

Abstract
Patients with chronic obstructive pulmonary disease (COPD)–pulmonary emphysema often develop locomotor muscle dysfunction, which entails reduced muscle mass and force-generation capacity and is associated with worse outcomes, including higher mortality. Myogenesis contributes to adult muscle integrity during injury–repair cycles. Injurious events crucially occur in the skeletal muscles of patients with COPD in the setting of exacerbations and infections, which lead to acute decompensations for limited periods of time, after which patients typically fail to recover the baseline status they had before the acute event. Autophagy, which is dysregulated in muscles from patients with COPD, is a key regulator of muscle stem-satellite- cells activation and myogenesis, yet very little research has so far mechanistically investigated the role of autophagy dysregulation in COPD muscles. Using a genetically inducible interleukin-13–driven pulmonary emphysema model leading to muscle dysfunction, and confirmed with a second genetic animal model, we found a significant myogenic dysfunction associated with the reduced proliferative capacity of satellite cells. Transplantation experiments followed by lineage tracing suggest that an intrinsic defect in satellite cells, and not in the COPD environment, plays a dominant role in the observed myogenic dysfunction. RNA sequencing analysis and direct observation of COPD mice satellite cells suggest dysregulated autophagy. Moreover, while autophagy flux experiments with bafilomycin demonstrated deacceleration of autophagosome turnover in COPD mice satellite cells, spermidine-induced autophagy stimulation leads to a higher replication rate and myogenesis in these animals. Our data suggest that pulmonary emphysema causes disrupted myogenesis, which could be improved with stimulation of autophagy and satellite cells activation, leading to an attenuated muscle dysfunction.
Keywords: pulmonary emphysema, satellite cells, autophagy, myogenesis, COPD
Patients with chronic obstructive pulmonary disease (COPD) often develop locomotor skeletal muscle dysfunction, which entails muscle atrophy and lower force-generation capacity (1, 2). COPD-associated muscle dysfunction, which occurs more often in emphysema than in chronic bronchitis phenotype (3), is strongly associated with higher mortality and other poor outcomes in these patients (4–6). Inferential models indicate that these associations persist after multivariably adjusting for the level of pulmonary disease and other covariables, suggesting that muscle dysfunction could partially contribute to the worse prognosis (1, 2).
In general, muscle function/force-generation capacity is determined by total mass and fiber’s metabolic properties (1). Myogenesis, which is muscle turnover driven by progenitor cells such as satellite cells (7), contributes to the maintenance of muscle mass and metabolic profile in the context of organ development, hypertrophy, and injury–repair cycles (8–10). Injurious events crucially occur in the skeletal muscles of patients with COPD in the setting of exacerbations and infections (11, 12), which lead to acute decompensations for limited periods of time, after which patients typically fail to recover the baseline status they had before the acute event (13, 14). Indeed, the frequency and severity of COPD exacerbations and infections powerfully associate with loss of muscle and lung integrity and with higher mortality over time (13–16). Strong evidence indicates that dysfunctional myogenesis contributes to muscle loss in nonprimarily muscular conditions such as aging and cancer (17–21), and recent clinical observations also suggest its role in COPD (18, 22–28). Moreover, accumulating evidence indicates that biological signals that are prevalent in COPD patients, such as hypoxia, hypercapnia, and smoking, regulate muscle response to injury as well (29–31). So far, almost no mechanistic research focused on skeletal myogenesis has been conducted on an animal model of COPD-induced skeletal muscle dysfunction. Such information could accelerate research aimed at improving muscle recovery in the setting of acute injurious events following COPD decompensation, with potential mortality benefits.
Autophagy, a major cellular homeostatic mechanism activated by environmental stress (32), contributes to myogenesis by supporting bioenergetic demands for the activation of satellite and other stem cells (33–36) and by preventing quiescence-to-senescence transition (17, 20, 37). While consistent clinical observations have revealed a profound autophagy dysregulation in skeletal muscles from patients with COPD (38–40), there is no clarity regarding the contribution of abnormal autophagy to the integrity of COPD muscles. Indeed, while impaired autophagy has been found to cause muscle loss (38), excessive autophagy appears to contribute to muscle wasting as well (41). So far, no mechanistic research has addressed the causal role of autophagy in the regulation of satellite cell-mediated myogenesis in the COPD setting.
Mechanistic research focused on COPD-driven locomotor muscle dysfunction requires an animal model that ideally should fulfill the following conditions: 1) be inducible, to minimize temporal confounders such as muscle development and age-related sarcopenia (42); 2) be robust enough, to reminisce the disease severity shown by the majority of patients with COPD with muscle dysfunction (43); 3) develop the muscle phenotype after, and not simultaneously with, the occurrence of pulmonary disease to reflect a secondary COPD comorbidity; 4) recapitulate multidimensional features observed in patients such as morphologic, metabolic, and functional aspects of muscle dysfunction (1, 44, 45); and 5) occur in the context of a pulmonary disease phenotype with airways obstruction, typical histologic changes and other features demonstrated by humans (46, 47).
In the present work, we took advantage of an established murine model of pulmonary emphysema-induced skeletal muscle dysfunction to interrogate the animal’s myogenic response. That animal, which highly reminisces the skeletal muscle phenotype demonstrated by patients with COPD (44–46, 48, 49), develops pulmonary emphysema in a transgene-driven inducible fashion, allowing control of the time-sensitive myogenic effects of chronic pulmonary disease. This animal model deliberately does not involve cigarette smoking, given that this signal has been recently shown to influence muscle response to injury (29), which could confound the specific effect of pulmonary emphysema on myogenesis. Moreover, features characterizing muscle dysfunction do not occur in this animal until the emphysema is established at 8 weeks after induction (44, 45), suggesting a similar trajectory demonstrated by patients with COPD (1). Our central hypothesis was that autophagy dysregulation contributes to inadequate muscle repair in COPD and that autophagy stimulation would attenuate that deficit.Some of the results of these studies have been previously reported in the form of a preprint (bioRxiv, [13 July 2021] https://doi.org/10.1101/2021.07.08.450201).
Methods
Animals
All the procedures involving animals were approved by the Albany Medical College Institutional Animal Care and Use Committee (IACUC protocol 18–10001), and animals were handled according to the National Institutes of Health guidelines. All methods were performed in accordance with the relevant guidelines and regulations, as stated by the Journal and public agencies.
COPD model (primary)
Experiments were conducted using CC10-rtTA-IL-13 (IL13TG) doxycycline-inducible transgenic mice that develop chronic lung remodeling reminiscent of pulmonary emphysema upon induction. CC10-rtTA-IL-13 heterozygote animals were bred to C57BL/6 wild-type (WT) mice to obtain hemizygous IL13TG experimental animals and homozygous IL13WT littermate controls. Both IL13TG (emphysema) and IL13WT (nonemphysema, used as control littermates) mice were provided doxycycline in the drinking water along with sucrose, starting at 5 weeks of age for a total of 17 weeks (∼22 weeks of age). Both male and female mice were used for the studies, and results are reported as aggregate data from both sexes. Because female mice have been shown to have higher muscle regenerative capacity (50), to prevent a sex-specific biasing, we normalized the COPD animal 5-ethynyl-2’-deoxyuridine (EdU) incorporation assay as the percentage of the WT same-sex counterpart and conducted the transplantation assays using same-sex donors and using, in the same receiving animal, one tibialis anterior (TA) muscle for COPD-donated cells and the contralateral TA for WT-donated cells. Food and water were accessible ad libitum, and a 12-hour light/dark cycle was maintained by our Animal Resource Facility. Samples were collected directly after euthanasia by cervical dislocation. The time elapsed between euthanasia and sample procurement never exceeded 3 minutes when methodologically feasible.
COPD model (validation)
Six lung–cell-derived MMP1 (matrix metalloproteinase-1) transgenic mice (22 weeks old), generated as previously described (51), were used directly upon import from Columbia University for specific characterization of muscle function readouts and EdU incorporation assay.
βActin-DsRed
Transgenic mice expressing red fluorescent protein (hence named “Red+/+”) variant DsRed.MST under the control of the chicken β-actin promoter coupled with the cytomegalovirus immediate-early (CMVie) enhancer (52) were purchased from the Jackson Laboratory (Stock No: 005,441) and used for transplantation experiments in the hemizygous form.
GFP-LC3
Mice expressing GFP (EGFP)–LC3 cassette inserted between the CAG promoter (CMVie enhancer and chicken β-actin promoter) on a C57BL background were purchased from RIKEN Bio-Resource Center (#BRC00806). Genotype was determined as previously reported and confirmed by direct visualization of the GFP puncta (53).
GFP-Pax7
An animal expressing an internal ribosome entry site (IRES)-CreERT2 fusion protein downstream the Pax7 stop codon (Jackson Lab, stock no: 017,763) was crossed with a loxP-flanked STOP cassette holding animal (Jackson Lab, stock no: 007,906) to obtain a double homozygous animal (Pax7-Cre+/+Lox-GFP+/+). The double homozygous animal was then crossed with the hemizygous IL13TG animal to obtain both IL13TG and IL13WT GFP–Pax7-reporting littermates for experiments (54).
Statistics
Data are expressed as the means ± SE unless otherwise indicated in figure legends. When results were compared with a reference value, we used a single-sample t test; when comparisons were performed between two groups, significance was evaluated by a Student’s t test; and when more than two groups were compared, ANOVA was used, followed by the Holm-Sidak test using GraphPad Prism software. Results were considered significant when P < 0.05.
Data Availability
The datasets generated and analyzed during the current study are available via accession number GSE180261.
Further information on materials and methods is available in the data supplement.
Results
Inducible Pulmonary Emphysema Causes Muscle Dysfunction and an Autophagy Dysregulation Signature Reminiscent of Patients with COPD
To define whether our selected animal model of genetically driven pulmonary emphysema/COPD is an adequate platform to investigate skeletal myogenesis, we provided doxycycline to induce IL13TG (COPD) mice and compared these with doxycycline-treated IL13WT (WT) counterparts. Lung macroscopy demonstrated evidence of hyperinflation as reflected by larger organ size, which was corroborated by microscopic examination of lung sections, which showed evidence of airspace enlargement in IL13TG compared with IL13WT animals (Figures 1A, 1B1, and 1B2). Oxygen saturation at room air demonstrated that IL13TG mice are hypoxemic as opposed to IL13WT animals, which are not (Figure 1C). Moreover, muscle force-generation capacity as measured by the in vivo grip strength test and ex vivo isolated extensor digitorum longus muscle contractility test demonstrated evidence of skeletal muscle dysfunction in IL13TG mice relative to WT counterparts (Figures 1D and 1E and Figures E1A and E1B in the data supplement). Surrogates of muscle atrophy such as muscle mass and the cross-sectional area (CSA) of individual fibers were also significantly reduced in IL13TG compared with IL13WT animals (Figures 1F and 1G–1I). Moreover, consistent with previously reported human data (40, 55), glycolytic (type IIb and IIx) fibers were more affected by muscle atrophy compared with type IIa fibers (Figure E1C). To define whether muscles from COPD mice demonstrate features of autophagy dysregulation, we interrogated a set of genes whose altered expressions have been previously reported in muscle biopsies from patients with COPD (38). IL13TG (COPD) mice demonstrated a similar transcript profile to that previously found in human studies (Figure 1J, see also reference [38]). Food intake, a major regulator of autophagy activation (56), was found not significantly different between WT and COPD animals (Figure E1D), which is also consistent with previous human data (1, 57). These results supported the rationale to further investigate the effect of autophagy on myogenesis using validated methods.
Figure 1.
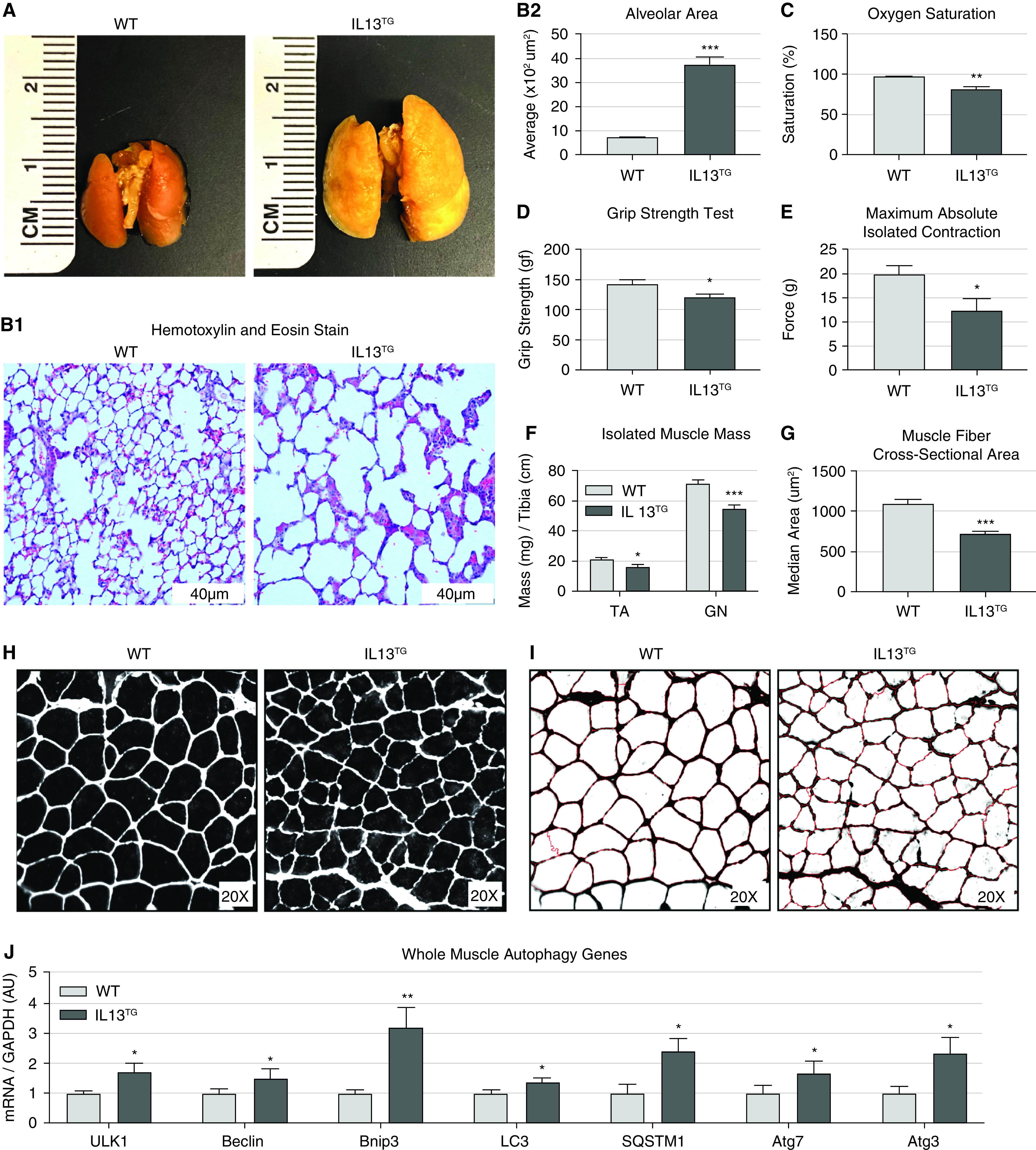
Transgenic IL13TG mice develop pulmonary emphysema followed by skeletal muscle dysfunction. (A) Lungs procured from fully induced animals exhibit a significant size increase consistent with hyperinflation. (B1) Hematoxylin and eosin staining of lung sections under magnification show damaged alveoli with increased size and irregular shape. Scale bars, 40 μm. (B2) Quantification of alveolar space in wild-type (WT) versus chronic obstructive pulmonary disease (COPD) IL13TG mice (n = 4). (C) Arterial hemoglobin oxygen saturation at room air is reduced in IL13TG mice (n = 5). (D) Emphysematous mice demonstrate reduced force-generation capacity as measured by four-limb grip strength (n = 5). (E) Isolated extensor digitorum longus (EDL) muscle stimulated maximum (absolute) contraction is reduced in IL13TG mice (n = 8). (F) Freshly isolated wet muscle mass of both tibialis anterior (TA) and gastrocnemius (GN) muscles is reduced in IL13TG mice (n = 8). (G and H) Extensor digitorum longus muscle sections from IL13TG animals demonstrate reduced fiber’s cross-sectional area (CSA) as measured automatically by CellProfiler software (n = 5). (I) CellProfiler output image files used for the CSA measurement of unbiased fibers show high fidelity with minimal methodological noise; CSA of average fiber is significantly reduced in IL13TG animals when compared with WT age-matched littermates (n = 5); error was determined by median values in this experiment. (J) Real-time PCR panel of autophagy genes in TA muscle shows a dysregulation profile similar to previous clinical data (n = 8). *P < 0.05, **P < 0.01, and ***P < 0.001. AU = arbitrary unit.
Pulmonary Emphysema Causes Suboptimal Myogenic Capacity
To investigate the myogenic response of IL13TG (COPD) mice in comparison with IL13WT (WT) counterparts, we conducted a two-wave barium chloride injury–repair assay (33), a method that has been previously found to better resolve the satellite cell’s contribution to myogenesis (58). At sequential time points after the second injury, animals were killed, and TA muscle histology was analyzed. COPD mice demonstrated a more disorganized early repair than WT littermates (Figure 2A1). While at two weeks after injury, both COPD and WT mice demonstrated recovery of fibers integrity, the CSA of the COPD mice was significantly reduced compared to the WT littermates (Figure 2A2). To further analyze the regenerative process, we interrogated the mRNA expression level of desmin, an intermediate filament protein highly expressed in immature muscle fibers during fetal life and regeneration (59), and the canonical surrogate of muscle differentiation embryonic myosin heavy chain (eMHC) (60) at 4 days after injury; and found them to be not different between the muscles of WT and COPD mice (Figure 2B). Moreover, we performed immunostaining analyses for these proteins and found no appreciable staining difference between WT and COPD mice muscle staining (Figure 2C).
Figure 2.

Emphysematous (COPD) mice demonstrate dysfunctional myogenesis. (A) Schematic of barium chloride treatment; TA muscles from WT and IL13TG animals were analyzed by hematoxylin and eosin (H&E) staining on Days 4, 8, 12, and 14 after injury. IL13TG TA muscle recovery at Day 14 as determined by CSA of fiber remains significantly below baseline muscle CSA when compared with the individual’s counter-lateral leg TA muscle CSA (n = 3). (B) Analysis of gene expression in pre- and fourth day post-injury TA muscle in WT and IL13TG animals. qRT-PCR revealed that expression of desmin and embryonic myosin heavy chain (eMHC) is not significantly affected by the genotype (n = 3). (C) Cryosections obtained from post-injury Day 4 TA muscles in WT and IL13TG animals and immunostained for desmin and eMHC demonstrate similar expression patterns among both genotypes. Scale bars, 200 μm. *P < 0.05.
Pulmonary Emphysema Leads to Reduced Proliferation but not Differentiation of Myogenic Satellite Cells
Satellite cells contribute majorly to muscle repair after injury (7). To participate in myogenesis, satellite cells undergo a few cycles of “symmetrical” cell division that maintain their undifferentiated phenotype, followed by an “asymmetrical” division that commits them to the myogenic lineage (8). To score the replicative capacity of satellite cells isolated from COPD versus WT mice, we conducted an EdU incorporation assay (33). This method, based on EdU incorporation to the DNA every time cells undergo a division cycle, has been previously calibrated to resolve the replicative capacity of satellite cells at 40 hours after their isolation (61). We observed that satellite cells isolated from COPD mice demonstrated a significantly lower replication capacity than WT mice (Figure 3A). To rule out the possibility that the overexpression of IL13 in our COPD mouse model contributed to the dysfunctional satellite cell replication independently of COPD per se, we used a second established animal model of COPD based on interstitial collagenase (MMP1) expression (51). These MMP1TG (COPD) mice, which also develop features of muscle dysfunction (Figures E2A and E2B), demonstrate a reduced EdU incorporation in comparison with MMP1WT (WT) counterparts (Figure 3B). As these MMP1TG mice validated the concept of dysfunctional satellite cell activation/replication in genetically induced pulmonary emphysema, the following experiments were conducted with IL13TG and IL13WT mice, which served as the primary model in this research.
Figure 3.

Pulmonary emphysema leads to reduced proliferation but not differentiation of myogenic satellite cells. (A) Fully induced IL13TG animals have significantly reduced satellite cell proliferation capacity as evaluated by 5-ethynyl-2’-deoxyuridine assay (n = 4). (B) Emphysematous MMP1TG mice show a significant reduction in myogenic cell proliferation (n = 4). (C) Integrin α7-positive cells isolated using magnetic-activated cell sorting (MACS) cell separation technique showed similar counts between IL13TG and WT animals (n = 4). (D) Pax7-GFP autofluorescent muscle stem cells counted in situ using automatic cell counting showed no significant difference between IL13TG and WT mice (n = 4). (E) qRT-PCR revealed that expression of myogenin, skeletal muscle actin, MyoD, Myf5, desmin, and eMHC is generally reduced in proliferating (Day 4) myotubes obtained from IL13TG versus WT mice animals, with MyoD, MyF5, and eMHC reaching statistical significance (n = 3). (F) Myoblasts obtained from IL13TG and WT mice and plated in equal numbers demonstrate similar differentiation when cultured for 5 days in differentiation media supplemented with 2% horse serum. Scale bars, 100 μm. (G) Myotubes obtained from IL13TG and WT mice and cultured in differentiation media (Day 4) were immunostained for desmin and skeletal myosin (My-32) and demonstrated similar expression patterns. Scale bars, 500 μm. *P < 0.05 and ***P < 0.001.
To define whether the COPD phenotype is associated with a reduced number of satellite cells, we quantified epitope-specific (α7-integrin positive)-isolated cells (62), which were not significantly different between COPD and WT phenotypes (Figure 3C). Given that isolation of satellite cells disrupts their niche and causes activation (8), we also scored the number of quiescent satellite cells in COPD and WT animals. As the transcription factor Pax7 is a canonical marker of quiescent satellite cells and is required for their normal function (58), we conducted an unbiased analysis of fluorescent Pax7-positive cells in uninjured muscle sections. To do that, we crossed the COPD and WT mice with an animal holding an inducible GFP reporter in satellite cells (IRES-CreERT2 cassette) inserted after the endogenous termination codon of the Pax-7 gene (54). This model, which captures ∼95% of Pax7-positive cells (54), was unbiasedly analyzed, accounting for more than 1,500 fibers per section. Fluorescent Pax7-positive cell numbers were not significantly different between COPD and WT mice (Figure 3D and Figure E3). To investigate the possibility of a major myoblast differentiation defect in COPD mice, we cultured myoblasts obtained from COPD and WT counterparts. Cells were then exposed to differentiation media to induce their fusion and myotube formation. We then conducted quantitative PCR experiments to determine mRNA expression levels of myogenic factors including myogenin, MyoD, Myf5, desmin, and eMHC and found these to be downregulated in COPD-obtained differentiating myotubes compared with WT counterparts. MyoD, Myf5, and eMHC downregulation in COPD reached statistical significance, although all the remaining markers trended similarly without reaching statistical significance (Figure 3E). To appreciate the transition from myoblast into myotubes, we followed cell differentiation serially on consecutive days and found that COPD and WT-obtained myoblast underwent an unremarkable transformation into myotubes (Figure 3F). Moreover, we performed immunofluorescence assays on the differentiating myoblasts using primary antibodies against desmin and the canonical marker of muscle differentiation myosin heavy chain 1 (My32) (59) (Figure 3G) and found them to be not differently expressed in myotubes obtained from WT versus COPD mice.
Taken together, these data suggest a suboptimal myogenic response in COPD animals, which is associated with a replicative dysfunction but not a reduced number or apparent defective differentiation of satellite cells.
COPD Leads to an Intrinsic Dysfunction of Satellite Cells
To define whether the described myogenic dysfunction is driven by an intrinsic satellite cell’s deficiency or instead by environmental factors determined by the COPD milieu, we conducted transplantation experiments with lineage tracing assays. To do that, we generated COPD and WT mice constitutively expressing red fluorescence protein (RFP) in skeletal muscle and satellite cells. These animals, resulting from the crossing of IL13TG and IL13WT mice with an RFP-expressing mouse under the control of the β-actin promoter (63), were used to obtain 50,000 satellite cells, which were then freshly transplanted into receiving animals with either COPD or WT phenotype (Figure 4A) (64). Given that freshly isolated satellite cells contribute to muscle repair by fusing with preexisting mature fibers or with other satellite cells (8, 64), the RFP-expressing fibers after muscle repair in this model are a specific surrogate of transplanted satellite cell-driven myogenesis (52). Two weeks after transplantation, mice were killed, and RFP-positive fibers were counted, indicating that IL13WT-derived cells were able to contribute to a larger number of fibers even when transplanted into a COPD animal, whereas IL13TG-derived satellite cells participated in neofibers formation in a substantially lower number even if transplanted into a healthy animal (Figure 4B). Phase-contrast images indicate that transplantation experiments do not prevent full repair of muscle integrity (Figure E4). These data suggest that cells donated from COPD animals carry an intrinsic defect that could contribute to dysfunctional myogenesis independently of the COPD muscle environment.
Figure 4.

Transplantation experiments with linage tracing reveal that cells donated from COPD mouse donors have altered myogenic capacity. (A) Cartoon illustrates transplant–experimental design. (B1) WT recipients receiving a donation of red-IL13TG cells in one TA muscle and red-WT cells in the counter-lateral TA muscle show a significantly reduced number of red muscle fibers after repair in IL13TG receiving leg when compared with the counter-lateral WT receiving leg (n = 4). (B2) Similar experiments using IL13TG recipients reveal significant reduction (n = 5). Scale bars, 100 μm. *P < 0.05 and **P < 0.01.
COPD Causes a Deacceleration of Autophagy Flux in Freshly Isolated Satellite Cells
To gain mechanistic insight into the regulation of satellite cell dysfunction in COPD, we freshly isolated post-injury cells and sorted them with fluorescence-activated cell sorting, as previously established (65); and processed them for RNA sequencing analysis. As satellite cells demonstrate a conspicuous reduction of cell division, we queried for ontology terms associated with that process and found that ∼75% of the captured genes belonging to cell cycle (GO:0007049) and cell division (GO:0051301) ontology terms were dysregulated in COPD in comparison with WT littermates. Moreover, given that autophagy plays a central role in satellite cell activation and symmetrical cell division (33, 34), we specifically queried for this process and found that ∼85% of the captured genes belonging to autophagy (GO:006914) and regulation of autophagy (GO:0010506) were downregulated in our analysis (Figure 5A, Figure E5, Tables E1, E2 and E5, and GEO accession number GSE180261). Expression level of Beclin1, which is a central regulator of autophagy (66) and has previously described as upregulated in skeletal muscle biopsies of COPD patients (38), was found increased in isolated satellite cells from COPD mice in comparison with WT littermates (Figures 5B and 5C). To directly investigate the potential role of autophagy dysregulation in COPD mice satellite cells, we crossed IL13TG and IL13WT mice with an animal expressing an enhanced GFP sequence fused with the LC3 family proteins, which retain constitutive fluorescence of autophagosomes after their formation until these fuse with lysosomes (67).
Figure 5.

Post-injury RNA sequencing of isolated muscle satellite cells shows dysregulation of multiple transcripts related to cell cycle and autophagy in the IL13TG animal compared with WT counterparts. (A) Heat map of gene expression shows the direction of differential gene expression in IL13TG versus WT cells (n = 5). Red indicates upregulated gene expression, and blue indicates downregulated gene expression. (B) Freshly isolated satellite cells from IL13TG and WT mice were lysed and sampled for western blot analysis, and membrane probed for anti-beclin1 antibody; ponceau red staining was used as a lane loading control. (C) Densitometric comparison of Beclin1 expression levels demonstrates upregulation of that product in IL13TG compared with WT mice (n = 4). (D) LC3-GFP/IL13TG reporter mice show an abnormal buildup of puncta compared with WT mice. Mean fluorescent intensity (MFI) generated by LC3-GFP puncta measured in 1,000 cells per animal show a significant increase in MFI in IL13TG compared with WT counterparts, indicating a dysfunctional autophagosome turnover (n = 4). After bafilomycin administration, the satellite cells of IL13TG mice retained the ability to further increase MFI but with a slower rate than WT counterparts (WT increase of 846.8 [±122.4]; COPD increase of 443.8 [±131.2]; genotype effect on MFI: P = 0.007; bafilomycin effect on MFI: P < 0.0001; interaction: P = 0.04). (E) High magnification image of freshly isolated satellite cells from IL13TG and WT mice without bafilomycin treatment demonstrate higher GFP puncta in IL13TG mice cells and cytosolic localization of green fluorescence. Nuclear compartment was stained with DAPI. *P < 0.05 and ***P < 0.001. Readers may view the uncut gels for B in the data supplement.
In this model, autophagosome mass can be unbiasedly scored in isolated cells by determining the mean fluorescence intensity (MFI) generated by GFP puncta (33, 53, 56). Satellite cells isolated from COPD and WT mice were then analyzed, which revealed an increased MFI in satellite cells obtained from COPD mice (Figure 5D, first two columns). These data strongly suggest dysregulation of autophagy in satellite cells from COPD mice, which is characterized by an increased number of autophagosomes. However, as the increase in puncta-driven fluorescence could be due to either an increase in autophagosome formation or instead by reduced turnover causing accumulation of unfused autophagosome (53, 56), these experiments did not reveal the relative acceleration of autophagy flux of COPD satellite cells in comparison WT counterparts. To address that specific point, we treated isolated satellite cells from LC3–GFP-expressing COPD and WT mice with the autophagy-flux inhibitor bafilomycin, a vacuolar-type H+-translocating ATPase inhibitor, which prevents lysosome degradation, thus increasing punctate GFP-LC3 exclusively when autophagy is active (20, 68).
These experiments indicated that while satellite cells from COPD mice retained active flux and thus the ability to build up autophagosomes, the rate of formation was significantly slower than the one demonstrated by the WT mice (WT increase of 846.8 [±122.4]; COPD increase of 443.8 [±131.2]; genotype effect on MFI: P = 0.007; bafilomycin effect on MFI: P < 0.0001; interaction: P = 0.04) (Figures 5D and 5E). As the baseline COPD of mice satellite cell MFI is elevated compared with WT counterparts, we entertained the possibility that the significantly slower buildup of GFP puncta in COPD-derived cells could be artifactually caused by a “roofing” effect, which would prevent further increase of MFI due to saturation of fluorescent recruitable units. To address that possibility, we analyzed the distribution of MFI of the entire cell population used to score the presented values and found that both COPD and WT-derived cells generate mean values below the maximal captured fluorescence, thereby ruling out the system’s saturation (Figure E6). Importantly, while autophagy flux has not been previously investigated in COPD muscles, a similar profile of autophagosome accumulation and Beclin1 upregulation has been previously found in locomotor muscle biopsies from COPD patients (38, 40).
Spermidine Administration Leads to an Improved Satellite Cell Replication Capacity and Myogenesis After Transplant
We then sought to determine whether pharmacologic autophagy induction can attenuate the replicative and myogenic deficits of satellite cells in genetically induced COPD. COPD mice are chronically debilitated (44); thus, to reduce the potential bias entailed by the administration of a toxic drug due to off-target effects, we chose the lower toxicity drug spermidine, which is an inhibitor of the autophagy antagonist acetyltransferase EP300 (69) and has been previously shown to improve myogenesis (20). To interrogate the effect of spermidine on COPD mice autophagy, we provided the drug in drinking water to LC3–GFP-expressing COPD and WT mice. Satellite cells were later isolated, and MFI generated by GFP puncta was scored, which demonstrated values not significantly different among COPD and WT mice, suggesting a normalization of autophagosome turnover (Figures 6A and 6B). Interestingly, while spermidine led to an increase in MFI WT satellite cells, it caused a slight but statistically significant reduction of it in the satellite cells of COPD animals, suggesting that spermidine likely caused an improvement in autophagosome-lysosome fusion, a step associated with a fluorescence reduction due to autophagy turnover (53) (Figures 6A and 6B). To define whether that effect correlated with a better replication capacity, we conducted an EdU incorporation assay. Spermidine administration led to an improvement of replication capacity of freshly isolated satellite cells from COPD mice, reflected by EdU incorporation, which became not significantly different from that of WT animals (Figure 6C). To determine if spermidine leads to a better myogenic potential of satellite cells, we provided it to COPD and WT RFP-expressing mice and then conducted transplantation experiments using freshly isolated satellite cells as previously described. Cells obtained from spermidine-provided COPD mice recovered myogenic potential as demonstrated by the development of red fibers in the receiving WT mice (Figure 6D, see transplantation cartoon). These data suggest that the autophagy-boosting drug spermidine improves the autophagosome turnover of satellite cells, replication capacity, and myogenic potential in an animal model of COPD. A summary of the described findings is presented in Figure 7.
Figure 6.
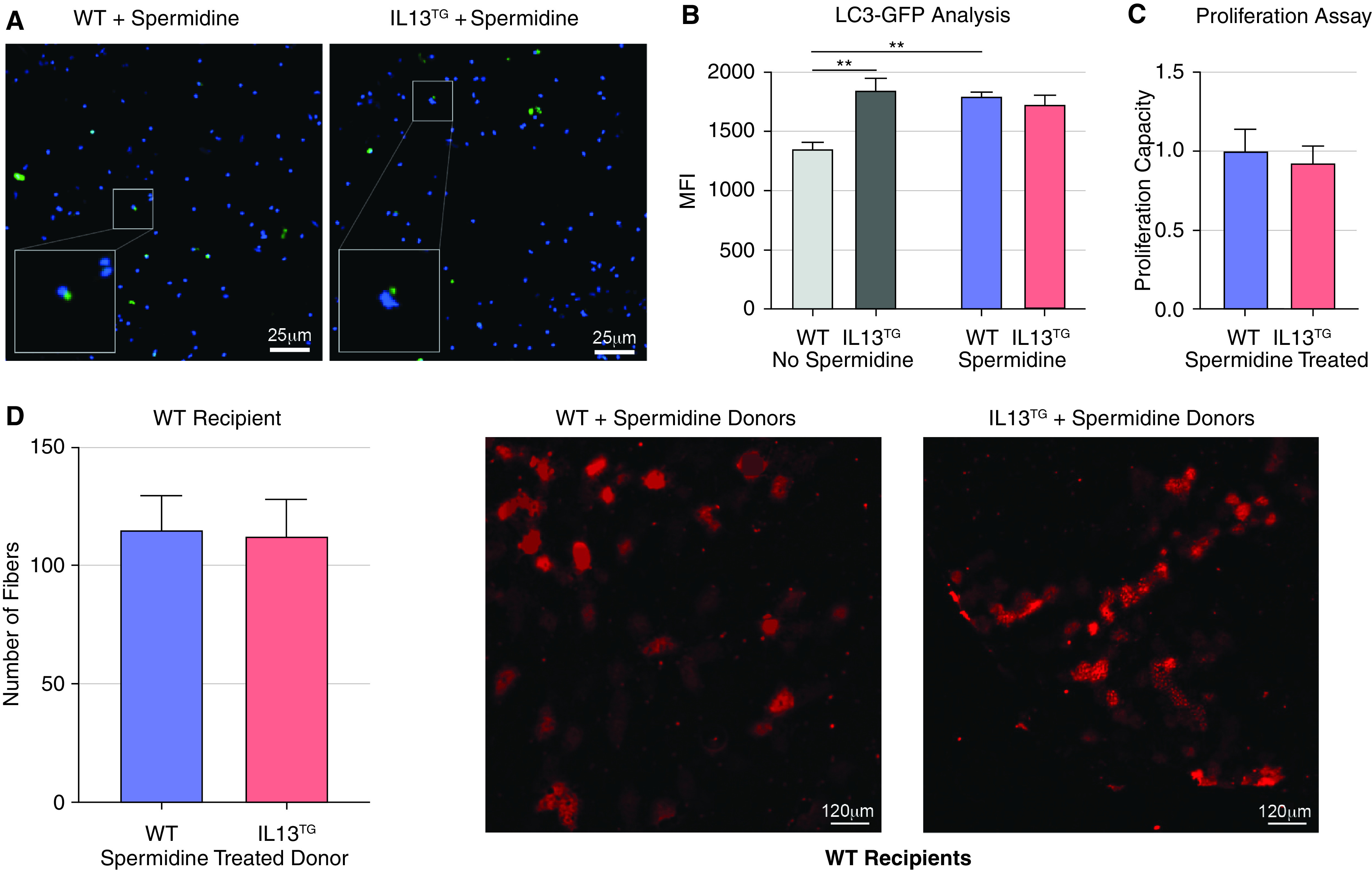
Spermidine-induced autophagy improves myogenesis in COPD mice. (A and B) LC3-GFP/IL13TG reporter mice show normalization of puncta buildup and MFI in isolated satellite cells after treatment with spermidine (n = 4). (C) 5-Ethynyl-2′-deoxyuridine proliferation assay shows no difference between IL13TG and WT animals after treatment with spermidine (n = 4). (D) Transplantation experiments in IL13WT recipients conducted as previously described showed no difference in the number of recovered red fibers when both IL13TG and IL13WT donors were treated with spermidine (n = 4). Scale bars, 120 μm. **P < 0.01. Readers may view the uncut gels for D in the data supplement.
Figure 7.

A proposed schematic model showing that the COPD lung phenotype leads to altered autophagy turnover, which triggers myogenic dysfunction and undermines muscle repair, contributing to muscle wasting over time.
Discussion
While autophagy dysregulation is conspicuous in locomotor skeletal muscles from COPD patients (38, 40), it is unclear if that abnormality plays a causal role in COPD myopathy (39). For instance, while autophagy activation leads to muscle integrity preservation (70), it has also been associated with muscle loss (41); and an optimal autophagic balance seems to be needed to maintain muscle mass (71). Myogenesis, which contributes to skeletal muscle integrity (58), depends in part on autophagy activation, which provides the bioenergetic needs for the activation of satellite cells and prevents their transition into senescence (17, 20, 33–37). We report here that myogenic capacity and replication of satellite cells are both reduced in genetically induced pulmonary emphysema. Although relatively unexplored in nonprimarily muscle diseases, myogenesis is emerging as an important mechanism regulating muscle integrity in COPD and other pulmonary conditions (18, 21–28). During an injurious event, satellite cells undergo an initial phase of activation followed by symmetrical replication, giving rise to similar daughter cells, which expand the pool of myogenic cells (72). After that, asymmetric division in an apical-basal orientation gives rise to two daughter cells, only one of which activates the transcription factor Myf5 and commits to myogenic differentiation (8). Our data suggest that it is the symmetrical cell division that is primarily disrupted in the COPD muscle response to injury. Intriguingly, mRNA levels of MyoD, Myf5, and eMHC were significantly downregulated in cultured COPD differentiating myotubes compared with WT counterparts. This finding did not correlate with an obvious morphologic alteration in the myoblast–myotube transition between genotypes and could represent a response to an abnormal cell division earlier in the activation process or to an abnormality not captured by the ex vivo myotube formation assays. Importantly, the disconnect between expressed mRNA products corresponding to the myogenic lineage, most notably MyoD, has long been characterized on other models (73). Future studies using more specific tools to interrogate asymmetrical cell division, such as the Myf5-YFP reported mouse (74), will be needed to address that point.
Myogenesis is highly influenced by the interaction between progenitor cells and their environment, including the muscle stem cell niche (8). Our combination of satellite cell transplantation and lineage tracing assays supported a dominant role of intrinsic stem cell dysfunction, which led to RNA sequencing analysis of these cells. As reduced satellite cell division is a feature of COPD mice, we queried for multiple genes that associate with the observed replicative dysfunction. Indeed, we found various genes related to cell division to be dysregulated in the COPD animal satellite cells. Moreover, as the replication of satellite cells has been extensively associated with autophagy, we queried and found multiple autophagy-associated genes to be dysregulated in the RNA sequencing analysis and investigated this process with specific surrogates. LC3 family proteins involving MAP1-LC3, GATE-16, and GABARAP, which are mammalian counterparts of yeast Atg8, are present on complete autophagosomes and degraded after autophagosome fusion with lysosomes (56, 75). We used a GFP-fused LC3-reporting animal to track autophagy turnover on freshly isolated satellite cells, which demonstrated an increased MFI in cells procured from the COPD animal in comparison with WT counterparts (53). That observation could represent either an acceleration of baseline autophagosome generation or reduced autophagosome–lysosome fusion, leading to an accumulation of GFP-LC3 signal. Remarkably, a similar signature of accumulated autophagosomes has been described in muscle biopsies from patients with COPD (38, 40). Moreover, in patients with COPD, the number of accumulated autophagosomes was found to be correlated to the severity of lung dysfunction as reflected by the reduction in the forced expiratory volume in the first second (38).
GFP-LC3 puncta indicates either enhancement of autophagosome formation (autophagy induction) or a decrease in autophagosome turnover. If autophagosome–lysosome fusion is blocked, GFP-LC3 puncta increases (53). In contrast, very rapid fusion of autophagosomes with lysosomes results in a smaller number of GFP-LC3 dots, which underestimates autophagic activity (53). Therefore, detection of puncta steady-state levels is not sufficient to determine autophagy dynamics, and analysis of autophagy flux is required to characterize the differences between WT and COPD genotypes. We conducted that analysis using the autophagy-flux inhibitor bafilomycin, a vacuolar-type H+-translocating ATPase inhibitor, which prevents lysosome degradation, thus increasing punctate GFP-LC3 exclusively when autophagy is active (20, 68). These experiments indicated that while satellite cells from COPD mice retained the ability to build up autophagosomes, the rate of formation was significantly slower than the one demonstrated by the WT mice.
To further determine whether autophagy deacceleration in satellite cells contributed to a reduced proliferative capacity and myogenic potential, we treated mice with spermidine, a drug known to upregulate autophagy flux and myogenesis (20, 37). Although spermidine induces autophagy via the inhibition of acetyltransferase EP300 (69, 76), and does not affect the phosphorylation of mTOR or its substrate ribosomal protein S6 kinase (77), the specific mechanism of spermidine-induced autophagy in COPD mice will be a matter of future research. Given that autophagy is also dysregulated in the muscle fiber, the beneficial effect of spermidine could, at least partially, occur indirectly via its effect on autophagosome turnover at the nonsatellite cell level. Future studies using a satellite cell’s specific autophagy knockout animal (20) will address whether autophagy integrity in the satellite cell compartment is required for spermidine or other drugs to cause improved myogenesis in pulmonary emphysema. Importantly, spermidine is remarkably less toxic and immunosuppressive than rapamycin (78), making it an attractive target to investigate its potential myogenic benefits in chronically debilitated COPD patients.
While most of the research conducted in the field of COPD-associated muscle dysfunction has so far focused on the conspicuous reduction of muscle mass (1, 4–6, 44, 45, 48, 79–83), to our knowledge, this is the first mechanistic study that addresses autophagy-driven myogenesis in the context of pulmonary emphysema. The fact that the MMP1 model of COPD, which is bred on a non-C57 background and develops the emphysema phenotype via different mechanisms (51), also demonstrates skeletal muscle dysfunction and reduced replication of satellite cells suggests that it is the pulmonary emphysema per se that drives the myogenic dysfunction. It is important to emphasize that the MMP1 holds a constitutive transgene, and thus developmental myogenesis could be disrupted before the COPD phenotype fully develops (47). Due to this important limitation, the MMP1 line serves only as a validation, but not the primary focus, of the explored mechanisms.
The upstream mechanism regulating dysfunctional autophagy in this model remains to be elucidated. IL13TG mice develop hypoxemia, and it has been recently reported that exposure of myoblasts to hypoxia leads to an increase in histone deacetylase C9 (HDAC9) expression, which deacetylates H3K9 of autophagy-related genes and thus epigenetically inhibits autophagosome formation (84). Low oxygen could also contribute to dysfunctional myogenesis via hypoxia-inducible factor 1α, which inhibits canonical Wnt signaling, thereby undermining satellite cell function (85). The COPD environment dysregulates the inflammatory microenvironment in skeletal muscle, which could impact the satellite cell’s niche, leading to an altered myogenic response (86, 87). Interestingly, while downregulation of muscle succinate dehydrogenase (SDH) has been consistently shown in COPD patients (88–90), we have recently reported that reduced expression of SDH subunit C contributes to muscle metabolic dysfunction in IL13TG mice (49). SDH dysfunction leads to succinate accumulation (49), which has recently been shown to influence the regulation of cyclin-dependent kinase 2-driven phosphorylation in satellite cells via succinate receptor SUCNR1 (91). As cyclin-dependent kinase 2 is a critical regulator of cell division, succinate could articulate metabolic and myogenic dysregulations in this context.
Importantly, the observed COPD-induced dysfunctional myogenesis demonstrates distinct features in comparison to other nonprimarily muscular diseases. For instance, cancer cachexia causes dysfunctional myogenesis via NF-kappa B-mediated Pax7 dysregulation, which is associated with an increased replication and abnormal differentiation of satellite cells (18), in contrast to our findings of reduced satellite cell replication. Also, while the number of myogenic cells in the COPD animal is not significantly different from the WT counterparts, dysfunctional myogenesis associated with advanced age is associated with a p16INK4a-regulated transition from quiescence to senescence with a reduction in the number of satellite cells (17). A recent report also suggests a reduced number of satellite cells in skeletal muscles from patients surviving critical illness and developing muscle wasting (22, 92).
The mechanism articulating autophagy deacceleration with reduced satellite cell replication and myogenesis in pulmonary emphysema also remains to be elucidated. Previous research strongly indicates that inhibition of autophagy suppresses ATP levels needed for the activation of satellite cells, which is partially rescued by exogenous pyruvate supply as an energy source (33). Moreover, autophagy is necessary to maintain the quiescent state of satellite cells, which is indispensable for their transition into an activated form upon muscle injury (20). The interaction between mitochondrial integrity and autophagy flux has been demonstrated in other models (35–37) and could also be relevant here. Future studies analyzing the substrate imbalances in COPD mouse satellite cells could provide relevant data in that regard.
We realize this study has the limitation of being based on a murine lung-specific conditional gene overexpression leading to pulmonary emphysema phenotype. While IL13 regulates the pathophysiology of pulmonary emphysema (93) and correlates with disease severity in that setting (94), IL13 is not described to cause muscle toxicity independently of the COPD phenotype. Very significantly, while IL4/IL13 signaling has been found to promote muscle trophism and not dysfunction (95, 96) at the expense of higher myogenic capacity, this COPD animal model demonstrates reduced satellite cell replication rate, suggesting that IL13 is unlikely to cause the reported myogenic phenotype. Indeed, this model does not demonstrate significant muscle loss until the pulmonary emphysema develops, at 8 weeks after the transgene induction start (44, 45). However, the possibility of muscle toxicity driven by IL13 independently of the pulmonary emphysema development cannot be fully excluded, as it has recently been reported on lung stem cell activity (97). Also, although, as previously mentioned, cigarette smoking was deliberately excluded from this model, we realize that most patients with advanced COPD are current or previous smokers, and, thus, the lack of this signal in the present research constitutes a limitation that can be addressed in future research.
Conclusions
Skeletal myogenesis is dysfunctional in animal models of COPD, which is associated with deaccelerated autophagy in satellite cells and attenuated by systemic administration of spermidine.
Footnotes
Supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under the award number HL130704 and HL160661 (A.J.); HL086936 (J.D’A.); HL049426 (H.A.S.); HL114501 (J.A.E.); and HL115813 and HL155558 (C.G.L.).
Author Contributions: J.B., L.A.D., D.V.S., T.C.K., and A.J. designed and performed experiments. J.B., C.E.V., J.D’A., C.G.L., J.A.E., H.A.S., and A.J. designed the experiments and wrote the current manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0351OC on March 14, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Jaitovich A, Barreiro E. Skeletal muscle dysfunction in chronic obstructive pulmonary disease. What we know and can do for our patients. Am J Respir Crit Care Med . 2018;198:175–186. doi: 10.1164/rccm.201710-2140CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maltais F, Decramer M, Casaburi R, Barreiro E, Burelle Y, Debigaré R, et al. ATS/ERS Ad Hoc Committee on Limb Muscle Dysfunction in COPD An official American Thoracic Society/European Respiratory Society statement: update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2014;189:e15–e62. doi: 10.1164/rccm.201402-0373ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vanfleteren LE, Spruit MA, Groenen M, Gaffron S, van Empel VP, Bruijnzeel PL, et al. Clusters of comorbidities based on validated objective measurements and systemic inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2013;187:728–735. doi: 10.1164/rccm.201209-1665OC. [DOI] [PubMed] [Google Scholar]
- 4. Marquis K, Debigaré R, Lacasse Y, LeBlanc P, Jobin J, Carrier G, et al. Midthigh muscle cross-sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2002;166:809–813. doi: 10.1164/rccm.2107031. [DOI] [PubMed] [Google Scholar]
- 5. Shrikrishna D, Patel M, Tanner RJ, Seymour JM, Connolly BA, Puthucheary ZA, et al. Quadriceps wasting and physical inactivity in patients with COPD. Eur Respir J . 2012;40:1115–1122. doi: 10.1183/09031936.00170111. [DOI] [PubMed] [Google Scholar]
- 6. Swallow EB, Reyes D, Hopkinson NS, Man WD, Porcher R, Cetti EJ, et al. Quadriceps strength predicts mortality in patients with moderate to severe chronic obstructive pulmonary disease. Thorax . 2007;62:115–120. doi: 10.1136/thx.2006.062026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol . 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev . 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu N, Garry GA, Li S, Bezprozvannaya S, Sanchez-Ortiz E, Chen B, et al. A Twist2-dependent progenitor cell contributes to adult skeletal muscle. Nat Cell Biol . 2017;19:202–213. doi: 10.1038/ncb3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Englund DA, Murach KA, Dungan CM, Figueiredo VC, Vechetti IJ, Jr, Dupont-Versteegden EE, et al. Depletion of resident muscle stem cells negatively impacts running volume, physical function, and muscle fiber hypertrophy in response to lifelong physical activity. Am J Physiol Cell Physiol . 2020;318:C1178–C1188. doi: 10.1152/ajpcell.00090.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med . 2008;359:2355–2365. doi: 10.1056/NEJMra0800353. [DOI] [PubMed] [Google Scholar]
- 12. Puthucheary ZA, Hart N. Skeletal muscle mass and mortality - but what about functional outcome? Crit Care . 2014;18:110. doi: 10.1186/cc13729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abdulai RM, Jensen TJ, Patel NR, Polkey MI, Jansson P, Celli BR, et al. Deterioration of limb muscle function during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2018;197:433–449. doi: 10.1164/rccm.201703-0615CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mason SE, Moreta-Martinez R, Labaki WW, Strand M, Baraghoshi D, Regan EA, et al. COPDGene Investigators COPDGene Investigators. Respiratory exacerbations are associated with muscle loss in current and former smokers. Thorax . 2021;76:554–560. doi: 10.1136/thoraxjnl-2020-215999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax . 2002;57:847–852. doi: 10.1136/thorax.57.10.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanabe N, Muro S, Hirai T, Oguma T, Terada K, Marumo S, et al. Impact of exacerbations on emphysema progression in chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2011;183:1653–1659. doi: 10.1164/rccm.201009-1535OC. [DOI] [PubMed] [Google Scholar]
- 17. Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature . 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- 18. He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, et al. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest . 2013;123:4821–4835. doi: 10.1172/JCI68523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Talbert EE, Cuitino MC, Ladner KJ, Rajasekerea PV, Siebert M, Shakya R, et al. Modeling human cancer-induced cachexia. Cell Rep . 2019;28:1612–1622.e4. doi: 10.1016/j.celrep.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, et al. Autophagy maintains stemness by preventing senescence. Nature . 2016;529:37–42. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- 21. Dial AG, Grafham GK, Monaco CMF, Voth J, Brandt L, Tarnopolsky MA, et al. Alterations in skeletal muscle repair in young adults with type 1 diabetes mellitus. Am J Physiol Cell Physiol . 2021;321:C876–C883. doi: 10.1152/ajpcell.00322.2021. [DOI] [PubMed] [Google Scholar]
- 22. Polkey MI, Griffiths MJ, Kemp PR. Muscle regeneration after critical illness: are satellite cells the answer? Am J Respir Crit Care Med . 2016;194:780–782. doi: 10.1164/rccm.201603-0633ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pomiès P, Rodriguez J, Blaquière M, Sedraoui S, Gouzi F, Carnac G, et al. Reduced myotube diameter, atrophic signalling and elevated oxidative stress in cultured satellite cells from COPD patients. J Cell Mol Med . 2015;19:175–186. doi: 10.1111/jcmm.12390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thériault ME, Paré ME, Maltais F, Debigaré R. Satellite cells senescence in limb muscle of severe patients with COPD. PLoS One . 2012;7:e39124. doi: 10.1371/journal.pone.0039124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sancho-Muñoz A, Guitart M, Rodríguez DA, Gea J, Martínez-Llorens J, Barreiro E. Deficient muscle regeneration potential in sarcopenic COPD patients: Role of satellite cells. J Cell Physiol . 2021;236:3083–3098. doi: 10.1002/jcp.30073. [DOI] [PubMed] [Google Scholar]
- 26. Kneppers AEM, Langen RCJ, Gosker HR, Verdijk LB, Lipovec NC, Leermakers PA, et al. Increased myogenic and protein turnover signaling in skeletal muscle of chronic obstructive pulmonary disease patients with sarcopenia. J Am Med Dir Assoc . 2017;18:637.e1–637.e11. doi: 10.1016/j.jamda.2017.04.016. [DOI] [PubMed] [Google Scholar]
- 27. Kneppers AEM, Haast RAM, Langen RCJ, Verdijk LB, Leermakers PA, Gosker HR, et al. Distinct skeletal muscle molecular responses to pulmonary rehabilitation in chronic obstructive pulmonary disease: a cluster analysis. J Cachexia Sarcopenia Muscle . 2019;10:311–322. doi: 10.1002/jcsm.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Menon MK, Houchen L, Singh SJ, Morgan MD, Bradding P, Steiner MC. Inflammatory and satellite cells in the quadriceps of patients with COPD and response to resistance training. Chest . 2012;142:1134–1142. doi: 10.1378/chest.11-2144. [DOI] [PubMed] [Google Scholar]
- 29. Chan SMH, Cerni C, Passey S, Seow HJ, Bernardo I, van der Poel C, et al. Cigarette smoking exacerbates skeletal muscle injury without compromising its regenerative capacity. Am J Respir Cell Mol Biol . 2020;62:217–230. doi: 10.1165/rcmb.2019-0106OC. [DOI] [PubMed] [Google Scholar]
- 30. Ceco E, Celli D, Weinberg S, Shigemura M, Welch LC, Volpe L, et al. Elevated CO2 levels delay skeletal muscle repair by increasing fatty acid oxidation. Front Physiol . 2021;11:630910. doi: 10.3389/fphys.2020.630910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chaillou T, Lanner JT. Regulation of myogenesis and skeletal muscle regeneration: effects of oxygen levels on satellite cell activity. FASEB J . 2016;30:3929–3941. doi: 10.1096/fj.201600757R. [DOI] [PubMed] [Google Scholar]
- 32. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med . 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 33. Tang AH, Rando TA. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J . 2014;33:2782–2797. doi: 10.15252/embj.201488278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fiacco E, Castagnetti F, Bianconi V, Madaro L, De Bardi M, Nazio F, et al. Autophagy regulates satellite cell ability to regenerate normal and dystrophic muscles. Cell Death Differ . 2016;23:1839–1849. doi: 10.1038/cdd.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature . 2017;543:205–210. doi: 10.1038/nature21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ma T, Li J, Xu Y, Yu C, Xu T, Wang H, et al. Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat Cell Biol . 2015;17:1379–1387. doi: 10.1038/ncb3256. [DOI] [PubMed] [Google Scholar]
- 37. García-Prat L, Muñoz-Cánoves P, Martinez-Vicente M. Dysfunctional autophagy is a driver of muscle stem cell functional decline with aging. Autophagy . 2016;12:612–613. doi: 10.1080/15548627.2016.1143211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo Y, Gosker HR, Schols AM, Kapchinsky S, Bourbeau J, Sandri M, et al. Autophagy in locomotor muscles of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2013;188:1313–1320. doi: 10.1164/rccm.201304-0732OC. [DOI] [PubMed] [Google Scholar]
- 39. Hussain SN, Sandri M. Role of autophagy in COPD skeletal muscle dysfunction. J Appl Physiol (1985) . 2013;114:1273–1281. doi: 10.1152/japplphysiol.00893.2012. [DOI] [PubMed] [Google Scholar]
- 40. Puig-Vilanova E, Rodriguez DA, Lloreta J, Ausin P, Pascual-Guardia S, Broquetas J, et al. Oxidative stress, redox signaling pathways, and autophagy in cachectic muscles of male patients with advanced COPD and lung cancer. Free Radic Biol Med . 2015;79:91–108. doi: 10.1016/j.freeradbiomed.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 41. Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP. Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J . 2011;25:99–110. doi: 10.1096/fj.10-164152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Evans WJ. What is sarcopenia? J Gerontol A Biol Sci Med Sci . 1995;50:5–8. doi: 10.1093/gerona/50a.special_issue.5. [DOI] [PubMed] [Google Scholar]
- 43. Kwan HY, Maddocks M, Nolan CM, Jones SE, Patel S, Barker RE, et al. The prognostic significance of weight loss in chronic obstructive pulmonary disease-related cachexia: a prospective cohort study. J Cachexia Sarcopenia Muscle . 2019;10:1330–1338. doi: 10.1002/jcsm.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Balnis J, Korponay TC, Vincent CE, Singer DV, Adam AP, Lacomis D, et al. IL-13-driven pulmonary emphysema leads to skeletal muscle dysfunction attenuated by endurance exercise. J Appl Physiol (1985) . 2020;128:134–148. doi: 10.1152/japplphysiol.00627.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Balnis J, Vincent CE, Jones AJ, Drake LA, Coon JJ, Lee CG, et al. Established biomarkers of chronic obstructive pulmonary disease reflect skeletal muscle integrity’s response to exercise in an animal model of pulmonary emphysema. Am J Respir Cell Mol Biol . 2020;63:266–269. doi: 10.1165/rcmb.2019-0439LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ, Jr, et al. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest . 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Campbell EJ. Animal models of emphysema: the next generations. J Clin Invest . 2000;106:1445–1446. doi: 10.1172/JCI11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Balnis J, Lee CG, Elias JA, Jaitovich A. Hypercapnia-driven skeletal muscle dysfunction in an animal model of pulmonary emphysema suggests a complex phenotype. Front Physiol . 2020;11:600290. doi: 10.3389/fphys.2020.600290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Balnis J, Drake LA, Vincent CE, Korponay TC, Singer DV, Lacomis D, et al. SDH subunit C regulates muscle oxygen consumption and fatigability in an animal model of pulmonary emphysema. Am J Respir Cell Mol Biol . 2021;65:259–271. doi: 10.1165/rcmb.2020-0551OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Deasy BM, Lu A, Tebbets JC, Feduska JM, Schugar RC, Pollett JB, et al. A role for cell sex in stem cell-mediated skeletal muscle regeneration: female cells have higher muscle regeneration efficiency. J Cell Biol . 2007;177:73–86. doi: 10.1083/jcb.200612094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. D’Armiento J, Dalal SS, Okada Y, Berg RA, Chada K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell . 1992;71:955–961. doi: 10.1016/0092-8674(92)90391-o. [DOI] [PubMed] [Google Scholar]
- 52. Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O. Type-1 pericytes participate in fibrous tissue deposition in aged skeletal muscle. Am J Physiol Cell Physiol . 2013;305:C1098–C1113. doi: 10.1152/ajpcell.00171.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mizushima N. Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol . 2009;452:13–23. doi: 10.1016/S0076-6879(08)03602-1. [DOI] [PubMed] [Google Scholar]
- 54. Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development . 2011;138:3625–3637. doi: 10.1242/dev.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fermoselle C, Rabinovich R, Ausín P, Puig-Vilanova E, Coronell C, Sanchez F, et al. Does oxidative stress modulate limb muscle atrophy in severe COPD patients? Eur Respir J . 2012;40:851–862. doi: 10.1183/09031936.00137211. [DOI] [PubMed] [Google Scholar]
- 56. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MdJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy . 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barreiro E, Jaitovich A. Skeletal muscle dysfunction in COPD: relevance of nutritional support and pulmonary rehabilitation. J Thorac Dis . 2018;10:S1330–S1331. doi: 10.21037/jtd.2018.05.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci USA . 2013;110:16474–16479. doi: 10.1073/pnas.1307680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu N, Nelson BR, Bezprozvannaya S, Shelton JM, Richardson JA, Bassel-Duby R, et al. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc Natl Acad Sci USA . 2014;111:4109–4114. doi: 10.1073/pnas.1401732111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schiaffino S, Rossi AC, Smerdu V, Leinwand LA, Reggiani C. Developmental myosins: expression patterns and functional significance. Skelet Muscle . 2015;5:22. doi: 10.1186/s13395-015-0046-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature . 2014;510:393–396. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sincennes MC, Wang YX, Rudnicki MA. Primary mouse myoblast purification using magnetic cell separation. Methods Mol Biol . 2017;1556:41–50. doi: 10.1007/978-1-4939-6771-1_3. [DOI] [PubMed] [Google Scholar]
- 63. Vintersten K, Monetti C, Gertsenstein M, Zhang P, Laszlo L, Biechele S, et al. Mouse in red: red fluorescent protein expression in mouse ES cells, embryos, and adult animals. Genesis . 2004;40:241–246. doi: 10.1002/gene.20095. [DOI] [PubMed] [Google Scholar]
- 64. Feige P, Rudnicki MA. Isolation of satellite cells and transplantation into mice for lineage tracing in muscle. Nat Protoc . 2020;15:1082–1097. doi: 10.1038/s41596-019-0278-8. [DOI] [PubMed] [Google Scholar]
- 65. Liu L, Cheung TH, Charville GW, Rando TA. Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat Protoc . 2015;10:1612–1624. doi: 10.1038/nprot.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nakashima K, Yakabe Y. AMPK activation stimulates myofibrillar protein degradation and expression of atrophy-related ubiquitin ligases by increasing FOXO transcription factors in C2C12 myotubes. Biosci Biotechnol Biochem . 2007;71:1650–1656. doi: 10.1271/bbb.70057. [DOI] [PubMed] [Google Scholar]
- 67. Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell . 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) (1) Autophagy . 2021;17:1–382. doi: 10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mariño G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell . 2014;53:710–725. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 70. Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab . 2009;10:507–515. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 71. Martyn JAJ, Kaneki M. Muscle atrophy and the sestrins. N Engl J Med . 2020;383:1279–1282. doi: 10.1056/NEJMcibr2003528. [DOI] [PubMed] [Google Scholar]
- 72. Feige P, Brun CE, Ritso M, Rudnicki MA. Orienting muscle stem cells for regeneration in homeostasis, aging, and disease. Cell Stem Cell . 2018;23:653–664. doi: 10.1016/j.stem.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu L, Cheung TH, Charville GW, Hurgo BM, Leavitt T, Shih J, et al. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep . 2013;4:189–204. doi: 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell . 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J . 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med . 2016;22:1428–1438. doi: 10.1038/nm.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Morselli E, Mariño G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol . 2011;192:615–629. doi: 10.1083/jcb.201008167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Madeo F, Bauer MA, Carmona-Gutierrez D, Kroemer G. Spermidine: a physiological autophagy inducer acting as an anti-aging vitamin in humans? Autophagy . 2019;15:165–168. doi: 10.1080/15548627.2018.1530929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Balnis J, Korponay TC, Jaitovich A. AMP-activated protein kinase (AMPK) at the crossroads between CO2 retention and skeletal muscle dysfunction in chronic obstructive pulmonary disease (COPD) Int J Mol Sci . 2020;21:E955. doi: 10.3390/ijms21030955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jaitovich A, Angulo M, Lecuona E, Dada LA, Welch LC, Cheng Y, et al. High CO2 levels cause skeletal muscle atrophy via AMP-activated kinase (AMPK), FoxO3a protein, and muscle-specific Ring finger protein 1 (MuRF1) J Biol Chem . 2015;290:9183–9194. doi: 10.1074/jbc.M114.625715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Korponay TC, Balnis J, Vincent CE, Singer DV, Chopra A, Adam AP, et al. High CO2 downregulates skeletal muscle protein anabolism via AMP-activated protein kinase α2-mediated depressed ribosomal biogenesis. Am J Respir Cell Mol Biol . 2020;62:74–86. doi: 10.1165/rcmb.2019-0061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jaitovich A, Dumas CL, Itty R, Chieng HC, Khan MMHS, Naqvi A, et al. ICU admission body composition: skeletal muscle, bone, and fat effects on mortality and disability at hospital discharge-a prospective, cohort study. Crit Care . 2020;24:566. doi: 10.1186/s13054-020-03276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jaitovich A, Khan MMHS, Itty R, Chieng HC, Dumas CL, Nadendla P, et al. ICU admission muscle and fat mass, survival, and disability at discharge: a prospective cohort study. Chest . 2019;155:322–330. doi: 10.1016/j.chest.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang Z, Zhang L, Zhou Y, Li L, Zhao J, Qin W, et al. Increase in HDAC9 suppresses myoblast differentiation via epigenetic regulation of autophagy in hypoxia. Cell Death Dis . 2019;10:552. doi: 10.1038/s41419-019-1763-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Majmundar AJ, Lee DS, Skuli N, Mesquita RC, Kim MN, Yodh AG, et al. HIF modulation of Wnt signaling regulates skeletal myogenesis in vivo. Development . 2015;142:2405–2412. doi: 10.1242/dev.123026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sin DD, Reid WD. Is inflammation good, bad or irrelevant for skeletal muscles in COPD? Thorax . 2008;63:95–96. doi: 10.1136/thx.2007.088575. [DOI] [PubMed] [Google Scholar]
- 87. Crul T, Spruit MA, Gayan-Ramirez G, Quarck R, Gosselink R, Troosters T, et al. Markers of inflammation and disuse in vastus lateralis of chronic obstructive pulmonary disease patients. Eur J Clin Invest . 2007;37:897–904. doi: 10.1111/j.1365-2362.2007.01867.x. [DOI] [PubMed] [Google Scholar]
- 88. Gosker HR, van Mameren H, van Dijk PJ, Engelen MP, van der Vusse GJ, Wouters EF, et al. Skeletal muscle fibre-type shifting and metabolic profile in patients with chronic obstructive pulmonary disease. Eur Respir J . 2002;19:617–625. doi: 10.1183/09031936.02.00762001. [DOI] [PubMed] [Google Scholar]
- 89. Konokhova Y, Spendiff S, Jagoe RT, Aare S, Kapchinsky S, MacMillan NJ, et al. Failed upregulation of TFAM protein and mitochondrial DNA in oxidatively deficient fibers of chronic obstructive pulmonary disease locomotor muscle. Skelet Muscle . 2016;6:10. doi: 10.1186/s13395-016-0083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jakobsson P, Jorfeldt L, Henriksson J. Metabolic enzyme activity in the quadriceps femoris muscle in patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 1995;151:374–377. doi: 10.1164/ajrccm.151.2.7842194. [DOI] [PubMed] [Google Scholar]
- 91. Reddy A, Bozi LHM, Yaghi OK, Mills EL, Xiao H, Nicholson HE, et al. pH-gated succinate secretion regulates muscle remodeling in response to exercise. Cell . 2020;183:62–75.e17. doi: 10.1016/j.cell.2020.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dos Santos C, Hussain SN, Mathur S, Picard M, Herridge M, Correa J, et al. MEND ICU Group RECOVER Program Investigators; Canadian Critical Care Translational Biology Group. Mechanisms of chronic muscle wasting and dysfunction after an intensive care unit stay. A pilot study. Am J Respir Crit Care Med . 2016;194:821–830. doi: 10.1164/rccm.201512-2344OC. [DOI] [PubMed] [Google Scholar]
- 93. Doyle AD, Mukherjee M, LeSuer WE, Bittner TB, Pasha SM, Frere JJ, et al. Eosinophil-derived IL-13 promotes emphysema. Eur Respir J . 2019;53:1801291. doi: 10.1183/13993003.01291-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lee JS, Rosengart MR, Kondragunta V, Zhang Y, McMurray J, Branch RA, et al. Inverse association of plasma IL-13 and inflammatory chemokines with lung function impairment in stable COPD: a cross-sectional cohort study. Respir Res . 2007;8:64. doi: 10.1186/1465-9921-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell . 2013;153:376–388. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Leavy O. Type 2 immunity: regenerating muscles the type 2 way. Nat Rev Immunol . 2013;13:395. doi: 10.1038/nri3460. [DOI] [PubMed] [Google Scholar]
- 97. Glisinski KM, Schlobohm AJ, Paramore SV, Birukova A, Moseley MA, Foster MW, et al. Interleukin-13 disrupts type 2 pneumocyte stem cell activity. JCI Insight . 2020;5:131232. doi: 10.1172/jci.insight.131232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and analyzed during the current study are available via accession number GSE180261.
Further information on materials and methods is available in the data supplement.