

Abstract
Urticeae s.l., a tribe of Urticaceae well-known for their stinging trichomes, consists of more than 10 genera and approximately 220 species. Relationships within this tribe remain poorly known due to the limited molecular and taxonomic sampling in previous studies, and chloroplast genome (CP genome/plastome) evolution is still largely unaddressed. To address these concerns, we used genome skimming data—CP genome and nuclear ribosomal DNA (18S-ITS1-5.8S-ITS2-26S); 106 accessions—for the very first time to attempt resolving the recalcitrant relationships and to explore chloroplast structural evolution across the group. Furthermore, we assembled a taxon rich two-locus dataset of trnL-F spacer and ITS sequences across 291 accessions to complement our genome skimming dataset. We found that Urticeae plastomes exhibit the tetrad structure typical of angiosperms, with sizes ranging from 145 to 161 kb and encoding a set of 110–112 unique genes. The studied plastomes have also undergone several structural variations, including inverted repeat (IR) expansions and contractions, inversion of the trnN-GUU gene, losses of the rps19 gene, and the rpl2 intron, and the proliferation of multiple repeat types; 11 hypervariable regions were also identified. Our phylogenomic analyses largely resolved major relationships across tribe Urticeae, supporting the monophyly of the tribe and most of its genera except for Laportea, Urera, and Urtica, which were recovered as polyphyletic with strong support. Our analyses also resolved with strong support several previously contentious branches: (1) Girardinia as a sister to the Dendrocnide-Discocnide-Laportea-Nanocnide-Zhengyia-Urtica-Hesperocnide clade and (2) Poikilospermum as sister to the recently transcribed Urera sensu stricto. Analyses of the taxon-rich, two-locus dataset showed lower support but was largely congruent with results from the CP genome and nuclear ribosomal DNA dataset. Collectively, our study highlights the power of genome skimming data to ameliorate phylogenetic resolution and provides new insights into phylogenetic relationships and chloroplast structural evolution in Urticeae.
Keywords: Urticaceae s.l., chloroplast structural evolution, phylogenomic, genome skimming, Urticaceae
Introduction
Urticaceae, commonly known as the nettle family, is a cosmopolitan group of plants comprising approximately 54 genera and ∼2,600 species circumscribed into six tribes (Boehmerieae Gaudich., Cecropiaceae Gaudich., Elatostemateae Gaudich., Forsskaoleae Gaudich., Parietarieae Gaudich., and Urticeae Lam. and DC.; Conn and Hadiah, 2009) with various distinct morphological characters (Stevens, 2017). For example, members of tribe Urticeae are well known for their stinging trichomes (Friis, 1993). Urticeae sensu Friis (1989) consists of 10 genera of vast economic importance as sources of fiber (Singh and Shrestha, 1988; Bodros and Baley, 2008; Gurung et al., 2012) medicine (Momo et al., 2006; Tanti et al., 2010; Luo et al., 2011; Benvenutti et al., 2020; Sharan Shrestha et al., 2020), and food (Di Virgilio et al., 2015; Mahlangeni et al., 2020). This generic circumscription of the Urticeae, however, was established prior to the era of molecular phylogenetics. With the advent of the molecular tools, classification within tribe Urticeae has received much attention, with both taxonomic and phylogenetic studies spurring realignments (Hadiah et al., 2008; Kim et al., 2015; Huang et al., 2019; Wells et al., 2021). Molecular analyses have led to the exclusion of Gyrotaenia and the inclusion of Touchardia, Poikilospermum and Zhengyia in the tribe; hence, Urticeae presently comprises 12 genera (Wu et al., 2013; Kim et al., 2015; Jin et al., 2019). Molecular phylogenetic studies have also been able to demonstrate the monophyly of this tribe as well as which genera are polyphyletic or monophyletic.
Although our understanding of evolutionary relationships of the tribe Urticeae has improved in recent years, some important nodes remain unresolved. For example, the phylogenetic position of Laportea remains contentious in previous studies. Wu et al. (2013), using seven combined markers from the mitochondrial, nuclear, and chloroplast genomes, recovered Laportea sister to a clade comprising Obetia-Urera-Touchardia and Poikilospermum, though with weak support (Figure 1A). Subsequent studies, however, have supported alternative, conflicting resolutions of Laportea (Figures 1B–D; Kim et al., 2015; Wu et al., 2018; Huang et al., 2019) probably due to the limited sampling. The placement of Poikilospermum also remains uncertain; although it has consistently been placed sister to Urera, support for this was either lacking (Figures 1A–C; Wu et al., 2013, 2018; Kim et al., 2015; Wells et al., 2021) or low (Figure 1D; Huang et al., 2019). The genus Hesperocnide, although supported as monophyletic in earlier studies, was recently recovered as polyphyletic by Huang et al. (2019), suggesting that further investigation of this genus may be required. Conflict concerning the placement of Girardinia further compounds taxonomic problems within Urticeae; several studies support its relationship with Dendrocnide-Discocnide, but without support (Figures 1A,B; Wu et al., 2013; Kim et al., 2015), while others (Wu et al., 2018; Huang et al., 2019) have recovered Girardinia sister to a clade comprising Dendrocnide-Discocnide-Laportea-Nanocnide-Zhengyia-Urtica-Hesperocnide, albeit also with low support (Figures 1C,D). These uncertainties around phylogenetic relationships within Urticeae are likely due to limited taxon or genic sampling in previous studies. Therefore, a broadly sampled phylogenomic study should offer useful framework for resolving these outstanding problems and guiding revised taxonomic treatments of the tribe.
FIGURE 1.

Alternative relationships of Urticeae based on combined loci data from previous analyses. (A) Wu et al. (2013): Maximum likelihood (ML)/Maximum parsimony (MP)/Bayesian inference (BI) analyses based on nuclear, chloroplast, and mitochondrial loci; (B) Kim et al. (2015): MP/BI tree inferred from chloroplast and nuclear DNA data; (C) Wu et al. (2018): ML/MP/BI tree inferred from nuclear, chloroplast, and mitochondrial data; (D) Huang et al. (2019): MP/BI analyses based on chloroplast and nuclear data. Numeric values besides each genus correspond to the total number of accessions sampled per genus and the number below each figure represents the total number (individual/species) of Urticeae samples used in each respective analysis. “*” indicates full support; “–“indicates no support in (A,C), support values of < 75 (MP) and < 0.95 (BI) in (B), and support values of < 50% (MP) and < 0.7 (BI) in (D).
Chloroplasts are ubiquitous organelles in plants with tractable attributes that make them highly suitable for use in phylogenetic and phylogeographic studies (Demenou et al., 2020; Silverio et al., 2021; Simmonds et al., 2021; Wang et al., 2021). In Urticaceae, whole chloroplast genomes have proven to be indispensable for sequence variation exploration (Wang et al., 2020b; Li et al., 2021). More broadly, studies of chloroplast genomes have been useful for understanding molecular evolutionary patterns of gene duplication, loss, rearrangement, and transfer across angiosperms (Yan et al., 2018; Do et al., 2020; Liu et al., 2020a; Oyebanji et al., 2020), though discordant relationships may be caused by plastid capture and other evolutionary processes.
For the present study, we sequenced and examined chloroplast genomes (CP genome/plastome) of the tribe Urticeae in order to explore plastome structural evolution in the tribe and to reconstruct the first-ever full plastome phylogeny for the tribe. Furthermore, we generated a robustly sampled dataset of Urticeae (comprising 291 accessions) aimed at reconstructing a more taxonomically rich phylogeny for the tribe. Specifically, we aimed to (1) characterize structural changes in Urticeae plastomes, (2) resolve deep relationships in the tribe using different data partitioning strategies, and (3) evaluate and update existing classifications for Urticeae in the light of our phylogenetic results based on both plastome and nuclear data.
Materials and Methods
Taxon Sampling
In this study, we sampled a total of 106 accessions, comprising 90 ingroup accessions (58 spp. in 12 genera) from the tribe Urticeae, plus 12 accessions (12 spp. in 11 genera) from other Urticaceae tribes and four (3 spp. in 3 genera) from outside the family as outgroups. These represent the genome skimming—CP genome and the nuclear ribosomal DNA (18S-ITS1-5.8S-ITS2-26S) dataset for the phylogenetic analyses (Supplementary Table 1). Of the 106 accessions, 57 representative accessions (each a different taxon) were selected for CP genome structural analyses. To produce a more comprehensive phylogenetic framework for the tribe Urticeae, we also generated a new two-locus dataset of 291 accessions (145 spp. in 26 genera) based on ITS and the trnL-F intergenic spacer. The ITS and the trnL-F intergenic spacer dataset was sampled based on maximum taxon data availability on NCBI database. Of the 291 accessions included, 187 sequences were obtained from NCBI GenBank while the remaining were newly sequenced for this study. Information on the plant material (collection localities and voucher specimen numbers) and the associated GenBank accessions are listed in Supplementary Table 1.
DNA Extraction and Sequencing
A modified cetyl trimethyl ammonium bromide (CTAB) protocol (Doyle and Doyle, 1987) was used to extract total DNA from both silica gel-dried leaves and herbarium samples. Genomic DNA from each sample was then assessed for quality and quantity using both a NanoDrop 2,000 spectrophotometer (Thermo Fisher Scientific, United States) and agarose gel electrophoresis before library preparation. The library was built using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England BioLabs) according to the manufacturer’s instructions. Sequencing was then done using the Illumina HiSeq X Ten platform, yielding 150 bp paired-end reads. For each individual, 2–4 Gb of clean data was generated.
Assembly and Annotation
SPAdes (Bankevich et al., 2012) was used for de novo assembly of all sequences using kmer length of 85–111 bp. For the CP genome, we visualized and filtered the newly assembled contigs to generate a complete circular genome in both Bandage v. 0.80 (Wick et al., 2015) and Geneious v. 8.1 (Kearse et al., 2012). The newly assembled sequences were annotated using the reference genome Debregeasia longifolia_MBD01 (MN18994) in the Plant Genome Annotation (PGA) platform (Qu et al., 2019), followed by manual curation of genes in Geneious to check if the start and stop codons are correct. Furthermore, for CP genomes, tRNAscan-SE v. 1.21 (Schattner et al., 2005) was used to further verify the tRNA genes with default settings. We used Chloroplot (Zheng et al., 2020) to generate the physical maps of the CP genomes.
Plastome Structural Variation Analyses
Patterns of Inverted Repeat Boundary Shifts and Inversion
We characterized the genomic features of the 57 unique plastomes, including their size, structure (SC and IR regions), protein coding (PCG) and other (tRNA and rRNA) genes, and GC content. The junctions between the IR and single copy (SC) regions were then compared and analyzed using Geneious v. 8.1 (Kearse et al., 2012). ProgressiveMAUVE (Darling et al., 2010) was used to detect gene rearrangements and inversions among Urticeae taxa with Elatostema parvum as the reference genome. Default settings were used in ProgressiveMAUVE to automatically calculate the seed weight (15), and calculate Locally Collinear Blocks (LCBs) with a minimum LCB score of 30,000.
Repeat Sequence Analyses
We searched for the occurrence and distribution of three types of repeats within the studied plastomes of the tribe Urticeae. First, the program REPuter (Kurtz et al., 2001) was used to identify dispersed repeat sequences (forward, reverse, complement, and palindromic) using the following constraint values: a hamming distance of 3, minimum repeat size of 30 bp, and a maximum computed repeat of 100. Second, the tandem repeats were identified using the online program Tandem Repeats Finder (Benson, 1999) with the alignment parameters match, mismatch, and indels set to 2, 7, and 7, respectively. For this analysis, the maximum period size and TR array size were limited to 500 and 2,000,000 bp, respectively, and the minimum alignment score for reporting repeats was set at 50. Third, we used a Perl-based microsatellite identification tool (MISA; Thiel et al., 2003) to search for simple sequence repeats (SSRs) (i.e., mono-, di-, tri-, tetra-, penta-, and hexanucleotide repeats) within Urticeae plastomes. The threshold values for this analysis were set at 10, 6, 5, 5, 5, and 5 for mono-, di-, tri-, tetra-, penta- and hexanucleotides, respectively.
Sequence Divergence Analyses
To illustrate interspecific sequence variation and gene organization of the entire plastomes across the 57 examined species, we used mVISTA with the shuffle-LAGAN mode (Frazer et al., 2004) and E. parvum as the reference genome. For the assessment of sequence divergence and exploration of highly variable chloroplast markers, a sliding window analysis was performed in DnaSP v. 6 (Rozas et al., 2017) to compute the nucleotide diversity (π) for all protein-coding (CDS) and non-coding (nCDS i.e., intron and intergenic spacer) regions. The step size was set to 300 bp, with a window length of 1,000 bp. The gene recovered to have the highest nucleotide diversity was then used to draw a phylogenetic tree to test the resolution of the identified barcode for our species.
Phylogenetic Inference
Phylogenetic analyses were conducted using different partitioning schemes from two datasets: the genome skimming [CP genome and the 18S-ITS1-5.8S-ITS2-26S (nrDNA) sequences] and two-locus (ITS and the trnL-F intergenic spacer) dataset. We extracted the coding (CDS) and non-coding (nCDS) regions from the CP genome to elucidate the phylogenetic utility of the different regions. This partitioning is important as both CDS and nCDS regions have been shown to exhibit distinct rates of nucleotide substitution (Wolfe et al., 1987; Jansen and Ruhlman, 2012). In total, six molecular data matrices were generated to explore the phylogenetic relationships of the tribe Urticeae, of which five were from the genome skimming dataset: (1) Whole chloroplast (CP) genomes, (2) CP coding regions (CDS), (3) CP non-coding regions (nCDS), (4) nuclear ribosomal DNA (nrDNA), and (5) combined whole CP genomes and nuclear ribosomal DNA (CP + nrDNA). The final matrix (6) sampled the two-locus dataset trnL-F intergenic spacer and ITS sequences (trnL-F + ITS) across expanded taxonomic sampling of 291 accessions.
Phylogenetic analyses were conducted using maximum likelihood (ML) and Bayesian inference (BI) methods in RAxML v. 8.2.11 (Stamatakis, 2014) and MrBayes v. 3.2 (Ronquist et al., 2012), respectively. Substitution models for all the datasets were first determined based on Akaike information criterion (AIC; Akaike, 1973) in jModelTest2 v. 2.1.7 (Darriba et al., 2012; Supplementary Table 2). Maximum likelihood analyses was done in RAxML using the bootstrap option of 1,000 replicates. For BI analyses, we performed two independent runs, each consisting of four Markov Chain Monte Carlo (MCMC) chains, and sampling of one tree every 1,000 generations for 1 million (CP, nCDS, and CP + nrDNA), 3 million (CDS), and 20 million (trnL-F + ITS and only nrDNA) generations. The convergence of the MCMC chains of each run was determined when the average standard deviation of split frequencies (ASDSF) achieved ≤ 0.01, and adequate mixing was based on the Effective Sample Sizes (ESS) values ≥ 200. Stationarity was assessed by checking if the plot of log-likelihood scores had plateaued in Tracer v1.7.1 (Rambaut et al., 2018). The first 25% of the sampled trees acquired from all the runs were discarded as burn-in, and consensus trees were constructed from the remaining trees to estimate posterior probabilities.
Results
Chloroplast Genome Organization
The plastomes of Urticeae species varied greatly in sequence length, ranging in size from 145,419 bp (Nanocnide lobata) to 161,930 bp (Laportea grossa) (Table 1). All exhibited a quadripartite structure typical of angiosperms (Figure 2A)—a pair of IRs (24,593–30,335 bp) separated by the LSC (77,955–84,521 bp) and SSC regions (16,500–19,838 bp). We observed a marginal difference in the GC content across the whole plastome (36.3–37.2%) and its elements — the IR (41.8–43.3%), LSC (33.8–34.7%), and SSC (29.6–31.1%) regions.
TABLE 1.
Summary of sizes of the whole Urticeae plastomes, and the three compartments.
| Species | Nucleotide length (bp) |
|||
| Genome | LSC | SSC | IR | |
| Dendrocnide basirotunda_J2078 | 152,646 | 83,433 | 18,229 | 25,492 |
| Dendrocnide meyenia_D7 | 152,621 | 83,430 | 18,149 | 25,521 |
| Dendrocnide sinuata_J7885 | 152,559 | 83,348 | 18,187 | 25,512 |
| Dendrocnide urentissima_D4 | 152,658 | 83,444 | 18,230 | 25,492 |
| Discocnide mexicana_W268 | 153,327 | 83,841 | 17,580 | 25,953 |
| Girardinia bullosa_A1 | 152,388 | 82,974 | 17,728 | 25,833 |
| Girardinia chingiana_G1 | 152,659 | 83,451 | 18,068 | 25,570 |
| Girardinia diversifolia_G56 | 152,979 | 83,636 | 18,127 | 25,608 |
| Girardinia formosana hayata_G3 | 152,596 | 83,364 | 18,056 | 25,588 |
| Girardinia suborbiculata subsp. grammata_G22 | 152,687 | 83,453 | 18,020 | 25,607 |
| Girardinia suborbiculata subsp. suborbiculata_G15 | 152,894 | 83,650 | 18,104 | 25,570 |
| Girardinia suborbiculata subsp. triloba_G19 | 152,874 | 83,516 | 18,142 | 25,608 |
| Hesperocnide tenella_W61 | 146,864 | 79,555 | 17,691 | 24,809 |
| Laportea aestuans_L30 | 153,521 | 82,883 | 16,500 | 27,609 |
| Laportea bulbifera_GLGE14842 | 149,436 | 81,759 | 17,859 | 24,909 |
| Laportea canadensis_W167 | 150,253 | 82,394 | 17,783 | 25,038 |
| Laportea cuspidata_L27 | 149,149 | 80,905 | 17,450 | 25,397 |
| Laportea decumana_L15 | 151,855 | 82,777 | 18,080 | 25,499 |
| Laportea grossa_L2 | 161,930 | 83,658 | 19,838 | 29,217 |
| Laportea medogensis_GLGE141037 | 150,196 | 82,385 | 17,759 | 25,026 |
| Laportea mooreana_L12 | 150,827 | 81,878 | 18,371 | 25,289 |
| Laportea ovalifolia_L14 | 153,659 | 82,193 | 16,596 | 27,435 |
| Nanocnide japonica_N3 | 145,970 | 78,396 | 17,300 | 25,137 |
| Nanocnide lobata_N6 | 145,419 | 77,955 | 17,258 | 25,103 |
| Obetia aldabrensis_W291 | 153,239 | 84,219 | 18,628 | 25,196 |
| Poikilospermum cordifolium_Poi7 | 153,801 | 84,436 | 18,617 | 25,374 |
| Poikilospermum lanceolatum_Poi8 | 153,879 | 84,521 | 18,618 | 25,370 |
| Poikilospermum naucleiflorum_Poi6 | 153,782 | 84,414 | 18,600 | 25,384 |
| Touchardia latifolia_T2 | 152,871 | 84,003 | 18,252 | 25,308 |
| Urera baccifera_Ur21 | 153,215 | 84,314 | 18,027 | 25,437 |
| Urera cameroonensis_Ur12 | 153,212 | 83,990 | 18,532 | 25,345 |
| Urera capitata_W143 | 153,771 | 84,297 | 18,626 | 25,424 |
| Urera cf cordifolia_Ur15 | 153,214 | 83,992 | 18,536 | 25,343 |
| Urera glabra_Ur17 | 152,663 | 83,499 | 18,502 | 25,331 |
| Urera hypselodendron_Ur16 | 153,212 | 84,007 | 18,515 | 25,345 |
| Urera oligoloba_Ur23 | 153,919 | 84,056 | 18,561 | 25,151 |
| Urera robusta_Ur19 | 153,198 | 84,017 | 18,491 | 25,345 |
| Urtica angustifolia_J3303 | 146,703 | 79,830 | 17,683 | 24,595 |
| Urtica ardens_GLGE152058 | 146,795 | 79,693 | 17,686 | 24,708 |
| Urtica atrichocaulis_S11193 | 146,717 | 79,884 | 17,633 | 24,600 |
| Urtica chamaedryoides_W162 | 146,455 | 79,304 | 17,701 | 24,725 |
| Urtica dioica subsp. xijiangensis_U41 | 147,935 | 79,627 | 17,530 | 25,389 |
| Urtica dioica_W174 | 146,928 | 80,052 | 17,676 | 24,600 |
| Urtica domingensis_W145 | 146,125 | 79,260 | 17,665 | 24,600 |
| Urtica hyperborea_J5455 | 147,898 | 79,748 | 17,588 | 25,281 |
| Urtica kioviensis_U24 | 146,725 | 79,855 | 17,666 | 24,602 |
| Urtica macrorrhiza_U50 | 146,747 | 79,886 | 17,661 | 24,600 |
| Urtica magellanica_U33 | 146,606 | 79,613 | 17,657 | 24,668 |
| Urtica mairei_J1664 | 146,790 | 79,689 | 17,685 | 24,708 |
| Urtica membranifolia_S13031 | 158,078 | 79,719 | 17,689 | 30,335 |
| Urtica morifolia_U200 | 146,755 | 79,643 | 17,690 | 24,711 |
| Urtica radicans_U21 | 146,667 | 79,819 | 17,662 | 24,593 |
| Urtica rupestris_U28 | 146,751 | 79,859 | 17,696 | 24,601 |
| Urtica sp_U19 | 147,508 | 79,069 | 17,669 | 25,385 |
| Urtica thunbergiana_J2498 | 146,846 | 79,667 | 17,711 | 24,734 |
| Urtica urens_W175 | 147,516 | 79,076 | 17,668 | 25,386 |
| Zhengyia shennongensis_Zh1 | 150,109 | 81,186 | 17,885 | 25,519 |
LSC, Large Single Copy; SSC, Small Single Copy; IR, Inverted Repeat.
FIGURE 2.

(A) Gene map of complete chloroplast genome of Girardinia bullosa (a typical representative of gene organization in Urticeae s.l. plastomes); (B) inset map showing the inverted orientation of trnN-GUU in clade 3C except for Discocnide mexicana; (C) inset map of the Urticeae plastome, showing the typical orientation of trnN-GUU. Genes inside and outside the outer circle are transcribed clockwise and counterclockwise, respectively.
A range of 110–112 unique genes was detected across these plastomes, including 76–78 PCGs, 30 tRNA genes, and 4 rRNA genes. The IR region had complete duplications for 7 tRNA genes, 6 PCGs, and 4 rRNA genes. Across all 57 plastomes, 15 genes had a single intron (atpF, ndhA, ndhB, petB, petD, rpl2, rpl16, rpoC1, rps16, trnA-UGC, trnG-UCC, trnI-GAU, trnK-UUU, trnL-UAA, and trnV-UAC), while two genes (clpP and ycf3) had two introns. The rps12 gene was spliced into two transcriptions, with one exon in the LSC and two in the IR. Notably, the rpl2 gene of Hesperocnide tenella and most Urtica taxa except for Urtica dioica subsp. xijiangensis_U41, Urtica dioica_J5488, Urtica hyperborea_J5455, Urtica sp_U19, and Urtica urens lacked an intron. Apart from the region containing an inverted trnN-GUU in five species (four Dendrocnide species and Laportea decumana; Figures 2B,C), no significant gene rearrangement was observed within the studied plastomes (Supplementary Figure 1A).
Inverted Repeat Expansion and Contraction
Comparison of the IR boundaries among the 57 plastomes from tribe Urticeae revealed varying expansion and contraction of the IRs (Figure 3A). Herein, we report only the functional genes located at the IR-SC boundaries. The LSC/IRb border was embedded in the rps19 gene (with 50–131 bp located within IRb) in 43 taxa. The remaining 14 species showed: an expansion in three species (rpl22 in the LSC—rps19 in the IRb); contraction (rps19 in the LSC—rpl2 in the IRb) of the IR in three species; the loss of the rps19 gene in eight species (rpl22 in the LSC—rpl2 in the IRb), causing variations in the boundary (Figure 3B). The IRb/SSC boundary generally fell within the ndhF gene (with 50–131 bp located at IRb), except in six species where the boundary was detected in the intergenic region of trnNGUU-ndhF (Figure 3B). We observed that the IRa/LSC boundary of most species lay within either the intergenic rpl2-trnHGUG or non-coding trnH-GUG regions, except for four species (Hesperocnide tenella_W61, Urtica chamaedryoides_W162, Urtica magellanica_U33, and Urtica morifolia_U200) in which the boundary was located within the intergenic region trnH-GUG—psbA (Figure 3B). The most conserved boundary across species was that of the SSC/IRa, which was always positioned within the ycf1 coding gene, which had a length of 195–3,054 bp overlapping into the IRa region (Figure 3B).
FIGURE 3.

(A) representative map showing expansions and contractions in the IR region; (B) comparison of the IR/SC junctions among 57 Urticeae plastomes. The genes around the borders are shown above or below the main line. LSC, Large Single Copy; SSC, Small Single Copy; IR (a and b), Inverted Repeat a and b.
Repeat Structure and Search for Simple Sequence Repeats
The 57 Urticeae plastomes showed a total of 6,274 repeats based on four classifications (Figure 4A and Supplementary Table 3). Generally, the most frequent repeat type was the SSR (2,919, 46.53%), followed by tandem (1,185, 18.89%), dispersed (1,140, 18.17%), and palindromic repeats (1,030, 16.42%) (Figure 4A). The distribution of the dispersed, tandem, and palindromic repeats varied between 25 (Nanocnide japonica_N3) and 124 (Discocnide mexicana_W268 and Zhengyia shennongensis_Zh1) (Figure 4B), and that of the number of SSRs ranged from 18 (Laportea cuspidata_L27) to 82 (Laportea grossa_L2) (Figure 4C). The majority of the SSRs were mononucleotides (2,627, 89.97%), with poly-A and poly-T SSR motifs being the two most frequent (Figure 4D and Supplementary Table 5). Dinucleotides, trinucleotides, tetranucleotides, pentanucleotides, and hexanucleotides accounted for 8.50, 1.27, 0.14, 0.03, and 0.07% of the SSR repeats, respectively (Figure 4C and Supplementary Table 4).
FIGURE 4.

(A) Chord diagram showing a connection between species and their corresponding repeat types (Dispersed, Palindromic, Tandem, and SSR). The tick marks beside each repeat type indicate the frequency of the number of repeats detected and their percentages, respectively; (B) frequency of tandem, palindromic, and dispersed repeats; (C) number of the six SSR(simple sequence repeats) nucleotides; (D) the seven most abundant SSR motifs in Urticeae plastomes. The values along the axis represent the abundance values.
Sequence Divergence Analysis
Pairwise comparison of divergent regions within the 57 Urticeae plastomes using mVISTA revealed very low intra- and inter-generic (Supplementary Figure 1B) sequence divergence across the plastomes. Moreover, nCDS regions were generally more divergent and had higher levels of variation than CDS regions (Supplementary Figures 1B, 2). For the CDS, the top five genes with the highest nucleotide diversity (π) values (all with π > 5%) were rpoc2, cemA, rpoA, rpl22, ccsA, and ycf1 (Supplementary Figure 2A). The most variable nCDS regions were the trnQ(UUG)—psbK, trnG(GCC)—trnfM(CAU), ycf3—trnS(GGA), cemA—petA, and ndhE—ndhG spacer regions, all with π > 10% (Supplementary Figure 2B). The ycf1 gene tree depicted highly resolved and supported relationships, owing to the gene’s high nucleotide diversity (Supplementary Figure 2C).
Phylogenetic Relationships
The sequence characteristics, tree diagnostic values, and the best-fit model determined by jModelTest for all datasets are given in Supplementary Table 2. The phylogenetic results presented here are based on both ML and BI analyses. The ML and BI analyses generated here generally had nearly identical topologies with few differences at the shallow nodes. Factors driving discrepancies between the ML and BI topologies have been previously reported (Huelsenbeck, 1995; Sullivan and Joyce, 2005; Som, 2014). Of those, the optimality criterion and specific hypotheses in the modeling of sequence evolution are parsimonious to explain the few discrepancies between the ML and BI topologies inferred from the same data matrix in our study. In most cases, the phylogenetic relationships inferred from ML were discussed because it has the most supporting shreds of evidence from the morphological affinities between the known species within the tribe Urticeae. The phylogenetic relationships constructed for each data matrix are further reported.
Chloroplast Data Analyses
The CDS, nCDS, and whole CP phylogenetic trees were largely identical in their topologies with only a few exceptions concerning the relationships of two clades 3F3I and 3F3II (Supplementary Figures 3A–CI). In the CDS data, these were sister to one another, hence formed a monophyletic clade 3F3 (Supplementary Figure 3A). However, in the whole CP dataset, 3F3I was sister to both 3F3II, and 3F4, while in nCDS dataset, 3F3II was sister to both 3F3I and 3F4 (Supplementary Figures 3B,CI). Nevertheless, it should be noted that the whole CP dataset generally had better support compared to both the CDS and nCDS datasets.
nrDNA Data Analysis
Regarding relationships between major clades in Urticeae, the results from the nrDNA dataset (Supplementary Figure 3CII) recovered almost congruent relationships with that of the whole CP dataset (Supplementary Figure 3CI), other than a few discrepancies in particular major clades and phylogenetic placement of some species. For instance, in the nrDNA phylogeny, clade 3D (Girardinia) was recovered as sister to clade 3C (Supplementary Figure 3CII), whereas in whole CP phylogeny, clade 3D was recovered as sister to a clade comprising subclades 3C, 3B, and 3A (Supplementary Figure 3CI). The sister relationships of clade 3G, and those within clade 3E-F also changed depending on the dataset examined. Moreover, we found slight differences in some shallower relationships between the whole CP and nrDNA phylogenies (e.g., the contradicting phylogenetic positions of Dendrocnide urentissima, Girardinia suborbiculata subsp. suborbiculata, etc.; Supplementary Figure 3C). These differences were, however, mostly restricted to areas of poor support, and the whole CP phylogeny was generally better supported than that of nrDNA.
Combined Whole Chloroplast Genome and nrDNA (CP + nrDNA) Analysis
Phylogenetic resolution and node support values were significantly improved by the combination of whole CP genome and nrDNA data (Figure 5). The phylogeny inferred from the combined data matrix was the best resolved and supported phylogenetic tree amongst all the other data matrices, and was more similar in topology to the three chloroplast data matrices (whole CP, CDS, and nCDS, regions) than the nrDNA one (Figure 5 and Supplementary Figures 3A–C). The monophyly of Urticeae was strongly supported (BS/PP = 100/1), with Elatostemeae as its sister tribe (Figure 5). Generally, the phylogeny was well resolved, with most nodes being strongly supported by both ML and BI analyses, except the placement of Zhengyia shennongensis (BS = 100 PP = “–“), the relationship between Urtica domingensis and Hesperocnide tenella (BS = “–“ PP = 1), and the relationship between Laportea aestuans and Laportea ovalifolia (BS = “–“ PP = 1) (Figure 5). Nine genera within Urticeae were recovered as monophyletic (Dendrocnide, Discocnide, Girardinia, Hesperocnide, Obetia, Nanocnide, Poikilospermum, Touchardia, and Zhengyia) and three as polyphyletic (Urtica, Laportea, and Urera), all with strong support. For ease of discussion, we sectioned Urticeae into six major clades, each with full bootstrap support; the names reflect the clade naming system of Wu et al. (2013). They include Clade 3A (Urtica, Hesperocnide, and Zhengyia), Clade 3B (Nanocnide and Laportea cuspidata), Clade 3C (Dendrocnide, Discocnide, and Laportea decumana), Clade 3D (Girardinia), and Clade 3G (Laportea). Clade 3E-F was recovered as sister to the rest of the Urticeae tribe with maximum support, and comprised Poikilospermum, Urera, Obetia, and Laportea. Within it, Poikilospermum (sub-clade 3F4) was recovered for the first time as a sister clade to Urera (sub-clade 3F3) with full support (Figure 5). Urera comprised three separate subclades within Clade 3E-F, each with strong support. Moreover, in this study Laportea was split into five different clades. Clade 3D (Girardinia) was also recovered for the first time as sister to a clade comprising 3A, 3B, and 3C, with full support.
FIGURE 5.

Phylogenetic relationships of Urticeae inferred from maximum likelihood (ML) and Bayesian inference (BI) based on combined complete plastome and nrDNA sequences. Numbers on the branch indicate clade classification (in purple) and ML_BS/BI_PP values (in black)—note that branches with no support values indicate both ML_BS ≥ 90 and BI_PP = 1.00; lastly, “*” indicate incongruence between ML and BI trees and “–” no support values. Representative images of genera within Urticeae s.l. are shown on the right. Photographs: (A–C,E,G,K) by Z.Y. Wu, (D,F) by C.A. Ogoma, (H) by U. Dreschel, (I) by C. Kunath, and (J) photographed by J. Cantley.
Combined Analysis of trnL-F + ITS
The tree topology from the analysis of the trnL-F and ITS dataset was largely congruent with the previously published phylogenies inferred from a small number of loci. Eight genera were strongly supported as monophyletic (i.e., Dendrocnide, Discocnide, Girardinia, Obetia, Nanocnide, Poikilospermum, Touchardia, and Zhengyia) while four genera were recovered as polyphyletic (i.e., Hesperocnide, Urtica, Laportea, and Urera). Hesperocnide was recovered here as polyphyletic (BS/PP > 90/0.90 and BS/PP < 90/0.90; Figure 6) as compared to the combined whole (CP + nrDNA) where it was retrieved as monophyletic with full bootstrap support (Figure 5). Moreover, most of the shallow nodes of trnL-F and ITS tree received lower bootstrap support (Figure 6) compared to the combined whole (CP + nrDNA) tree, in which nearly all the nodes were fully supported.
FIGURE 6.
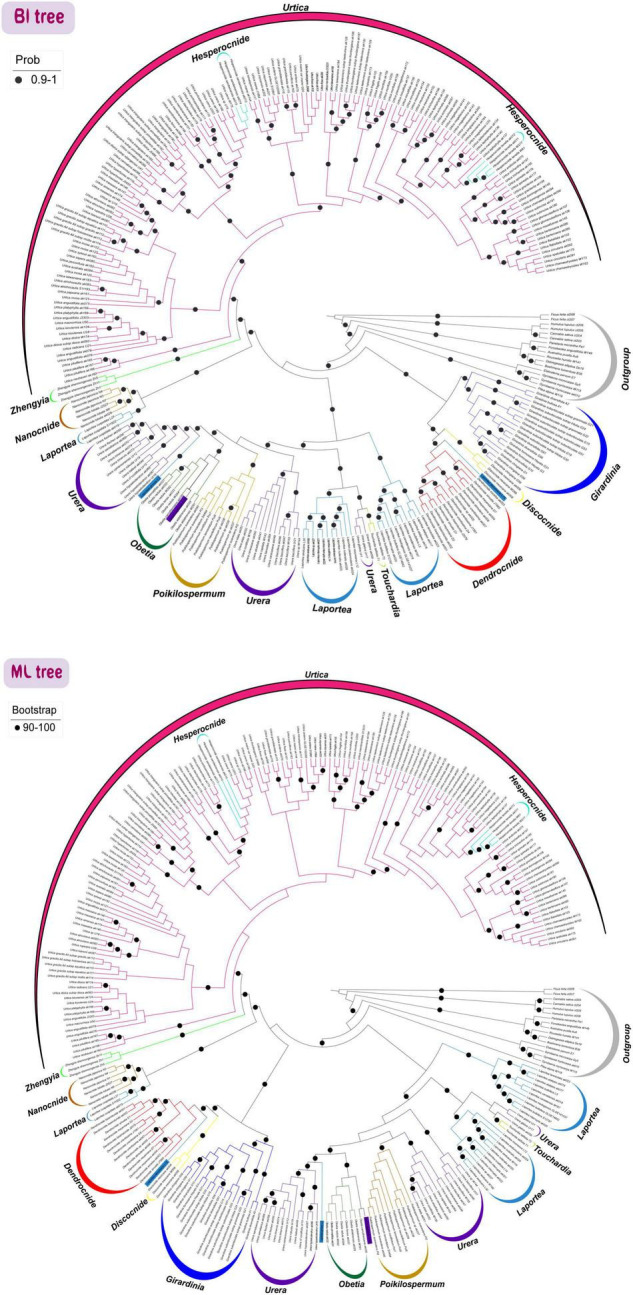
Phylogenetic relationships of Urticeae tribe inferred from maximum likelihood (ML) and Bayesian inference (BI) based on trnL-F intergenic spacer and ITS (trnL-F + ITS) loci sequences. Support values indicated by black circle show ML_BS ≥ 90 and BI_PP ≥ 0.90.
Discussion
Plastome Structural Evolution
All 57 Urticeae CP genomes examined are quadripartite but varied in size. The observed range was consistent with chloroplast genome sizes of angiosperms (Zhang et al., 2021) and the few existing sequenced plastomes of Urticaceae (Wang et al., 2020b; Li et al., 2021), which range between 120 and 180 kb. Of the plastomes in our study, Laportea grossa had the largest genome, while Nanocnide lobata had the smallest, implying that CP genomes in Urticaceae are structurally different. Also, the number of PCGs in the Urticeae plastomes in our study (76–78) was comparable with the typical range for angiosperm plastomes (70–88 genes) (Wicke et al., 2011). Likewise, we found congruence with the range of GC content previously reported in other plastomes of Urticaceae, e.g., Pilea mollis (36.72%; Li et al., 2021), Elatostema dissectum (36.2%; Fu et al., 2019), Droguetia iners (36.9%), and Debregeasia elliptica (36.4%) (Wang et al., 2020b). Generally, the GC content had no significant phylogenetic implication in our study. Moreover, consistent with previous studies (Li et al., 2020, 2021; Dong et al., 2021), the GC content was higher in the IR than in the SC. The GC inequality perhaps also plays a significant factor in the conservatism of the IR region compared to the SC regions (Li et al., 2020).
Among the genes present in our Urticeae plastomes, rpl2 was noteworthy, considering that 18 of the examined species had no introns for this gene. Intron loss has been widely documented in angiosperm plastomes: e.g., Avena sativa (rpoC1 intron loss; Liu et al., 2020b), Cicer arietinum (rps12 and clpP intron losses; Jansen et al., 2008), Lagerstroemia (rpl2 intron loss; Gu et al., 2016), and Asteropeiaceae + Physenaceae (rpl2 intron loss; Yao et al., 2019). Another notable structural change found here was an inversion of the trnN-GUU gene, which is a synapomorphy of the clade 3C, except for the clade’s basal species Discocnide mexicana (Figure 2B). Gene inversions have also been detected in many angiosperm plastomes, including those of Poaceae (Guisinger et al., 2010), Styracaceae (Yan et al., 2018), Orchidaceae (Uncifera acuminata; Liu et al., 2020a), and Adoxaceae (Wang et al., 2020a). The latter, involving the inversion of the ndhF gene in Adoxaceae, is relevant to our study since it involves only one gene that also borders the inverted gene in our study (trnN-GUU). Typically, plastome inversions are deemed highly valuable in phylogenetics owing to their relative rarity, easily determined homology, and easily inferred state polarity (Cosner et al., 1997; Dugas et al., 2015; Schwarz et al., 2015). Despite some significant research efforts regarding the intramolecular recombination between dispersed short inverted/direct repeats and tRNA genes (Cosner et al., 1997; Haberle et al., 2008; Sloan et al., 2014), the cause of inversions in plant genomes remains unclear.
Our analyses showed that IR expansion and contraction vary across Urticeae, and lack taxonomic utility at a broader scale. Mostly, the SC/IR borders are relatively conserved among angiosperm plastomes and usually located within the rps19 or ycf1gene (Downie and Jansen, 2015), even though it is assumed that IR expansion or contraction is accompanied by the shift of genes located in the IR/SC boundary (Zhu et al., 2016). Similar IR/SC changes are also evident in other Urticaceae plastomes (Wang et al., 2020b; Li et al., 2021). Changes in the IR/SC junctions have been considered one of the main drivers of the size diversity in the CP genomes of higher plants (Ma et al., 2013; Yang et al., 2016; Yan et al., 2018; Xue et al., 2019). Notably, we found the loss of the rps19 gene to be the most parsimonious explanation for the diversification of the genes bordering the IR/LSC in the eight plastomes examined from the genus Urtica—(U. ardens_GLGE152058, U. dioica subsp. xijiangensis_U41, U. domingensis_W145, U. hyperborea_J5455, U. mairei_J1664, U. membranifolia_S13031, U. morifolia_U200, and U. thunbergiana_J2498; Figure 3A).
We detected several repeat types within the sampled plastomes of tribe Urticeae, among which SSRs were the most frequent, accounting for 46.53% of the repeats (Figure 4A). The most abundant SSRs were mononucleotide homopolymers, particularly poly−A and T motifs (Figure 4D and Supplementary Table 5). This phenomenon of A/T motif abundance has also been reported in Pilea (Li et al., 2021) and Debregeasia (Wang et al., 2020b) species, and might occur because the A/T motifs are more frequently dynamic compared to G/C (Li et al., 2020). Generally, it is presumed that repeat sequences are closely connected with a vast number of indels; therefore, the more abundant they are, the greater the nucleotide diversity (McDonald et al., 2011). Hence, the chloroplast repeat sequences could be potential sources of variation for evolutionary studies, and population genetics (Xue et al., 2012). We also found higher nucleotide diversity in the nCDS than in the CDS regions, consistent with findings from other taxa (Jansen and Ruhlman, 2012; Huang et al., 2014). Although the nucleotide content of chloroplast genomes is usually relatively stable, with a highly conserved gene structure (Jansen et al., 2005; Ravi et al., 2008; Wicke et al., 2011), mutation hotspots still exist within it (Zhang et al., 2021). We detected a total of 11 hypervariable loci in both CDS and nCDS regions (Supplementary Figure 2) that could be potentially used as DNA barcodes in future studies of this group. Among them was the locus ycf1, which was also reported in previous Urticaceae studies (Wang et al., 2020b; Li et al., 2021) as a highly variable locus with great taxonomic utility. Moreover, a study by Dong et al. (2015) reinforces this view, and recommemnds ycf1 as a suitable plastid barcode for land plants. Indeed, our ycf1 phylogenetic tree (Supplementary Figure 2C) is consistent with the above studies, especially with regard to the high resolution and support level. Therefore, we suggest that ycf1 represents a highly useful molecular marker, not just for tribe Urticeae, but likely for the entire family. Presently, DNA barcodes are widely used in species identification, resource management, and studies of phylogeny and evolution (Gregory, 2005; Liu et al., 2019).
Phylogenetic Relationships of Urticeae
Phylogenetic Relationships Based on Genome Skimming (CP Genome + nrDNA) Data
The combined matrix (CP genome + nrDNA) yielded a well-supported phylogeny and resolved many relationships of the tribe Urticeae depite the topological difference in clades 3(D, 3G, and E-F), between the two separate datasets (Supplementary Figure 3C). This resolution shown by the combined matrix may be ascribed to the greater number of phylogenetically informative plastid sites (Supplementary Table 2). Moreover, it could be due to a weak phylogenetic signal in the nrDNA that agrees and complements the signal of the CP matrix. However, beyond some major conflicts, the individual CP and nrDNA trees are generally in agreement with most conflicting relationships pertaining to poorly supported areas of the phylogeny, although we did not perform follow-up analyses to identify what this means for different parts of the tree. Cases of topological dissimilarity are often reported in phylogenetic studies (Wendel and Doyle, 1998; reviewed by Degnan and Rosenberg, 2009). This phenomenon can be best explained by a number of factors including differences in taxon sampling, incomplete lineage sorting, hybridization/introgression, paralogy, gene duplication and/or loss, and horizontal gene transfer (Degnan and Rosenberg, 2006; Naciri and Linder, 2015; Lin et al., 2019; Nicola et al., 2019). Hence, as more samples become available, future studies should investigate the factors responsible for the observed conflicting relationships within the Urticeae.
Our study represents the first phylogeny of the tribe Urticeae based on a broad sampling of both CP genomes and nrDNA sequences. Importantly, we clarify which of the Urticeae genera are strongly supported as monophyletic or polyphyletic (Figure 5). Compared to previous studies based on a limited number of genes (Hadiah et al., 2008; Deng et al., 2013; Wu et al., 2013, 2018; Kim et al., 2015; Grosse-Veldmann et al., 2016; Huang et al., 2019; Wells et al., 2021), we exploited the utility of whole CP genomes for resolving phylogenetic relationships in Urticeae, and also revealed the most informative sites and regions across the plastome. Our results proved to be largely consistent with most of the recently established phylogenetic relationships of Urticeae based on a range of 3–7 selected marker regions (Wu et al., 2013, 2018; Kim et al., 2015; Huang et al., 2019; Wells et al., 2021). In general, however, our data improved resolution throughout Urticeae compared with previous studies, with almost all nodes being fully supported, especially those previously known to be problematic. Four of the most important new phylogenetic insights generated by the current study are discussed below.
First, the sister relationship of Girardinia has been contentious. Girardinia had been resolved as sister to Dendrocnide-Discocnide based on chloroplast, mitochondrial, and nuclear data (Wu et al., 2013), and using ITS, rbcL, and trnL-F regions (Kim et al., 2015), but without support in either case. Subsequently, using expanded taxon sampling and five markers from both the nuclear and CP genomes, the sister relationship of Girardinia to Dendrocnide-Discocnide-Laportea-Nanocnide-Zhengyia-Urtica-Hesperocnide was resolved, but with limited support (Wu et al., 2018; Huang et al., 2019). Our results support this latter relationship but with maximum support (BS/PP = 100/1), for the first time.
Second, our molecular phylogeny of the “Urera alliance clade” (this study clade 3E-F) corroborated the generic delimitation and subdivisions of the “Urera clade” from Wells et al. (2021), and showed two clades of Laportea (which they did not examine) as also a member (Figure 5). Their division of the paraphyletic Urera into three genera was strongly supported here: these were Urera s.s. (our Clade 3F3), Scepocarpus (entirely African; our clade 3F1, which also includes Laportea grossa), and an expanded Touchardia (part of clade 3E, that includes Urera glabra from Hawaii and three species of Laportea as per our study). Our data suggests that the two Laportea clades should hence be fully examined and considerations made as to whether to subsume them within the resurrected Scepocarpus and the expanded Touchardia.
Third, previous studies (Kim et al., 2015; Wu et al., 2018; Huang et al., 2019) have typically resolved Laportea into three clades. For instance, Kim et al. (2015) recovered three Laportea clades corresponding to sections Laportea Gaudich. (L. alatipes, L. bulbifera, L. canadensis, L. lanceolata), Sceptrocnide (Maxim.) C. J. Chen (L. cuspidata), and Fleurya (Gaudich.) Chew [L. aestuans (L.) Chew, L. interrupta, L. ruderalis (G. Forst.) Chew], consistent with the sectional classification of Wang and Chen (1995). Our analysis, however, resolved Laportea into five major clades. Moreover, we found that L. aestuans was polyphyletic: one subgroup was sister to L. mooreana with full support and the other was sister to L. ovalifolia with support of BS/PP = –/1. The latter relationship was detected by Wu et al. (2018) but without support. However, other studies found different relationships: L. aestuans as sister to L. interrupta, and L. ruderalis with full support according to Kim et al. (2015), or sister to L. ruderalis and L. peduncularis with support of MP/PP = 96/1 according to Huang et al. (2019). These discrepancies likely reflect differences in taxon and molecular sampling—with a wider sampling of populations, L. aestuans might comprise more than two unrelated clades. While additional study on Laportea is clearly needed, the current study provides one of the most comprehensive phylogenetic perspectives on this little-studied genus. Future investigations should, however, employ more extensive molecular data across the entire phylogenetic spectrum of Laportea to further clarify its relationships and the number of lineages.
Finally, our analysis resolved the sister relationship between Poikilospermum and Urera previously obtained by Huang et al. (2019), but replacing their modest support (BS/PP = 65/0.89) with full support (BS/PP = 100/1) for the first time.
Comparison Between Genome Skimming (CP Genome + nrDNA) and Two-Locus (trnL-F + ITS) Phylogeny
In our study, the trees inferred from both the CP genome + DNA and the two-locus dataset (trnL-F + ITS) provided full support for the monophyly of Urticeae. However, the CP genome + nrDNA tree presented a higher percentage of fully supported nodes compared with that of the two-locus tree (Figures 5, 6). This underscores the importance of genome-scale datasets for resolving major recalcitrant relationships.
The most notable finding from our two-locus phylogenetic analysis was the reconstruction of Hesperocnide as polyphyletic, consistent with Huang et al. (2019). Our current CP genome + nrDNA analysis and prior molecular studies, however, recovered Hesperocnide as monophyletic (Kim et al., 2015), with a close relationship to Urtica (Sytsma et al., 2002; Hadiah et al., 2008; Deng et al., 2013; Wu et al., 2013; Kim et al., 2015). The polyphyletic results from the two-locus tree can be ascribed to the sampling of members of the second species that were absent in the plastome analysis. Consequently, Wu et al. (2013) suggested that Hesperocnide be subsumed in the genus Urtica, since these two genera show some morphological similarities. However, owing to this equivocality about the phylogeny of Hesperocnide, we suggest a more rigorous examination of this genus to fully validate its status.
Conclusion and Future Directions
Our study provides important novel insights on Urticeae phylogeny and plastome evolution. The detailed comparative analyses show that Urticeae plastomes exhibit striking differences in genome size, gene number, inversions, intron loss, sequence repeats, and IR/SC boundaries. These kinds of variations will be useful for studies on molecular marker discovery, population genetics, and phylogeny. Resolving the enigmatic relationships within tribe Urticeae has, to date, been a daunting task due to the paucity of genomic resources for the clade. Our study is the first to report phylogenetic relationships in Urticeae based on a broad sampling of whole plastome sequences. This dataset allowed for resolution of several recalcitrant branches (e.g., the relationship of Poikilospermum to Urera, the sister relationship of Girardinia, etc.) that were ambiguous in previous studies. Although our taxon sampling was sufficient to resolve relationships among the major clades in the tribe, additional sampling of particular genera (e.g., Laportea) and species (e.g., Laportea aestuans and Hesperocnide sandwicensis) would further refine our understanding of phylogenetic relationships in Urticeae. Building on the solid framework established here, future studies with even greater taxonomic and genomic sampling could contribute to a better understanding of the diversification patterns in Urticeae in relation to climatic, biogeographic, and ecological factors.
Data Availability Statement
The datasets presented in this study can be accessed at NCBI GenBank; the list of accessions can be found in Supplementary Table 1.
Author Contributions
Z-YW, D-ZL, JL, and CO conceptualized the study. Z-YW, JL, RM, AM, and YZ collected the samples. OO and CO conducted the analyses. CO and Z-YW drafted the manuscript. Z-YW, CO, GS, MW, OO, RM, D-ZL, and AM revised the manuscript. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are really grateful to Qi Chen and Ruo-Nan Wang for their great assistance during the data analysis. We also immensely appreciate the following herbaria for providing access to study specimens: Kunming Institute of Botany (KUN), Royal Botanical Gardens, Kew (K), University of Florida Herbarium (FLAS), and Royal Botanical Gardens, Edinburgh (E). We thank the Royal Botanic Gardens Kew for providing some of the DNA materials. This work was facilitated by the Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences (CAS).
Funding
Funding for this project was supported by the National Natural Science Foundations of China (31970356, 42171071, and 41971071), CAS’ Youth Innovation Promotion Association (2019385), the Key Research Program of Frontier Sciences, CAS (ZDBS-LY-7001), the Top-notch Young Talents Projects of Yunnan Provincial “Ten Thousand Talents Program” (YNWR-QNBJ-2020-293 and YNWR-QNBJ-2018-146), and CAS Strategic Priority Research Program (XDB31000000). MW was supported by the Postdoctoral International Exchange Program of the Office of China Postdoctoral Council, and the Postdoctoral Targeted Funding and Postdoctoral Research Fund of Yunnan Province.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.870949/full#supplementary-material
(A) Mauve alignment showing gene arrangements within the studied 57 Urticeae plastomes (length indicated above). Large colored boxes represent the gene blocks and the colored lines indicates linear position of different genes in the plastome. (B) Comparison of 57 Urticeae CP genomes using mVISTA, with the E. parvum genome as the reference. The y-axis represents the percent identity within 50–100%. Gray arrows indicate the direction of gene transcription. Blue blocks indicate conserved genes, while red blocks indicate conserved non-coding sequences (CNS).
Variable sites in homologous regions of the 57 sampled plastomes from Urticeae. The y-axis represent the nucleotide diversity (Pi) of each window, and x-axis is the position of the midpoint of each window used in the Sliding window analysis. (A) Coding regions. (B) Non-coding regions. (C) The ycf1 gene tree depicting highly resolved and supported relationships achieved by the identified barcode.
(A) Phylogenetic relationships of Urticeae tribe inferred from maximum likelihood (ML) and Bayesian inference (BI) based on CP coding (CDS) regions. Support values above the branches are maximum likelihood bootstrap support values (ML_BS)/Bayesian posterior probabilities (BI_PP)—note: branches with no support values indicate both ML_BS ≥ 90 and BI_PP = 1.00—whereas “*” indicate incongruence between ML and BI trees. Major clades of Urticeae s.l. are indicated on the right, respectively. CDS, chloroplast coding region. (B) Phylogenetic relationships of Urticeae tribe inferred from maximum likelihood (ML) and Bayesian inference (BI) based on CP non-coding (nCDS) regions. Support values above the branches are maximum likelihood bootstrap support (ML_BS)/Bayesian posterior probability (BI_PP)—note that branches with no support values indicate both ML_BS ≥ 90 and BI_PP = 1.00—whereas “*” indicate incongruence between ML and BI trees. Major clades of Urticeae s.l. are indicated on the right, respectively. nCDS, chloroplast non-coding region. (C) Phylogenetic relationships of Urticeae tribe inferred from maximum likelihood (ML) and Bayesian inference (BI) based on integrated CP genome and nrDNA trees. Support values above the branches are maximum likelihood bootstrap support (ML_BS)/Bayesian posterior probability (BI_PP)—note that branches with no support values indicate both ML_BS ≥ 90 and BI_PP = 1.00—whereas “*” indicate incongruence between ML and BI trees. Major clades of Urticeae s.l. are indicated on the right, respectively. CP, Complete chloroplast genome; nrDNA, nuclear ribosomal DNA (18S-ITS1-5.8S-ITS2-26S).
References
- Akaike H. (1973). “Information theory as an extension of the maximum likelihood principle,” in Second International Symposium on Information Theory, eds Petrov B. N., Csaki F. (Budapest: Akademiai Kiado; ), 267–281. [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27 573–580. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenutti R. C., Dalla Vecchia C. A., Locateli G., Serpa P. Z., Lutinski J. A., Rodrigues Junior S. A., et al. (2020). Gastroprotective activity of hydroalcoholic extract of the leaves of Urera baccifera in rodents. J. Ethnopharmacol. 250:112473. 10.1016/j.jep.2019.112473 [DOI] [PubMed] [Google Scholar]
- Bodros E., Baley C. (2008). Study of the tensile properties of stinging nettle fibres (Urtica dioica). Mater. Lett. 62 2143–2145. 10.1016/j.matlet.2007.11.034 [DOI] [Google Scholar]
- Conn B. J., Hadiah J. T. (2009). Nomenclature of tribes within the Urticaceae. Kew Bull. 64 349–352. 10.1007/s12225-009-9108-4 [DOI] [Google Scholar]
- Cosner M. E., Jansen R. K., Palmer J. D., Downie S. R. (1997). The highly rearranged chloroplast genome of Trachelium caeruleum (Campanulaceae): multiple inversions, inverted repeat expansion and contraction, transposition, insertions/deletions, and several repeat families. Curr. Genet. 31 419–429. 10.1007/s002940050225 [DOI] [PubMed] [Google Scholar]
- Darling A. E., Mau B., Perna N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D., Taboada G. L., Doallo R., Posada D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. 10.1038/nmeth.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J. H., Rosenberg N. A. (2006). Discordance of species trees with their most likely gene trees. PLoS Genet. 2:e68. 10.1371/journal.pgen.0020068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J. H., Rosenberg N. A. (2009). Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24 332–340. 10.1016/j.tree.2009.01.009 [DOI] [PubMed] [Google Scholar]
- Demenou B. B., Migliore J., Heuertz M., Monthe F. K., Ojeda D. I., Wieringa J. J., et al. (2020). Plastome phylogeography in two African rain forest legume trees reveals that Dahomey Gap populations originate from the Cameroon volcanic line. Mol. Phylogenet. Evol. 150:106854. 10.1016/j.ympev.2020.106854 [DOI] [PubMed] [Google Scholar]
- Deng T., Kim C., Zhang D. G., Zhang J., Li Z. M., Nie Z. L., et al. (2013). Zhengyia shennongensis: a new bulbiliferous genus and species of the nettle family (Urticaceae) from central China exhibiting parallel evolution of the bulbil trait. Taxon 62:89. 10.1002/tax.621008 [DOI] [Google Scholar]
- Di Virgilio N., Papazoglou E. G., Jankauskiene Z., Di Lonardo S., Praczyk M., Wielgusz K. (2015). The potential of stinging nettle (Urtica dioica L.) as a crop with multiple uses. Ind. Crops Prod. 68 42–49. 10.1016/j.indcrop.2014.08.012 [DOI] [Google Scholar]
- Do H. D. K., Kim C., Chase M. W., Kim J. H. (2020). Implications of plastome evolution in the true lilies (monocot order Liliales). Mol. Phylogenet. Evol. 148:106818. 10.1016/j.ympev.2020.106818 [DOI] [PubMed] [Google Scholar]
- Dong W., Liu Y., Xu C., Gao Y., Yuan Q., Suo Z., et al. (2021). Chloroplast phylogenomic insights into the evolution of Distylium (Hamamelidaceae). BMC Genom. 22:293. 10.1186/s12864-021-07590-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong W., Xu C., Li C., Sun J., Zuo Y., Shi S., et al. (2015). ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5, 8348. 10.1038/srep08348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downie S. R., Jansen R. K. (2015). A comparative analysis of whole plastid genomes from the Apiales: expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst. Bot. 40 336–351. 10.1600/036364415x686620 [DOI] [Google Scholar]
- Doyle J. J., Doyle J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19 11–15. [Google Scholar]
- Dugas D. V., Hernandez D., Koenen E. J. M., Schwarz E., Straub S., Hughes C. E., et al. (2015). Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 5:16958. 10.1038/srep16958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer K. A., Pachter L., Poliakov A., Rubin E. M., Dubchak I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32 W273–W279. 10.1093/nar/gkh458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friis I. (1989). “The Urticaceae: a systematic review,” in Evolution, Systematics, and Fossil History of the Hamamelidae, eds Crane P. R., Blackmore S. (Oxford: Clarendon Press; ), 285–308. [Google Scholar]
- Friis I. (1993). “Urticaceae,” in The Families and Genera of Vascular Plants, eds Kubitzki K., Rohwer J. G., Bittrich V. (Berlin: Springer-Verlag; ), 612–630. 10.1007/978-3-662-02899-5_76 [DOI] [Google Scholar]
- Fu L. F., Xin Z. B., Wen F., Li S., Wei Y. G. (2019). Complete chloroplast genome sequence of Elatostema dissectum (Urticaceae). Mitochondrial DNA B 4 838–839. 10.1080/23802359.2019.1567292 [DOI] [Google Scholar]
- Gregory T. R. (2005). DNA barcoding does not compete with taxonomy. Nature 434:1067. 10.1038/4341067b [DOI] [PubMed] [Google Scholar]
- Grosse-Veldmann B., Nürk N. M., Smissen R., Breitwieser I., Quandt D., Weigend M. (2016). Pulling the sting out of nettle systematics – a comprehensive phylogeny of the genus Urtica L. (Urticaceae). Mol. Phylogenet. Evol. 102 9–19. 10.1016/j.ympev.2016.05.019 [DOI] [PubMed] [Google Scholar]
- Gu C., Tembrock L. R., Johnson N. G., Simmons M. P., Wu Z. (2016). The complete plastid genome of Lagerstroemia fauriei and loss of rpl2 intron from Lagerstroemia (Lythraceae). PLoS One 11:e0150752. 10.1371/journal.pone.0150752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guisinger M. M., Chumley T. W., Kuehl J. V., Boore J. L., Jansen R. K. (2010). Implications of the plastid genome sequence of typha (typhaceae, poales) for understanding genome evolution in poaceae. J. Mol. Evol. 70 149–166. 10.1007/s00239-009-9317-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung A., Flanigan H., Ghimeray A., Bista R., Gunrung O. (2012). Traditional knowledge of processing and use of the himalayan giant nettle (Girardinia diversifolia (Link) Friis) among the gurungs of sikles, Nepal. Ethnobot. Res. App. 10 167–174. 10.1234/era.v10i0.622 [DOI] [Google Scholar]
- Haberle R. C., Fourcade H. M., Boore J. L., Jansen R. K. (2008). Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA genes. J. Mol. Evol. 66 350–361. 10.1007/s00239-008-9086-4 [DOI] [PubMed] [Google Scholar]
- Hadiah J. T., Conn B. J., Quinn C. J. (2008). Infra-familial phylogeny of Urticaceae, using chloroplast sequence data. Aust. Syst. Bot. 21 375–385. 10.1071/Sb08041 [DOI] [Google Scholar]
- Huang H., Shi C., Liu Y., Mao S. Y., Gao L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. 10.1186/1471-2148-14-151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Deng T., Moore M. J., Wang H., Li Z., Lin N., et al. (2019). Tropical asian origin, boreotropical migration and long-distance dispersal in nettles (Urticeae, Urticaceae). Mol. Phylogenet. Evol. 137 190–199. 10.1016/j.ympev.2019.05.007 [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J. P. (1995). Performance of phylogenetic methods in simulation. Syst. Biol. 44 17–48. 10.2307/2413481 [DOI] [Google Scholar]
- Jansen R. K., Raubeson L. A., Boore J. L., dePamphilis C. W., Chumley T. W., Haberle R. C., et al. (2005). Methods for obtaining and analyzing whole chloroplast genome sequences. Meth. Enzymol. 395 348–384. 10.1016/S0076-6879(05)95020-9 [DOI] [PubMed] [Google Scholar]
- Jansen R. K., Ruhlman T. A. (2012). “Plastid genomes of seed plants,” in Genomics of Chloroplasts and Mitochondria, eds Bock R., Knoop V. (Dordrecht: Springer; ), 103–126. 10.1007/978-94-007-2920-9_5 [DOI] [Google Scholar]
- Jansen R. K., Wojciechowski M. F., Sanniyasi E., Lee S. B., Daniell H. (2008). Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol. Phylogenet. Evol. 48 1204–1217. 10.1016/j.ympev.2008.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X. F., Zhang J., Lu Y. F., Yang W. W., Chen W. J. (2019). Nanocnide zhejiangensis sp. nov. (Urticaceae: Urticeae) from Zhejiang Province, East China. Nord. J. Bot. 37:e02339. 10.1111/njb.02339 [DOI] [Google Scholar]
- Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28 1647–1649. 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C., Deng T., Chase M., Zhang D. G., Nie Z. L., Sun H. (2015). Generic phylogeny and character evolution in Urticeae (Urticaceae) inferred from nuclear and plastid DNA regions. Taxon 64 65–78. 10.12705/641.20 [DOI] [Google Scholar]
- Kurtz S., Choudhuri J. V., Ohlebusch E., Schleiermacher C., Stoye J., Giegerich R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29 4633–4642. 10.1093/nar/29.22.4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Tang J., Zeng S., Han F., Yuan J., Yu J. (2021). Comparative plastid genomics of four Pilea (Urticaceae) species: insight into interspecific plastid genome diversity in Pilea. BMC Plant Biol. 21:25. 10.1186/s12870-020-02793-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Dong Y., Liu Y., Yu X., Yang M., Huang Y. (2020). Comparative analyses of Euonymus chloroplast genomes: genetic structure, screening for loci with suitable polymorphism, positive selection genes, and phylogenetic relationships within Celastrineae. Front. Plant Sci. 11:593984. 10.3389/fpls.2020.593984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. Y., Hao Y. J., Li J. H., Fu C. X., Soltis P. S., Soltis D. E., et al. (2019). Phylogenomic conflict resulting from ancient introgression following species diversification in Stewartia s.l. (Theaceae). Mol. Phylogenet. Evol. 135 1–11. 10.1016/j.ympev.2019.02.018 [DOI] [PubMed] [Google Scholar]
- Liu D. K., Tu X. D., Zhao Z., Zeng M. Y., Zhang S., Ma L., et al. (2020a). Plastid phylogenomic data yield new and robust insights into the phylogeny of Cleisostoma–Gastrochilus clades (Orchidaceae, Aeridinae). Mol. Phylogenet. Evol. 145:106729. 10.1016/j.ympev.2019.106729 [DOI] [PubMed] [Google Scholar]
- Liu Q., Li X., Li M., Xu W., Schwarzacher T., Heslop-Harrison J. S. (2020b). Comparative chloroplast genome analyses of Avena: insights into evolutionary dynamics and phylogeny. BMC Plant Biol. 20:406. 10.1186/s12870-020-02621-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Chang E. M., Liu J. F., Huang Y. N., Wang Y., Yao N., et al. (2019). Complete chloroplast genome sequence and phylogenetic analysis of Quercus bawanglingensis Huang, Li et Xing, a vulnerable oak tree in China. Forests 10:587. 10.3390/f10070587 [DOI] [Google Scholar]
- Luo X., Li L. L., Zhang S. S., Lu J. L., Zeng Y., Zhang H. Y., et al. (2011). Therapeutic effects of total coumarins from Urtica dentata hand on collagen-induced arthritis in Balb/c mice. J. Ethnopharmacol. 138 523–529. 10.1016/j.jep.2011.09.050 [DOI] [PubMed] [Google Scholar]
- Ma J., Yang B., Zhu W., Sun L., Tian J., Wang X. (2013). The complete chloroplast genome sequence of Mahonia bealei (Berberidaceae) reveals a significant expansion of the inverted repeat and phylogenetic relationship with other angiosperms. Gene 528 120–131. 10.1016/j.gene.2013.07.037 [DOI] [PubMed] [Google Scholar]
- Mahlangeni N. T., Moodley R., Jonnalagadda S. B. (2020). Nutritional value, antioxidant and antidiabetic properties of nettles (Laportea alatipes and Obetia tenax). Sci. Rep. 10:9762. 10.1038/s41598-020-67055-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald M. J., Wang W. C., Huang H. D., Leu J. Y. (2011). Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 9:e1000622. 10.1371/journal.pbio.1000622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momo C. E., Oben J. E., Tazoo D., Dongo E. (2006). Antidiabetic and hypolipidaemic effects of a methanol/methylene-chloride extract of Laportea ovalifolia (Urticaceae), measured in rats with alloxan-induced diabetes. Ann. Trop. Med. Parasitol. 100 69–74. 10.1179/136485906X78517 [DOI] [PubMed] [Google Scholar]
- Naciri Y., Linder H. P. (2015). Species delimitation and relationships: the dance of the seven veils. Taxon 64 3–16. 10.12705/641.24 [DOI] [Google Scholar]
- Nicola M. V., Johnson L. A., Pozner R. (2019). Unraveling patterns and processes of diversification in the South Andean-Patagonian Nassauvia subgenus Strongyloma (Asteraceae, Nassauvieae). Mol. Phylogenet. Evol. 136 164–182. 10.1016/j.ympev.2019.03.004 [DOI] [PubMed] [Google Scholar]
- Oyebanji O., Zhang R., Chen S. Y., Yi T. S. (2020). New insights into the plastome evolution of the Millettioid/Phaseoloid clade (Papilionoideae, Leguminosae). Front. Plant Sci. 11:151–151. 10.3389/fpls.2020.00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X. J., Moore M. J., Li D. Z., Yi T. S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15:50. 10.1186/s13007-019-0435-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Drummond A. J., Xie D., Baele G., Suchard M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67 901–904. 10.1093/sysbio/syy032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi V., Khurana J. P., Tyagi A. K., Khurana P. (2008). An update on chloroplast genomes. Plant Syst. Evol. 271 101–122. 10.1007/s00606-007-0608-0 [DOI] [Google Scholar]
- Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Hohna S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61 539–542. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J., Ferrer-Mata A., Sanchez-DelBarrio J. C., Guirao-Rico S., Librado P., Ramos-Onsins S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34 3299–3302. 10.1093/molbev/msx248 [DOI] [PubMed] [Google Scholar]
- Schattner P., Brooks A. N., Lowe T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33 W686–W689. 10.1093/nar/gki366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz E. N., Ruhlman T. A., Sabir J. S. M., Hajrah N. H., Alharbi N. S., Al-Malki A. L., et al. (2015). Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. J. Syst. Evol. 53 458–468. 10.1111/jse.12179 [DOI] [Google Scholar]
- Sharan Shrestha S., Sut S., Ferrarese I., Barbon Di Marco S., Zengin G., De Franco M., et al. (2020). Himalayan nettle Girardinia diversifolia as a candidate ingredient for pharmaceutical and nutraceutical applications-phytochemical analysis and in vitro bioassays. Molecules 25:1563. 10.3390/molecules25071563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverio R. M. A. V., Vieira L. D. N., Antonio de Baura V., Balsanelli E., Maltempi, de Souza E., et al. (2021). Plastid phylogenomics of Pleurothallidinae (Orchidaceae): conservative plastomes, new variable markers, and comparative analyses of plastid, nuclear, and mitochondrial data. PLoS One 16:e0256126. 10.1371/journal.pone.0256126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds S. E., Smith J. F., Davidson C., Buerki S. (2021). Phylogenetics and comparative plastome genomics of two of the largest genera of angiosperms, Piper and Peperomia (Piperaceae). Mol. Phylogenet. Evol. 163:107229. 10.1016/j.ympev.2021.107229 [DOI] [PubMed] [Google Scholar]
- Singh S. C., Shrestha R. (1988). Girardinia diversifolia (Urticaceae), a non-conventional fiber resource in Nepal. Econ. Bot. 42 445–447. [Google Scholar]
- Sloan D. B., Triant D. A., Forrester N. J., Bergner L. M., Wu M., Taylor D. R. (2014). A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol. Phylogenet. Evol. 72 82–89. 10.1016/j.ympev.2013.12.004 [DOI] [PubMed] [Google Scholar]
- Som A. (2014). Causes, consequences and solutions of phylogenetic incongruence. Brief. Bioinform. 16 536–548. 10.1093/bib/bbu015 [DOI] [PubMed] [Google Scholar]
- Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens P. F. (2017). Angiosperm Phylogeny Website. Version 14, July 2017. Available online at: http://www.mobot.org/MOBOT/research/APweb (accessed November 9, 2021). [Google Scholar]
- Sullivan J., Joyce P. (2005). Model selection in phylogenetics. Annu. Rev. Ecol. Evol. Syst. 36 445–466. 10.1146/annurev.ecolsys.36.102003.152633 [DOI] [Google Scholar]
- Sytsma K. J., Morawetz J., Pires J. C., Nepokroeff M., Conti E., Zjhra M., et al. (2002). Urticalean rosids: circumscription, rosid ancestry, and phylogenetics based on rbcL, trnL-F, and ndhF sequences. Am. J. Bot. 89 1531–1546. 10.3732/ajb.89.9.1531 [DOI] [PubMed] [Google Scholar]
- Tanti B., Buragohain A., Gurung L., Kakati D., Das A. K., Borah S. P. (2010). Assessment of antimicrobial and antioxidant activities of Dendrocnide sinuata (Blume) Chew leaves-a medicinal plant used by ethnic communities of North East India. IJNPR 1 17–21. [Google Scholar]
- Thiel T., Michalek W., Varshney R. K., Graner A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106 411–422. 10.1007/s00122-002-1031-0 [DOI] [PubMed] [Google Scholar]
- Wang R. N., Milne R. I., Du X. Y., Liu J., Wu Z. Y. (2020b). Characteristics and mutational hotspots of plastomes in Debregeasia (Urticaceae). Front. Genet. 11:729. 10.3389/fgene.2020.00729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. X., Liu H., Moore M. J., Landrein S., Liu B., Zhu Z. X., et al. (2020a). Plastid phylogenomic insights into the evolution of the Caprifoliaceae s.l. (Dipsacales). Mol. Phylogenet. Evol. 142:106641. 10.1016/j.ympev.2019.106641 [DOI] [PubMed] [Google Scholar]
- Wang J. H., Moore M. J., Wang H., Zhu Z. X., Wang H. F. (2021). Plastome evolution and phylogenetic relationships among Malvaceae subfamilies. Gene 765:145103. 10.1016/j.gene.2020.145103 [DOI] [PubMed] [Google Scholar]
- Wang W. T., Chen C. J. (1995). Flora Reipublicae Popularis Sinicae, Vol. 23. Beijing: Science Press. [Google Scholar]
- Wells T., Maurin O., Dodsworth S., Friis I., Cowan R., Epitawalage N., et al. (2021). Combination of Sanger and target-enrichment markers supports revised generic delimitation in the problematic ‘Urera clade’ of the nettle family (Urticaceae). Mol. Phylogenet. Evol. 158:107008. 10.1016/j.ympev.2020.107008 [DOI] [PubMed] [Google Scholar]
- Wendel J. F., Doyle J. J. (1998). “Phylogenetic incongruence: window into genome history and molecular evolution,” in Molecular Systematics of Plants II, eds Soltis P. S., Soltis D. E., Doyle J. J. (Boston: Kluwer Academic Publishing; ), 265–296. 10.1007/978-1-4615-5419-6_10 [DOI] [Google Scholar]
- Wick R. R., Schultz M. B., Zobel J., Holt K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31 3350–3352. 10.1093/bioinformatics/btv383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S., Schneeweiss G. M., dePamphilis C. W., Muller K. F., Quandt D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76 273–297. 10.1007/s11103-011-9762-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe K. H., Li W. H., Sharp P. M. (1987). Rates of nucleotide substitution vary greatly among plant, mitochondrial, chloroplast, and nuclear DNAs. PNAS. 84 9054–9058. 10.1073/pnas.84.24.9054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. Y., Liu J., Provan J., Wang H., Chen C. J., Cadotte M. W., et al. (2018). Testing Darwin’s transoceanic dispersal hypothesis for the inland nettle family (Urticaceae). Ecol. Lett. 21 1515–1529. 10.1111/ele.13132 [DOI] [PubMed] [Google Scholar]
- Wu Z. Y., Monro A. K., Milne R. I., Wang H., Yi T. S., Liu J., et al. (2013). Molecular phylogeny of the nettle family (Urticaceae) inferred from multiple loci of three genomes and extensive generic sampling. Mol. Phylogenet. Evol. 69 814–827. 10.1016/j.ympev.2013.06.022 [DOI] [PubMed] [Google Scholar]
- Xue J., Wang S., Zhou S. L. (2012). Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae). Am. J. Bot. 99 e240–e244. 10.3732/ajb.1100547 [DOI] [PubMed] [Google Scholar]
- Xue S., Shi T., Luo W., Ni X., Iqbal S., Ni Z., et al. (2019). Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic. Res. 6:89. 10.1038/s41438-019-0171-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M., Fritsch P. W., Moore M. J., Feng T., Meng A., Yang J., et al. (2018). Plastid phylogenomics resolves infrafamilial relationships of the Styracaceae and sheds light on the backbone relationships of the Ericales. Mol. Phylogenet. Evol. 121 198–211. 10.1016/j.ympev.2018.01.004 [DOI] [PubMed] [Google Scholar]
- Yang Y., Zhou T., Duan D., Yang J., Feng L., Zhao G. (2016). Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 7:959. 10.3389/fpls.2016.00959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G., Jin J. J., Li H. T., Yang J. B., Mandala V. S., Croley M., et al. (2019). Plastid phylogenomic insights into the evolution of Caryophyllales. Mol. Phylogenet. Evol. 134 74–86. 10.1016/j.ympev.2018.12.023 [DOI] [PubMed] [Google Scholar]
- Zhang X. F., Landis J. B., Wang H. X., Zhu Z. X., Wang H. F. (2021). Comparative analysis of chloroplast genome structure and molecular dating in Myrtales. BMC Plant Biol. 21:219. 10.1186/s12870-021-02985-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S., Poczai P., Hyvonen J., Tang J., Amiryousefi A. (2020). Chloroplot: an online program for the versatile plotting of organelle genomes. Front. Genet. 11:576124. 10.3389/fgene.2020.576124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A., Guo W., Gupta S., Fan W., Mower J. P. (2016). Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 209 1747–1756. 10.1111/nph.13743 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Mauve alignment showing gene arrangements within the studied 57 Urticeae plastomes (length indicated above). Large colored boxes represent the gene blocks and the colored lines indicates linear position of different genes in the plastome. (B) Comparison of 57 Urticeae CP genomes using mVISTA, with the E. parvum genome as the reference. The y-axis represents the percent identity within 50–100%. Gray arrows indicate the direction of gene transcription. Blue blocks indicate conserved genes, while red blocks indicate conserved non-coding sequences (CNS).
Variable sites in homologous regions of the 57 sampled plastomes from Urticeae. The y-axis represent the nucleotide diversity (Pi) of each window, and x-axis is the position of the midpoint of each window used in the Sliding window analysis. (A) Coding regions. (B) Non-coding regions. (C) The ycf1 gene tree depicting highly resolved and supported relationships achieved by the identified barcode.
(A) Phylogenetic relationships of Urticeae tribe inferred from maximum likelihood (ML) and Bayesian inference (BI) based on CP coding (CDS) regions. Support values above the branches are maximum likelihood bootstrap support values (ML_BS)/Bayesian posterior probabilities (BI_PP)—note: branches with no support values indicate both ML_BS ≥ 90 and BI_PP = 1.00—whereas “*” indicate incongruence between ML and BI trees. Major clades of Urticeae s.l. are indicated on the right, respectively. CDS, chloroplast coding region. (B) Phylogenetic relationships of Urticeae tribe inferred from maximum likelihood (ML) and Bayesian inference (BI) based on CP non-coding (nCDS) regions. Support values above the branches are maximum likelihood bootstrap support (ML_BS)/Bayesian posterior probability (BI_PP)—note that branches with no support values indicate both ML_BS ≥ 90 and BI_PP = 1.00—whereas “*” indicate incongruence between ML and BI trees. Major clades of Urticeae s.l. are indicated on the right, respectively. nCDS, chloroplast non-coding region. (C) Phylogenetic relationships of Urticeae tribe inferred from maximum likelihood (ML) and Bayesian inference (BI) based on integrated CP genome and nrDNA trees. Support values above the branches are maximum likelihood bootstrap support (ML_BS)/Bayesian posterior probability (BI_PP)—note that branches with no support values indicate both ML_BS ≥ 90 and BI_PP = 1.00—whereas “*” indicate incongruence between ML and BI trees. Major clades of Urticeae s.l. are indicated on the right, respectively. CP, Complete chloroplast genome; nrDNA, nuclear ribosomal DNA (18S-ITS1-5.8S-ITS2-26S).
Data Availability Statement
The datasets presented in this study can be accessed at NCBI GenBank; the list of accessions can be found in Supplementary Table 1.