

Abstract
The genome-wide association study (GWAS)-identified asthma susceptibility risk alleles on chromosome 17q21 increase the expression of ORMDL3 (ORMDL sphingolipid biosynthesis regulator 3) in lung tissue. Given the importance of epithelial integrity in asthma, we hypothesized that ORMDL3 directly impacted bronchial epithelial function. To determine whether and how ORMDL3 expression impacts the bronchial epithelium, in studies using both primary human bronchial epithelial cells and human bronchial epithelial cell line, 16HBE (16HBE14o-), we assessed the impact of ORMDL3 on autophagy. Studies included: autophagosome detection by electron microscopy, RFP-GFP-LC3B to assess autophagic activity, and Western blot analysis of autophagy-related proteins. Mechanistic assessments included immunoprecipitation assays, intracellular calcium mobilization assessments, and cell viability assays. Coexpression of ORMDL3 and autophagy-related genes was measured in primary human bronchial epithelial cells derived from 44 subjects. Overexpressing ORMDL3 demonstrated increased numbers of autophagosomes and increased levels of autophagy-related proteins LC3B, ATG3, ATG7, and ATG16L1. ORMDL3 overexpression promotes autophagy and subsequent cell death by impairing intracellular calcium mobilization through interacting with SERCA2. Strong correlation was observed between expression of ORMDL3 and autophagy-related genes in patient-derived bronchial epithelial cells. Increased ORMDL3 expression induces autophagy, possibly through interacting with SERCA2, thereby inhibiting intracellular calcium influx, and induces cell death, impairing bronchial epithelial function in asthma.
Keywords: autophagy, calcium mobilization, asthma, human bronchial epithelium, cell death
Clinical Relevance
A segment of chromosome 17q21 that includes ORMDL3 (ORMDL sphingolipid biosynthesis regulator 3) is the genetic region most consistently replicated in genome-wide association studies of childhood asthma. We demonstrate that increased expression of ORMDL3 promotes autophagy in human bronchial epithelial cells by inhibiting the intracellular calcium pump SERCA. This work provides a new mechanism through which the airway epithelium may be predisposed to cellular damage, representing a novel unappreciated mechanism by which 17q21 genetic variants confer asthma susceptibility.
Genome-wide association studies (GWASs) have revealed a common haplotype on chromosome 17q21 to be the most widely reproducible asthma-susceptibility locus identified to date (1, 2). Common risk alleles that are in complete linkage disequilibrium with each other increase asthma susceptibility by 21–56% in all major world populations, regardless of ancestry (3, 4). The alleles reside on a preserved haplotype, and it has been repeatedly demonstrated that the asthma risk haplotype regulates the expression of a cluster of genes on 17q21 in a strand-specific manner by expression quantitative trait locus mapping (1, 5) and allelic imbalance assays (6).
The two 17q21 genes under the strongest genetic influence of the asthma-susceptibility regulatory haplotype are GSDMB (Gasdermin B) and the ORMDL3 (ORMDL sphingolipid biosynthesis regulator 3), with ∼20% of the population variances of both genes’ expression attributable to this locus. This effect is observed broadly across all tissues where the two genes are naturally coexpressed, most consistently in asthma relevant tissues and cell types, including CD4+ lymphocytes (5, 7) and the lung (8, 9). The asthma risk alleles increase the expression of both genes, implying that increased expression of ORMDL3 and/or GSDMB confers asthma risk. Guided by these observations, Miller and colleagues demonstrated that ubiquitous overexpression of ORMDL3 in a transgenic mouse model leads to asthma-like histopathologic and physiologic changes in the lung, including features of airway smooth muscle and glandular hyperplasia, increased airway hyperresponsiveness, and evidence of early airway remodeling (10). These findings were observed even in the absence of allergen sensitization or challenge, and before any inflammatory cell infiltration, highlighting the importance of local pulmonary ORMDL3 expression in asthma pathogenesis. Furthermore, Ormdl3-null mice are protected from developing fungal-induced allergic airway disease; this protection is lost by restoration of Ormdl3 expression in the bronchial epithelium (BE) alone (11, 12).
How ORMDL3 overexpression confers asthma risk in airway epithelial cells is not fully understood. As recently reviewed (13), ORMDL3 is implicated in multiple processes, including sphingolipid metabolism (14), the endoplasmic reticulum unfolded protein response (UPR) (15), and concentration of intracellular calcium [Ca2+]i homeostasis (15). Alluded to by others (11, 12), one of the most reproducible findings from our initial studies of ORMDL3 function was the observation that ORMDL3 induces autophagy when overexpressed in BE (reported herein). Because the autophagy pathway has recently been implicated in multiple pulmonary diseases, including chronic obstructive pulmonary disease, acute lung injury, and asthma (16–19), we set out to determine whether and how ORMDL3 regulates autophagy in human BE. Herein we provide evidence, for the first time, that ORMDL3 promotes autophagy in human BE, and this in turn leads to increased cell death. We further demonstrate that this process is mediated through the physical interaction of ORMDL3 with SERCA2 and subsequent disruption of intracellular calcium mobilization.
Some of the results of these studies have been previously reported in the form of abstracts (20, 21).
Methods
Quantitative Assessment of Autophagy
Human bronchial epithelial cell line, 16HBE (16HBE14o-) was seeded on four-chamber culture slides (Falcon, 354114) at a density of 20,000 cells per chamber; 6 μl Autophagy Sensor (Premo Autophagy Tandem Sensor RFP (red fluorescent protein)-GFP-LC3B Kit, Thermo Fisher Scientific, P36239) was added to each chamber. After 30 hours, cells were treated with or without 20 μM chloroquine for an additional 16 hours. Cell images were obtained using an Olympus FV-1000 Confocal Microscope (Harvard Medical School Neurobiology Imaging Facility). The number of cells positive for both sensor transduction and autophagy was counted blinded to experimental conditions. Cells containing more than five puncta or with puncta accumulations were considered positive for autophagy.
Calcium Flux Assessment
16HBE cells were seeded on confocal dishes and loaded with calcium-sensitive Fluo-4 AM dye (Invitrogen, F14201), Pluronic F-127 (Invitrogen, P36400), and probenecid (Invitrogen, P3000MP) for 45 minutes of incubation before measurement. [Ca2+]i was measured with a temperature- and humidity-controlled Andor Revolution Spinning Disk Microscope (Harvard Medical School Neurobiology Imaging Facility) to maintain physiological conditions (5% CO2, 37°C, and humidity). Images were captured every 3 seconds and digitized using MetaMorph Imaging Software (Molecular Devices). Fluorescence intensities were analyzed using ImageJ.
Gene Expression Correlation Analysis
Genome-wide gene expression data were generated by microarray in BE brushing samples from 44 subjects participating in the Asthma BRIDGE (Asthma BioRepository for Integrative Genomic Exploration) study (see Table E1 in the online supplement), as previously described (7). Expression profiles were derived using the Illumina HT-12 v4 Expression or HumanRef8 v2 BeadChip platforms (Illumina, Inc.) (22). Autophagy-related genes selected for the analysis included ATG7, ATG12, and ATG16L1. Correlation between these genes and ORMDL3 gene expression was performed using linear regression models, as implemented with the R package “limma,” after adjusting for age, sex, race, and processing batch. Differential expression analyses were adjusted for age, sex, race, and the first two principal components of gene expression.
Additional methods are described in the online supplement, including ORMDL3 overexpression, CRISPR-associated protein 9 (Cas9) ORMDL3 gene knockout (KO), lentiviral-based ORMDL3 stable overexpression, transmission electron microscopy sample preparation, Western blotting, immunoprecipitation, immunofluorescence staining, lactate dehydrogenase (LDH) measurement, cell proliferation rate measurement, and statistical methods.
Results
ORMDL3 Promotes Autophagy in Human Bronchial Epithelial Cells
We first examined the subcellular morphologic changes after overexpression of ORMDL3 in 16HBE cells by electron microscopy. Compared with vector-transfected controls, cells transiently overexpressing ORMDL3 demonstrated an increased number of double-membrane structures resembling autophagosomes (Figures 1A and E1A). Similar structures were also identified in 16HBE cells with stable ORMDL3 overexpression (Figures 1B and E1B). Consistently, ORMDL3 overexpression resulted in increased levels of autophagy-related proteins ATG3, ATG5, ATG7, ATG16L1, and Beclin-1, as well as the autophagosome protein LC3B in both primary normal human bronchial epithelial (NHBE) cells (Figures 1C, 1D, and E1C) and 16HBE cells (Figure E1D), suggesting activation of the autophagy pathway on overexpression of ORMDL3. We next measured autophagy using a tandem RFP-GFP-LC3B lentiviral sensor (23), where LC3B-positive autophagosomes were indicated by coexpression of GFP and RFP (in yellow), whereas autolysosomes were indicated by the expression of RFP (in red) only (Figure 1E). Compared with control cells, 16HBE cells overexpressing ORMDL3 exhibited a greater percentage of autophagy-positive cells (P < 0.05) (Figure 1F).
Figure 1.
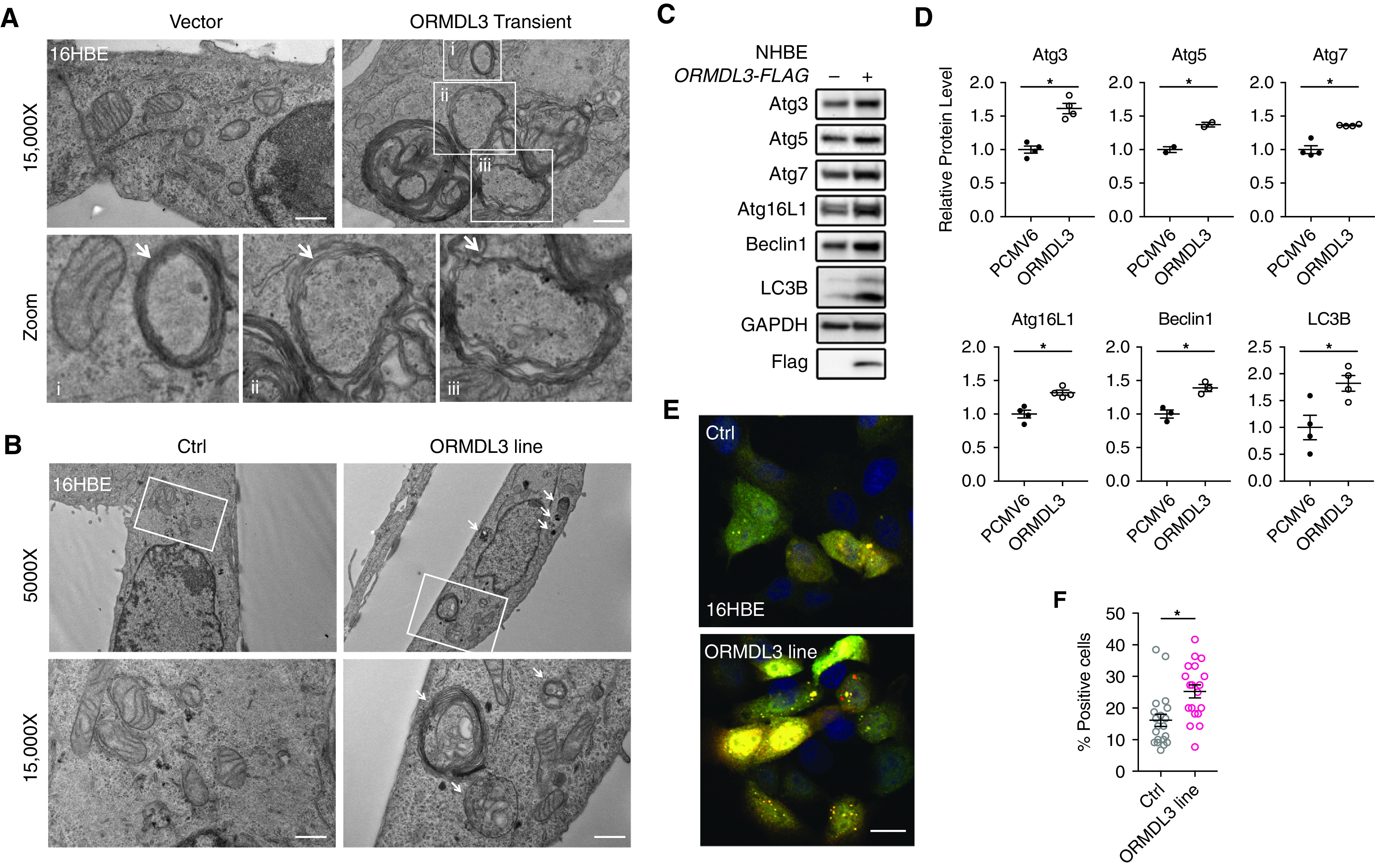
ORMDL3 (ORMDL sphingolipid biosynthesis regulator 3) overexpression promotes autophagy in primary normal human bronchial epithelial (NHBE) and human bronchial epithelial cell line, 16HBE (16HBE14o-) cells. (A and B) Representative electron microscopy (EM) images from two biological replicates show increased number of autophagosomes (white arrows) in 16HBE cells transiently (A) or stably (B) transfected with ORMDL3. Scale bars, 500 nm. (i–iii) Zoom images from B ORMDL3 overexpression group. (C) Blots and protein abundance of autophagy markers measured by Western blot in primary NHBE cells transfected with either empty vector or Flag-tagged human ORMDL3. (D) Western blots were quantified by ImageJ. Means ± SEM are from two to four biological replicates. *P < 0.05, unpaired t test. (E) Confocal images and (F) quantification of autophagosomes by RFP (red fluorescent protein)-GFP-LC3B tandem sensor in 16HBE cells. Yellow and red puncta denote LC3B-positive autophagosomes and autolysosomes, respectively. Scale bar, 20 μm. Means ± SEM shown from 20 image views of control (Ctrl) and 10 image views from each of the two overexpression lines. Cells with five or more puncta or with puncta accumulations are defined as positive. *P < 0.05, unpaired t test. ATG = autophagy related; ATG16L1 = autophagy related 16 like 1.
Decreased ORMDL3 Inhibits Autophagy in Human Bronchial Epithelial Cells
Complementing our gain-of-function studies, we next sought to determine whether inhibition of ORMDL3 constrains epithelial cell autophagy. First, siRNA-mediated silencing of ORMDL3 (Figures E2A and E2B) resulted in decreased levels of the autophagy-related proteins ATG3, ATG5, ATG7, and ATG16L1, as well as reduced levels of the autophagosome marker LC3B, in both 16HBE cells (Figure E2C) and primary NHBE cells (Figures 2A and 2B). Furthermore, compared with wild-type (WT) control cells, CRISPR-Cas9-generated ORMDL3 16HBE KO cells infected with the RFP-GFP-LC3B viral sensor showed a reduction of autophagy-positive cells at basal levels (P < 0.05) (Figures 2C, 2D, and E2D) and had fewer autophagosome aggregates (Figures 2C and 2E) with chloroquine treatment, a potent inhibitor of autophagosome–lysosome fusion, than condition-matched WT cells. Taken together, ORMDL3 promotes autophagy in human BE cells.
Figure 2.
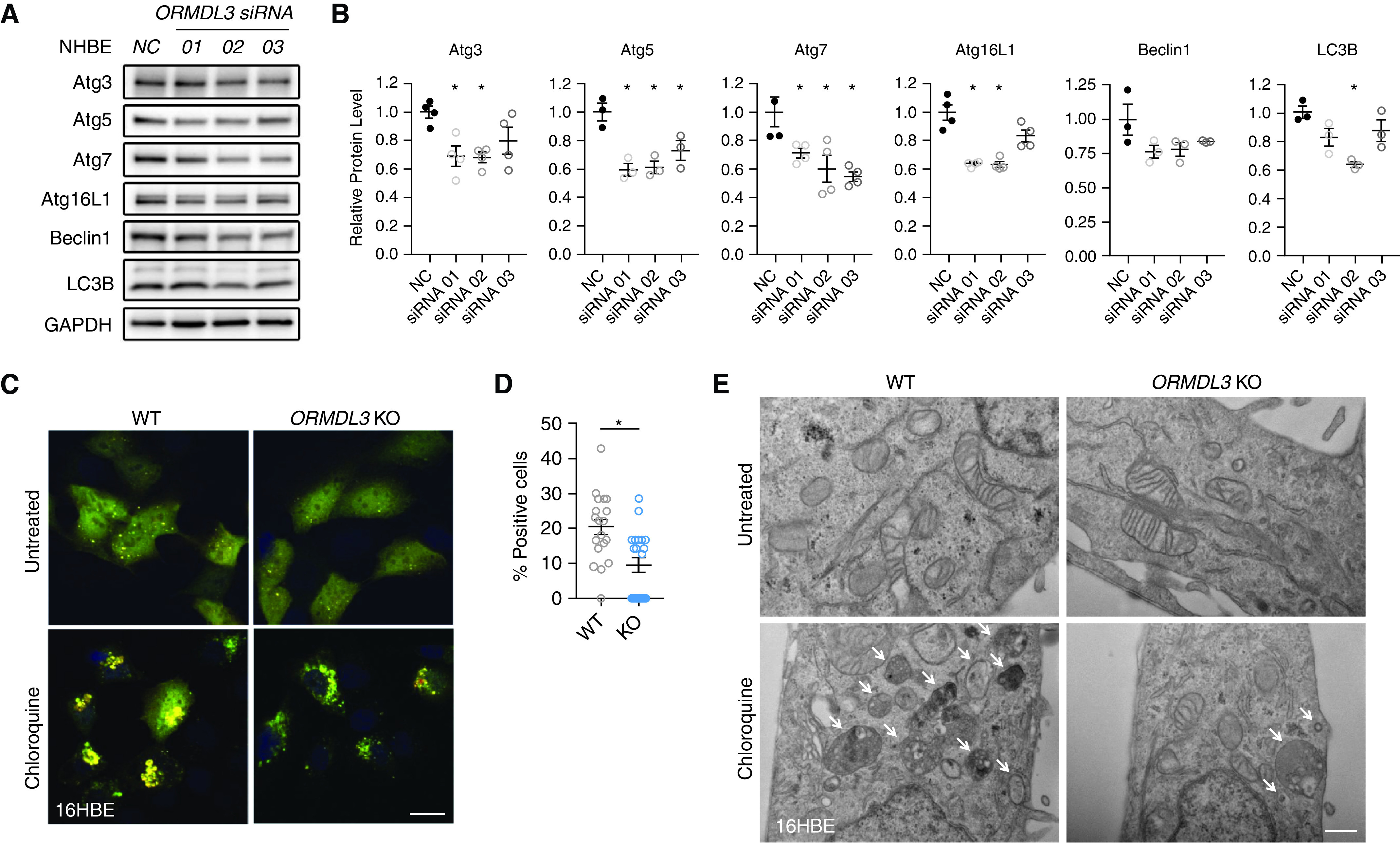
ORMDL3 knockdown inhibits autophagy in primary NHBE and 16HBE cells. (A) Detection, and (B) quantification of the autophagy genes by Western blot in primary NHBE cells transfected with anti-ORMDL3 siRNA or negative control (NC). Means ± SEM from three biological replicates with three distinct siRNAs. *P < 0.05, unpaired t test. (C) Representative confocal images of autophagosomes as shown by the RFP-GFP-LC3B tandem sensor in wild-type (WT) and CRISPR-associated protein 9 generated ORMDL3 knockout (KO) 16HBE cells (ORMDL3 KO) treated with or without chloroquine (20 μM) for 16 hours. Scale bar, 20 μm. (D) Quantification of the percentage of autophagy-positive cells at basal levels. *P < 0.05 compared with WT by unpaired t test. Means ± SEM shown from 10 images from each of the two control or KO lines. (E) Representative electron microscopy pictures from two independent experiments in WT or ORMDL3 KO 16HBE stable cells in the presence or absence of chloroquine (20 μM for 16 h). Scale bar, 500 nm. Arrowheads = autophagosomes.
ORMDL3 Interacts with SERCA2, Thereby Promoting Autophagy in Human Bronchial Epithelial Cells
Although previous studies have implicated ORMDL3 in Beclin-1–mediated autophagy in endothelial cells (17) and B cells (16), the mechanisms by which ORMDL3 may regulate autophagy in the airway epithelium is unclear. Given that ORMDL3 can trigger the UPR (11, 12), which itself may induce autophagy (24), we assessed whether the increased autophagy observed in our cellular models was accompanied by UPR activation. UPR signaling activation is mediated by three paralleled endoplasmic reticulum membrane transducers, PERK (PRKR-like endoplasmic reticulum kinase), ATF6 (activating transcription factor 6), and IRE1 (inositol-requiring kinase 1) (25). Despite evident activation of IRE1α signaling as supported by increased protein levels of IRE1α and phospho-JNK after treatment of the endoplasmic reticulum stressor thapsigargin (Tg), neither mRNA nor protein levels of UPR targets altered (Figures E3B and E3D–E3F) after overexpression or knockdown of ORMDL3 (Figures E3A and E3C), suggesting that UPR induction is unlikely a primary mechanism responsible for ORMDL3-regulated autophagy in BE.
To determine the molecular mechanisms by which ORMDL3 promotes autophagy, we screened for ORMDL3 interacting proteins by affinity purification followed by protein mass spectrometry using Flag-tagged ORMDL3 as bait in two epithelial cell types (16HBE and HEK-293). After subtraction of background protein sequences detected in vector control– transfected 16HBE and HEK-293 cells, we identified 181 proteins that uniquely interact with ORMDL3 in both epithelial cell lines. Of these, one of the most abundant proteins (⩾10 unique protein sequences) identified was SERCA2 (gene name ATP2A2), a sarco/endoplasmic reticulum Ca2+-ATPase previously shown to inhibit autophagy (26, 27). We confirmed that ORMDL3 interacted with SERCA2 by co-immunoprecipitation assay (Figure 3A) and that ORMDL3 and SERCA2 colocalized in the cytosol (Figure 3B). Given very limited affinity purification–mass spectrometry evidence of protein–protein interaction of ORMDL3 with other major autophagy proteins, we focused subsequent studies to test the hypothesis that ORMDL3 may promote epithelial autophagy via interacting with SERCA2.
Figure 3.
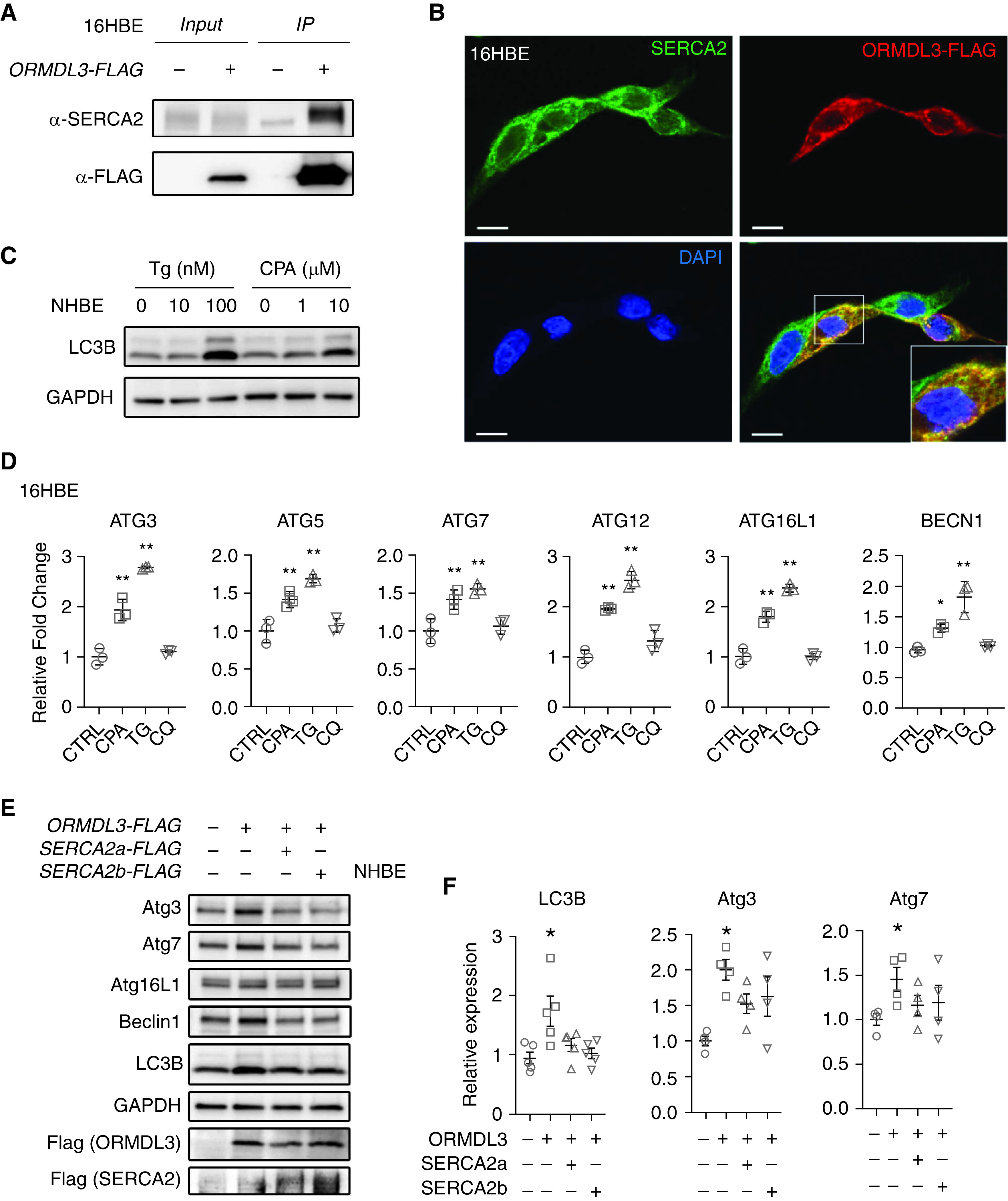
SERCA2 mediates ORMDL3-induced autophagy. (A) SERCA2 was immunoprecipitated by Flag-tagged ORMDL3 (Flag-IP) in 16HBE cells. Three independent repeats were performed. (B) Representative immunofluorescence from three independent experiments of the expression and colocalization of ORMDL3 (anti-Flag antibody, red) and SERCA2 (anti-SERCA2 antibody, green) in 16HBE cells transfected with Flag-tagged ORMDL3 plasmid. Nuclei were counterstained by DAPI (blue). Scale bars, 20 μm. (C) Detection of autophagy marker LC3B by Western blot in primary NHBE cells treated with SERCA2 inhibitors, thapsigargin (Tg, 10 nM and 100 nM) or cyclopiazonic acid (CPA, 1 μM and 10 μM) for 16 hours. Two independent experiments were performed. (D) 16HBE cells were treated with CPA (20 μM), Tg (100 nM), or chloroquine (CQ) (20 μM) for 20 hours. RNA levels were detected by qRT-PCR experiments. Mean ± SD shown for three independent experiments. *P < 0.05 and **P < 0.01. Statistical methods: ordinary one-way ANOVA and Dunnett’s multiple comparisons. (E) Representative Western blot images and (F) quantification of autophagy gene expression after overexpressing ORMDL3 with or without SERCA2a/SERCA2b co-overexpression in primary NHBE cells. Means ± SEM shown are from four biological replicates per condition. *P < 0.05 compared with control by one-way ANOVA followed by unpaired t test.
To test this, we first assessed the impact of pharmacologic inhibition of SERCA2 on autophagy activation. Herein, SERCA2 inhibitors cyclopiazonic acid (CPA) or Tg that disrupt Ca2+ homeostasis (28) were used to inhibit the function of SERCA2. The treatment of Tg or CPA resulted in increased protein levels of LC3B (Figure 3C) in primary NHBE cells and increased mRNA levels of autophagy-related genes, including ATG3, ATG5, ATG7, ATG12, ATG16L1, and Beclin-1, in 16HBE cells (Figure 3D). We next assessed whether SERCA2 modulated ORMDL3-induced autophagy. Interestingly, cotransfection of ORMDL3 with either SERCA2a or SERCA2b completely abrogated ORMDL3-mediated autophagy in primary NHBE cells (Figure 3E). These studies together suggest that ORMDL3 promotes bronchial epithelial autophagy through inhibitory interaction with SERCA2.
ORMDL3 Modulates [Ca2+]i Mobilization Possibly through SERCA2 in Human Bronchial Epithelial Cells
Given that SERCA is responsible for calcium transport from the cytosol into the sarcoplasmic reticulum, we evaluated the impact of ORMDL3 on intracellular calcium ([Ca2+]i) flux. In Ca2+-free media, treatment of epithelial cells with ATP results in rapid induction of [Ca2+]i release followed by a prolonged recovery phase of Ca2+ translocation from the cytosol into the sarco/endoplasmic reticulum (Figures 4A and 4B). 16HBE cells overexpressing ORMDL3 led to significant (at least 25% longer) delays in [Ca2+]i recovery compared with controls (Figures 4C, 4D, and E4A). Conversely, ORMDL3 deficiency resulted in a more rapid recovery of Ca2+ translocation compared with controls (Figures 4E, 4F, and E4B). More importantly, silencing of SERCA2 in ORMDL3-deficient cells partially rescued these Ca2+ recovery rates compared with WT cells (Figures 4E and 4F). These results cumulatively support ORMDL3 as a regulator of BE [Ca2+]i signaling, possibly through SERCA2.
Figure 4.

ORMDL3 modulates concentration of intracellular calcium [Ca2+]i mobilization by inhibiting SERCA2 in 16HBE cells. (A) Fluo-4 dye fluorescence signals indicating calcium impulse recorded in 16HBE cells before and after treatment with ATP (10 μM). (B) Representative time-series recordings of Fluo-4 AM fluorescence intensities of calcium concentration in WT 16HBE cells after ATP stimulation. (C) Cytosolic Ca2+ decay plots in Ctrl (black) and ORMDL3-overexpressing (ORMDL3, red) 16HBE cells. Means ± SEM from seven biological repeats performed per group. Lines denote the time-dependent recovery of the mean fluorescence intensity expressed as the percentage of peak cytosolic Ca2+ intensity. (D) Bar graphs of time to cytosolic Ca2+ clearance corresponding to C. (E) Cytosolic Ca2+ decay plots in WT (black), ORMDL3 KO (blue) 16HBE cells, and ORMDL3 KO transfected with SERCA2 siRNA (KO + siSERCA2, purple). Means ± SEM from four to nine biological repeats. (F) Seconds needed for cytosolic Ca2+ clearance in 16HBE WT, ORMDL3 KO, and ORMDL3 KO transfected with SERCA2 siRNA corresponding to E. *P < 0.05. One-way ANOVA followed by unpaired t test was used for statistical analysis.
Increased ORMDL3 Level Promotes Cell Death in Human Bronchial Epithelial Cells
Airway epithelial damage is a pathologic feature of asthma in both children and adults and is correlated with airway hyperresponsiveness and other measures of asthma severity (29, 30). Recognizing that autophagy can, under different contexts, be either beneficial (promoting prosurvival signals) or detrimental (accelerating cell death) (31), we set out to determine the impact of ORMDL3 overexpression on 16HBE survival and cell proliferation. Compared with control lines, 16HBE cells overexpressing ORMDL3 demonstrated increased cell death, as indicated by an ∼1.5-fold increase in LDH release (Figure 5A). This increase was partially alleviated by treatment with CDN1163, an allosteric SERCA activator, indicating that ORMDL3-induced cell damage was inhibited by SERCA2 activation (Figure 5A). In contrast, ORMDL3-induced cell death was unlikely mediated by apoptosis, as supported by minimal changes of cleaved caspase-3 and PARP (Figure 5C). Furthermore, extracellular LDH concentrations were decreased in ORMDL3-deficient 16HBE cells compared with WT, and treatment with SERCA2 inhibitors CPA and Tg reversed these effects (Figure 5B). Meanwhile, we observed minimal impacts of ORMDL3 on cell proliferation, as shown by overexpression or depletion of ORMDL3 in 16HBE lines (Figures E5A and E5B).
Figure 5.
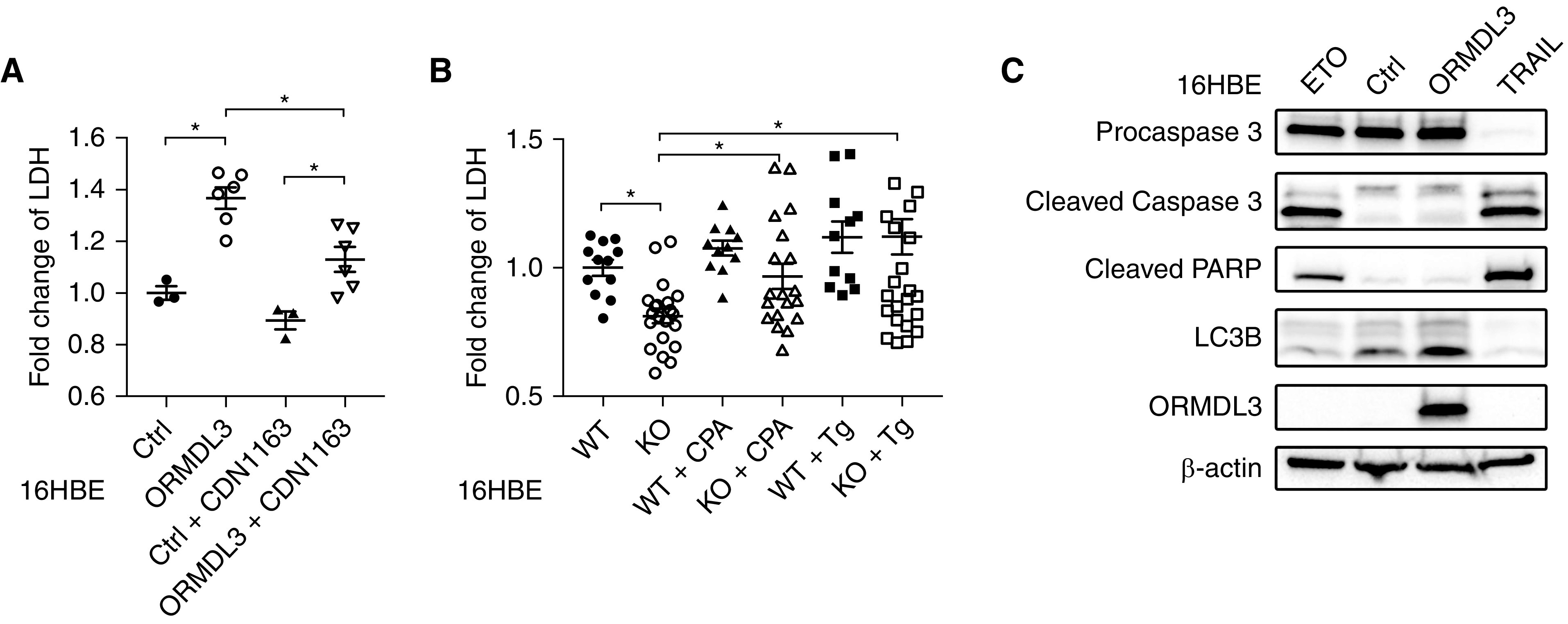
ORMDL3 regulates the cell viability of 16HBE cells. (A) The relative fold change of lactate dehydrogenase (LDH) release in ORMDL3-overexpressing (ORMDL3) cells compared with Ctrl 16HBE lines in the presence or absence of SERCA activator CDN1163 (1 μM) for 16 hours. Means ± SD were from three biological repeats with six technical repeats for each group. (B) The relative fold change of LDH in ORMDL3-deficient (KO) and WT 16HBE cells in the presence or absence of SERCA2 inhibitors CPA (20 μM) or Tg (20 nM) for 16 hours. Means ± SD were from two biological repeats, six technical repeats for each group. *P < 0.05. One-way ANOVA followed by unpaired t test was used for statistical analysis. (C) Apoptosis markers including cleaved Caspase 3 and cleaved PARP were detected in 16HBE cells with various treatments or transfections. Lanes 1 and 4 are from cells treated with etoposide (ETO, 10 μM, 24 h) and TRAIL (100 nM, 6 h), respectively, two inducers of apoptosis, as positive controls. Lanes 2 and 3 are control and ORMDL3-overexpressing lines, respectively. Two independent experiments were performed.
ORMDL3 Correlates with Autophagy Gene Expression in Human Primary Bronchial Epithelial Cells
In multiple 16HBE cell single colonies with stable overexpression or KO of ORMDL3, we observed significant correlation between levels of ORMDL3 and autophagy genes across all single colonies of stable lines (Figure E6). To corroborate our in vitro observations with in vivo evidence of a relationship between ORMDL3 and autophagy, we examined the correlation of ORMDL3 expression to that of known autophagy-related genes in human BE samples obtained by bronchoscopy brushings from 44 subjects from Asthma BRIDGE (27 cases, 17 control subjects) (22). As shown in Figure 6, expression of autophagy-related genes ATG7 and ATG12 showed significant correlation with expression of ORMDL3, suggesting the activation of the autophagy pathway in human BE cells. However, we found no correlation between autophagy gene expression and clinical asthmatic characteristics, possibly because of small sample size.
Figure 6.
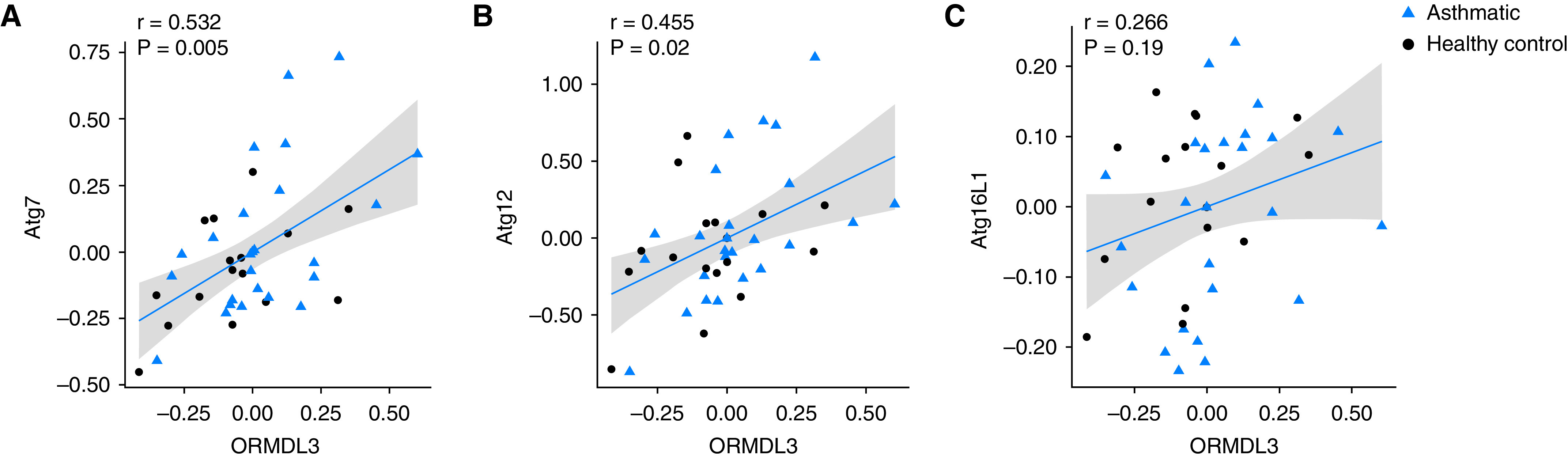
The correlation of ORMDL3 and autophagy gene expression in human primary bronchial epithelial cells from brushing samples. Scatterplots of linear correlation between expression of ORMDL3 gene and autophagy-related genes—(A) ATG7, (B) ATG12, and (C) ATG16L1—detected in microarray from bronchial epithelial cells obtained from bronchial brushing samples from 44 individuals (27 patients with asthma and 17 control subjects). 95% confidence intervals of correlation are shown in gray.
Discussion
Discovery of the 17q21 association with asthma risk is a convincing example of how hypothesis-free genetic approaches like GWAS can lead to new understandings of disease and reveal previously unappreciated pathobiology. The coupling of GWASs and expression quantitative trait locus mapping studies revealed that genetic risk for asthma at 17q21 was conferred by increased expression of two genes with no prior links to asthma—ORMDL3 and GSDMB—and animal models confirmed that overexpression of each recapitulated cardinal features of asthma (10, 32). Subsequent work had demonstrated that ORMDL3 may impart these phenotypes by influencing a variety of asthma relevant biochemical and cellular processes, including sphingolipid metabolism (14) and the UPR (15). In this study, we implicate another autophagy pathway by demonstrating its regulation by ORMDL3 in human BE cells. Inhibition of endogenous ORMDL3 expression reduces basal rates of autophagy in 16HBE cells and primary NHBE, whereas ORMDL3 overexpression alone (i.e., in the absence of additional cellular stress) induces autophagy.
Autophagy has been increasingly recognized to play a central role in the pathogenesis of various lung diseases, including chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis, and asthma (18, 33). In asthma, autophagy has been firmly implicated in multiple processes within the immune compartment (34), contributing to the programming of the innate immune response (35), the establishment of atopic phenotypes (35, 36), and granulocytic inflammation (37). More evidence has emerged on human genetic analysis in individuals with asthma. Some studies indicate genetic variants in autophagy genes are significantly associated with lung function in subjects with asthma (38, 39). The function of autophagy in the BE is also receiving considerable attention in asthma. Increased expression of autophagy-related genes in bronchial epithelial cells has been repeatedly observed in individuals with asthma and consistently correlated with markers of airway remodeling (40), and morphologic evidence of bronchial epithelial autophagy in asthma has been reported (38, 41, 42).
In our study, we demonstrate that ORMDL3’s regulation of autophagy in the BE is possibly mediated through SERCA2- and ORMDL3-mediated regulation of [Ca2+]i flux. These latter findings are supported by complementary observations: 1) that ORMLD3 overexpression delayed [Ca2+]i recovery in the BE; and 2) that SERCA2 abrogates ORMDL3-mediated autophagy; and 3) that pharmacologic activation of SERCA2 attenuates induction of cell death by ORMDL3. Last, gene expression profiling in BE cell samples derived from patients with asthma confirmed the strong correlation of ORMDL3 expression with that of autophagy-related genes. Together with prior demonstration that increased ORMDL3 expression impairs BE barrier function (43), these findings strongly support that the asthma susceptibility gene ORMDL3 on chromosome 17q21 enhances autophagy that promotes epithelial cell damage and asthma risk.
Autophagy can be activated after UPR induction in response to the accumulation of aggregated misfolded proteins (44), including through IRE1-mediated feedback loops (44, 45). Despite this and prior observations that ORMDL3 expression in human airway epithelial cell A549 increases expression of the UPR pathway transcription factor ATF6 (11), we found no evidence that ORMDL3-induced autophagy involves UPR pathway activation. Rather, the absence of increased expression of markers of UPR activation in our models, combined with our subsequent implication of SERCA-mediated calcium flux, supports a UPR-independent mechanism.
In addition to confirming the importance of [Ca2+]i flux in the regulation of autophagic processes (46, 47), our findings that ORMDL3-induced BE cell autophagy is mediated through interaction with SERCA2 are consistent with studies implicating ORMDL3–SERCA interactions in other asthma-related processes, including airway remodeling and airway hyperresponsiveness. Overexpression of ORMDL3 induces the expression of SERCA2b (11) in BE and promotes airway remodeling (10). ORMDL3 also induces increased SERCA2b expression in smooth muscle, with resulting increases smooth muscle proliferation and contractility (48), features that promote airway hyperreactivity. Together with our data, these findings implicate the ORMDL3–SERCA2 cross-talk as an important regulator of three cellular processes central to the pathobiology of asthma: epithelial damage, airway remodeling, and airway smooth muscle dysfunction.
One potential limitation of our work is that the level of ORMDL3 overexpression achieved in our experimental models was greater than that conferred by natural genetic variation. Although results of experiments performed under supraphysiologic conditions must be interpreted cautiously, three important corroborative lines of evidence suggest our observations are both reliable and generalizable. First, we found that the expression of numerous autophagy-related genes was correlated with that of ORMDL3 in unadulterated patient-derived bronchial-brush BE samples. Second, the results of our overexpression studies are corroborated by the complementary set of siRNA and CRISP-Cas9 KO studies, which consistently demonstrated that ORMDL3 knockdown reduced endogenous autophagy activity. Last, our findings are consistent with previously reported observations in nonepithelial cell types, including B-lymphocytes (16) and endothelial cells (17), in which inhibition or overexpression of ORMDL3 respectively resulted in reduction or augmentation in basal levels of autophagy (as measured by reduced LC3-II and Beclin-1 protein expression).
In summary, we have shown that ORMDL3 regulates BE cell autophagy and that this process is mediated by SERCA2-facilitated [Ca2+]i mobilization. These findings provide further evidence that the chromosome 17q21 locus confers asthma risk through multiple complementary mechanisms. In addition to the as-of-yet unclear functions of GSDMB, genetic regulation of ORMDL3 alone impacts a wide range of asthma-relevant processes in multiple asthma-relevant cell types. This perhaps explains how the genetic associations of 17q21 with asthma risk are so robust and readily observable in diverse populations across the globe, as differences in genetic background or environmental exposures are overcome by the pleiotropic effects of this important locus. This pleiotropy also argues for the prioritization of ORMDL3 for therapeutic targeting, as inhibition of this key regulator could, ideally, simultaneously impact multiple clinical features of asthma.
Acknowledgments
Acknowledgment
The authors thank Dr. Dieter Gruenert from the University of California San Francisco for generously providing the 16HBE cell line and the Chicago Asthma BRIDGE team for providing the bronchial epithelial cell brushings for gene expression analysis (C. Ober, PI).
Footnotes
Supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health grants R01 HL123546 and RC2 HL101543 (B.A.R.), K01 HL127265 (D.C.C.-C.), and R33 HL120794, P01HL132825, and R01HL127200 (X.Z.).
Author Contributions: X.Z. and B.A.R. designed and supervised the overall study. F.G. and Y.H. designed, conducted, and analyzed the experimental work. L.Z., D.T., and L.L. conducted RNA extraction, RT-PCR, and quantitative PCR experimental work. D.C.C.-C. and P.K. performed the primary bronchial epithelial cells gene expression analyses. F.G., Y.H., D.C.C.-C, B.D.L., X.Z., and B.A.R. contributed to manuscript preparation.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0305OC on March 30, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature . 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 2. Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE, et al. Mexico City Childhood Asthma Study (MCAAS); Children’s Health Study (CHS) and HARBORS study; Genetics of Asthma in Latino Americans (GALA) Study, Study of Genes-Environment and Admixture in Latino Americans (GALA2) and Study of African Americans, Asthma, Genes & Environments (SAGE); Childhood Asthma Research and Education (CARE) Network; Childhood Asthma Management Program (CAMP); Study of Asthma Phenotypes and Pharmacogenomic Interactions by Race-Ethnicity (SAPPHIRE); Genetic Research on Asthma in African Diaspora (GRAAD) Study Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet . 2011;43:887–892. doi: 10.1038/ng.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Galanter J, Choudhry S, Eng C, Nazario S, Rodríguez-Santana JR, Casal J, et al. ORMDL3 gene is associated with asthma in three ethnically diverse populations. Am J Respir Crit Care Med . 2008;177:1194–1200. doi: 10.1164/rccm.200711-1644OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rogers AJ, Raby BA, Lasky-Su JA, Murphy A, Lazarus R, Klanderman BJ, et al. Assessing the reproducibility of asthma candidate gene associations, using genome-wide data. Am J Respir Crit Care Med . 2009;179:1084–1090. doi: 10.1164/rccm.200812-1860OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharma S, Zhou X, Thibault DM, Himes BE, Liu A, Szefler SJ, et al. A genome-wide survey of CD4(+) lymphocyte regulatory genetic variants identifies novel asthma genes. J Allergy Clin Immunol . 2014;134:1153–1162. doi: 10.1016/j.jaci.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Verlaan DJ, Berlivet S, Hunninghake GM, Madore AM, Larivière M, Moussette S, et al. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am J Hum Genet . 2009;85:377–393. doi: 10.1016/j.ajhg.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kothari PH, Qiu W, Croteau-Chonka DC, Martinez FD, Liu AH, Lemanske RF, Jr, et al. Asthma BioRepository for Integrative Genomic Exploration (Asthma BRIDGE) Consortium Role of local CpG DNA methylation in mediating the 17q21 asthma susceptibility gasdermin B (GSDMB)/ORMDL sphingolipid biosynthesis regulator 3 (ORMDL3) expression quantitative trait locus. J Allergy Clin Immunol . 2018;141:2282–2286.e6. doi: 10.1016/j.jaci.2017.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hao K, Bossé Y, Nickle DC, Paré PD, Postma DS, Laviolette M, et al. Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genet . 2012;8:e1003029. doi: 10.1371/journal.pgen.1003029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science . 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miller M, Rosenthal P, Beppu A, Mueller JL, Hoffman HM, Tam AB, et al. ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J Immunol . 2014;192:3475–3487. doi: 10.4049/jimmunol.1303047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miller M, Tam AB, Cho JY, Doherty TA, Pham A, Khorram N, et al. ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proc Natl Acad Sci USA . 2012;109:16648–16653. doi: 10.1073/pnas.1204151109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Löser S, Gregory LG, Zhang Y, Schaefer K, Walker SA, Buckley J, et al. Pulmonary ORMDL3 is critical for induction of Alternaria-induced allergic airways disease. J Allergy Clin Immunol . 2017;139:1496–1507.e3. doi: 10.1016/j.jaci.2016.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miller M, Broide DH. Why is ORMDL3 on chromosome 17q21 highly linked to asthma? Am J Respir Crit Care Med . 2019;199:404–406. doi: 10.1164/rccm.201810-1941ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, et al. Orm family proteins mediate sphingolipid homeostasis. Nature . 2010;463:1048–1053. doi: 10.1038/nature08787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cantero-Recasens G, Fandos C, Rubio-Moscardo F, Valverde MA, Vicente R. The asthma-associated ORMDL3 gene product regulates endoplasmic reticulum-mediated calcium signaling and cellular stress. Hum Mol Genet . 2010;19:111–121. doi: 10.1093/hmg/ddp471. [DOI] [PubMed] [Google Scholar]
- 16. Dang J, Bian X, Ma X, Li J, Long F, Shan S, et al. ORMDL3 facilitates the survival of splenic B cells via an ATF6α-endoplasmic reticulum stress-Beclin1 autophagy regulatory pathway. J Immunol . 2017;199:1647–1659. doi: 10.4049/jimmunol.1602124. [DOI] [PubMed] [Google Scholar]
- 17. Ma X, Qiu R, Dang J, Li J, Hu Q, Shan S, et al. ORMDL3 contributes to the risk of atherosclerosis in Chinese Han population and mediates oxidized low-density lipoprotein-induced autophagy in endothelial cells. Sci Rep . 2015;5:17194. doi: 10.1038/srep17194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Racanelli AC, Kikkers SA, Choi AMK, Cloonan SM. Autophagy and inflammation in chronic respiratory disease. Autophagy . 2018;14:221–232. doi: 10.1080/15548627.2017.1389823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Y, Liu J, Zhou JS, Huang HQ, Li ZY, Xu XC, et al. MTOR suppresses cigarette smoke-induced epithelial cell death and airway inflammation in chronic obstructive pulmonary disease. J Immunol . 2018;200:2571–2580. doi: 10.4049/jimmunol.1701681. [DOI] [PubMed] [Google Scholar]
- 20. Hao Y, Guo F, Kothari P, Levy BD, Zhou X, Raby BA. ORMDL3 and autophagy of the airway epithelium: an important mechanism underlying asthma pathogenesis? [abstract] Am J Respir Crit Care Med . 2019;199:A2830. [Google Scholar]
- 21. Guo F, Kothari P, Levy BD, Zhou X, Raby BA. The asthma susceptibility gene ORMDL3 regulates autophagy in human bronchial epithelial cells [abstract] Am J Respir Crit Care Med . 2018;197:A1204. [Google Scholar]
- 22. Croteau-Chonka DC, Qiu W, Martinez FD, Strunk RC, Lemanske RF, Jr, Liu AH, et al. Asthma BioRepository for Integrative Genomic Exploration (Asthma BRIDGE) Consortium Gene expression profiling in blood provides reproducible molecular insights into asthma control. Am J Respir Crit Care Med . 2017;195:179–188. doi: 10.1164/rccm.201601-0107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy . 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 24. Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol . 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature . 2016;529:326–335. doi: 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- 26. Wong VK, Li T, Law BY, Ma ED, Yip NC, Michelangeli F, et al. Saikosaponin-d, a novel SERCA inhibitor, induces autophagic cell death in apoptosis-defective cells. Cell Death Dis . 2013;4:e720. doi: 10.1038/cddis.2013.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mauvezin C, Nagy P, Juhász G, Neufeld TP. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun . 2015;6:7007. doi: 10.1038/ncomms8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sehgal P, Szalai P, Olesen C, Praetorius HA, Nissen P, Christensen SB, et al. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J Biol Chem . 2017;292:19656–19673. doi: 10.1074/jbc.M117.796920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barbato A, Turato G, Baraldo S, Bazzan E, Calabrese F, Panizzolo C, et al. Epithelial damage and angiogenesis in the airways of children with asthma. Am J Respir Crit Care Med . 2006;174:975–981. doi: 10.1164/rccm.200602-189OC. [DOI] [PubMed] [Google Scholar]
- 30. Laitinen LA, Heino M, Laitinen A, Kava T, Haahtela T. Damage of the airway epithelium and bronchial reactivity in patients with asthma. Am Rev Respir Dis . 1985;131:599–606. doi: 10.1164/arrd.1985.131.4.599. [DOI] [PubMed] [Google Scholar]
- 31. Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol . 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Das S, Miller M, Beppu AK, Mueller J, McGeough MD, Vuong C, et al. GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. Proc Natl Acad Sci USA . 2016;113:13132–13137. doi: 10.1073/pnas.1610433113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haspel JA, Choi AM. Autophagy: a core cellular process with emerging links to pulmonary disease. Am J Respir Crit Care Med . 2011;184:1237–1246. doi: 10.1164/rccm.201106-0966CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature . 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu Y, Eissa NT. Autophagy in innate and adaptive immunity. Proc Am Thorac Soc . 2010;7:22–28. doi: 10.1513/pats.200909-103JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu JN, Suh DH, Trinh HK, Chwae YJ, Park HS, Shin YS. The role of autophagy in allergic inflammation: a new target for severe asthma. Exp Mol Med . 2016;48:e243. doi: 10.1038/emm.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Choi GE, Yoon SY, Kim JY, Kang DY, Jang YJ, Kim HS. Autophagy deficiency in myeloid cells exacerbates eosinophilic inflammation in chronic rhinosinusitis. J Allergy Clin Immunol . 2018;141:938–950.e12. doi: 10.1016/j.jaci.2017.10.038. [DOI] [PubMed] [Google Scholar]
- 38. Poon AH, Chouiali F, Tse SM, Litonjua AA, Hussain SN, Baglole CJ, et al. Genetic and histologic evidence for autophagy in asthma pathogenesis. J Allergy Clin Immunol . 2012;129:569–571. doi: 10.1016/j.jaci.2011.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martin LJ, Gupta J, Jyothula SS, Butsch Kovacic M, Biagini Myers JM, Patterson TL, et al. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS One . 2012;7:e33454. doi: 10.1371/journal.pone.0033454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McAlinden KD, Deshpande DA, Ghavami S, Xenaki D, Sohal SS, Oliver BG, et al. Autophagy activation in asthma airways remodeling. Am J Respir Cell Mol Biol . 2019;60:541–553. doi: 10.1165/rcmb.2018-0169OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu T, Liu Y, Miller M, Cao L, Zhao J, Wu J, et al. Autophagy plays a role in FSTL1-induced epithelial mesenchymal transition and airway remodeling in asthma. Am J Physiol Lung Cell Mol Physiol . 2017;313:L27–L40. doi: 10.1152/ajplung.00510.2016. [DOI] [PubMed] [Google Scholar]
- 42. Li W, Wu Y, Zhao Y, Li Z, Chen H, Dong L, et al. MTOR suppresses autophagy-mediated production of IL25 in allergic airway inflammation. Thorax . 2020;75:1047–1057. doi: 10.1136/thoraxjnl-2019-213771. [DOI] [PubMed] [Google Scholar]
- 43. Yang R, Tan M, Xu J, Zhao X. Investigating the regulatory role of ORMDL3 in airway barrier dysfunction using in vivo and in vitro models. Int J Mol Med . 2019;44:535–548. doi: 10.3892/ijmm.2019.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci . 2013;70:2425–2441. doi: 10.1007/s00018-012-1173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Böck J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature . 2013;503:272–276. doi: 10.1038/nature12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chandrachud U, Walker MW, Simas AM, Heetveld S, Petcherski A, Klein M, et al. Unbiased cell-based screening in a neuronal cell model of Batten disease highlights an interaction between Ca2+ homeostasis, autophagy, and CLN3 protein function. J Biol Chem . 2015;290:14361–14380. doi: 10.1074/jbc.M114.621706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy . 2015;11:1437–1438. doi: 10.1080/15548627.2015.1066957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen J, Miller M, Unno H, Rosenthal P, Sanderson MJ, Broide DH. Orosomucoid-like 3 (ORMDL3) upregulates airway smooth muscle proliferation, contraction, and Ca2+ oscillations in asthma. J Allergy Clin Immunol . 2018;142:207–218.e6. doi: 10.1016/j.jaci.2017.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]