the mouse subcutaneous tumor model has a limited value for mechanistic and translational studies of liver cancer immunotherapy, as indicated by data presented here. Furthermore, our results also suggest that the poor response of liver cancer to ICIs is not due to low immunogenicity of liver tumors or low expression of PD-L1 on tumor cells. Rather, it is due to the unique immunotolerant microenvironment in the liver, which can be remedied by concerted activation of innate and adaptive immune functions.
In conclusion, this study, together with the previous data,(23) demonstrates a principle and feasibility of a combinatorial immunotherapy to treat and even eradicate primary and metastasized liver tumors.
Supplementary Material
A). The total CD45+ cells per gram of tumor tissue in subcutaneous tumors (Sub T) or liver tumors (Liv T).
B-C). The percentages of CD4 T cells (B) and CD8 T cells (C) in total CD45+ cells in subcutaneous or liver tumors. Each group was indicated at the bottom of the bars.
D). The absolute number of Treg cells in each gram of tumor tissues. Left Y-axis signifies Sub T; right Y-axis signifies Liv T.
E-F). The percentages of B cells (E), and NK cells (F) in total CD45+ cells.
Samples of Sub T and Liv T tissues were collected from 3 individual experiments. Statistical analysis: One-way ANOVA or Kruskal-Wallis. * p<0.05, **p<0.01, ***p<0.001
The livers and spleens were pictured at day 11, 13, and 15 after intrasplenic tumor cell transplantation. Black arrows indicate the spleen tumor, and white arrows show the liver tumor. Scale bar: 5 mm.
A). The gross views of the liver and spleen at day 17.
B-D). The percentages of CD4 (B) and Treg (C) and CD8 T cells (D) in the whole live at day 17. Groups were indicated at the bottom of the bar, n = 4–10. Statistical analysis: One-way ANOVA or Kruskal-Wallis. ****p<0.001.
The mice were injected with luciferase-labeled MC38 tumor cells at day 60 after the first inoculation. The IVIS spectrum was taken at 1 hour, 1, 3, and 10 days after the second tumor cell injection. The 2nd IST: mice first received MC38 cells at day 0, and were treated with PolyIC on day 7, 9, 11, 13, and 15, and αPD-L1 on day 10, 12, and 14. MC38-Luc cells were injected 60 days later. IST Ctrl: mice were first injected with PBS at day 0, and MC38-Luc cells were injected 60 days later. Total Flux = the radiance (photons/sec). n = 4 for each group.
A). The gross photos of the mice at day 17. The mice injected with tumor cells were treated with PBS or the Combo plus PBS liposome or clodronate liposome (C.L.). Each group presents two individuals.
B). The flow cytometry of each group. Combo+C.L-1 represented Combo-treated mice with successful macrophage depletion. No monocyte-derived macrophages (MoMF, Ly6C+ F4/80+) or Kupffer cells (Ly6C− F4/80+) were detected. The gross photo of the liver with tumors showed below. Combo+C.L-2 represented a failed macrophage depletion. The MoMFs were not depleted with the liver photo shown below.
A). The experimental scheme.
B). The gross and H&E staining photos of livers in mice without or with treatment of anti-IFNα receptor (αIFNαR1) antibody.
C). The survival curves of the mice. n = 5 in each group. Kaplan-Meier, Log-Rank (Mantel-Cox) test.
D-E). The liver weights (D) and liver to body weight ratios (E) in each group. Statistical analysis: unpaired t-test. NS: not significant.
The scheme of gating strategy in FACS analysis of data presented in Figures 2, 4, 5, and 6 for lymphocytes (A), and Figures 3, 5, and 6 for myeloid cells (B).
Acknowledgements
We thank our lab members for helpful discussion.
Financial Support:
This work was supported by NIH grants (R01CA236074 and R01CA239629, P01AG073084) to G.S.F., and R37CA239072 to E.H.
List of Abbreviations:
- polyIC
polyinosinic:polycytidylic acid
- MDSC
myeloid-derived suppressor cells
- HCC
hepatocellular carcinoma
- CRC
colorectal cancer
- GzmB
granzyme B
- Tcm
central memory T cell
- Tem
effector memory T cell
- MoMF
monocyte-derived macrophages
- IST
intrasplenic transplantation
- cDCs
classical dendritic cells
Footnotes
Conflicts of interest
The authors declare no conflict of interest.
References
- 1.Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med 2018;50:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 2015;125:3384–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–1736. [DOI] [PubMed] [Google Scholar]
- 4.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002;99:12293–12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robert C, Ribas A, Hamid O, Daud A, Wolchok JD, Joshua AM, Hwu WJ, et al. Durable Complete Response After Discontinuation of Pembrolizumab in Patients With Metastatic Melanoma. J Clin Oncol 2018;36:1668–1674. [DOI] [PubMed] [Google Scholar]
- 7.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015;161:205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2021;7:6. [DOI] [PubMed] [Google Scholar]
- 9.Feng GS, Hanley KL, Liang Y, Lin X. Improving the Efficacy of Liver Cancer Immunotherapy: The Power of Combined Preclinical and Clinical Studies. Hepatology 2021;73 Suppl 1:104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, Verslype C, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol 2018;19:940–952. [DOI] [PubMed] [Google Scholar]
- 11.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yau T, Kang YK, Kim TY, El-Khoueiry AB, Santoro A, Sangro B, Melero I, et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol 2020;6:e204564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, Kudo M, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med 2020;382:1894–1905. [DOI] [PubMed] [Google Scholar]
- 14.Doherty DG. Immunity, tolerance and autoimmunity in the liver: A comprehensive review. J Autoimmun 2016;66:60–75. [DOI] [PubMed] [Google Scholar]
- 15.Kubes P, Jenne C. Immune Responses in the Liver. Annu Rev Immunol 2018;36:247–277. [DOI] [PubMed] [Google Scholar]
- 16.Wong YC, McCaughan GW, Bowen DG, Bertolino P. The CD8 T-cell response during tolerance induction in liver transplantation. Clin Transl Immunology 2016;5:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsilimigras DI, Brodt P, Clavien PA, Muschel RJ, D’Angelica MI, Endo I, Parks RW, et al. Liver metastases. Nat Rev Dis Primers 2021;7:27. [DOI] [PubMed] [Google Scholar]
- 18.Manfredi S, Lepage C, Hatem C, Coatmeur O, Faivre J, Bouvier AM. Epidemiology and management of liver metastases from colorectal cancer. Ann Surg 2006;244:254–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hackl C, Neumann P, Gerken M, Loss M, Klinkhammer-Schalke M, Schlitt HJ. Treatment of colorectal liver metastases in Germany: a ten-year population-based analysis of 5772 cases of primary colorectal adenocarcinoma. BMC Cancer 2014;14:810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nordlinger B, Sorbye H, Glimelius B, Poston GJ, Schlag PM, Rougier P, Bechstein WO, et al. Perioperative FOLFOX4 chemotherapy and surgery versus surgery alone for resectable liver metastases from colorectal cancer (EORTC 40983): long-term results of a randomised, controlled, phase 3 trial. Lancet Oncol 2013;14:1208–1215. [DOI] [PubMed] [Google Scholar]
- 21.Lumish MA, Cercek A. Immunotherapy for the treatment of colorectal cancer. J Surg Oncol 2021;123:760–774. [DOI] [PubMed] [Google Scholar]
- 22.Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, Diaz LA Jr. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol 2019;16:361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wen L, Xin B, Wu P, Lin CH, Peng C, Wang G, Lee J, et al. An Efficient Combination Immunotherapy for Primary Liver Cancer by Harmonized Activation of Innate and Adaptive Immunity in Mice. Hepatology 2019;69:2518–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J, Liao R, Wang G, Yang BH, Luo X, Varki NM, Qiu SJ, et al. Preventive Inhibition of Liver Tumorigenesis by Systemic Activation of Innate Immune Functions. Cell Rep 2017;21:1870–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly(I:C). Adv Drug Deliv Rev 2008;60:805–812. [DOI] [PubMed] [Google Scholar]
- 26.Heijstek MW, Kranenburg O, Borel Rinkes IH. Mouse models of colorectal cancer and liver metastases. Dig Surg 2005;22:16–25. [DOI] [PubMed] [Google Scholar]
- 27.Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, Madore J, et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019;35:238–255 e236. [DOI] [PubMed] [Google Scholar]
- 28.Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol 2008;83:1136–1144. [DOI] [PubMed] [Google Scholar]
- 29.Kenkel JA, Tseng WW, Davidson MG, Tolentino LL, Choi O, Bhattacharya N, Seeley ES, et al. An Immunosuppressive Dendritic Cell Subset Accumulates at Secondary Sites and Promotes Metastasis in Pancreatic Cancer. Cancer Res 2017;77:4158–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol 2012;22:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, Greten TF, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008;135:234–243. [DOI] [PubMed] [Google Scholar]
- 32.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 2014;124:687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 2014;211:781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A). The total CD45+ cells per gram of tumor tissue in subcutaneous tumors (Sub T) or liver tumors (Liv T).
B-C). The percentages of CD4 T cells (B) and CD8 T cells (C) in total CD45+ cells in subcutaneous or liver tumors. Each group was indicated at the bottom of the bars.
D). The absolute number of Treg cells in each gram of tumor tissues. Left Y-axis signifies Sub T; right Y-axis signifies Liv T.
E-F). The percentages of B cells (E), and NK cells (F) in total CD45+ cells.
Samples of Sub T and Liv T tissues were collected from 3 individual experiments. Statistical analysis: One-way ANOVA or Kruskal-Wallis. * p<0.05, **p<0.01, ***p<0.001
The livers and spleens were pictured at day 11, 13, and 15 after intrasplenic tumor cell transplantation. Black arrows indicate the spleen tumor, and white arrows show the liver tumor. Scale bar: 5 mm.
A). The gross views of the liver and spleen at day 17.
B-D). The percentages of CD4 (B) and Treg (C) and CD8 T cells (D) in the whole live at day 17. Groups were indicated at the bottom of the bar, n = 4–10. Statistical analysis: One-way ANOVA or Kruskal-Wallis. ****p<0.001.
The mice were injected with luciferase-labeled MC38 tumor cells at day 60 after the first inoculation. The IVIS spectrum was taken at 1 hour, 1, 3, and 10 days after the second tumor cell injection. The 2nd IST: mice first received MC38 cells at day 0, and were treated with PolyIC on day 7, 9, 11, 13, and 15, and αPD-L1 on day 10, 12, and 14. MC38-Luc cells were injected 60 days later. IST Ctrl: mice were first injected with PBS at day 0, and MC38-Luc cells were injected 60 days later. Total Flux = the radiance (photons/sec). n = 4 for each group.
A). The gross photos of the mice at day 17. The mice injected with tumor cells were treated with PBS or the Combo plus PBS liposome or clodronate liposome (C.L.). Each group presents two individuals.
B). The flow cytometry of each group. Combo+C.L-1 represented Combo-treated mice with successful macrophage depletion. No monocyte-derived macrophages (MoMF, Ly6C+ F4/80+) or Kupffer cells (Ly6C− F4/80+) were detected. The gross photo of the liver with tumors showed below. Combo+C.L-2 represented a failed macrophage depletion. The MoMFs were not depleted with the liver photo shown below.
A). The experimental scheme.
B). The gross and H&E staining photos of livers in mice without or with treatment of anti-IFNα receptor (αIFNαR1) antibody.
C). The survival curves of the mice. n = 5 in each group. Kaplan-Meier, Log-Rank (Mantel-Cox) test.
D-E). The liver weights (D) and liver to body weight ratios (E) in each group. Statistical analysis: unpaired t-test. NS: not significant.
The scheme of gating strategy in FACS analysis of data presented in Figures 2, 4, 5, and 6 for lymphocytes (A), and Figures 3, 5, and 6 for myeloid cells (B).