

Abstract
Measles virus (MeV) infection remains a significant public health threat despite ongoing global efforts to increase vaccine coverage. As eradication of MeV stalls, and vulnerable populations expand, effective antivirals against MeV are in high demand. Here, we describe the development of an antiviral peptide that targets the MeV fusion (F) protein. This antiviral peptide construct is composed of a carbobenzoxy-d-Phe-l-Phe-Gly (fusion inhibitor peptide; FIP) conjugated to a lipidated MeV F C-terminal heptad repeat (HRC) domain derivative. Initial in vitro testing showed high antiviral potency and specific targeting of MeV F-associated cell plasma membranes, with minimal cytotoxicity. The FIP and HRC-derived peptide conjugates showed synergistic antiviral activities when administered individually. However, their chemical conjugation resulted in markedly increased antiviral potency. In vitro mechanistic experiments revealed that the FIP–HRC lipid conjugate exerted its antiviral activity predominantly through stabilization of the prefusion F, while HRC-derived peptides alone act predominantly on the F protein after its activation. Coupled with in vivo experiments showing effective prevention of MeV infection in cotton rats, FIP–HRC lipid conjugates show promise as potential MeV antivirals via specific targeting and stabilization of the prefusion MeV F structure.
Keywords: measles, fusion protein, fusion inhibitor, lipopeptide, antiviral therapy
Graphical Abstract
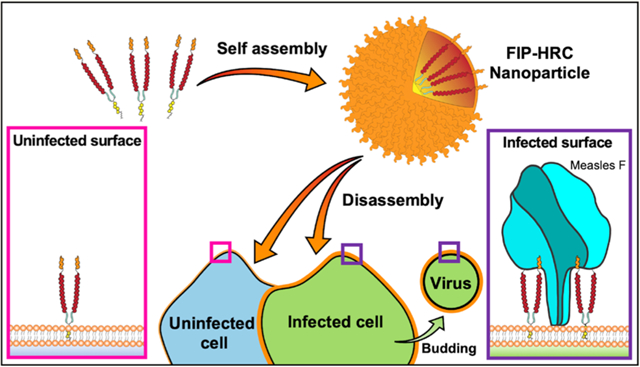
Measles virus (MeV) infection remains a significant public health threat despite ongoing global efforts to increase vaccine coverage.1 Obstacles to vaccination practice and policy have led to an overall increase in MeV incidence. Natural MeV infection exhibits tropism for CD150-expressing immune cells in early stages of infection, allowing entry into lymphocytes, dendritic cells, or macrophages present in the respiratory tract.2–4 Ongoing replication in these cells as they circulate to draining lymph nodes can lead to viremia coupled with significant immunosuppression. Late infection occurs through the basolateral membranes of nectin-4-expressing epithelial cells, mediating re-entry into the respiratory tract and facilitating aerosol transmission. While over 100 000 deaths are directly related to MeV infection each year, the virus’s ability to suppress immune effector function and reduce humoral and cellular immune memory for other pathogens significantly increases the disease burden.2,4–6 With no effective antiviral regimen currently available for MeV, insufficient coverage (≥95% as recommended by the WHO) within a given population will lead to a rise in incidence in vulnerable populations. MeV has been undergoing a global resurgence; estimated yearly global measles deaths rose from 89 780 in 2016 to 207 500 in 2019. The situation may worsen since the SARS-CoV-2 pandemic has led to reduced routine childhood vaccination coverage and may expose more vulnerable individuals to infection.7
Without a licensed antiviral drug, a significant proportion of cases will remain unavoidable.8–11
Inhibiting virion entry by targeting the associated viral proteins is an attractive means of generating antiviral compounds that could mitigate the spread of early infection as well as prevent transmission. MeV has two surface glycoproteins, hemagglutinin (H) and fusion (F) protein, that mediate viral entry into host cells.12,13 H is responsible for cell attachment through surface receptors and for F protein activation; the activated F protein mediates viral–host membrane fusion at the plasma membrane.
The F protein exists in a homotrimeric state, with each monomer made up of a globular head domain (F1) and helical stalk domain (F2). Upon activation, the F protein undergoes a conformational change that exposes a hydrophobic fusion peptide otherwise buried in the prefusion state of the protein. After insertion of the fusion peptide into the host membrane, the F protein trimer forms an extended transient intermediate structure. The inherent instability of this conformation forces the intermediate to fold unto itself, forming a 6-helix bundle structure made up of the heptad repeat domains of each monomer, driven by complementary interactions between the C-terminal and N-terminal heptad repeat domains (HRC and HRN, respectively) as the F protein reaches its stable, postfusion state. This transition also enables the viral and host membranes to come into proximity with one another, allowing for lipid mixing and subsequent fusion.
Targeting the intermediate state of the fusion protein is one of our antiviral approaches. We have previously demonstrated that dimeric peptides derived from a consensus sequence of the MeV HRC domain can inhibit viral entry, fusion, and spread in vitro as well as prevent infection in vivo.14–17 The addition of a lipid moiety also leads to nanoparticle assembly, which increases peptides’ in vivo biodistribution and facilitates passive targeting.16
The addition of a lipid moiety to anchor the peptide to the host cell membrane led to a significant increase in the potency. We recently applied a similar strategy (dimerization and lipidation) to HRC domains derived from SARS-CoV-2 spike protein and successfully blocked SARS-CoV-2 transmission in vivo.18 This promising result is highly relevant to the current COVID-19 pandemic and indicates the broad applicability of this technology.
The HRC-derived peptides complement the respective HRN domains of MeV F proteins in the intermediate state, thus inhibiting the transition of F into its postfusion state and subsequent viral–host membrane fusion. However, peptides and small molecules that stabilize the prefusion state of MeV F have also been described.19 In particular, carbobenzoxy-d-Phe-l-Phe-Gly (fusion inhibitor peptide; FIP) was crystallized in a complex with prefusion MeV F and was shown to bind a hydrophobic pocket in F1 proposed to be involved in H-mediated F activation.20
For respiratory syncytial virus (RSV), several small molecules that prevent F refolding have been identified, and one of these has been assessed in a clinical trial.21 Mechanistic studies suggest an association between the small molecules and the RSV F protein.22 FIP and other compounds that bind to MeV F’s hydrophobic pocket, such as N-(3-cyanophenyl)-2-phenylacetamide (compound 3g in ref 19), inhibit viral entry in vitro by interacting with the prefusion state of the F protein, binding to the virus before it reaches the target cell. This mechanism is in distinct contrast to HRC peptides, which interact with the F protein only during its activation process (i.e., after the virus has attached to the cell membrane). We hypothesized that conjugating FIP to MeV F HRC-derived sequences would result in targeted HRC peptides that are specifically directed to the viral particle or the site of infection, thereby increasing the antiviral activity. Although we found this general hypothesis to be correct, we also discovered that the mechanism of action of the inhibitor was in large part due to its stabilizing effect on prefusion F.
RESULTS AND DISCUSSION
Combining FIP and HRC into a Single Structure Significantly Increases in Vitro and in Vivo Antiviral Activity.
Based on our previous finding that peptide dimerization and lipidation improve antiviral activity, we designed a small library of conjugates based on the FIP, HRC, and FIP–HRC peptides (Figure 1 and Figures S1–S12). These cysteine-terminated peptides were modified with a flexible bis-maleimide–PEG11 linker (PEG11), a maleimide–PEG4–cholesterol (PEG4–chol) linker, and a bis(maleimide–PEG4)2–cholesterol linker to give a total of nine conjugates. These monomeric and dimeric constructs were used to evaluate the effect of FIP, lipidation, and dimerization (Figure 1 and Table S1) on antiviral activity. An initial screen for fusion inhibition utilized a β-galactosidase complementation assay between cells that express MeV glycoproteins and cells that express the MeV receptors nectin-4. This screen confirmed our previous findings that dimeric peptides conjugated to cholesterol were more potent than their monomeric counterparts in inhibiting cell-to-cell fusion (Figure 1 and Table S2). The [FIP–HRC–PEG4]2–chol was more potent than the monomer with cholesterol and [FIP–HRC]2–PEG11 (i.e., dimer without cholesterol, see Table S1). Adding FIP to the HRC domain resulted in more potent fusion inhibition for all conjugates tested (see Table S2 for the inhibitory activity of the other peptides shown in Figure 1; Figure S13 shows the absence of toxicity). Interestingly, the most dramatic improvement in potency upon conjugation of FIP to the HRC sequence was observed for the dimeric cholesterol-containing compounds. [HRC–PEG4]2–chol had an IC50 of 250 nM while the [FIP–HRC–PEG4]2–chol was an order of magnitude more potent with an IC50 of 20 nM (Figure 1). The IC50 of [FIP–PEG4]2–chol was 300 nM, demonstrating that the enhanced potency depends on both FIP and HRC.
Figure 1.
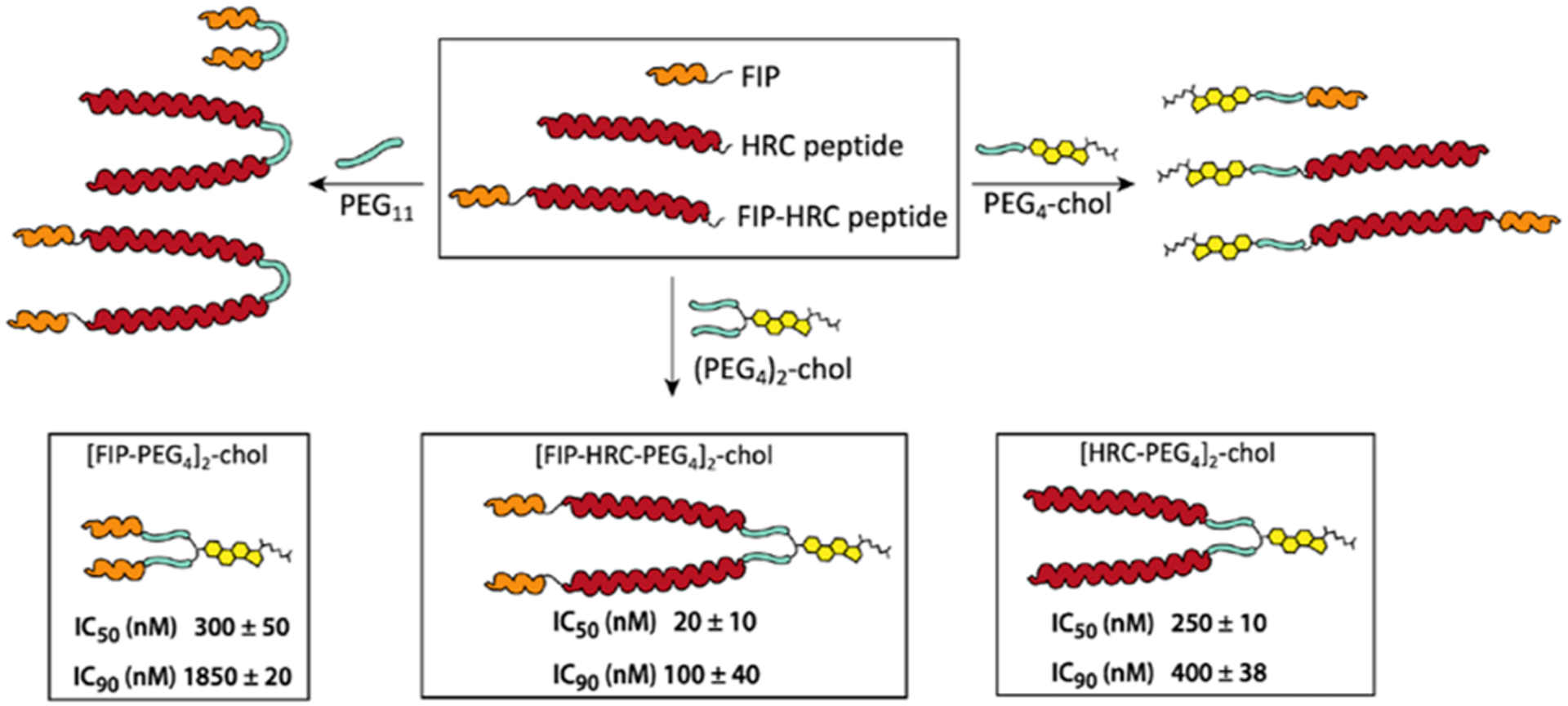
MeV fusion inhibitor peptides: design and structural features. IC50 and IC90 values for fusion inhibition by [FIP–PEG4]2–chol, [HRC–PEG4]2–chol, and [FIP–HRC–PEG4]2–chol. HEK 293T cells transiently transfected with nectin-4 and the omega reporter subunit of β-gal (“target cells”) were incubated with cells coexpressing viral glycoproteins (H and F) and the alpha reporter subunit of β-gal (“effector cells”) in the presence or absence of inhibitory peptides. In the absence of peptides, fusion between the target and effector cells permits reconstitution of β-galactosidase activity, quantified using the luminescence-based kit, Galacto-Star β-galactosidase reporter gene (ThermoFisher). The data presented are the means of three independent experiments (±SE).
The in vitro fusion assay is a faithful predictor of the spread of the virus in vivo, and potency in this fusion assay correlates with in vivo efficacy.15,17 Under the experimental conditions used here, in cells expressing both viral receptors, the [FIP–HRC–PEG4]2–chol IC50 and IC90 values were significantly lower than the [HRC–PEG4]2–chol IC50 and IC90 values (Table 1). The [FIP–PEG4]2–chol peptide is significantly less potent when both viral receptors are present.
Table 1.
Inhibition of MeV Fusiona
| Nectin-4 | CD150 | |||
|---|---|---|---|---|
| peptide | IC50 (nM) | IC90 (nM) | IC50 (nM) | IC90 (nM) |
| [FIP–PEG4]2–chol | 300 ± 50 | 1850 ± 20 | 510 ± 360 | 927 ± 60 |
| [HRC–PEG4]2–chol | 250 ± 10 | 400 ± 38 | 100 ± 65 | 700 ± 30 |
| [FIP–HRC–PEG4]2–chol | 20 ± 10 | 100 ± 40 | 26 ± 12 | 125 ± 10 |
IC50 and IC90 values for fusion inhibition. HEK 293T cells transiently transfected with nectin-4 or CD150 and the omega reporter subunit of β-gal (“target cells”) were incubated with cells coexpressing viral glycoproteins (H and F) and the alpha reporter subunit of β-gal (“effector cells”) in the presence or absence of inhibitory peptides. In the absence of peptides, fusion between the target and effector cells permits reconstitution of β-galactosidase activity, quantified using the luminescence-based kit, Galacto-Star β-galactosidase reporter gene (ThermoFisher). The data presented are the means of three independent experiments (±SE).
Most strikingly, pretreatment of cotton rats with 1 mg/kg [FIP–HRC–PEG4]2–chol before viral challenge (Figure 2) resulted in undetectable viral titers in the lungs, in contrast to the massive infection in untreated animals. This dosage is 5 times lower than that previously required for [HRC–PEG4]2–chol efficacy.15,16 Together, these data demonstrate the ability of [FIP–HRC–PEG4]2–chol to inhibit MeV in vitro and in vivo.
Figure 2.
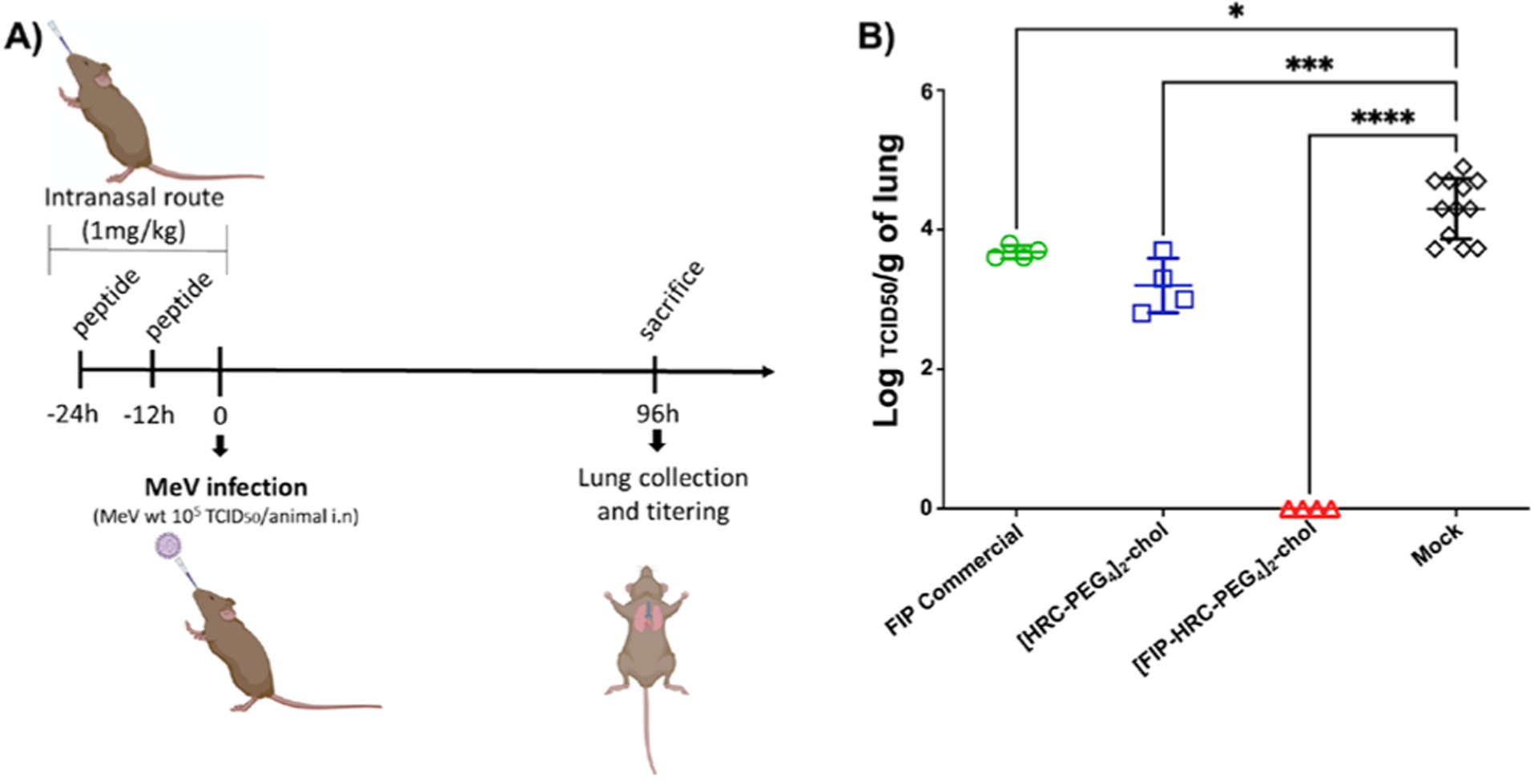
Intranasal administration of FIP–HRC protects cotton rats from MeV infection. (A) Cotton rats (n = 4/12) were infected intranasally with wt MeV. Treatment was given intranasally in 100 μL (1 mg/kg dose of peptides), 24 and 12 h before infection. Control animals were treated with vehicle. The animals were euthanized 4 days postinfection. (B) Viral titration of lung homogenates showed that [FIP–HRC–PEG4]2–cholesterol is the most potent inhibitor. ****p < 0.0001, *** p < 0.0005, * p < 0.05 (one-way ANOVA, multiple comparison).
[FIP–HRC–PEG4]2–chol Self-Assembles in Nanoparticles that Target Measles Fusion Protein in Its Prefusion State.
The magnitude of the enhanced potency of the [FIP–HRC–PEG4]2–chol peptide was unanticipated, and we sought to decipher its basis. Previous studies have shown that the addition of a lipid moiety to HRC-derived peptides facilitates self-assembly and integration onto the cell surface.16,23,24 To assess the effect of FIP on peptide–lipid self-assembly, we evaluated the colloidal behavior of the peptide–lipid conjugates in aqueous solution via dynamic light scattering (DLS). DLS studies showed that both [FIP–HRC–PEG4]2–chol and [HRC–PEG4]2–chol self-assemble into small nanoparticles (Figure S14). We have shown that [HRC–PEG4]2–chol nanoparticles disassemble nonspecifically on the cell membrane and that the disassembly is critical for antiviral activity in vitro and in vivo.16,23 Here, we asked whether the FIP component enhances MeV F protein targeting by the [FIP–HRC–PEG4]2–chol nanoparticles. Cultured monolayer cells (HEK 293T) expressing MeV prefusion F were incubated with [FIP–HRC–PEG4]2–chol, [FIP–HRC]2–PEG11, and [HRC–PEG4]2–chol and assessed using fluorescent microscopy (Figure 3). Flow cytometry analysis revealed that [HRC–PEG4]2–chol targets the cell membrane in a nonspecific manner, i.e., irrespective of the amount of prefusion F protein present on the cell surface. The membrane targeting is attributable to the lipid moiety, not the MeV F HRC-derived sequence present in the peptide.16,24 [FIP–HRC–PEG4]2–chol, in contrast, targets the cells that express MeV prefusion F in a specific, F-dependent manner (linear correlation, Figure 3 and Figure S15). A linear correlation between the amount of peptide and prefusion F present on the cell surface was also observed for the [FIP–HRC]2-PEG11 (i.e., the dimer peptide without lipid), suggesting that FIP is the element responsible for specific targeting to cells expressing MeV F protein.
Figure 3.
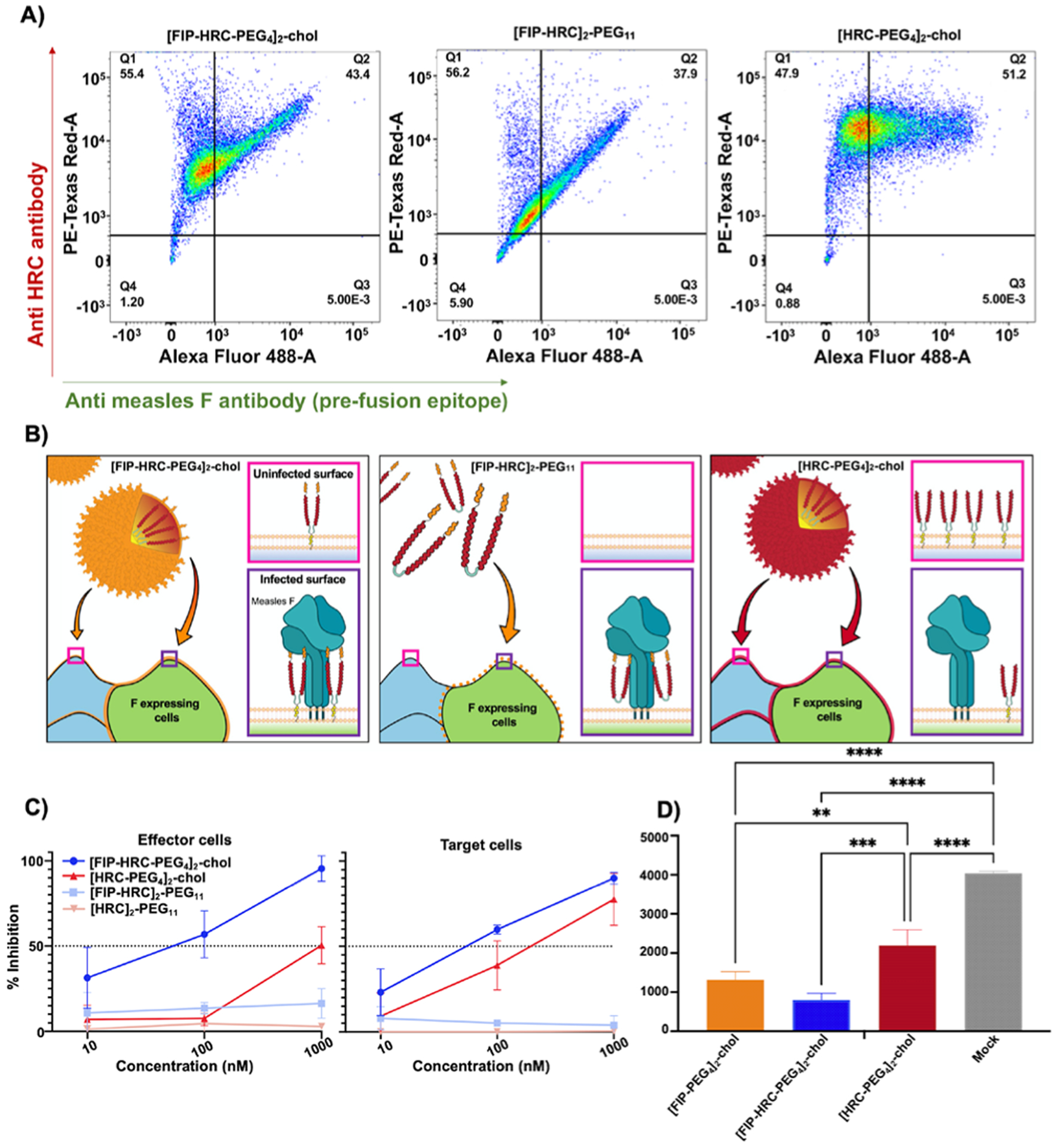
FIP drives HRC domain localization to MeV F expressing cells. (A) HEK 293T cells expressing MeV F were incubated with [FIP–HRC–PEG4]2–chol, [FIP–HRC]2–PEG11, and [HRC–PEG4]2–chol peptides (1000 nM) for 60 min at 37 °C. MeV F protein was detected using a mouse monoclonal antibody that recognizes the prefusion epitope. The HRC peptide was detected using a specific rabbit polyclonal serum. Double staining with antimouse Alexa Fluor 488 (green, x-axis) and antirabbit Alexa Fluor 594 (red, y-axis) was performed to assess colocalization. Data are from a representative experiment (n = 3). For [FIP–HRC–PEG4]2–chol, the HRC signal is proportional to the F signal, suggesting that this peptide preferentially bound to F-expressing cells. For [FIP–HRC]2–PEG11 (without cholesterol), colocalization of prefusion F and HRC signals is also observed. When lipid is present but FIP is missing, [HRC–PEG4]2–chol inserts on cells without any specificity for F-expressing cells. (B) Proposed model for the interactions of the 3 peptides with cells. [FIP–HRC–PEG4]2–chol is shown forming a particle that partially targets F-expressing cells where it inserts and interacts with F (bottom) but also inserts into cells that do not express F (top). [FIP–HRC]2–PEG11 does not form particles but also preferentially targets F-expressing cells. Finally, [HRC–PEG4]2–chol forms particles and inserts into cells whether they express F (bottom) or not (top). (C) HEK 293T cells expressing F, H, and the alpha reporter subunit of β-gal (“effector” cells) were incubated for 1 h at 37 °C with [FIP–HRC-PEG4]2–chol, [HRC–PEG4]2–chol, [FIP–HRC]2–PEG11, and [HRC]2–PEG11 at the indicated concentrations (x-axis, graph on left). After 1 h, the cells were washed with DPBS to remove unbound peptide, and then, the cells were incubated with cells expressing CD150 receptor (“target” cells). The same process was repeated with the “target” cells incubated with peptide and mixed with “effector” cells (graph on right). A quantitative cell–cell fusion assay based on β-galactosidase (β-gal) complementation was performed to measure fusion after 6 h; results are means of three independent experiments (±SE). When peptides were incubated with effector cells the difference between [FIP–HRC–PEG4]2–chol and [HRC–PEG4]2–chol lipopeptides was statistically significant [two-way multiple comparisons (ANOVA), p < 0.0001. When peptides were incubated with target cells, the difference between [FIP–HRC–PEG4]2–chol and [HRC–PEG4]2–chol lipopeptides was statistically significant [two-way multiple comparisons (ANOVA), p < 0.05]. (D) Assessment of virucidal activity. MeV was incubated with 1 μM of the indicated peptides or DMSO for 1 h at 37 °C. The resulting virus-compound solutions were diluted 1:100 in Opti-Mem and used to infect Vero-SLAM cells for a plaque reduction assay. Pretreatment of the virus (10 000 PFU) with FIP–HRC reduced plaques (entry) by 80%, and FIP lipopeptides reduced plaques by 70%. For HRC, plaques were reduced by 45%. The results are the means of three replicas (±SD). ****p < 0.0001, *** p < 0.0005, ** p < 0.005 (one-way ANOVA, multiple comparisons).
To determine whether the FIP–HRC peptides exhibit antiviral efficacy specifically when they insert on effector membranes, cells expressing viral glycoproteins (F and H) were incubated for 1 h at 37 °C with [FIP–HRC–PEG4]2–chol, [FIP–HRC]2–PEG11, [HRC–PEG4]2–chol, and [HRC]2–PEG11 peptides (at the indicated concentrations as shown in Figure 3C). After 1 h, the cells were washed and incubated with cells that express the CD150 receptor. To determine whether, in contrast, the activity is exerted mainly on the target membrane (as we have shown for simply HRC-based lipopeptides25), cells expressing the CD150 receptor (“target” cells) were treated as described above. A quantitative cell–cell fusion assay based on β-galactosidase (β-gal) complementation showed that fusion was reduced after incubating [FIP–HRC–PEG4]2–chol (blue line) with either effector or target cells, even when the peptide was removed before the assay. The [FIP–HRC]2–PEG11 (light blue line) showed some inhibition only after pretreatment with effector cells. The [HRC–PEG4]2–chol peptide (red line) acted predominantly after its incubation on target cells, as expected, and [HRC]2–PEG11 (light red line) did not inhibit the fusion process. As evidence for specificity of the mechanism, we found that MeV peptides do not inhibit RSV fusion, while a small molecule that stabilizes RSV F does so (Figure S16, green line).26
Using live virus in a direct assay of virucidal activity, we showed that both [FIP–HRC–PEG4]2–chol and [FIP–PEG4]2–chol lipopeptides inactivate the live virus (Figure 3D). We show that pretreatment of the virus (10 000 PFU) with FIP–HRC or FIP lipopeptides at a concentration of 1 μM for 1 h had an irreversible effect on infectivity. Viral entry was reduced by 70% with FIP and 80% with FIP–HRC after this treatment, despite the subsequent 100× dilution of peptide prior to the assay. HRC had a minor effect on infectivity and reduced viral entry by 45%. For both [FIP–HRC–PEG4]2–chol and [HRC–PEG4]2–chol, the IC50 for the plaque reduction assay is ~10 nM. However, [FIP–HRC–PEG4]2–chol is virucidal vs. live virus, while [HRC–PEG4]2–chol inhibition was consistent with activity at the final (diluted) concentration. For [FIP–PEG4]2–chol, the preincubation led to a 70% reduction of infectivity, although the IC50 for this peptide is ~70 nM, further confirming its virucidal activity.
To further explore how FIP targets F, we incubated cells with [FIP–HRC]2–PEG11 (without lipid) with or without 3g, a small molecule that binds to MeV F’s hydrophobic pocket (N-(3-cyanophenyl)-2-phenylacetamide, termed 3g in Figure S17).19 The [FIP–HRC]2–PEG11 peptide colocalizes with F, but the colocalization is disrupted by compound 3g (either by coincubation or sequential incubation).
[FIP–HRC–PEG4]2–chol Stabilizes F in Its Prefusion State.
To further examine the mechanism by which the FIP–HRC peptides exert their antiviral activity (Figure 4), we investigated whether they act before or after F activation, using an assay we had developed to distinguish the various stages of F activation (Figure 4A,B, see also Figure S18A).27 For these experiments, a chimeric receptor binding protein with a MeV H stalk and HPIV3 HN head domain was used (H-HN),28,29 to permit sialic acid-bearing receptors on red blood cells to serve as receptors, and the experimental use of zanamivir to regulate this interaction.28,29 Cells coexpressing H-HN and MeV F were mixed with sialic acid receptor-bearing red blood cells (RBCs), in the presence or absence of the FIP–HRC peptides. After this incubation, zanamivir was added at a dose that completely blocks HN–sialic acid receptor interaction. We determined the amount of target RBCs present in three distinct groups: (i) released into the medium by zanamivir, indicating that they had been attached only through H-HN, not by F insertion into the target cell membrane27,30 (such release would mean that the peptide acts on MeV prior to activation); (ii) not released by zanamivir, indicating that F had been activated and inserted into the target RBC membrane (this population—quantified by lysing the RBCs—indicates that the peptide acts after F activation); and finally, (iii) RBCs that had undergone fusion (quantified by cell lysis) and the F activation process that proceeded past the transitional intermediate to achieve fusion (these indicate that the F was not inhibited by any added compound). The [HRC–PEG4]2–chol and [FIP–HRC–PEG4]2–chol act on different steps of entry. While [HRC–PEG4]2–chol acted after F-activation (note, RBCs retained), [FIP–HRC–PEG4]2–chol acted before F-activation (RBCs released by zanamivir) (Figure 4A). These results suggest that [FIP–HRC–PEG4]2–chol functions in a manner similar to FIP alone, which has been shown to work by stabilizing MeV F in its prefusion state.20,31
Figure 4.
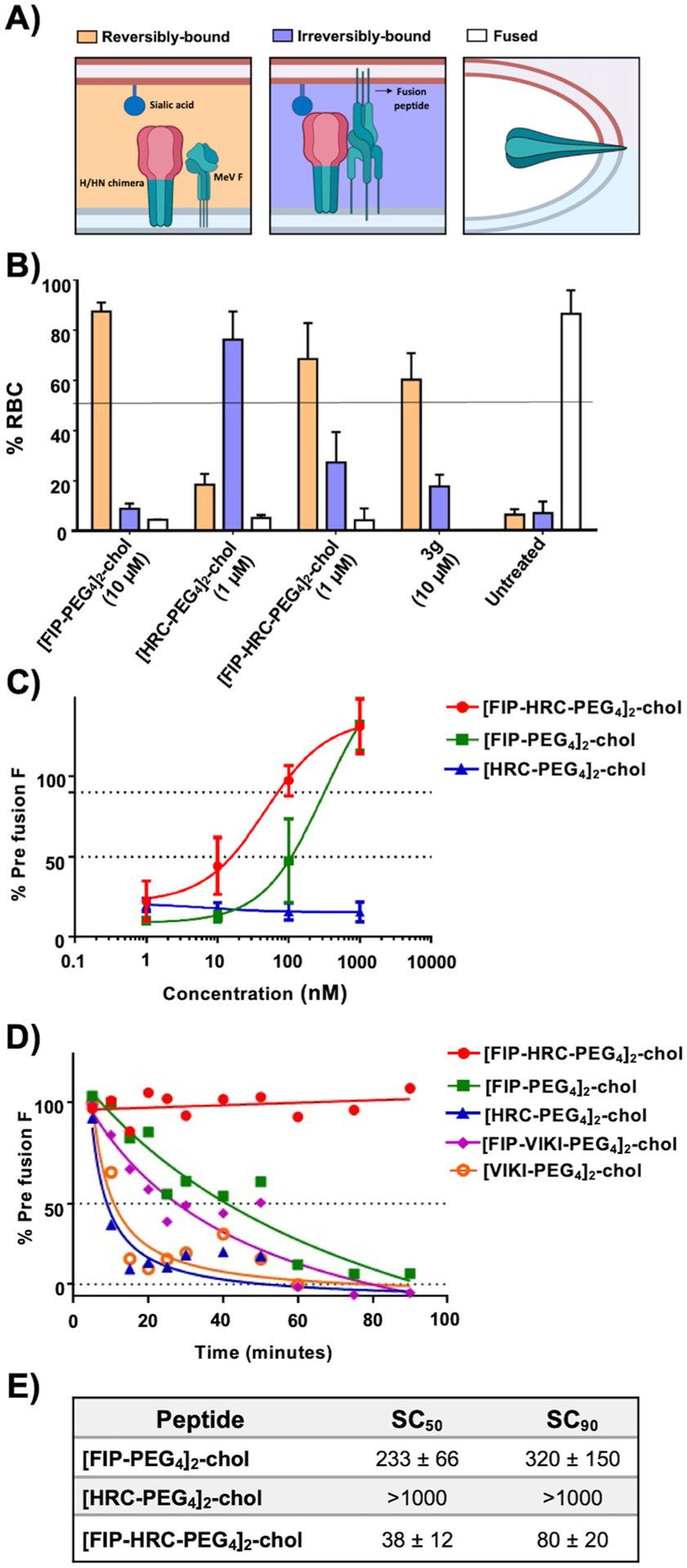
[FIP–HRC–PEG4]2–chol stabilizes F in its prefusion state. (A) Schematic illustration of the mechanistic assay. The panel shows a chimeric receptor binding protein with a MeV H stalk (green) and HPIV3 HN head (red) domain; the HPIV3 HN head permits the use of sialic acid-bearing receptors on red blood cells as receptors. RBCs were reversibly bound by HN–receptor interaction (orange) when the peptide acts before F activation, irreversibly bound (violet) when the peptide acts after F has been activated and inserted into the target RBC membrane, or fused (white) when the peptide does not inhibit fusion. (B) [FIP–HRC–PEG4]2–chol peptide prevents F activation. HEK 293T cells coexpressing H-HN T193A (a chimeric binding protein containing the MeV stalk and the HPIV3 head that binds sialic acid receptors and triggers MeV F) and MeV F (S262R, an easily activated F) were allowed to bind to sialic acid at 4 °C. Upon transfer to 37 °C, media containing the indicated compound or peptide (1 μM) were added for 60 min, and then, 10 mM zanamivir was added to release the RBCs that were reversibly bound (i.e., bound only by H-HN and not by F insertion). RBCs that are reversibly bound by HN–receptor interaction (orange), irreversibly bound by F insertion (violet), or fused (white) were quantified. The ordinate values are means (±SE) of results from triplicate experiments. [HRC–PEG4]2–chol blocks fusion after F insertion into the target cell (irreversibly bound, blue). [FIP–HRC–PEG4]2–chol blocks at the prefusion state (reversibly bound, orange). 3g is a small molecule that stabilizes the F in its prefusion state (reversibly bound RBC, orange). Zanamivir released all of the RBCs when added at the beginning of the 37 °C incubation. (C) [FIP–HRC–PEG4]2–chol stabilizes the measles F in its prefusion state. HEK 293T cells expressing MeV F were incubated overnight at 37 °C. The cells were then placed at 55 °C for 10 min in the presence of increasing concentrations of the indicated peptides. The cells were then incubated at 4 °C with prefusion conformation-specific mouse monoclonal antibodies (mAbs). Secondary antimouse antibodies conjugated with Alexa 488 were used for detection. Stained cells were identified using a cell analyzer high-content image system. The values on the y-axis indicate the percentage of positive cells compared to untreated cells and represent the percentage of conformational antibody binding (reflecting the percentage of F in the prefusion state). The values are means (±SE) of results from three experiments. (D) Table showing the concentration at which the prefusion epitope is 50% (“stable concentration50” or SC50) and 90% (“stable concentration90” or SC90) with respect to the samples that were not incubated. The [FIP–HRC–PEG4]2–cholesterol peptide is the most effective at stabilizing F in a prefusion state. (E) [FIP–HRC–PEG4]2–chol stabilizes the measles F in its prefusion state over time. HEK 293T cells expressing wt MeV F were incubated overnight at 37 °C. The cells were incubated in the presence of the indicated peptide at the concentration of 500 nM at the indicated time at 55 °C. Afterward, the cells were incubated with prefusion conformation-specific mouse mAb, and antimouse Ab conjugated with Alexa 488 was used for detection. Stained cells were identified using a cell analyzer high-content image system. The values on the y-axis indicate the percentage of positive cells compared to untreated cells and represent the percentage of conformational antibody binding (reflecting the percentage of F in a prefusion state).
This finding was bolstered by experiments that quantified the stability of MeV F in its prefusion state, showing that [FIP–HRC–PEG4]2–chol increases the thermal stability of prefusion MeV F significantly more than [FIP–PEG4]2–chol (Figure 4E). [FIP–HRC–PEG4]2–chol required a lower concentration than [FIP–PEG4]2–chol to stabilize F (Figure 4C,D) and, at a given concentration (500 nM), stabilized F for longer (Figure 4E). As expected, no stabilization was observed for [HRC–PEG4]2–chol despite its being significantly more potent than FIP at inhibiting viral fusion.
The contribution of the MeV-specific sequence to inhibition was assessed with the use of a human parainfluenza type 3-derived peptide (“VIKI”) that inhibits measles fusion by interacting with the MeV F HRN.15 [FIP–VIKI–PEG4]2–chol was similar in potency and F stabilization properties to the [FIP–PEG4]2–chol alone, indicating that the potency of [FIP–HRC–PEG4]2–chol results from the specific combination of FIP and the MeV peptide sequence and cannot be substituted.
While these results suggest that [FIP–HRC–PEG4]2–chol acts by stabilizing MeV F in its prefusion state, we asked whether the HRC component of the compound may also block F’s refolding after activation and prevent subsequent fusion. For this purpose, we used a variant MeV F that cannot interact with FIP because the FIP binding pocket in prefusion F is altered.20,31,32 This F protein was also inhibited by [FIP–HRC–PEG4]2–chol peptide (Figure S18B) indicating that, in the absence of FIP inhibition, the HRC component of the molecule is active.
FIP and HRC into a Single Structure: Beyond Synergism of FIP and HRC.
The [FIP–PEG4]2–chol, [HRC–PEG4]2–chol, and [FIP–HRC–PEG4]2–chol data show that the [FIP–HRC–PEG4]2–chol peptides’ antiviral properties are not simply the sum of the [FIP–PEG4]2–chol and [HRC–PEG4]2–chol activities. We performed an isobologram analysis to determine whether [FIP–HRC–PEG4]2–chol’s increased potency is the result of synergy between [FIP–PEG4]2–chol and [HRC–PEG4]2–chol. An isobologram evaluates the nature of interaction between two drugs at any given effect level (e.g., IC50), after the IC50 of the two drugs has been calculated in dose dependence studies. Here, the two drugs are [HRC–PEG4]2–chol and [FIP–PEG4]2–chol, respectively. The results are plotted as (IC50, [HRC–PEG4]2–chol, 0) and (0, IC50, [FIP–PEG4]2–chol) on a two-coordinate plot. The line connecting these two points is termed the line of additivity (Figure 5, green line). Any point on this line would define a combination of the two drugs needed to generate the IC50 if both acted independently in the mixture. Next, the IC50 of specific combinations of the two drugs (e.g., at 1:1, 1:2, and 1:5 ratios) is calculated and included in the same plot (Figure 5, black data points). Synergy, additivity, or antagonism corresponds to the data points located below, on, or above the line, respectively. Our studies revealed that [HRC–PEG4]2–chol and [FIP–PEG4]2–chol, in combination, exhibit a synergistic relationship. The addition of both compounds at different ratios results in potencies beyond a simple additive mixture of their activities (Figure 5). The IC50 values of [FIP–PEG4]2–chol and [HRC–PEG4]2–chol peptides are 300 and 250 nM, respectively, when used independently. At a 1:1 combination, an additive interaction would have produced an IC50 value of 135 nM for both [FIP–PEG4]2–chol and [HRC–PEG4]2–chol, respectively (interpolation from the green line in Figure 5). However, experimental data showed an IC50 value of 60 nM for both conjugates, indicating synergism. Furthermore, the conjugated version of the peptide (i.e., [FIP–HRC–PEG4]2–chol) transcended the observed synergism with an IC50 of 20 nM (Figure 5, blue data point). To explain this enhanced potency, we propose a model based on the findings shown so far (Figure 3B). In this model, [FIP–PEG4]2–chol, [HRC–PEG4]2–chol, and [FIP–HRC–PEG4]2–chol peptides form particles that self-assemble in solution and integrate on the cell membrane. While [HRC–PEG4]2–chol peptides distribute equally on all of the cell membranes, both [FIP–PEG4]2–chol and [FIP–HRC–PEG4]2–chol peptides appear to insert specifically on cell membranes presenting the F protein in its prefusion sate. Since [FIP–PEG4]2–chol and [FIP–HRC–PEG4]2–chol peptides act by stabilizing F in its prefusion state, localizing next to F would be ideal.
Figure 5.
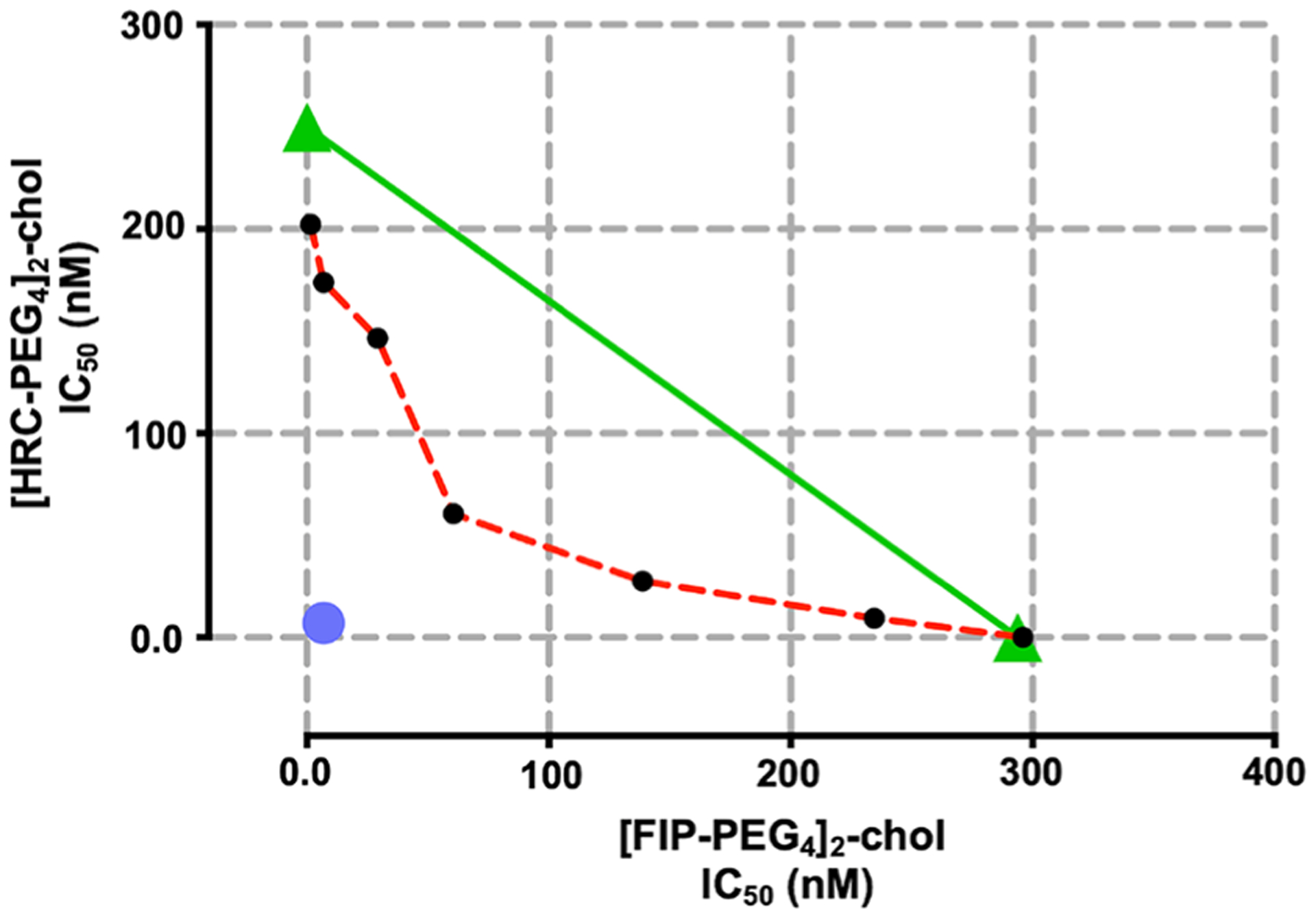
[FIP–PEG4]2–chol and [HRC–PEG4]2–chol synergize to inhibit fusion. Isobologram analysis of FIP + HRC peptide, where the green diagonal line represents the line of additivity, and the experimental data points, represented by dots below, on, or above the line, indicate synergy, additivity, or antagonism, respectively. The red dotted line is the curve generated from contributions of FIP and HRC in different ratios. The blue dot represents the IC50 of [FIP–HRC–PEG4]2–chol. The data are from three experiments.
CONCLUSION
We show that a targeted synthetic peptide–lipid conjugate, [FIP–HRC–PEG4]2–chol, potently inhibits MeV viral entry in vitro and in vivo by stabilizing MeV F in its prefusion conformation, while maintaining the HRC-specific antiviral properties. We have worked with similar lipid tagged peptides for the past 10 years,25,33–35 and they are stable at room temperature as lyophilized powders for several years. These peptides show little to no toxicity in vitro and are safe to use in vivo.
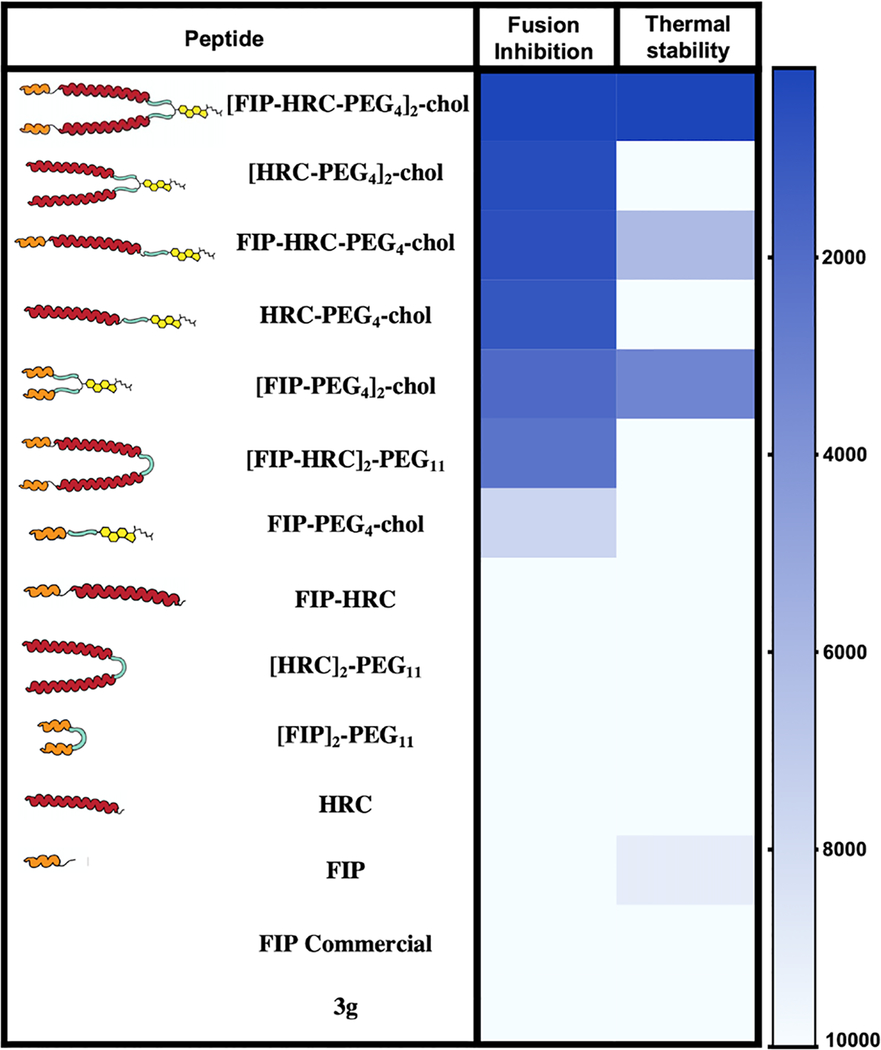
The [FIP–HRC–PEG4]2–chol described here is the most potent fusion inhibitor against measles that we have identified. Dimerization and lipidation dramatically increased potency. We discovered that the combination of FIP and HRC into a single lipidated dimeric molecule leads to a better antiviral activity than the synergistic activity of the individual inhibitors added together. The addition of FIP not only conferred F targeting properties to the self-assembling HRC lipid conjugated peptides (as expected) but also dramatically altered the mechanism of action, an unexpected result (for a comparison of the properties of all of the FIP, HRC, and FIP–HRC combinations, see Table 2). Future experiments will determine how easily resistance can be acquired and, if resistance is elicited, whether there is a fitness cost associated with resistance. We have shown that F proteins that are resistant to small molecule inhibitors31,32 can still be inactivated by HRC peptides that act by a different mechanism, blocking refolding of the prehairpin intermediate14 (see also Figure S18B), and we propose that resistance to FIP–HRC may be more difficult to elicit because of this secondary blockage.
Table 2.
Functional Properties of the FIP, HRC, and FIP–HRC Peptides in Fusion and Stability Assaysa
|
Fusion inhibition and thermal stabilization displayed as colors ranging from blue (high inhibition and high thermal stabilization) to white (low inhibition and low thermal stabilization) as shown in the heatmap key (in nM). Data are from at least three independent experiments.
As discussed above, several small F-stabilizing molecules for both MeV and RSV have been described. In future studies, we will assess combinations of these small molecules with the virus-specific HRC-derived peptides. Success in these studies would indicate that the findings have broader applicability and point to an additional mechanism of action of HRC-derived peptides.
To explain the added potency of FIP and HRC when combined into a single structure, compared to when they are simply mixed, the FACS data shown in Figure 3A and the model in Figure 3B provide a working hypothesis. FIP caused selected incorporation of the lipid tagged peptides on cells expressing F (that represent infected cells). This model also provides an explanation for the difference between the IC50 and IC90 for [FIP–PEG4]2–chol and [HRC–PEG4]2–chol peptides. These peptides have similar IC50 (300 vs 250 nM) but significantly different IC90 (1850 vs 400 nM). [FIP–PEG4]2–chol is effective at a relatively lower concentration because it specifically targets F but is less potent at blocking F activation and therefore requires a higher concentration to block 100%. When [FIP–PEG4]2–chol and [HRC–PEG4]2–chol are added together, they are synergistic since they work on two sequential aspects of the fusion process. However, it is only when they are in a single structure that the F targeting properties of the FIP and the higher potency endowed by the HRC combine to full effect.
The molecular interaction between FIP–HRC and the F protein—how the HRC region interacts with F to enhance FIP’s stabilization effect—remains to be elucidated. Several attempts at cocrystallization with the currently available soluble F proteins have failed.20 It is possible that the FIP–HRC binds to, or induces, a prefusion state of the F that is not available in the disulfide bond constrained soluble F protein that was used in the cocrystallization attempts. Future cryoelectron microscopy analysis may make it possible to address the structural interactions.
In September 2019, a National Institute of Allergy and Infectious Diseases measles workshop evaluated the state of research on measles pathogenesis and antivirals. Besides passive immunotherapy, only two measles-specific antiviral strategies had been tested in vivo15,16,36—our [HRC–PEG4]2–chol peptide and a polymerase inhibitor.36 The need for additional antiviral strategies was emphasized. The [FIP–HRC–PEG4]2–chol peptide described here functions via a pathway distinct from other fusion inhibitors. We propose that it not only will block infection prophylactically but will also prevent transmission among individuals. We hypothesize that the virus shed by an infected individual will be prebound by the inhibitors, decreasing the infectious viral load that can be transmitted. We predict that such an inhibitor-bound virus may even induce a protective immune response, turning infected hosts into possible vehicles of vaccination. We will address this hypothesis in future transmission studies.
MATERIAL AND METHODS
Plasmids.
The genes of MeV IC323 or B3 hemagglutinin, H/HN chimera, RSV F, fusion, nectin 4, and CD150 (or SLAM) proteins were codon optimized, synthesized, and sub cloned into the mammalian expression vector pCAGGS.
Cells.
HEK 293T (human kidney epithelial), Vero, and Vero-SLAM/CD150 (African green monkey kidney) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies; Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Life Technologies; Thermo Fisher Scientific) and antibiotics at 37 °C in 5% CO2. The Vero-SLAM/CD150 culture media were supplemented with 1 mg/mL Geneticin (Thermo Fisher Scientific).
β-Galactosidase (β-Gal) Complementation-Based Fusion Assay.
The β-Gal complementation-based fusion assay was performed as previously described. Briefly, HEK 293T cells transiently transfected with nectin 4 or CD150 receptor-bearing cells expressing the omega subunit of ß-Gal are mixed with cells coexpressing glycoprotein F and H and the alpha subunit of ß-Gal, and cell fusion leads to alpha–omega complementation, in the presence or absence of fusion inhibitory peptides. Fusion is stopped by lysing the cells, and after the addition of the substrate (the Tropix Galacto-Star chemiluminescent reporter assay system, Applied Biosystems), fusion is quantified on a Tecan M1000PRO microplate reader.
Cell Surface Staining with Conformation-Specific Anti-F mAbs.
HEK 293T cells transiently transfected with viral glycoprotein constructs were incubated overnight at 37 °C in complete medium (DMEM, 10% FBS). 20 h post-transfection, cells were transferred to the indicated temperatures for the times indicated in the figures. Thereafter, cells were incubated with mouse monoclonal antibodies (mAbs) that specifically detect MeV F in its prefusion conformation37 (1:1000) for 1 h on ice. Cells were washed with PBS and incubated for 1 h on ice with Alexa-488 antimouse secondary antibody (1:500; Life Technologies), washed with PBS, and fixed for 10 min on ice with 4% paraformaldehyde (PFA) with a 1:1000 dilution of DAPI (4′,6-diamidino-2-phenylindole; Thermo Fisher) for 60 min. Plates were washed; 0.01% sodium azide was added, and plates were imaged with an InCell analyzer. Percentages of antibody-bound cells were determined using CellProfiler software.
Flow Cytometry Analysis.
Flow cytometry was performed on a LSRFortessa FACS instrument with BD FACSDivaTM software at Department of Microbiology & Immunology-Flow Cytometry Core—Columbia University. HEK 293T cells expressing MeV F, after incubation with [FIP–HRC–PEG4]2–chol, [FIP–HRC]2–PEG11, or [HRC–PEG4]2–chol peptides (1 μM) for 60 min at 37 °C, were double stained with antimouse Alexa Fluor 488 and antirabbit Alexa Fluor 594. For FITC (or Alexa Fluor 488) registration, the cells were excited with 488 nm, and the fluorescence was detected with a BP 525/50 filter. For PE-Texas Red (Alexa Fluor 594) registration, the cells were excited with 561 nm, and the fluorescence was detected with a BP582/15 filter.
In Vivo Experiments.
Cotton Rats.
Inbred cotton rats (Sigmodon hispidus) were purchased from Envigo, Inc., Indianapolis. Both male and female cotton rats aged 5–7 weeks were used. For i.n. infection, 105 TCID50 of MeV (strain WTFb) was inoculated intranasally to isoflurane-anesthetized cotton rats in a volume of 100 μL. Peptide treatment was performed in the same way at indicated time points. 4 days after infection, the animals were euthanized by CO2 inhalation, and their lungs were collected and weighed. Lung tissue was minced with scissors and homogenized with a glass dounce homogenizer. Serial 10-fold dilutions of supernatant fluids were assessed on Vero SLAM cells for the presence of infectious virus in a 48-well plate using the cytopathic effect (CPE) as the end point. Plates were scored for CPE microscopically after 7 days, and the TCID50 was calculated.
RBC Fusion Assay.
RBC fusion assays were performed on HEK 293T cells transiently expressing MeV H_Y17H HPIV3_T193A chimera and MeV F (S262R). Cell monolayers were washed three times with serum-free medium, placed at 4 °C with 1% RBCs in DMEM for 30 min, and then treated with different concentrations of peptide (1000, 100, 10, and 1 nm) and placed at 37 °C. Zanamivir was added at a final concentration of 10 mM; the cells were incubated at 4 °C and rocked, and the liquid phase was collected in V-bottomed 96-well plates for the measurement of reversibly bound RBCs. The cells were then incubated at 4 °C with ACK-Lysing buffer (ThermoFisher A10492–01), and the liquid phase was collected in V-bottomed 96-well plates for the measurement of irreversibly bound RBCs. The cells were then lysed in lysis buffer and transferred to flat-bottom 96-well plates for quantification of fused RBCs. The amount of RBCs in each of the three compartments (reversibly bound, irreversibly bound, and fused) was determined by the measurement of absorption at 410 nm using a Tecan M1000PRO microplate reader.
Cell Toxicity Assay.
HEK 293T cells were incubated at 37 °C in the presence or absence of the indicated peptides at several concentrations up to 5 μM. The peptides were added to the media, and the cells were incubated a 37 °C. Cell viability was determined after 24 h using the Vybrant MTT cell proliferation assay kit according to the manufacturer’s guidelines. Cycloheximide (CHE) was used as a positive control. Absorbance was read at 540 nm using a Tecan M1000PRO microplate reader.
Virucidal Assay.
Aliquots of MeV viral preparations38 were incubated for 1 h at 37 °C in Opti-Mem supplemented with the indicated peptide or DMSO. After incubation, the resulting virus-compound solutions were diluted 1:100 in Opti-Mem and used to infect Vero-SLAM cells to determine their infectivity by a plaque reduction assay.
Statistical Analysis.
All analyses were performed in GraphPad Prism5 software. ANOVA was used for cotton rat experiments
Chemicals and Peptides.
N-(3-Cyanophenyl)-2-phenylacetamide (also known as 3g) was commercially acquired from ZereneX Molecular Limited. The purity of 3g was tested by high-pressure liquid chromatography (HPLC) and shown to be >95% pure.
Peptides were purchased from Shanghai Ruifu Chemical Co., Ltd. Bromoacetyl cholesterol and bismaleimide cholesterol derivatives were custom-made by Charnwood Molecular, Ltd. Dimethyl sulfoxide (DMSO), tetrahydrofuran (THF), and N,N-diisopropylethylamine (DIPEA) were purchased from Sigma-Aldrich. HPLC purification was performed on a 1100 Series Agilent HPLC system equipped with a UV diode array detector and a fraction collector using a reverse phase (RP) Phenomenex Jupiter C4 LC column 300 Å (150 × 21.2 mm, particle size 5 μm). MALDI-TOF analysis was performed on a Bruker UltrafleXtreme MALDI-TOF-TOF instrument.
JNJ-678 (RSV inhibitor) was commercially acquired from MCE (MedChemExpress).
Supplementary Material
ACKNOWLEDGMENTS
We thank J. S. Orange and S. G. Kernie for their contributions to this study. This work was supported by funding from the National Institutes of Health (AI119762, AI121349, NS105699, and NS091263) to M.P. and the Sharon Golub Fund at Columbia University Vagelos College of Physicians and Surgeons.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.1c02057.
List of MeV peptides and their modifications; HPLC traces and mass spec analysis of selected MeV peptides; MeV peptides’ toxicity; Dynamic light scattering (DLS) analysis of MeV peptides; FIP–HRC peptide targeting MeV F expressing cells; antiviral activity of FIP–HRC and HRC peptides on RSV-mediated fusion; localization of [FIP–HRC]2–PEG11 peptide in HEK 293T cells; [FIP–HRC–PEG4]2–chol prevents F activation by interaction with the HRC domain; reaction scheme showing the synthesis of the MeV peptides; inhibitory activity of MeV peptides; and F stabilization properties of MeV peptides (PDF)
List of materials with sources and identifiers (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsnano.1c02057
The authors declare the following competing financial interest(s): A.M., C.A.A., and M.P. are listed as inventors of [FIP-HRC-PEG4]2-chol peptide on a provisional patent application covering findings reported in this manuscript.
Contributor Information
Francesca T. Bovier, Center for Host–Pathogen Interaction, Columbia University Vagelos College of Physicians and Surgeons, New York, New York 10032, United States; Department of Pediatrics, Columbia University Vagelos College of Physicians and Surgeons, New York, New York 10032, United States; Department of Experimental Medicine, University of Campania “Luigi Vanvitelli”, 81100 Caserta, Italy;.
Ksenia Rybkina, Center for Host–Pathogen Interaction, Columbia University Vagelos College of Physicians and Surgeons, New York, New York 10032, United States; Department of Pediatrics, Columbia University Vagelos College of Physicians and Surgeons, New York, New York 10032, United States.
Sudipta Biswas, Center for Host–Pathogen Interaction, Columbia University Vagelos College of Physicians and Surgeons, New York, New York 10032, United States; Robert Frederick Smith School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, New York 14853, United States.
Olivia Harder, Department of Veterinary Biosciences, College of Veterinary Medicine, The Ohio State University, Columbus, Ohio 43210, United States.
Tara C. Marcink, Center for Host–Pathogen Interaction, Columbia University Vagelos College of Physicians and Surgeons, New York, New York 10032, United States; Department of Pediatrics, Columbia University Vagelos College of Physicians and Surgeons, New York, New York 10032, United States
Stefan Niewiesk, Department of Veterinary Biosciences, College of Veterinary Medicine, The Ohio State University, Columbus, Ohio 43210, United States.
Anne Moscona, Center for Host–Pathogen Interaction, Columbia University Vagelos College of Physicians and Surgeons, New York, New York 10032, United States; Department of Pediatrics, Department of Microbiology & Immunology, and Department of Physiology & Cellular Biophysics, Columbia University Vagelos College of Physicians and Surgeons, New York, New York 10032, United States.
Christopher A. Alabi, Robert Frederick Smith School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, New York 14853, United States;.
Matteo Porotto, Center for Host–Pathogen Interaction, Columbia University Vagelos College of Physicians and Surgeons, New York, New York 10032, United States; Department of Pediatrics, Columbia University Vagelos College of Physicians and Surgeons, New York, New York 10032, United States; Department of Experimental Medicine, University of Campania “Luigi Vanvitelli”, 81100 Caserta, Italy;.
REFERENCES
- (1).Melegaro A Measles Vaccination: No Time to Rest. Lancet Glob Health 2019, 7 (3), e282–e283. [DOI] [PubMed] [Google Scholar]
- (2).Lemon K; de Vries RD; Mesman AW; McQuaid S; van Amerongen G; Yuksel S; Ludlow M; Rennick LJ; Kuiken T; Rima BK; Geijtenbeek TB; Osterhaus AD; Duprex WP; de Swart RL Early Target Cells of Measles Virus after Aerosol Infection of Non-Human Primates. PLoS Pathog. 2011, 7 (1), No. e1001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tatsuo H; Ono N; Tanaka K; Yanagi Y Slam (Cdw150) Is a Cellular Receptor for Measles Virus. Nature 2000, 406 (6798), 893–7. [DOI] [PubMed] [Google Scholar]
- (4).de Vries RD; Mesman AW; Geijtenbeek TB; Duprex WP; de Swart RL The Pathogenesis of Measles. Curr. Opin. Virol 2012, 2 (3), 248–55. [DOI] [PubMed] [Google Scholar]
- (5).Muhlebach MD; Mateo M; Sinn PL; Prufer S; Uhlig KM; Leonard VH; Navaratnarajah CK; Frenzke M; Wong XX; Sawatsky B; Ramachandran S; McCray PB Jr.; Cichutek K; von Messling V; Lopez M; Cattaneo R Adherens Junction Protein Nectin-4 Is the Epithelial Receptor for Measles Virus. Nature 2011, 480 (7378), 530–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mina MJ; Kula T; Leng Y; Li M; de Vries RD; Knip M; Siljander H; Rewers M; Choy DF; Wilson MS; Larman HB; Nelson AN; Griffin DE; de Swart RL; Elledge SJ Measles Virus Infection Diminishes Preexisting Antibodies That Offer Protection from Other Pathogens. Science 2019, 366 (6465), 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Durrheim DN; Andrus JK; Tabassum S; Bashour H; Githanga D; Pfaff G A Dangerous Measles Future Looms beyond the Covid-19 Pandemic. Nat. Med 2021, 27 (3), 360–361. [DOI] [PubMed] [Google Scholar]
- (8).Dabbagh A; Patel MK; Dumolard L; Gacic-Dobo M; Mulders MN; Okwo-Bele JM; Kretsinger K; Papania MJ; Rota PA; Goodson JL Progress toward Regional Measles Elimination - Worldwide, 2000–2016. MMWR Morb Mortal Wkly Rep 2017, 66 (42), 1148–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Patel MK; Orenstein WA Classification of Global Measles Cases in 2013–17 as Due to Policy or Vaccination Failure: A Retrospective Review of Global Surveillance Data. Lancet Glob Health 2019, 7 (3), e313–e320. [DOI] [PubMed] [Google Scholar]
- (10).Griffin DE Measles Immunity and Immunosuppression. Curr. Opin. Virol 2021, 46, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Griffin DE Measles Virus Persistence and Its Consequences. Curr. Opin. Virol 2020, 41, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hashiguchi T; Maenaka K; Yanagi Y Measles Virus Hemagglutinin: Structural Insights into Cell Entry and Measles Vaccine. Front. Microbiol 2011, 2, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Plattet P; Alves L; Herren M; Aguilar HC Measles Virus Fusion Protein: Structure, Function and Inhibition. Viruses 2016, 8 (4), 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mathieu C; Ferren M; Jurgens E; Dumont C; Rybkina K; Harder O; Stelitano D; Madeddu S; Sanna G; Schwartz D; Biswas S; Hardie D; Hashiguchi T; Moscona A; Horvat B; Niewiesk S; Porotto M Measles Virus Bearing Measles Inclusion Body Encephalitis-Derived Fusion Protein Is Pathogenic after Infection via the Respiratory Route. J. Virol 2019, 93 (8), e01862–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mathieu C; Huey D; Jurgens E; Welsch JC; DeVito I; Talekar A; Horvat B; Niewiesk S; Moscona A; Porotto M Prevention of Measles Virus Infection by Intranasal Delivery of Fusion Inhibitor Peptides. J. Virol 2015, 89 (2), 1143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Figueira TN; Palermo LM; Veiga AS; Huey D; Alabi CA; Santos NC; Welsch JC; Mathieu C; Horvat B; Niewiesk S; Moscona A; Castanho M; Porotto M In Vivo Efficacy of Measles Virus Fusion Protein-Derived Peptides Is Modulated by the Properties of Self-Assembly and Membrane Residence. J. Virol 2017, 91 (1), e01554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Welsch JC; Talekar A; Mathieu C; Pessi A; Moscona A; Horvat B; Porotto M Fatal Measles Virus Infection Prevented by Brain-Penetrant Fusion Inhibitors. J. Virol 2013, 87 (24), 13785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).de Vries RD; Schmitz KS; Bovier FT; Predella C; Khao J; Noack D; Haagmans BL; Herfst S; Stearns KN; Drew-Bear J; Biswas S; Rockx B; McGill G; Dorrello NV; Gellman SH; Alabi CA; de Swart RL; Moscona A; Porotto M Intranasal Fusion Inhibitory Lipopeptide Prevents Direct-Contact SARS-CoV-2 Transmission in Ferrets. Science 2021, 371, 1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sun A; Prussia A; Zhan W; Murray EE; Doyle J; Cheng LT; Yoon JJ; Radchenko EV; Palyulin VA; Compans RW; Liotta DC; Plemper RK; Snyder JP Nonpeptide Inhibitors of Measles Virus Entry. J. Med. Chem 2006, 49 (17), 5080–92. [DOI] [PubMed] [Google Scholar]
- (20).Hashiguchi T; Fukuda Y; Matsuoka R; Kuroda D; Kubota M; Shirogane Y; Watanabe S; Tsumoto K; Kohda D; Plemper RK; Yanagi Y Structures of the Prefusion Form of Measles Virus Fusion Protein in Complex with Inhibitors. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (10), 2496–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).DeVincenzo JP; Whitley RJ; Mackman RL; Scaglioni-Weinlich C; Harrison L; Farrell E; McBride S; Lambkin-Williams R; Jordan R; Xin Y; Ramanathan S; O’Riordan T; Lewis SA; Li X; Toback SL; Lin SL; Chien JW Oral Gs-5806 Activity in a Respiratory Syncytial Virus Challenge Study. N. Engl. J. Med 2014, 371 (8), 711–22. [DOI] [PubMed] [Google Scholar]
- (22).Battles MB; Langedijk JP; Furmanova-Hollenstein P; Chaiwatpongsakorn S; Costello HM; Kwanten L; Vranckx L; Vink P; Jaensch S; Jonckers TH; Koul A; Arnoult E; Peeples ME; Roymans D; McLellan JS Molecular Mechanism of Respiratory Syncytial Virus Fusion Inhibitors. Nat. Chem. Biol 2016, 12 (2), 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Figueira TN; Mendonca DA; Gaspar D; Melo MN; Moscona A; Porotto M; Castanho M; Veiga AS Structure-Stability-Function Mechanistic Links in the Anti-Measles Virus Action of Tocopherol-Derivatized Peptide Nanoparticles. ACS Nano 2018, 12 (10), 9855–9865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Figueira TN; Freire JM; Cunha-Santos C; Heras M; Goncalves J; Moscona A; Porotto M; Salome Veiga A; Castanho MA Quantitative Analysis of Molecular Partition towards Lipid Membranes Using Surface Plasmon Resonance. Sci. Rep 2017, 7, 45647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Porotto M; Yokoyama CC; Palermo LM; Mungall B; Aljofan M; Cortese R; Pessi A; Moscona A Viral Entry Inhibitors Targeted to the Membrane Site of Action. J. Virol 2010, 84 (13), 6760–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Roymans D; Alnajjar SS; Battles MB; Sitthicharoenchai P; Furmanova-Hollenstein P; Rigaux P; Berg JVD; Kwanten L; Ginderen MV; Verheyen N; Vranckx L; Jaensch S; Arnoult E; Voorzaat R; Gallup JM; Larios-Mora A; Crabbe M; Huntjens D; Raboisson P; Langedijk JP; Ackermann MR; McLellan JS; Vendeville S; Koul A Therapeutic Efficacy of a Respiratory Syncytial Virus Fusion Inhibitor. Nat. Commun 2017, 8 (1), 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Porotto M; Murrell M; Greengard O; Doctor L; Moscona A Influence of the Human Parainfluenza Virus 3 Attachment Protein’s Neuraminidase Activity on Its Capacity to Activate the Fusion Protein. J. Virol 2005, 79 (4), 2383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Talekar A; Moscona A; Porotto M Measles Virus Fusion Machinery Activated by Sialic Acid Binding Globular Domain. J. Virol 2013, 87 (24), 13619–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jurgens EM; Mathieu C; Palermo LM; Hardie D; Horvat B; Moscona A; Porotto M, Measles Fusion Machinery Is Dysregulated in Neuropathogenic Variants. mBio 2015, 6 (1), e02528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Greengard O; Poltoratskaia N; Leikina E; Zimmerberg J; Moscona A The Anti-Influenza Virus Agent 4-Gu-Dana (Zanamivir) Inhibits Cell Fusion Mediated by Human Parainfluenza Virus and Influenza Virus Ha. J. Virol 2000, 74 (23), 11108–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ha MN; Delpeut S; Noyce RS; Sisson G; Black KM; Lin LT; Bilimoria D; Plemper RK; Prive GG; Richardson CD Mutations in the Fusion Protein of Measles Virus That Confer Resistance to the Membrane Fusion Inhibitors Carbobenzoxy-D-Phe-L-Phe-Gly and 4-Nitro-2-Phenylacetyl Amino-Benzamide. J. Virol 2017, 91 (23), e01026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Doyle J; Prussia A; White LK; Sun A; Liotta DC; Snyder JP; Compans RW; Plemper RK Two Domains That Control Prefusion Stability and Transport Competence of the Measles Virus Fusion Protein. J. Virol 2006, 80 (3), 1524–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Pessi A; Langella A; Capito E; Ghezzi S; Vicenzi E; Poli G; Ketas T; Mathieu C; Cortese R; Horvat B; Moscona A; Porotto M A General Strategy to Endow Natural Fusion-Protein-Derived Peptides with Potent Antiviral Activity. PLoS One 2012, 7 (5), No. e36833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lee KK; Pessi A; Gui L; Santoprete A; Talekar A; Moscona A; Porotto M Capturing a Fusion Intermediate of Influenza Hemagglutinin with a Cholesterol-Conjugated Peptide, a New Antiviral Strategy for Influenza Virus. J. Biol. Chem 2011, 286 (49), 42141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Porotto M; Rockx B; Yokoyama CC; Talekar A; Devito I; Palermo LM; Liu J; Cortese R; Lu M; Feldmann H; Pessi A; Moscona A Inhibition of Nipah Virus Infection in Vivo: Targeting an Early Stage of Paramyxovirus Fusion Activation During Viral Entry. PLoS Pathog. 2010, 6 (10), No. e1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Krumm SA; Yan D; Hovingh ES; Evers TJ; Enkirch T; Reddy GP; Sun A; Saindane MT; Arrendale RF; Painter G; Liotta DC; Natchus MG; von Messling V; Plemper RK An Orally Available, Small-Molecule Polymerase Inhibitor Shows Efficacy against a Lethal Morbillivirus Infection in a Large Animal Model. Sci. Transl. Med 2014, 6 (232), 232ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mathieu C; Ferren M; Harder O; Bovier FT; Marcink TC; Predella C; Angius F; Drew-Bear J; Dorrello NV; Greninger AL; Moscona A; Niewiesk S; Horvat B; Porotto M Single-Chain Variable Fragment Antibody Constructs Neutralize Measles Virus Infection in Vitro and in Vivo. Cell. Mol. Immunol 2021, 18, 1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mathieu C; Bovier FT; Ferren M; Lieberman NAP; Predella C; Lalande A; Peddu V; Lin MJ; Addetia A; Patel A; Outlaw V; Corneo B; Dorrello NV; Briese T; Hardie D; Horvat B; Moscona A; Greninger AL; Porotto M Molecular Features of the Measles Virus Viral Fusion Complex That Favor Infection and Spread in the Brain. mBio 2021, 12, No. e0079921. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.